
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpanding the chemical space of peptides via biocompatible tryptophan C7-arylation
Lei
Liu†
ab,
Yanyang
Zhao†
a,
Yiming
Su
a,
Boning
Wang
a,
Yue
Xiong
b,
Tianhang
Wang
a,
Xiude
Hua
bc,
Yonghao
Ye
*bc,
Zhuangzhi
Shi
 *a and
Huan
Wang
*a
*a and
Huan
Wang
*a
aState Key Laboratory of Coordination Chemistry, Chemistry and Biomedicine Innovation Center of Nanjing University, Jiangsu Key Laboratory of Advanced Organic Materials, School of Chemistry and Chemical Engineering, Nanjing University, No. 163 Xianlin Ave, Nanjing, 210093, China. E-mail: wanghuan@nju.edu.cn; shiz@nju.edu.cn
bState Key Laboratory of Agricultural and Forestry Biosecurity, State & Local Joint Engineering Research Center of Green Pesticide Invention and Application, College of Plant Protection, Nanjing Agricultural University, Nanjing 210095, PR China. E-mail: yeyh@njau.edu.cn
cKey Laboratory of Integrated Management of Crop Diseases and Pests, Ministry of Education, Nanjing 210095, PR China
First published on 15th January 2026
Abstract
Late-stage functionalization of peptides and amino acids is a powerful strategy for modulating biological activity and enabling targeted molecular imaging, offering a promising route for expanding the chemical space of peptides. Here, we report a Rh-catalyzed, P(III)-directed C7-selective arylation of tryptophan residues using a removable N-PtBu2 auxiliary. This method exhibits broad substrate scope, excellent regioselectivity, and high functional group tolerance, enabling efficient and modular derivatization of tryptophan-containing amino acids and peptides. The resulting C7-arylated Trp derivatives serve as fluorogenic probes with environment-sensitive, turn-on fluorescence suitable for wash-free imaging of bacterial cells. Moreover, incorporation of these modified residues into antimicrobial peptides significantly enhanced antifungal activity against Aspergillus fumigatus, achieving up to 49-fold improvement over the parent peptide. By enabling this biocompatible tryptophan C7-arylation, this work establishes a versatile platform for peptide diversification and therapeutic peptide engineering.
Introduction
Peptides, as essential scaffolds in biological systems, have garnered increasing attention in therapeutic development and molecular imaging due to their high specificity, biocompatibility, and structural tunability.1 Chemical modification serves as a powerful strategy to optimize their bioactivity and pharmacokinetic properties, with metal-catalyzed late-stage functionalization emerging as a particularly attractive approach to diversify peptide structures while preserving native function.2Among proteinogenic amino acids, tryptophan (Trp) presents unique opportunities for selective modification owing to its distinctive reactivity and intrinsic fluorescence.3 In natural products and bioactive peptides, C7-arylated Trp residues often play pivotal roles in structural conformation, molecular recognition, and biological activity, as exemplified by Chloropeptin I and AL-471 (specific inhibitors of HIV gp120-CD4 interaction) and TMC-95A–B (HIV therapeutic candidates targeting 20S proteasome inhibition) (Fig. 1A).4 The synthesis of these compounds has attracted significant attention, with the Hoveyda,5 Boger,6 Zhu,7 Danishefsky,8 and Hirama9 groups achieving total syntheses of cyclic peptides Chloropeptin I and TMC-95A–B via coupling reactions, where tryptophan C7-arylation served as the key step. While the Hoveyda group successfully constructed the C7-arylated tryptophan motif in Chloropeptin I through palladium-mediated Stille coupling in 2003,5 this method suffered from limitations including stoichiometric palladium usage and prerequisite preparation of aryl iodides/stannanes, resulting in cumbersome and inefficient syntheses (Fig. 1B). Mowever, these methods face limitations such as poor atom economy, the need for pre-installed leaving groups, and challenges in achieving catalytic processes within complex biomolecular systems. Consequently, the development of more efficient and facile methodologies remains an ongoing pursuit.
 | ||
| Fig. 1 Late-stage Trp(C7) arylation of peptides via P(III)-directed Rh-catalysis. (A) Representative bioactive compounds containing C7 arylated Trp motifs. (B) Synthesis of key intermediate of Chloropeptin I by Pd-Mediated Stille Coupling. (C) This work: a general approach for the C7 arylation of Trp in peptides. | ||
Prized for their ability to rapidly generate chemical complexity, C–H activation and functionalization reactions have enabled a paradigm shift in synthetic chemistry. In this context, metal-catalyzed C–H activation has significantly streamlined the syntheses of bioactive complex molecules.10 While C7-arylation of simple indole substrates has been reported,11 the lack of efficient catalytic strategies for achieving highly regioselective C7-arylation of tryptophan-containing peptides under biocompatible conditions remains a critical bottleneck, severely limiting the development of innovative tryptophan-modified peptide therapeutics. Despite recent advances in Trp functionalization at the C2, C4, and N1 positions,12 selective arylation at the Trp(C7) position remains particularly challenging due to the inert nature of the C7–H bond, the relatively low reactivity of aryl electrophiles compared to C2 or C4 positions, and competing side reactions that complicate site selectivity.13
Herein, we report a biocompatible system that overcomes these long-standing limitations and achieves late-stage C7 arylation of Trp residues in both amino acids and peptides (Fig. 1C). In addition to the PtBu2 directing group installed at the Trp(N) to ensure regioselectivity, we employed a Rh catalyst pre-ligated with an intrinsic phosphine ligand in combination with an external P-based small-molecule additive. This design proved crucial for enhancing the catalytic efficiency in peptide substrates. This method utilizes simple and readily available bromoaryl compounds as aryl donors and is compatible with both solution-phase and solid-phase peptide synthesis (SPPS), providing access to structurally diverse Trp-modified peptides. Furthermore, we demonstrate its utility in constructing environmentally sensitive Trp-derived fluorescent probes and in enhancing the bioactivity of antimicrobial peptides through strategic C7 functionalization. This method not only broadens the substrate scope and enhances reaction efficiency but also provides a potentially general solution for achieving selective C7 arylation in complex peptide contexts.
Results and discussion
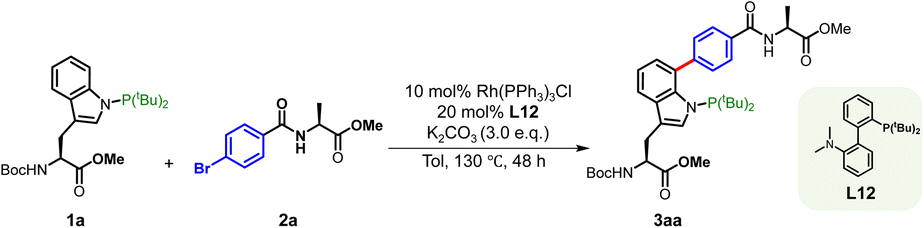
We initiated our study with BocNH-(N-PtBu2)Trp-CO2Me (1a) and aryl bromide-modified alanine derivative 2a, employing Rh(PPh3)3Cl (6 mol%) as the catalyst and K2CO3 as the base in toluene at 120 °C for 24 h, which afforded C7-selective adduct 3aa in ∼6% yield (Table S1, entry 1). NMR analysis confirmed that the arylation occurred selectively at the C7 position of the Trp residue (Fig. S1). Screening a series of mono- and bidentate ligands revealed Ligand 12 (L12) as optimal, improving the yield to 30% (Tables S1 and S2, entry 9). Subsequent optimization of reaction parameters, including temperature, reaction time, solvent, catalyst and ligand loading, further improved the efficiency, delivering up to 80% yield (Table 1, entry 1, entries 7–13). Increasing the catalyst loading to 15 mol% gave only marginal improvement (Table S1, entry 13). Other catalysts, such as [Rh(cod)Cl]2 and [Rh(coe)2Cl]2, proved much less effective (Table 1, entries 2 and 3), and control experiments confirmed the essential role of rhodium catalysis (Table 1, entry 4). Alteration of the base failed to improve the reaction efficiency (Table 1, entries 5 and 6). The optimized conditions were thus established as: 10 mol% Rh(PPh3)3Cl, 3.0 eq. K2CO3, 20 mol% L12, with toluene as solvent at 130 °C for 48 h.|
|
||
|---|---|---|
| Entry | Variation from the “standard conditions” | Yield of 3aab (%) |
| a Conditions: 1a (0.10 mmol), 2a (0.30 mmol), 20 mol% L12, K2CO3(0.3 mmol), 10 mol% catalyst, solvent (1.0 mL), at 130 °C under Ar atmosphere for 48 h. b Isolated yields are reported. | ||
| 1 | None | 80 |
| 2 | [Rh(cod)Cl]2 instead of Rh(PPh3)3Cl | 5 |
| 3 | [Rh(coe)2Cl]2 instead of Rh(PPh3)3Cl | Trace |
| 4 | Without Rh(PPh3)3Cl | — |
| 5 | Using NaOAc instead of K2CO3 | 10 |
| 6 | Using tBuOLi instead of K2CO3 | — |
| 7 | 6 mol% Rh(PPh3)3Cl, without L12 | 12 |
| 8 | 6 mol% Rh(PPh3)3Cl, 10 mol% L12 | 30 |
| 9 | 6 mol% Rh(PPh3)3Cl, 20 mol% L12 | 65 |
| 10 | 24 h | 40 |
| 11 | 120 °C | 32 |
| 12 | DME as the solvent | 57 |
| 13 | PhCl as the solvent | 73 |
Next, we evaluated the substrate scope of the P(III)-directed Trp(C7) arylation (Fig. 2). Using Trp derivative 1a and simple aryl bromides as model substrates, we found that L12 could be omitted without loss of efficiency. A wide range of aryl bromides proved suitable, including ortho-, meta-, and para-substituted derivatives, affording products 3ab–3ai in 72–92% yields. Lipidation is an important modification of peptides and proteins.14 Notably, a para-aliphatic aryl bromide (2j) delivered the lipidated product 3aj in 79% yield, highlighting the utility of this method for site-specific lipid installation in Trp(C7) (Fig. 2, entry 3aj). Functional handles such as N-heterocycles (methylindole, quinoline), photoreactive groups (benzophenone, 9-fluorenone), and polycyclic aromatics were also well tolerated, giving 3ak–3aw in 69–90% yields. Importantly, the protocol accommodated fluorescent motifs, with bromo-BODIPY (2x) and bromo-tetraphenylethylene (2y) furnishing 3ax and 3ay in 88% and 87% yields, respectively. To address the potential epimerization issue during the reaction, substrates 1a and 1a′ were synthesized and subjected to reactions with aryl bromide 2i. Results showed that crude reaction mixtures of 1a and 1a′ gave distinct retention times when analyzed by HPLC (Fig. S2), indicating that the stereochemical integrity was retained and no epimerization occurred under the reaction conditions. These results underscore the broad compatibility of the method with structurally diverse and functionalized aryl bromides.
 | ||
| Fig. 2 N-P(III)tBu2-directed Rh-catalyzed Trp(C7)-selective arylation of substrate 1a. Conditions: [a] 1a (0.1 mmol), 2x (0.3 mmol), K2CO3(0.3 mmol), 10 mol% Rh(PPh3)3Cl, solvent (1.0 mL), at 120 °C under Ar atmosphere for 48 h. [b] 130 °C. Isolated yields are reported. Fluorescent emission spectra of 3ay and TPE (10 µM) were recorded in H2O/dioxane mixtures upon excitation at 325 nm. | ||
Vendrell and Lavilla et al. have shown that direct C2-conjugation of BODIPY to tryptophan affords environmentally sensitive fluorophores suitable for imaging membrane interactions.15 The C7-BODIPY-conjugated tryptophan derivative 3ax exhibited the characteristic absorption at 498 nm and emission at 520 nm of BODIPY fluorophores (Fig. 2 and S3). Notably, 3ax showed a substantial enhancement in fluorescence emission efficiency under hydrophobic conditions, with a 12-fold increase in emission intensity at 515 nm when the solvent was changed from H2O/dioxane (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) to pure dioxane. A second example compound 3ay, featuring C7-conjugation of tetraphenylethylene (TPE) to tryptophan, introduced a distinct substitution pattern that significantly enhanced the aggregation-induced emission (AIE) effect compared to the parent TPE molecule (Fig. 2). A pronounced fluorescence turn-on was observed even at low water fractions in H2O/DMSO mixtures, highlighting the sensitivity of conjugate 3ay to changes in solvent polarity and aggregation state. Collectively, these findings highlight both the synthetic utility of the P(III)-directed Rh-catalyzed Trp(C7) arylation strategy and the potential of site-selective fluorophore installation to create unnatural amino acids with tunable photophysical properties.
:5) to pure dioxane. A second example compound 3ay, featuring C7-conjugation of tetraphenylethylene (TPE) to tryptophan, introduced a distinct substitution pattern that significantly enhanced the aggregation-induced emission (AIE) effect compared to the parent TPE molecule (Fig. 2). A pronounced fluorescence turn-on was observed even at low water fractions in H2O/DMSO mixtures, highlighting the sensitivity of conjugate 3ay to changes in solvent polarity and aggregation state. Collectively, these findings highlight both the synthetic utility of the P(III)-directed Rh-catalyzed Trp(C7) arylation strategy and the potential of site-selective fluorophore installation to create unnatural amino acids with tunable photophysical properties.
The robustness of this arylation method prompted us to explore its application in the site-selective modification of Trp(C7) in short peptides (Fig. 3A). To access the required peptide substrates, we leveraged Fmoc- or Boc-protected N-P(tBu)2-Trp as a versatile building block compatible with both liquid-phase and solid-phase peptide synthesis, and subsequently incorporated this residue into target sequences through standard peptide assembly protocols. Notably, in these peptide-based reactions, the presence of ligand L12 was indispensable for productive arylation. Using aryl bromide-modified alanine 2z as the arylation reagent, Trp-containing dipeptides 1b–1j were all converted into the corresponding Trp(C7)-arylated peptide-Ala conjugates in good yields (entries 3bz–3jz). A broad range of side chains, including protected Tyr, Ser, Lys, Trp, His, Asp, Glu, unprotected Met and Phe were well tolerated in this protocol with no observable methionine oxidation. The position of the Trp(N-PtBu2) residue in peptides exerted minimal influence, as illustrated by the efficient generation of 3kz–3mz. We further applied this method in the Trp(C7) modification of tri-, tetra- and pentapeptides with 2z, affording modified products 3nz–3pz in 50–63% isolated yields. To further assess whether arylation compromises stereochemical integrity, we used dipeptide 3fz as a model compound. The arylation reaction mixture of compound 3fz was subjected to Marfey's reagent (FDLA) derivatization and analyzed by LC-MS. Results showed that the amino-acid configuration is well retained during the reaction, with no detectable epimerization (Fig. S4). We next applied this method to solid-phase peptide synthesis (SPPS) (Fig. 3B and S5). BocNH-(N-PtBu2)Trp-COOH was synthesized as a building block and incorporated into model hexapeptide P1 following standard SPPS protocols. Direct on-resin C7-arylation of the resin-supported P1 was conducted under standard conditions. The resulting peptide product was cleaved and deprotected with 95% TFA, followed by HPLC purification, affording product P3ai in 9% overall isolated yield. This result showed that the reaction was feasible on resin, further demonstrating the utility of this method.
 | ||
| Fig. 3 In solution and on-resin Trp(C7) arylation of peptides. (A) N-PtBu2-enabled Rh-catalyzed arylation of di, tri-, tetra- and pentapeptides at the C7 position of Trp residues. Conditions: 1x (0.1 mmol), 2z (0.3 mmol), K2CO3 (0.3 mmol), 20 mol% L12, 10 mol% Rh(PPh3)3Cl, solvent (1.0 mL), 48 h, at 130 °C under Ar. (B) On-resin Trp(C7) arylation of peptides. Conditions: P1 (0.1 mmol), 2i (0.3 mmol), K2CO3 (0.3 mmol), 20 mol% L12, 10 mol% Rh(PPh3)3Cl, solvent (3.0 mL), 48 h, at 120 °C under Ar. Isolated yields are reported. | ||
Crosslinking amino acids and peptides represents a powerful approach for generating oligomers and expanding their structural and functional diversity, as exemplified by aryl–aryl crosslinks in dimeric peptide natural products.16 Inspired by this, we next explored our method for crosslinking of amino acids using di- and tribromoaryl linkers (Fig. 4). Dibromobenzene derivatives 3a–3e efficiently generated Trp–Trp dimers in good yields (Fig. 4, entries 4aa–4ae). Other linkers, including dibromo-substituted terphenyl, naphthalene, 1-phenyl-4-(4-phenylphenyl)benzene, fluorescent chrysene derivatives and aliphatic substituted bromoaryl compounds also delivered Trp dimers in moderate to good yields (entries 4af–4aj). Remarkably, trimerization could be realized with 1,3,5-tribromobenzene and 1,3,5-tris(4-bromophenyl)benzene, affording the Trp trimers 4ak and 4al in 41% and 45% yield, respectively. These findings underscore the broad versatility of this arylation strategy in constructing structurally complex peptide architectures.
 | ||
| Fig. 4 Crosslinking of Trp via C7 arylation. Standard conditions: 1a (0.6–0.9 mmol), 3x (0.1 mmol), K2CO3 (0.3 mmol), 20 mol% L12, 15 mol% Rh(PPh3)3Cl, solvent (1.0 mL), 48 h, at 130 °C under Ar. Isolated yields are reported. | ||
To address the challenge of removing the N-PtBu2 directing group from Trp(C7) arylation products, we established tailored deprotection protocols for both amino acid and peptide substrates (Fig. 5). Treatment with TBAF at 80 °C for 12 hours afforded the deprotected Trp(C7)-arylated amino acids and peptides in good yields, as demonstrated by 5i′ and 5j′ in 66% and 71% yields, respectively (Fig. S6). This procedure demonstrated excellent compatibility with common N- and C-terminal protecting groups, including N-terminal triflyl (Tf) groups and C-terminal methyl esters. For Trp(C7)-arylated peptides synthesized via SPPS, the N-PtBu2 protecting group can be removed concomitantly under standard global deprotection conditions by extending the reaction time (Fig. 5 and S7, entry 6c).
 | ||
| Fig. 5 Deprotection of the P(III) directing group. Removal of the N-PtBu2 directing group via global deprotection of peptides synthesized from SPPS. TFA cleavage cocktail (TFA/TIPS/H2O = 95/2.5/2.5, v/v/v), 5 h, at 30 °C. | ||
With the methodology established, we applied our Trp(C7) arylation platform to synthesize functional Trp derivatives and peptides. Fluorescent amino acids are valuable tools for non-invasive labeling of peptides and proteins.3a,b,17 While native Trp 5a absorbs at 280 nm and emits around 350 nm, modification of its indole chromophore through conjugation with alkenes, aryl groups, fluorophores and other substituents can significantly enhance its photophysical properties.15d,18 Motivated by this, we combined Trp C–H modification strategies, including our C7 arylation, to design derivatives with tailored fluorescence. Compound 5b was synthesized from TfNH-Trp-CO2Me(5a) via Pd-catalyzed C4 olefination (Fig. 6A). Compared to native Trp, the UV-vis absorption spectrum of 5b was significantly red-shifted with absorption extending to 408 nm (Fig. 6B and S8). Sequential C7 arylation (N-P(III)(tBu)2-directed Rh catalysis), C2 arylation, and C4 alkenylation furnished 5d (Fig. 6A). The additional arylation further red-shifted the absorption edge to 460 nm (Fig. 6B and S8). Compound 5d exhibited an emission maximum at 500 nm, corresponding to a Stokes shift of 140 nm (Fig. S9). Encouraged by these results, we synthesized a series of Trp derivatives (5b–5f) bearing C2 and C7 arylations in combination with C4 olefination (Fig. S8). All tryptophan derivatives demonstrated excellent stability when stored at room temperature and under daylight (Fig. S10).
 | ||
| Fig. 6 Synthesis of tryptophan fluorescent molecules by N-PtBu2-enabled Rh-catalyzed arylation of Trp. (A) Sequential modification of Trp via C–H activation methods. (B) UV-vis absorption spectra of 5a–5d (10 µM) in MeCN. (C) Fluorescence-fold of 5d were determined in H2O/dioxane mixtures (0.1:9.9 to 0:100, v/v) at a concentration of 5 µM, with excitation at 355 nm. fw(V%) represents the volumetric fraction of water. (D) Fluorescence and brightfield microscopy images of E. coli after labelling with 5g-labelled antimicrobial peptide (25 mM) (excitation at 488 nm). Scale bar: 2 µm. | ||
Fluorescence studies in H2O/dioxane mixtures, simulating hydrophobic and hydrophilic microenvironments, revealed pronounced environmental sensitivity of 5b–5f (Fig. S11). Notably, 5c exhibited a 296-fold fluorescence enhancement when the solvent was changed from water to 99% dioxane, whereas 5b increased only 7.8-fold, highlighting the critical role of C7 arylation in modulating environmental responsiveness. Similarly, compound 5d displayed a 172-fold fluorescence enhancement under the same conditions, underscoring the potential of these derivatives for wash-free imaging applications (Fig. 6C and S11). Comparison across derivatives demonstrates that the site of modification strongly influences photophysical properties of Trp: C7 arylation consistently imparts enhanced environmental sensitivity and fluorescence turn-on effects, while C2 and C4 modifications primarily tune absorption maxima. This site-specific tuning enables the rational design of unnatural Trp derivatives with distinct and predictable optical behaviors, providing a versatile platform for the development of fluorescent probes and functional peptides.
Next, we employed compound 5d as a fluorophore for live-cell fluorescence imaging, taking advantage of its 460 nm absorption and strong fluorescence turn-on response in hydrophobic environments. Through SPPS, amino acid 5g (a derivative of 5d) was incorporated into the amphiphilic, bacterial membrane-targeting peptide PAF26 to generate the labeled peptide 5h (Fig. 6D and S12).19 Compound 5g displayed excellent chemical stability and was fully compatible with standard SPPS protocols.
To evaluate imaging performance, various concentrations of 5h were incubated with E. coli before analyzed by flow cytometric, and 25 µM was identified as optimal (Fig. S13). Owing to its environment-responsive turn-on behavior, 5h enabled direct visualization of bacterial cells without the need for washing steps. Real-time, wash-free imaging of E. coli cultures was successfully achieved using confocal laser scanning microscopy (CLSM), delivering high signal-to-noise ratios (Fig. 6D). These results demonstrate that the C7-arylated fluorescent Trp derivative can be used to label bacterial-targeting peptides without compromising their function, while also providing a practical platform for wash-free imaging in live cell environments.
Lipidation and arylation have proven to be robust and effective strategy for improving the therapeutic potential of peptide therapeutics.20 We next evaluated the function of various C7-modified Trp derivatives by installing them into the antifungal hexapeptide PAF26. Six PAF26 analogs (6a–6f) bearing distinct modified Trp residues were synthesized via Rh-catalyzed arylation (Fig. 7 and S14). Growth-inhibition assays against drug-resistant, marine-derived Aspergillus fumigatus revealed clear structure–activity relationships. Introduction of N-P(tBu)2 alone (compound 6a) produced only marginal enhancement relative to PAF26 (IC50 = 89.2 µM vs. 97.9 µM). Incorporation of an aryl substituent lacking the alkyl chain (6b) or a phenyl–alkyl lipid tail without N-P(tBu)2 (6c) afforded moderate improvements (IC50 = 78.4 and 73.7 µM, respectively), indicating that neither organophosphorus substitution nor lipidation alone is sufficient to substantially boost antifungal activity. In striking contrast, dual modification with both N-P(tBu)2 and a phenyl–alkyl chain (residue 3aj, compound 6d) resulted in a dramatic activity increase (IC50 = 2.0 µM), representing ∼49-fold higher potency than native PAF26 and ∼35-fold stronger inhibition than lipidated analog 6c. These comparisons demonstrate that lipidation and phosphoramidate substitution act cooperatively to strengthen antifungal activity—consistent with reports that organophosphorus motifs can favorably influence bioactivity.21 Together, the SAR trends across these Trp derivatives highlight the utility of C7-selective Trp arylation for the systematic modulation of peptide function, providing a versatile platform for antifungal peptide optimization and bioactive peptide probe development.
 | ||
| Fig. 7 Antimicrobial activities of PAF26 derivatives against Aspergillus fumigatus. | ||
Conclusions
In conclusion, we have developed a robust, regioselective Rh(I)-catalyzed C7 arylation of tryptophan via a removable N-PtBu2 directing group, enabling late-stage modification of amino acids and peptides. This versatile platform accommodates diverse aryl bromides and peptide scaffolds, facilitating the synthesis of structurally and functionally diverse Trp derivatives. The approach supports the design of environmentally responsive fluorophores with large Stokes shifts for wash-free imaging, as well as lipidated peptide analogues with enhanced antifungal activity. A key advance of this study is the identification of electron-rich, sterically hindered P(III) ligands as critical promoters of Rh-catalyzed Trp(C7) arylation in peptide substrates. These ligands likely suppress coordination interference from peptide amide bonds and side chains while facilitating oxidative addition and reductive elimination, thereby enhancing reaction efficiency. The rate-accelerating effect of bulky phosphine ligands has been validated in related Pt/Pd-catalyzed C–H activation systems.22 Collectively, these results highlight the potential of site-selective Trp(C7) functionalization as a powerful strategy to expand the chemical space of peptides, enabling the rational design of functional probes and bioactive peptides with tailored photophysical and biological properties.Author contributions
H. W. and Z. S. initiated and directed this study. L. W., Y. Z. and Y. S. carried out the design, chemical synthesis and characterization of amino acids and peptides. B. W., Y. X. and T. W. performed antibiotic experiments. All authors participated in the data analysis and manuscript preparation.Conflicts of interest
There are no conflicts to declare.Data availability
Supplementary information (SI): data for this article, including experimental procedures, and characterization data of all the substrates and products, are available in the supplementary information of the manuscript. See DOI: https://doi.org/10.1039/d5sc08312e.Acknowledgements
This work is supported by NSF of China (Grant 22325702), National Key R&D Program of China (2025YFA0922700), the Natural Science Foundation of Jiangsu Province (BK20253010, BK20232020 and BG2025036), Fundamental and Interdisciplinary Disciplines Breakthrough Plan of the Ministry of Education of China (JYB2025XDXM507), and Yachen Foundation of Nanjing University.References
- (a) M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309–325 CrossRef CAS PubMed; (b) L. Wang, N. Wang, W. Zhang, X. Cheng, Z. Yan, G. Shao, X. Wang, R. Wang and C. Fu, Signal Transduction Targeted Ther., 2022, 7, 48 Search PubMed; (c) Y. Han, Y. Zhang, H. Li, Z. Ma and Y. Wang, MedComm, 2025, 6, e70287 CrossRef CAS PubMed; (d) Q. Yang, Z. Hu, H. Jiang, J. Wang, H. Han, W. Shi and H. Qian, Chin. J. Nat. Med., 2025, 23, 31–42 CAS; (e) S. Guo, J. Wang, Q. Wang, J. Wang, S. Qin and W. Li, Heliyon, 2024, 10, e26009 Search PubMed; (f) X. Ji, A. L. Nielsen and C. Heinis, Angew. Chem., Int. Ed., 2024, 63, e202308251 Search PubMed; (g) P. Servatius, L. Junk and U. Kazmaier, Synlett, 2019, 30, 1289–1302 Search PubMed.
- (a) H.-R. Tong, B. Li, G. Li, G. He and G. Chen, CCS Chem., 2021, 3, 1797–1820 Search PubMed; (b) J. Rodríguez and M. Martínez-Calvo, Chem.–Eur. J., 2020, 26, 9792–9813 CrossRef PubMed; (c) D. G. Rivera, G. M. Ojeda-Carralero, L. Reguera and E. V. Van der Eycken, Chem. Soc. Rev., 2020, 49, 2039–2059 RSC; (d) W. Wang, M. M. Lorion, J. Shah, A. R. Kapdi and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 14700–14717 CrossRef CAS; (e) A. Schischko, N. Kaplaneris, T. Rogge, G. Sirvinskaite, J. Son and L. Ackermann, Nat. Commun., 2019, 10, 3553 CrossRef.
- (a) Z. Cheng, E. Kuru, A. Sachdeva and M. Vendrell, Nat. Rev. Chem., 2020, 4, 275–290 CrossRef CAS; (b) A. T. Krueger and B. Imperiali, ChemBioChem, 2013, 14, 788–799 CrossRef CAS; (c) Y. Chen and M. D. Barkley, Biochemistry, 1998, 37, 9976–9982 CrossRef CAS; (d) F. A. Serrano and E. Yukl, FASEB J., 2022, 36, R3984 CrossRef; (e) J. T. Vivian and P. R. Callis, Biophys. J., 2001, 80, 2093–2109 Search PubMed.
- (a) H. Gouda, K. Matsuzaki, H. Tanaka, S. Hirono, S. Ōmura, J. A. McCauley, P. A. Sprengeler, G. T. Furst and A. B. Smith, J. Am. Chem. Soc., 1996, 118, 13087–13088 CrossRef CAS; (b) J. Kohno, Y. Koguchi, M. Nishio, K. Nakao, M. Kuroda, R. Shimizu, T. Ohnuki and S. Komatsubara, J. Org. Chem., 2000, 65, 990–995 Search PubMed; (c) O. Martí-Marí, B. Martínez-Gualda, S. de la Puente-Secades, A. Mills, E. Quesada, R. Abdelnabi, L. Sun, A. Boonen, S. Noppen, J. Neyts, D. Schols, M.-J. Camarasa, F. Gago and A. San-Félix, J. Med. Chem., 2021, 64, 10027–10046 CrossRef PubMed.
- H. Deng, J.-K. Jung, T. Liu, K. W. Kuntz, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2003, 125, 9032–9034 Search PubMed.
- J. Garfunkle, F. S. Kimball, J. D. Trzupek, S. Takizawa, H. Shimamura, M. Tomishima and D. L. Boger, J. Am. Chem. Soc., 2009, 131, 16036–16038 CrossRef CAS PubMed.
- Y. Jia, M. Bois-Choussy and J. Zhu, Org. Lett., 2007, 9, 2401–2404 CrossRef CAS PubMed.
- S. Lin and S. J. Danishefsky, Angew. Chem., Int. Ed., 2001, 40, 1967–1970 CrossRef CAS.
- M. Inoue, H. Sakazaki, H. Furuyama and M. Hirama, Angew. Chem., Int. Ed., 2003, 42, 2654–2657 CrossRef CAS.
- (a) P. Tao and Y. Jia, Sci. China: Chem., 2016, 59, 1109–1125 Search PubMed; (b) I. Bakanas, R. F. Lusi, S. Wiesler, J. Hayward Cooke and R. Sarpong, Nat. Rev. Chem., 2023, 7, 783–799 CrossRef CAS; (c) S. Sengupta and G. Mehta, Org. Biomol. Chem., 2020, 18, 1851–1876 RSC.
- (a) X. Qiu, P. Wang, D. Wang, M. Wang, Y. Yuan and Z. Shi, Angew. Chem., Int. Ed., 2019, 58, 1504–1508 CrossRef CAS PubMed; (b) C. B. Kelly, R. Padilla-Salinas, W. Chen, M. Muuronen and J. Balsells, Adv. Synth. Catal., 2024, 366, 2044–2055 Search PubMed.
- (a) N. Kaplaneris, T. Rogge, R. Yin, H. Wang, G. Sirvinskaite and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 3476–3480 CrossRef CAS PubMed; (b) Z. Ruan, N. Sauermann, E. Manoni and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 3172–3176 CrossRef CAS; (c) Z. Bai, C. Cai, W. Sheng, Y. Ren and H. Wang, Angew. Chem., Int. Ed., 2020, 59, 14686–14692 CrossRef CAS PubMed; (d) L. Liu, X. Fan, B. Wang, H. Deng, T. Wang, J. Zheng, J. Chen, Z. Shi and H. Wang, Angew. Chem., Int. Ed., 2022, 61, e202206177 Search PubMed.
- J. Wen and Z. Shi, Acc. Chem. Res., 2021, 54, 1723–1736 Search PubMed.
- (a) H. Jiang, X. Zhang, X. Chen, P. Aramsangtienchai, Z. Tong and H. Lin, Chem. Rev., 2018, 118, 919–988 CrossRef CAS PubMed; (b) P. Kurtzhals, S. Ostergaard, E. Nishimura and T. Kjeldsen, Nat. Rev. Drug Discovery, 2023, 22, 59–80 Search PubMed.
- (a) L. Mendive-Tapia, C. Zhao, A. R. Akram, S. Preciado, F. Albericio, M. Lee, A. Serrels, N. Kielland, N. D. Read, R. Lavilla and M. Vendrell, Nat. Commun., 2016, 7, 10940 CrossRef CAS PubMed; (b) L. Mendive-Tapia, L. Miret-Casals, N. D. Barth, J. Wang, A. de Bray, M. Beltramo, V. Robert, C. Ampe, D. J. Hodson, A. Madder and M. Vendrell, Angew. Chem., Int. Ed., 2023, 62, e202302688 CrossRef CAS; (c) L. Mendive-Tapia, R. Subiros-Funosas, C. Zhao, F. Albericio, N. D. Read, R. Lavilla and M. Vendrell, Nat. Protoc., 2017, 12, 1588–1619 CrossRef CAS; (d) N. Kaplaneris, J. Son, L. Mendive-Tapia, A. Kopp, N. D. Barth, I. Maksso, M. Vendrell and L. Ackermann, Nat. Commun., 2021, 12, 3389 CrossRef CAS PubMed.
- K. A. D'Angelo, C. K. Schissel, B. L. Pentelute and M. Movassaghi, Science, 2022, 375, 894–899 Search PubMed.
- F. de Moliner, F. Nadal-Bufi and M. Vendrell, Curr. Opin. Chem. Biol., 2024, 80, 102458 Search PubMed.
- (a) W. Wang, M. M. Lorion, O. Martinazzoli and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 10554–10558 CrossRef CAS PubMed; (b) T. Oyama, L. Mendive-Tapia, V. Cowell, A. Kopp, M. Vendrell and L. Ackermann, Chem. Sci., 2023, 14, 5728–5733 RSC; (c) A. Kopp, T. Oyama and L. Ackermann, Chem. Commun., 2024, 60, 5423–5426 Search PubMed; (d) H. Qianzhu, E. H. Abdelkader, A. P. Welegedara, E. Habel, N. Paul, R. L. Frkic, C. J. Jackson, T. Huber and G. Otting, Angew. Chem., Int. Ed., 2024, 64, e202421000 CrossRef PubMed.
- A. Munoz, B. Lopez-Garcia, E. Perez-Paya and J. F. Marcos, Biol. Res. Commun., 2007, 354, 172–177 CAS.
- (a) T. Rounds and S. K. Straus, Int. J. Mol. Sci., 2020, 21, 9692 CrossRef CAS PubMed; (b) L. Zhang and G. Bulaj, Curr. Med. Chem., 2012, 19, 1602–1618 Search PubMed.
- M. Vorácová, M. Zore, J. Yli-Kauhaluoma and P. Kiuru, Bioorg. Med. Chem., 2023, 96, 117512 Search PubMed.
- (a) C. F. Bender, W. B. Hudson and R. A. Widenhoefer, Organometallics, 2008, 27, 2356–2358 Search PubMed; (b) X. Huang, K. W. Anderson, D. Zim, L. Jiang, A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 6653–6655 Search PubMed; (c) W. A. Moradi and S. L. Buchwald, J. Am. Chem. Soc., 2001, 123, 7996–8002 Search PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |