
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DeepPath: overcoming data scarcity for protein transition pathway prediction using physics-based deep learning
Yui Tik Pang†
 a,
Lixinhao Yang†b,
Katie M. Kuoa and
James C. Gumbart*ab
a,
Lixinhao Yang†b,
Katie M. Kuoa and
James C. Gumbart*ab
aSchool of Physics, Georgia Institute of Technology, Atlanta, GA 30332, USA. E-mail: gumbart@physics.gatech.edu
bSchool of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA 30332, USA
First published on 5th May 2026
Abstract
The structural dynamics of proteins play a crucial role in their function, yet many current deep learning methods chiefly yield high-resolution static snapshots of single conformations, with dynamics captured indirectly or at limited resolution unless paired with simulations or experiments. We present DeepPath, a physics-guided deep learning framework that rapidly predicts realistic protein transition pathways at atomistic resolution. Unlike conventional supervised learning approaches, DeepPath employs generative active learning (GAL) to iteratively refine its predictions, leveraging molecular mechanical force fields as oracles to guide pathway generation. We validated DeepPath on four biologically relevant test cases: AdK opening/closing, SHP2 activation, CdiB H1 expulsion, and BAM-complex gating, with DeepPath reproducing key transient interactions observed in previous studies. Notably, DeepPath also predicted an intermediate between the BAM inward- and outward-open states that closely aligns with an experimentally observed hybrid-barrel structure (TM-score = 0.91). This work highlights the potential of GAL for protein structure prediction.
Introduction
Proteins are fundamental to a plethora of biological processes, with their functions intrinsically linked to their three-dimensional structures. Accurate predictions of these structures have long been a central challenge in computational biology. The advent of deep learning techniques has revolutionized this field, particularly with the development of AlphaFold2 (AF2),1 swiftly followed by tools such as RoseTTAFold,2 ESMFold,3 AF3 (ref. 4) and others.5–10 These methods enable the accurate prediction of static protein structures from amino acid sequences, providing researchers with invaluable tools to investigate protein structures, functions, and interactions at an atomistic level.11–15 However, proteins are not static entities; they are dynamic molecules that undergo conformational changes essential for their biological functions. Capturing these transitions, not just endpoint structures, remains a key challenge.Protein dynamics are essential to enable multiple functional states, ligand interactions, and participation in complex cellular pathways.16–19 Insights into these conformational changes are vital not only for basic research, but also for practical applications like drug discovery. Understanding conformational flexibility can guide the rational design of therapeutic molecules for efficacy and selectivity.20,21 MD simulations are traditionally the gold standard for studying protein dynamics in silico. They offer atomistic insights into molecular movements based on thermodynamic principles.22 However, these simulations are computationally demanding. Even with state-of-the-art supercomputers, MD typically achieves only nanoseconds to microseconds per day,23–25 far short of the millisecond-to-second timescales required for many biological processes. Enhanced sampling techniques26–31 and recent machine learning (ML) integrations32–36 aim to address this limitation, but they often remain computationally expensive and require highly specific input settings for different systems.
Building on the success of ML methods in static protein structure prediction, researchers have explored ways to leverage these tools to generate more diverse conformations.37–41 For example, masking coevolutionary signals in multiple sequence alignment (MSA) inputs38–40 or introducing alanine mutations37,42 were shown to induce some degree of conformational diversity in the generated structures.42 Other strategies include retraining models with additional structures derived from long-timescale MD simulations,43 adding diffusion-based or flow-matching-based auxiliary networks,41,44 or designing tailored architectures and training data for a smaller subset of proteins.45–47 BioEmu, which integrates several of these strategies, demonstrated the ability to generate ensembles of alternative backbone conformations for small proteins.48 These methods have achieved varying degrees of success, but none of them have obtained MD-level of diversity and structural resolution. A persistent challenge shared by all is the limited availability of training data for dynamical protein structures, which can hinder generalization, particularly for systems lacking prior dynamic data.48
In this paper, we experiment with a high-potential yet underexplored strategy called Generative Active Learning (GAL). Unlike traditional supervised learning, which passively learns from static, labeled datasets, GAL actively drives the learning process by iteratively generating candidates and refining its prediction based on feedback from oracles (e.g., quantum chemistry simulations, docking scores, or physics-based models).49,50 This closed-loop optimization enables models to efficiently navigate vast design spaces, focusing computational and experimental resources on the most relevant regions. GAL has already been applied to areas like molecular chemistry49,51,52 and materials science53 for novel compound designs. These applications shared an important feature with the protein dynamics problem, where the available training data is still orders of magnitude short from filling in the vast configuration space, but well-developed physics-based models are available to predict the likelihood of reaching different states.
Here, we developed DeepPath, a novel deep learning architecture that leverages GAL to learn the transition pathways between two given protein structures by interacting with molecular mechanics (MM) force fields rather than from pre-computed pathway data. At the core of DeepPath is a two-module design: a generative-adversarial-network-(GAN)-based Explorer operating in reduced coordinates proposes transition pathways, while a dedicated Structure Builder reconstructs all-atom conformations, balancing computational speed and precision. To evaluate its capabilities, we tested DeepPath on a number of biologically relevant protein systems that exemplify key challenges in protein dynamics and large conformational changes. These systems range from soluble to membrane proteins, and from non-linear domain motion to substrate secretion. Specifically, we examined four protein transitions: domain opening/closing of adenylate kinase (AdK), activation of Src homology 2-containing protein tyrosine phosphatase-2 (SHP2), contact-dependent growth inhibition B (CdiB) helix 1 expulsion, and opening of the β-barrel assembly machinery (BAM) complex. We show that DeepPath efficiently explored the conformational spaces across all test cases, generating low-energy transition pathways within hours of training. Its predictions not only match existing data but also reveal novel conformations, broadening our understanding of these systems. Overall, these results underscore the potential of GAL as a scalable approach for predicting dynamic protein structures with all-atom resolution, even in the absence of large training datasets.
Results
Model overview
DeepPath introduces a GAL framework uniquely designed to overcome the scarcity of transition-state structural data for individual proteins. Instead of relying on static, precompiled datasets or pool-based active learning, DeepPath generates novel intermediates on the fly and refines them via interaction with a molecular mechanics (MM) energy minimizer as an “oracle”. This per-protein GAL loop, guided by force field energetics, allows DeepPath to autonomously expand its own training data, improving accuracy with each iteration without ever requiring a known pathway. To initiate the pathway exploration, short MD simulations are conducted of each of the input end states. These initial training data need not cover any of the transition states, but rather they give clues to the network about the relative mobility of each part of the protein. Then, DeepPath starts generating its own training data iteratively by constructing potential intermediate structures and validating their energies via an MM energy minimizer, slowly broadening the range of generated conformations while ensuring these structures remain realistic. Through this iterative process, DeepPath searches different possible transition pathways, expanding its training database until no alternative pathways with lower energies can be found.The architecture of DeepPath is modular by design, comprising two distinct but synergistic neural network components: the Explorer and the Structure Builder (Fig. 1a). The Explorer operates in a reduced coordinate space of carefully selected pairwise distances, enabling efficient exploration of the conformational landscape. It leverages a progressive GAN architecture,54,55 allowing coarse-to-fine pathway refinement while progressively increasing resolution over training iterations. The Structure Builder is a custom feed-forward network trained to reconstruct all-atom Cartesian coordinates from the Explorer's outputs. The choice of the reduced coordinate set is versatile, provided it can effectively differentiate between distinct protein conformations. To ensure the model's generality and physical interpretability, we have selected all residue–residue pairwise distances that show significant variation beyond a predetermined threshold as our reduced coordinates for this implementation.
 | ||
| Fig. 1 DeepPath overview. (a) GAL routine for training DeepPath. First, short MD equilibrium simulations are performed at each of the end state to generate some initial training data. This data is utilized to train the two modules of DeepPath, the Explorer and Structure Builder, which together produce all-atom structures along tentative transition pathways. Selected generated structures are refined with their corresponding energies determined using the MM energy minimizer. These refined structures are added to the training database to re-train DeepPath and gradually improve the reliability of the transition pathways generated as the training cycle continues. (b) The Explorer is a modified progressive GAN, with a combination of critics that work together to ensure the paths generated are realistic and energetically feasible. (c) Training duration for 20 iterations of GAL using one GPU for three protein test cases: CdiB, SHP2, and the BAM complex. | ||
The modular design of DeepPath is crucial to its success. The use of a reduced coordinate set isolates the Explorer from the high-dimensional coordinate space, which is characterized by a complex, uneven energy landscape that complicates the exploration and optimization of conformations. This separation of exploration from reconstruction avoids the pitfalls of directly learning in high-dimensional atomic spaces, a common source of instability in deep generative models for novel protein conformation generation. Additionally, this modular approach simplifies the overall network architecture, reduces the volume of training data required, and enables rapid production of valid transition pathways.
From our testing, DeepPath generates reliable transition pathways within 20 GAL iterations, which takes only a few hours on a single GPU for an average-sized protein (Fig. 1c) and achieves approximately 80% of the energy reduction observed at full convergence. For example, DeepPath computed the expulsion process of the H1 helix from the CdiB β-barrel, which has 371 residues, in less than 4.5 hours. DeepPath also proved scalable by accurately predicting the transition pathway between the inward-and outward-open states of the 1794-residue BAM complex, albeit requiring a longer computational time of 66.7 hours. A breakdown of the training time reveals that the majority of training cost increment is allocated to the training of the Structure Builder, the largest network within DeepPath. The use of the frame aligned point error (FAPE)1 in its loss function contributes to a computational complexity that scales quickly with the size of the protein, increasing from merely 2.1 hours for CdiB to 63.4 hours for the BAM complex. The time spent on energy minimization also increases with the complexity and size of the protein. In contrast, the training time for the Explorer remains largely constant across different protein sizes, highlighting the efficiency achieved through the use of reduced coordinate sets.
Explorer
The core innovation of DeepPath resides in the Explorer, a GAN responsible of exploring the vast protein conformational space and constructing plausible transition pathways between the two specified input end states. These pathways are represented as a series of pairwise distance arrays (x) that encode each intermediate state along the pathway. We chose a progressive GAN as the fundamental architecture for the Explorer as we found its incremental nature to be highly compatible with our GAL protocol (Fig. 1b). The progressive GAN grows the network by starting the training at a low resolution, then adds new layers that model increasingly fine details as training progresses. We adopted this design for the Explorer by first training it to generate transition pathways represented by only four intermediate states, and then adding new layers to the generator and doubling the path resolution every five GAL training iterations until a sufficient resolution is reached. This staged learning approach allows the network to first capture a broad overview of the energy landscape before fine-tuning the pathways to minimize energy more effectively, balancing global exploration with local optimization.Usually, GANs are composed of a generator and a critic, with the generator trained to generate outputs that are deemed realistic by the critic, and the critic trained to distinguish the generated outputs from the real ones. These two networks are trained against each other to improve each other's performance. Importantly, DeepPath trains the Explorer's generator not against ground-truth pathways but rather against a trio of specialized critics: a Wasserstein critic enforcing realism (CWGAN), an energy critic that approximates the MM-energy landscape (Cenergy), and a geometric path continuity critic (Cpath). This multi-critic training strategy56 enables stable generation of physically plausible transitions without requiring example pathways – a major departure from traditional supervised models. Together, the loss function for the generator is:
 | (1) |
Outside of providing guidance for the generator, the CWGAN and Cenergy also serve another important purpose in the GAL process. Due to the high computational cost of performing energy minimization (compared to the rest of the training loop), only a fraction of generated intermediate structures were energy minimized and had their energies evaluated. Drawing parallels to the active learning criterion “query-by-committee”,58 we pick structures that obtain conflicting verdicts from CWGAN and Cenergy. Specifically, if CWGAN predicts the structure to be unrealistic but Cenergy predicts it to be low energy, the structure is sent to have its energy evaluated. Conceptually, this criteria encourages structures that are rated novel and low-energy to be evaluated and added to the database, iteratively improving the training data quality and eventually leading to the generation of the most plausible transition pathways.
Together, these architectural choices allow DeepPath to generalize across protein sizes, topologies, and environments, from soluble domains to membrane-embedded β-barrels. The GAL loop ensures that DeepPath adapts to each protein individually, while the modular neural-network framework preserves interpretability and physical plausibility at every stage of generation.
Test case 0: adenylate kinase
We first tested the network on adenylate kinase (AdK), a well-studied benchmark system whose open-closed conformational transition has been extensively characterized in molecular simulations. The AdK transition is commonly described using the distances between the LID-CORE and AMPbd-CORE domain centers, and both the associated free-energy landscapes59 and representative transition pathways60 in this space have been reported in previous work. Following these studies, we projected the DeepPath-generated intermediates onto the two-dimensional space defined by the LID-CORE and AMPbd-CORE distances. In this representation, the predicted pathway follows the same curved transition manifold observed in conventional simulation trajectories60 (Fig. S1), rather than a simple linear interpolation between the open and closed states. Importantly, the ordering of domain motions along the DeepPath pathway is also consistent with one of the transition trends previously reported for apo AdK: the AMPbd (NMP-binding) domain begins to open prior to the LID domain. Because the DeepPath test case also corresponds to the apo form of AdK, this sequential motion is consistent with previous simulation studies suggesting that the smaller and more flexible AMPbd domain tends to initiate opening motions prior to, or in concert with, the larger LID domain.61,62Test case 1: SHP2
To evaluate DeepPath in a real-world scenario, we applied it to a protein target characterized by a significant domain shift that is crucial to its pathogenic behavior. Src homology-2 domain-containing protein tyrosine phosphatase-2 (SHP2) is a protein target that has been implicated in cancer but was long considered undruggable.63,64 In a healthy cell, SHP2 typically remains in its closed state and only transitions into its open state upon binding with its partner protein. In cancerous cells, SHP2 can go from the closed to open conformation spontaneously, without binding of the partner protein.65,66 In addition, the protein sometimes has point mutations that bias it to the open state.67,68 The overactive SHP2 in the open conformation results in cancerous cell proliferation, tumor invasion, and metastasis.69,70 While there have been numerous small molecules developed against SHP2, none are FDA-approved, and all are designed to target the closed state.66,71 By understanding the transition from the closed to the open state, more effective therapeutics can be designed to inhibit this conformational transition and, thus, the pathogenesis of cancer.69 These conformational changes are particularly interesting for assessing whether DeepPath can generate pathways that are not strictly linear but instead rotational, and whether it can prevent the N-SH2 domain from colliding with the PTP domain. We were particular interested in the E76A variant due to the partially open state that had been previously reported.67DeepPath was initially trained on two 100 ns equilibrium MD trajectories, one originating from the inactive state and the other from the active state. Due to the short length of these trajectories, only a limited conformational space near each of the end states was sampled (Fig. 2c). Following the initial training, DeepPath was trained for 50 iterations using the GAL protocol. In each iteration, DeepPath evaluated the validity of selected structures it generated by determining whether their MM potential energy, computed by the energy minimizer, fell below an adaptive threshold. Once 32 valid structures were identified, they were incorporated into a database as part of the training dataset. Over the whole course of the GAL process, a total of 1600 new structures were added to the training set.
 | ||
| Fig. 2 DeepPath predicted the transition paths between SHP2 active and inactive states. (a) The predicted transition pathway of SHP2 highlights the rotational movement of the N-SH2 domain (yellow) around the PTP domain (gray), accompanied by a rotation of the C-SH2 domain (blue). (b) The average potential energy of the top-five generated pathways decreases as training proceeds. (c) Training structures generated at different stages, projected onto the principal components of their reduced representations. Initial equilibrium simulations are shown in the top panel, GAL iterations 1–10 are shown in the middle panel, and iterations 11–20 in the bottom panel. The dotted square highlights the gap in the pathway that occurred early in training, which was successfully filled after 15 iterations of GAL training. (d) Close-up views highlights the challenge of generating structures in the region where the pathway initially breaks. Top: Before training, DeepPath predicts a structure with steric clashes around the A236–K244 loop (black) from the PTP domain. Middle: After 15 iterations of GAL, DeepPath lifts the C-SH2 domain, creating space for the A236–K244 loop to maneuver under the linker between the C-SH2 and N-SH2 domains, resolving the major steric clash between the A236–K244 loop and the C-SH2 domain. Bottom: At convergence, DeepPath completely resolves the steric clash issues, preventing the C-terminus from hooking into the A236–K244 loop. | ||
Fig. 2a shows the SHP2 transition pathway predicted by DeepPath. Consistent with previous studies,65,66 DeepPath identified a pivotal movement of the N-SH2 domain, which underwent a hinge-like swing around the PTP domain during the transition between the active and inactive states. Simultaneously, DeepPath predicted a rotation of the C-SH2 domain, positioned on top of the PTP domain. Interestingly, despite the large overall root-mean-square-deviation (RMSD) between the active and inactive states, the RMSD of each individual domain remains low throughout the predicted pathway (Fig. S2). This suggests that DeepPath preserved the local structural integrity of SHP2 during the transition, avoiding distortions that could compromise its functional state. Moreover, along the predicted transition pathway, DeepPath identified an intermediate conformation that closely resembles the partially open state reported by Tao et al.,67 notably recovering a critical contact between residues Y63 and E508 (Fig. S3).
Examining the progression of DeepPath during the GAL protocol, we observed that the average potential energy of the top-five generated pathways decreases gradually during the training and stabilizes at approximately −18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 920 kcal mol−1 (Fig. 2b). This drop in energy can be explained by analyzing the structures generated by DeepPath during the training process. Early in the training, DeepPath showed an “understanding” that the N-SH2 domain must rotate around the PTP domain to transition between the active and inactive states. However, it initially lacked the ability to determine the optimal distance between the N-SH2 and PTP domains, resulting in steric clashes and higher energy barriers. Specifically, a loop (residues A236–K244) from the PTP domain clashed with both the N-SH2 and C-SH2 domains (Fig. 2d). When examining the validated structures in the training database, we noticed an early gap in the pathway, where all of the generated structures within the gap failed to obtain potential energies below the required threshold (Fig. 2c). However, DeepPath successfully generated valid structures that bridged this gap by the 14th iteration. It continued to produce more low-energy structures in subsequent iterations, eventually filling the gap entirely. The final predicted pathway reveals that DeepPath adjusted the C-SH2 domain, lifting it to create more space for the A236–K244 loop to pass underneath the linker between the C-SH2 and N-SH2 domains (Fig. 2d).
920 kcal mol−1 (Fig. 2b). This drop in energy can be explained by analyzing the structures generated by DeepPath during the training process. Early in the training, DeepPath showed an “understanding” that the N-SH2 domain must rotate around the PTP domain to transition between the active and inactive states. However, it initially lacked the ability to determine the optimal distance between the N-SH2 and PTP domains, resulting in steric clashes and higher energy barriers. Specifically, a loop (residues A236–K244) from the PTP domain clashed with both the N-SH2 and C-SH2 domains (Fig. 2d). When examining the validated structures in the training database, we noticed an early gap in the pathway, where all of the generated structures within the gap failed to obtain potential energies below the required threshold (Fig. 2c). However, DeepPath successfully generated valid structures that bridged this gap by the 14th iteration. It continued to produce more low-energy structures in subsequent iterations, eventually filling the gap entirely. The final predicted pathway reveals that DeepPath adjusted the C-SH2 domain, lifting it to create more space for the A236–K244 loop to pass underneath the linker between the C-SH2 and N-SH2 domains (Fig. 2d).
To assess the robustness of the DeepPath predictions, we examined the sensitivity of the results to the simulation setup used as the oracle. First, we repeated the SHP2 analysis using the AMBER ff14SB/TIP3P force field in addition to the CHARMM-based setup described above (Fig. S4).72 The projections in the latent coordinate space show that the generated structures progressively populate the same transition manifold as training proceeds, with the final pathways qualitatively similar between the two force fields. Although the AMBER-based runs required more iterations for the manifold to become continuous, this difference may reflect known variations in secondary-structure stability between force fields. In particular, the ff14SB/TIP3P combination tends to over-stabilize secondary structures,73 which could reduce hinge flexibility during early exploration.
We further investigated the role of the length of the initial MD trajectories used to generate the training data by repeating the analysis with trajectories of 20, 40, 60, and 80 ns (Fig. S5). When projected onto the same latent space defined by the 100 ns dataset (Fig. 2c), the 20 ns and 40 ns runs do not recover a well-defined transition manifold. In contrast, the 60 ns and 80 ns trajectories produce clear and continuous manifolds that closely resemble that obtained from the full 100 ns training trajectory. These results suggest that the quality of the learned transition manifold is influenced by the diversity of configurations sampled in the initial MD trajectories.
Test case 2: CdiB
DeepPath's application extends beyond proteins with multiple stable conformations. In this section, we demonstrate its capability to study the expulsion of a domain from a transmembrane channel. The contact-dependent growth inhibition (CDI) system is a mechanism utilized by Gram-negative bacteria to suppress the growth of neighboring bacterial cells to reduce competition.74 As a member of the Two-Partner Secretion (TPS) family, the CDI system consists of two components: the CdiA toxin and the CdiB transporter. Here, we focused on CdiB, a β-barrel protein that inserts into the bacterial outer membrane, allowing CdiA to pass through its lumen and be displayed on the cell surface.75–78 However, in its resting state, this β-barrel is occluded by an N-terminal α-helix (H1); upon activation, H1 exits into the periplasm to enable CdiA secretion.75,79–81We used DeepPath to generate the exit pathway of the H1 helix and compared our results with a previous study81 that explored this process using steered MD (SMD) and replica exchange umbrella sampling (REUS). In the resting state, the upper half of H1 was tightly packed within the barrel lumen's narrower, more constricted region, limiting its movement. As expulsion progresses, H1 gradually slides downward, freeing itself from these constraints, and makes transient interactions with residues in the lower section of the β-barrel (Fig. 3b). Further examination of the trajectories revealed a particularly prominent interaction between H1 and the β4–β5 region.
 | ||
| Fig. 3 Exit pathway of the N-terminal α-helix (H1) in CdiB as predicted by (a) DeepPath and (b) REUS simulations from a previous study.81 DeepPath efficiently generated exit pathways that closely match the REUS-derived pathway, despite requiring significantly less computational time. Notably, DeepPath accurately captured the key transient interaction between H1 and β4–β5 during expulsion. | ||
Here, we assessed whether we can use DeepPath to recover this pathway with only the resting structure known. To construct the active state, we shifted H1 downward by 35 Å using VMD,82 positioning it entirely within the periplasmic space. Two 10 ns equilibrium MD simulations were run, one starting from the resting state and one from the modeled active state. Additionally, to aid DeepPath in generating an initial guess, a series of modeled intermediate structures were generated by incrementally displacing H1 downward at an interval of 0.35 Å. These additional structures underwent energy minimization but were not subjected to further simulations.
The H1 exit pathway predicted by DeepPath after 50 iterations of GAL training highly resembles the one identified by the 2-µs aggregated REUS simulations (Fig. 3). The model predicted that H1 initially straightened itself slightly as it exits the narrow upper section of the β-barrel before moving downward, where it established transient interactions with the periplasmic end of β4–β5. Finally, it detached H1 from the β-barrel, reaching the modeled active conformation. Over the course of training, the interaction energy between H1 and the β-barrel gradually decreased (Fig. 4a). This trend suggests that DeepPath systematically explored alternative orientations of H1 and identified those leading to more favorable interactions. Additionally, when comparing the DeepPath output to the short equilibrium MD simulations, the DeepPath intermediates revealed distinct interactions between H1 and the β-barrel that are not present in the training data (Fig. S6).
 | ||
| Fig. 4 Diversity and energy refinement of DeepPath-generated H1 exit pathways. (a) Evolution of the interaction energy between H1 and the β-barrel over GAL training iterations shows an overall reduction in energy as DeepPath refined its predicted pathways. (b) PCA of the generated pathways based on the C-α coordinates of H1, with the β-barrel aligned. Each point represents an intermediate conformation, colored according to pathway clusters identified via path similarity analysis (PSA). (c) Representative conformations from each pathway cluster, illustrating structural variability in the periplasmic region while maintaining key contacts with the β-barrel. | ||
Beyond reproducing the H1 exit pathway, DeepPath demonstrated the ability to generate diverse pathway predictions, particularly in regions where structural diversity does not come with an energy penalty. As observed in the REUS simulations, once H1 exited the β-barrel, its unconstrained portion could adopt a wide range of conformations. DeepPath also captured this change of flexibility. Looking at an ensemble of 256 transition pathways generated after training, all pathways were highly consistent when H1 was still confined within the barrel. However, as more residues emerged into the periplasmic space, the diversity among the generated pathways greatly increased while still maintaining key interactions, such as those between H1 and β4–β5 (Fig. 4b and c).
To further assess the extent of this diversity, we applied path similarity analysis (PSA)83 to cluster the pathway ensemble generated by DeepPath. We identified four distinct pathway clusters using a cluster distance cutoff of 28 in Ward criteria, indicating multiple viable exit pathways predicted by DeepPath. These clustering results align with the principal component analysis (PCA) of the H1 C-α coordinates, where the four pathway groups well-separated along the first two principal components (Fig. 4b). Notably, despite the structural variation, the average potential energies among all pathway clusters remained between −12077 ± 7 to −12072 ± 6 kcal mol−1, confirming that the pathway diversity was achieved without sacrificing structural integrity (Table S1). These findings suggest that DeepPath's prediction extends beyond a single transition pathway and instead approximates a transition tube, encompassing a diverse set of thermodynamically accessible routes at finite temperature.
Test case 3: the BAM complex
To assess the scalability of DeepPath, we applied it to the BAM complex from Escherichia coli, a large multi-protein system with nearly 2000 total residues. The BAM complex consists of the transmembrane BamA β-barrel and the accessory proteins BamB–E. It plays a crucial role in the biogenesis of outer-membrane proteins (OMPs) in Gram-negative bacteria by facilitating their folding and insertion into the outer membrane.84–86 BAM adopts two distinct conformations: the inward-open and outward-open states.87–91 In the inward-open state, the lumen of BamA's β-barrel is accessible from the periplasm, allowing nascent OMPs to enter. In contrast, the outward-open state closes this passage while separating the first (β1) and last (β16) strands of the β-barrel, forming the lateral gate (LG), an entry point for nascent OMPs to integrate into the membrane. Recent structural studies revealed that during OMP insertion, BAM transiently forms a hybrid-barrel intermediate, where the substrate OMP uses BamA's β1 strand as a scaffold for folding.92–96 Here, we used DeepPath to predict the transition pathways between the inward- and outward-open states absent a substrate, training it solely on structural data from two 500 ns equilibrium MD simulations initiated from these two endpoint structures. Strikingly, DeepPath's predicted transition pathway revealed an intermediate conformation in agreement with an experimentally resolved hybrid-barrel structure of BAM interacting with EspP, a substrate OMP.92From the DeepPath-predicted transition pathway, it is clear that conformational changes in BamA's β-barrel are tightly coupled with a counterclockwise rotation of the periplasmic domains (Fig. 5a). Between the inward- and outward-open states, DeepPath predicted a two-stage transition in the β-barrel: (1) LG opening and (2) base narrowing. During the first half of the transition, the β1 strand moved in sync with the rotation of the periplasmic domains. This motion quickly broke the backbone hydrogen bonds between β1 and β16 in the inward-open state, initiating LG opening. As β1 continued to straighten and peel away from β16, the LG reached its maximum opening width after the periplasmic domains rotated by approximately 15° (Fig. 5b). In the second half, the periplasmic domain rotation correlated with the narrowing of the β-barrel's base. The transition was complete once the periplasmic domains rotated by approximately 40° in total, at which point the base of the β-barrel constricted, fully sealing the periplasmic opening and the outward-open state was stabilized.
 | ||
| Fig. 5 DeepPath-generated transition pathway of the BAM complex from the inward- to outward-open state and comparison to the string method. (a) Snapshots from the final iteration of DeepPath results showcasing the angular opening of the lateral gate (LG, orange), the movement of the P5 domain from left to right, and the counter-clockwise rotation of the periplasmic domains BamB–E as the LG opened. (b and c) Evolution of the transition pathways as represented by the LG angle and periplasmic domain rotational angle. (b) Top-1 pathways predicted by DeepPath. (c) Pathways obtained using SMwST, depicted for both inward-to-outward-open (forward) and outward-to-inward-open (backward) transitions. (d) The DeepPath-predicted “double-open” structure (green) overlapped with the cryo-EM structure of BAM/EspP hybrid-barrel intermediate (gray for BAM and orange for EspP; PDB: 7RJ5). Only shared residues of BAM were shown. (e) Comparison of the β-barrel structure predicted by DeepPath and SMwST during transition. (f) C-α RMSD between the predicted β-barrels and the hybrid-barrel experimental structure along the predicted transition pathways by DeepPath and SMwST. | ||
The DeepPath prediction suggests that BAM adopts an intermediate “double-open” state, where the BamA β-barrel lumen is accessible from both the periplasmic space and the membrane simultaneously. This observation is reminiscent of the hybrid-barrel intermediate, where both the LG and the periplasmic entrance must open to allow the nascent substrate OMP to pass through the BamA lumen and reach the LG as folding proceeds. To test whether the DeepPath-generated double-open structure corresponds to such an intermediate, we compared it to an experimentally resolved BAM/EspP hybrid-barrel intermediate structure (PDB: 7RJ5).92 Despite the absence of the substrate OMP in DeepPath's model, the alignment of the BamA β-barrel is remarkably high (Fig. 5d). The C-α RMSD between the predicted and experimental structures measured only 3.19 Å with a TM-score of 0.86. This structural agreement also extends to the membrane-inserting hydrophobic loops. Even without explicitly modeled membrane lipids, DeepPath correctly oriented the membrane-inserting hydrophobic loops, proving its capability to infer and preserve structural constraints purely from MD training data. Across all shared residues of the BAM complex, the C-α RMSD was 4.35 Å with a TM-score of 0.91. This demonstrates that DeepPath accurately captured the global structure, including the relative domain arrangement of the accessory proteins BamB–E (Fig. 5d).
As a comparison, we also applied the string method with swarms of trajectories (SMwST)97 to compute the same inward-to-outward-open transition pathway of the BAM complex. To generate the initial pathway estimates, we first ran two separate targeted MD (TMD) simulations: one pulling BAM from the inward- to outward-open state (forward) and another pulling it from the outward- to inward-open state (backward). These TMD-derived pathways were then used to seed two independent SMwST calculations. Each string was discretized into 51 equally spaced images, and 440–640 ps simulations were run per image, per iteration to evaluate the local energy gradients and update the pathway. The forward and backward strings were optimized for 300 and 437 iterations, respectively, accumulating to a total simulation time of 14.5 µs.
Overall, SMwST made significant progress in smoothing out abrupt transitions and reducing fluctuations, but major structural defects inherited from the original TMD pathways remained. Specifically, the forward TMD struggled to break the existing interactions that lock the LG in the inward-open state, while the backward TMD delayed widening the base of the β-barrel until the final stages of the pathway. Even after hundreds of SMwST iterations, remnants of these defects persisted in the optimized pathways. Notably, the forward and backward strings follow markedly different routes. In the forward string, the LG remains closed until the periplasmic domain has rotated for more than 30°, whereas in the backward string, LG closure occurred before any periplasmic domain rotation (Fig. 5c). Additionally, the forward transition retained a pronounced kink in the β1–β2 region, a distortion carried over from the initial TMD trajectory (Fig. 5e). Most importantly, neither the forward nor backward SMwST-generated pathways predicted the “double-open” state that DeepPath did. In fact, all intermediate structures generated by SMwST exhibited higher β-barrel C-α RMSD against the experimental hybrid-barrel structure (PDB: 7RJ5) than the starting end states, whereas DeepPath's predictions consistently achieved lower RMSD than the starting structures (Fig. 5f). This highlights DeepPath's superior predictive power in uncovering unseen intermediate structures accurately.
Comparing the optimization process of DeepPath with that of SMwST, we found that DeepPath converged much faster, both in terms of iterations required and computational cost. A key advantage of DeepPath is that, unlike traditional simulation-based methods, it is not constrained by directionality. Regardless of the simulation methods used to seed SMwST, the process must begin from one state and apply biasing forces to drive the system toward the other state. This approach unavoidably introduces distortions and human bias into the generated pathway. Once introduced, it would require hundreds of nanoseconds or even microseconds of equilibrium simulations to relax the structures and eliminate the defects, making it challenging for SMwST to recover physically realistic intermediates.
In contrast, DeepPath generates all intermediate conformations independently and simultaneously, without relying on biasing forces. Instead, it explores conformational space using a neural-network-driven approach that leverages the protein's intrinsic degrees of freedom and iterative energy evaluations, producing physically plausible initial guesses that require minimal structural corrections. Beyond better initial structure generation, DeepPath also demonstrated a strong ability to repair secondary structure distortions over optimization iterations. In early iterations, we observed twisting and breaking of native hydrogen bonds between BamA's β1 and β2, as well as disruptions in the secondary structure of extracellular loop 1. However, as the active learning iterations progressed, DeepPath gradually corrected these defects, ultimately producing secondary structures that align with the stable input conformations (Fig. S7). This refinement is accompanied by a systematic increase in β-strand content and a reduction of non-β assignments, with most residues either retaining or adopting β-strand character.
Discussion
In this work, we introduce DeepPath, a GAL framework that constructs atomistic protein transition pathways by synthesizing and evaluating new training data on the fly, guided by physics-based energy evaluations. Unlike previous methods that require extensive protein structure datasets, DeepPath self-generates both its training set and predictions, representing a novel paradigm in protein structure modeling. DeepPath's core innovation lies in its combination of generative modeling, active learning, and physics-based oracles in a closed-loop framework. At each iteration, new structures are proposed by a GAN, scored by a set of critic networks, and refined using MM minimization. Through repeated structural refinement and exploration based on this energy evaluation, DeepPath gradually builds up the training dataset and efficiently produces atomic-resolution transition pathways within hours.DeepPath demonstrates how GAL can be leveraged to exploit the physical and chemical knowledge embedded in MM force fields, training deep learning models to generate novel protein conformations even when curated examples of intermediates are scarce, which is a common bottleneck for models trained primarily on static structures or MD-derived sets. To achieve GAL, the Explorer has two additional critics alongside the standard discriminator network of GANs, namely the Energy and the Path Critics. Since there is a trio of critics, a balance of their loss weights is important for efficient learning and conformational space exploration. We found the ratio CWGAN:Cenergy:Cpath = 1:1:10 quite effective for transitions with higher energy barriers (SHP2 and BAM), and 1:1:2 for cases that requires more pathway diversity (AdK and CdiB). The introduction of the Energy Critic network also enabled quick energy landscape exploration by minimizing expensive force-field energy computations and avoiding instability caused by direct energy-based optimization, a known limitation in earlier force-field-coupled generative models.98 However, the current implementation only evaluates protein potential energies and accounts for environmental factors using implicit solvent rather than explicitly modeling water molecules or membrane lipids. Future work could address this limitation by extending the Structure Builder module to construct other biomolecules including water, lipids, and nucleic acids, enabling more accurate computation of interaction energies with the environment.
One key architectural feature of DeepPath is its modular separation between conformational exploration and atomistic structure reconstruction. The GAN-based Explorer operates in a reduced coordinate space defined by pairwise C-α distances, enabling efficient global sampling of conformational space without directly navigating the high-dimensional and rugged all-atom landscape. The Structure Builder then reconstructs all-atom models from these reduced representations using physically motivated loss functions that enforce geometric plausibility, including FAPE, backbone bond length regularization, and steric clash penalties. This design allows DeepPath to integrate essential structural constraints into both training and inference while maintaining scalability across systems of varying size and topology; this degree of flexibility is not typically afforded by coordinate-space generative models. Together, these architectural decisions underlie DeepPath's ability to outperform traditional sampling methods in both efficiency and predictive power. While traditional approaches such as the string method required over 14.5 µs of simulation for the BAM complex, DeepPath produced realistic transition pathways in just 66 hours on a single GPU, matching an experimentally determined intermediate structure. This efficiency enables routine application to large, complex, or poorly characterized systems.
To assess generality, we evaluated DeepPath across four mechanistically distinct processes: conformational transition (AdK), domain rotation (SHP2), membrane-embedded helix expulsion (CdiB), and large multimeric-complex remodeling (BAM). In all cases, DeepPath identified physically plausible pathways, recovered known intermediates, and in the case of BAM, predicted a previously observed “double-open” hybrid-barrel conformation92 with a TM-score of 0.91. This was especially significant given the absence of the substrate OMP in DeepPath's input and output. Notably, all four systems were modeled using the same architecture and training strategy with minor adjustments, underscoring DeepPath's ability to generalize without system-specific retraining or architectural tuning. Beyond accuracy, DeepPath also demonstrated the ability to generate diverse transition pathways. In the CdiB test case, pathway clustering via PSA and PCA revealed four distinct transition route families and highlighted DeepPath's capacity to explore multiple low-energy transition pathways. While DeepPath successfully captures electrostatically driven transitions, its current implementation underrepresents entropic contributions and hydrophobic interactions. For instance, in the CdiB test case, hydrophobic contacts evident in the extended (10 µs) REUS simulations were not fully recovered. While current limitations stem from the simplified energy model, the modular nature of DeepPath provides a flexible foundation for incorporating more sophisticated scoring functions or oracle models in future iterations.
Although DeepPath is currently formulated to generate pathways between two predefined end states, in practice the method can still be applied when only one experimentally determined structure is available. In such cases, a plausible or approximate second state can be constructed to guide the exploration. DeepPath can then identify physically reasonable intermediates along the transition manifold before approaching the imposed endpoint. This behavior was observed in the CdiB system, where meaningful intermediate conformations were recovered even when the final configuration was only approximately specified. As expected, the reliability of the later stages of the pathway depends on how closely the hypothesized endpoint reflects the true target conformation.
It is important to note that DeepPath is designed to complement, not replace, MD simulations and enhanced sampling techniques, which remain the gold standard for capturing thermodynamic and kinetic properties. By rapidly generating plausible protein transition pathways, DeepPath can help guide MD sampling toward relevant regions and accelerate convergence in free energy calculations. From a physical perspective, the pathways generated by DeepPath can be interpreted as low-energy transition routes on the underlying potential energy landscape, obtained by combining geometric learning with iterative energy evaluation from the molecular-mechanics oracle. Related ideas have also emerged in approaches that reconstruct conformational pathways from cryo-EM classification or elastic network models, where the resulting motions correspond to dominant collective transitions in the system.99,100 Beyond its role in accelerating simulations, DeepPath also shows promise for drug discovery applications. Understanding conformational dynamics is critical for designing drugs that target allosteric sites or stabilize specific functional states of proteins. For example, many kinase inhibitors and molecular chaperone modulators rely on exploiting transient conformational states.101–104 DeepPath helps to identify these states, aiding in the development of more selective and effective therapeutics.
Beyond transition pathways, the GAL framework presented here could be extended to a variety of biomolecular structure prediction challenges, including ligand-induced conformational changes, protein folding pathways, and RNA structure prediction. These problems face an even greater scarcity of high-quality structural data, making GAL particularly valuable for efficiently navigating large conformational spaces and generating low-energy structures. The GAL framework introduced in this work effectively integrates ML with MM force fields, enabling structural prediction beyond the scope of the available training dataset. Future extensions of this approach could incorporate diffusion models or flow-based generative architectures, improving the ability to generate diverse structural ensembles, and better capturing the full conformational landscape of biomolecules beyond the current state-of-the-art supervised models.
Methods
Reduced coordinate sets
To lower GPU memory costs and increase exploration efficiency, the Explorer operated in a reduced coordinate space, while the Structure Builder converted the Explorer's outputs into all-atom structures in Cartesian coordinates of all heavy atoms. The reduced coordinates were defined as C-α distances between residue pairs that were in contact in one end state but not in the other. This set of residue pairs was identified using the following procedure: the pairwise C-α distances between all residues were calculated using MDAnalysis105,106 on the MD trajectories from both end states. Residue pairs were selected if their C-α distances fell below ϵA in at least one frame and exceeded ϵB in at least one other frame. For SHP2 and BAM, ϵA and ϵB were set to 6 Å and 15 Å, respectively. For CdiB, ϵA and ϵB were set to 7 Å and 15 Å, respectively.Explorer architecture
The Explorer is a generative adversarial network (GAN) with a generator and multiple critics. The generator's input is a pair of reduced coordinate sets, representing the end-state structures set for the pathway. Its output is a series of reduced coordinate sets, each representing an intermediate structure, and collectively they connect to form a predicted transition pathway. To facilitate this output structure, we adopted the architecture of a progressive GAN,54,55 with the output size doubling after each neural network block within the generator, i.e., the first block produces a pathway with four intermediate states based on the two end states provided; and the second block builds on top of it and creates a pathway with eight intermediate states; and so on. The training procedure was also progressive: during the first five active learning iterations, only the first block was trained; in the next five iterations, the first two blocks were trained; and so forth. In this study, the maximum number of blocks used was four, with the final block added at the 16th active learning iteration. This resulted in an output size of 32 intermediate structures, after which no further blocks were added. The generator was trained to reduce the total loss calculated by all the critics (eqn (1)).The Explorer consists of three critics: WGAN, Energy, and Path Critics. To ensure every point on the pathway is being optimized, the WGAN and Energy Critics evaluate the loss based on 32 intermediate structures sampled at random intervals along an interpolated pathways formed by linear interpolating between the generated structures. Path Critic evaluates the pathway quality loss using the generated structures directly without interpolation.
The WGAN Critic (CWGAN) is a multi-layer perceptron (MLP) with eight hidden layers, each containing 128 neurons with LeakyReLU activations. Its input is a single intermediate structure in reduced coordinates, and it outputs a “realism” score for the input structure. The loss function for this critic follows the WGAN with gradient penalty,57 with an extra regularization term 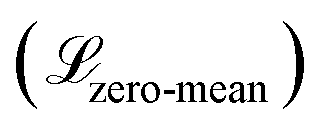 to enforce a zero-centered score distribution:
to enforce a zero-centered score distribution:
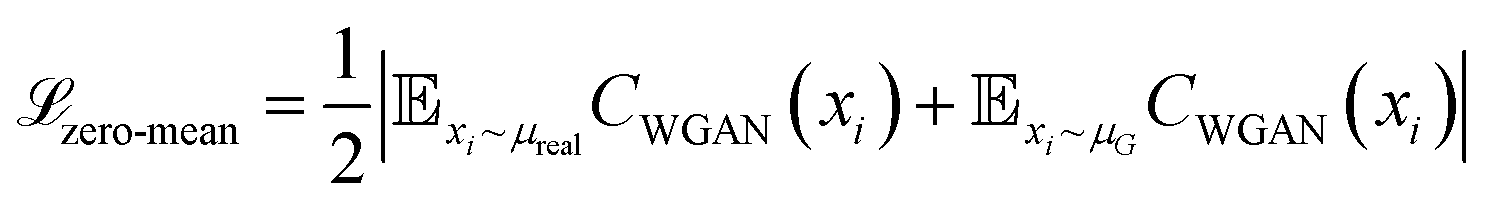 | (2) |
The WGAN Critic is trained together with the generator.
The Energy Critic (Cenergy) is an MLP with two hidden layers, with 128 and 64 neurons, respectively, and ReLU activations. Its input is also a single intermediate structure in reduced coordinates, and it outputs the probability for the input structure to fall into each of the ten energy bins. We found predicting energy labels to be more tolerant to data noise, compared to predicting the energy value directly. Categorical cross-entropy was used as the loss function. It is trained independent of the generator. To make the Energy critic robust against out-of-distribution samples produced by the generator, we employed ensemble knowledge distillation. Three independent teacher models were first trained on ground-truth labels from randomly drawn subsets of the energy-labeled structures. Then, soft pseudo-labels were produced on all generated structure using this ensemble of teacher models. Finally, a student model was trained on a mix of ground-truth labels and soft labels:
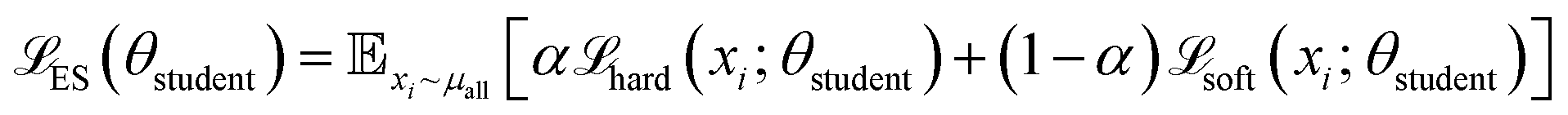 | (3) |
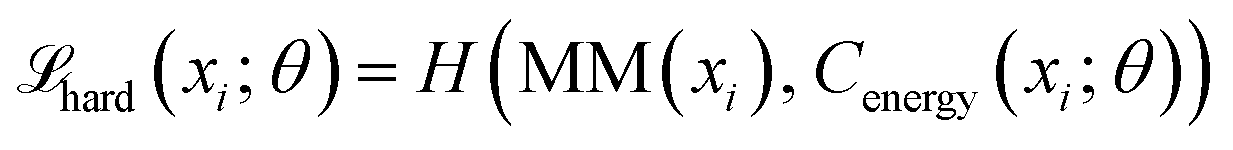 | (4) |
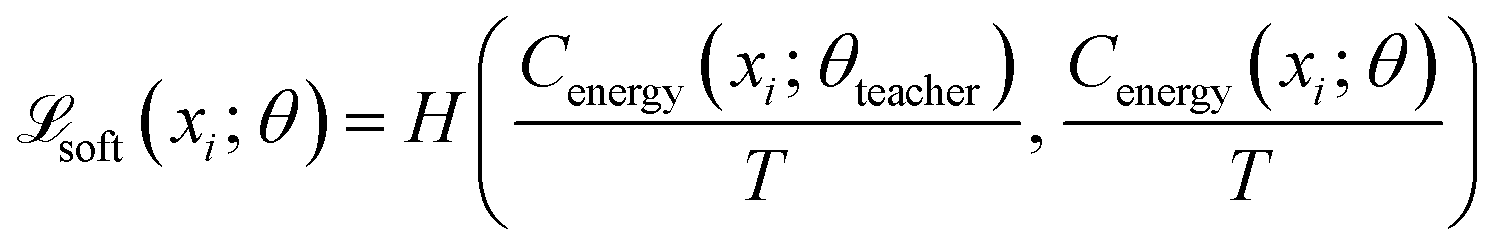 | (5) |
The Path Critic is algorithmic-based and does not contain any trainable parameters. It computes the mean squared error (MSE) between the vector difference of adjacent structures in reduced coordinate space generated by the generator and the baseline one formed by linear interpolation between the two end states:
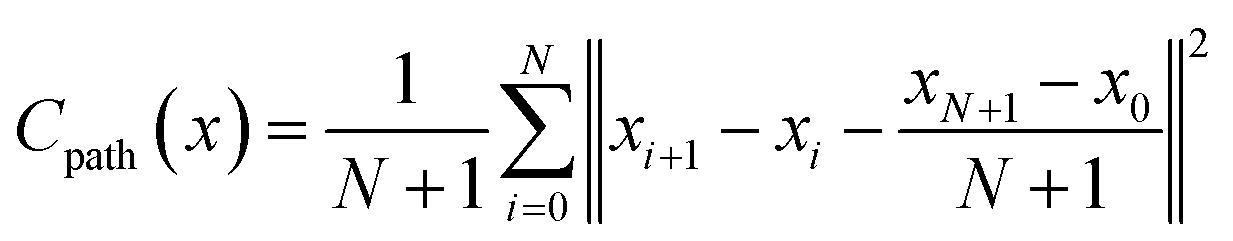 | (6) |
Structure Builder architecture
The Structure Builder is a custom feed-forward network that converts structures from reduced coordinates to all-atom Cartesian coordinates. It consists of a repeating block (Algorithm 1) that progressively optimizes the so-called backbone frames and the dihedral angles of the sidechains, a reduced protein representation that has been proven effective in AF2.1 The block is repeated five times. In the end, the Cartesian coordinates of all heavy atoms are computed from the backbone frames and sidechain rotation components generated by the last block.
The training loss for Structure Builder is based on both the intermediate outputs of each repeating block and the final structure it predicts. During the initial training, the loss consists of frame-aligned point error (FAPE) of all predicted intermediate backbone frames 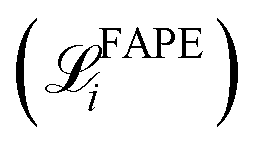 and the final all-atom structures
and the final all-atom structures 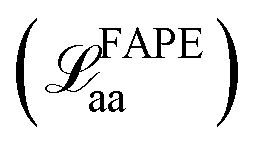 , and MSE of the cosine and sine of all predicted sidechain angles
, and MSE of the cosine and sine of all predicted sidechain angles  . In subsequent training inside the active learning loop, three extra terms are added. First, a flat bottom L1-loss on the amide bond length
. In subsequent training inside the active learning loop, three extra terms are added. First, a flat bottom L1-loss on the amide bond length  to penalize bonds that deviate from the theoretical length (1.334 Å) by more than 0.05 Å. Second, a flat bottom potential that penalizes atoms that getting closer than 1.2 Å from each other
to penalize bonds that deviate from the theoretical length (1.334 Å) by more than 0.05 Å. Second, a flat bottom potential that penalizes atoms that getting closer than 1.2 Å from each other  . Optionally, a sigmoid of the final all-atom structure RMSD
. Optionally, a sigmoid of the final all-atom structure RMSD  can be added to enforce the global domain arrangement. Among the four test cases, it is only turned on when generating the BAM complex transition paths. The total losses during the two stages are:
can be added to enforce the global domain arrangement. Among the four test cases, it is only turned on when generating the BAM complex transition paths. The total losses during the two stages are:
 | (7) |
 | (8) |
The learning rates during the initial and subsequent trainings are 0.001 and 0.0001, respectively.
Generative active learning training procedure
DeepPath was trained using a generative active learning (GAL) framework to refine its prediction of transition pathways, minimizing the reliance on pre-existing datasets. The training procedure is described as follows:1. Initial training: a set of initial structural training data was prepared (see DeepPath initial training set generation). These structures were then energy-minimized and translated into reduced representations. The Structure Builder was trained on the minimized structures until it can reproduce the protein domain arrangements at both end states with reasonable accuracy. It took 1000, 500, and 2000 training epochs for SHP2, CdiB and the BAM complex, respectively. The Energy Critic was trained on the potential energies of the minimized structures for 1000 epochs without knowledge distillation.
2. Explorer GAN training: the generator and WGAN Critic of the Explorer were trained jointly in an adversarial manner (see Explorer architecture). The generator was trained for 50 to 2000 epochs, with the number of epochs reducing as the training database grows in size. For each epoch the generator was trained, the WGAN critic was trained for two epochs.
3. Candidate structures generation and selection: a batch of 256 tentative transition pathways were generated by the Explorer. Intermediate structures were sampled and selected from these pathways with two selection processes. First, intermediate structures with high predicted energies from the pathway with the lowest total predicted energy were selected. This is to help DeepPath close gaps that might exist in its generated pathways. Second, intermediate structures predicted to be low energy by Energy Critic but deemed non-realistic by WGAN Critic were selected. This strategy draws parallels to the “query-by-committee” active learning selection criteria58 to encourage sampling of structures DeepPath is uncertain of. A total of 32 structures were selected in each training loop for SHP2 and CdiB, while 24 structures were selected for the BAM complex.
4. Oracle consultation: the selected intermediate structures were converted into all-atom structures by Structure Builder. These structures were then energy minimized through three steps. First, the backbone atoms were locked in place and only the sidechain atoms were minimized. Next, the structures were minimized again with the backbone locks removed but replaced with harmonic restraints on specific pairwise C-α distances. These included the distances forming the reduced coordinate space in the Explorer to ensure the newly generated structures adhere to the learned transition pathway. Hence, the equilibrium positions of these restraints were set to match the reduced coordinates originally output by the Explorer. In addition, harmonic restraints were used on C-α distances that were deemed constant (below 9 Å and has standard deviation below 0.4 Å throughout the original MD trajectories) to ensure existing residue–residue interactions were retrained. The force constant of these restraints was set to 20 kcal mol−1 Å−2. Finally, the structures were minimized once again without any restrictions. The potential energies of the final minimized structures were recorded. If their energies were lower than the median energy plus one MAD of the existing structures in the training database, the structures were accepted. However, if fewer than half of the final minimized structures fulfilled this limit, the half of the structures with the lowest energies were accepted instead. The accepted structures were added to the training database.
5. Retraining Structure Builder and Energy Critic: the Structure Builder and Energy Critic were retrained using the updated database. The Structure Builder was trained for 5 to 50 epochs, decreasing as the database grows. The Energy Critic was trained using ensemble knowledge distillation (see Explorer architecture). First, each of the three teacher models was trained for 2000 to 10000 epochs. Then, the student model was trained for 1000 to 3000 epochs.
6. Iterations and convergence: steps 2–5 were repeated until no significantly lower-energy pathways and no novel conformations were identified. Such criteria were reached after 50 GAL iterations for SHP2 and CdiB and after 55 iterations for the BAM complex.
All the energy evaluation and minimization within the GAL iterations were performed using OpenMM 7.7.107 The CHARMM36m protein force field108 was used for SHP2 and the BAM complex, and CHARMM36 (ref. 109) was used for CdiB. In addition, for SHP2 and CdiB, Onufriev-Bashford-Case GBSA model110 with the GBOBCII parameters was used to implicitly model solvents, with the solute and solvent dielectrics set to 1.0 and 78.5, respectively. An implicit solvent model was not used for the BAM complex. The reported training time was based on our testing using a single NVIDIA GeForce RTX 4090 for the SHP2 and CdiB and an NVIDIA A100 80GB PCIe for the BAM complex. The main user-provided inputs and the key training parameters used in DeepPath are summarized in Table S2. All models are trained with Adam optimizer111 using the default settings from Tensorflow 2.14 (ref. 112) unless otherwise specified.
Protein model constructions
For SHP2, the inactive and active states were built from publicly available structures. The inactive state was built from PDB ID: 4DGP while the active state was built from PDB ID: 6CRF. Missing loops and residues were added from the AlphaFold predicted structure. To build the mutant E76A, the mutate function in VMD was implemented through Psfgen. SHP2 was built in a TIP3P waterbox with dimensions 100 Å × 100 Å × 100 Å and 0.15 M NaCl.The CdiB and the BAM complex models were prepared and adopted directly from previous studies.81,92
Molecular dynamics (MD) simulations
MD simulations were performed using NAMD 2.12–2.14 (ref. 113) and NAMD 3.0b.23 The CHARMM36m protein force field108 was used for the SHP2 simulations. The CHARMM36 protein109 and lipid force fields114 were used for the CdiB and BAM complex simulations. All simulations used TIP3P water.115 A uniform 4 fs time step was employed through the use of hydrogen mass repartitioning (HMR).116,117 Long-range electrostatics were calculated at each time step using the particle-mesh Ewald method.118 A short-range cutoff for Lennard-Jones interactions was set at 12 Å, with a switching function beginning at 10 Å. Bonds involving hydrogen atoms were constrained to their equilibrium lengths, employing the SETTLE algorithm for water molecules and the SHAKE algorithm for all others. Unless otherwise specified, the temperature and pressure were fixed at 310 K and 1 atm, respectively, using Langevin dynamics and piston,119 respectively.Following TMD, additional constraint simulations were run to ensure the systems fully reached the target structures and the two pathways' end states were identical. The C-α atoms of the BAM complex were constrained to the outward-open state (PDB: 5LJO) and inward-open state (PDB: 5D0O) starting from the last frame of the forward and backward TMDs, respectively. The constraining force constant was gradually increased from 0.2 kcal mol−1 Å−2 to 15 kcal mol−1 Å−2 over 17.4 ns of simulation time.
Frames were extracted from the TMD simulations to form the initial strings. Each string was composed of 51 equally-spaced images, putting each neighboring image roughly 1.5 Å apart from each other. The two end images of both forward and backward strings were originally fixed at the first- and last-frame structures from their respective TMD simulations. At iterations 56 and 117 of the forward and backward strings, respectively, the end-state structures were changed to those from the additional constraint simulations.
SMwST requires users to define the collective variables (CVs) that describe the conformation change along the transition pathway being studied. To minimize human intervention, we selected Cartesian coordinates of every other C-α atom in the BAM complex as the CVs. An adaptation of SMwST for high-dimensional CVs from Ma and Schulten120 was used in this study. The forward and backward strings were optimized for 300 and 437 iterations, respectively, accumulating to a total simulation time of 14.5 µs.
At each iteration, 20 replicas (swarms) of short trajectories (20 ps) were launched from each image. The average drift among these trajectories was computed, and the image is updated accordingly. A smoothing algorithm was employed to redistribute the images such that they remain equidistant from each other.120 A short constraint simulation (40–240 ps) was run to pull the closest swarm trajectories' last frame to the new designated location of each image.
Author contributions
J. C. G. and Y. T. P. conceptualized the project. Y. T. P., L. Y., and K. M. K. curated and analyzed the data. All authors contributed to writing, reviewing, and editing the draft.Conflicts of interest
Y. T. P., K. M. K., and J. C. G. are co-inventors on a provisional patent application (18/441606 submitted by the Georgia Institute of Technology) covering the methodological advances described in this article. They are also stockholders of Atomistic Insights, a company that aims to develop the inventions described in this manuscript. The remaining author declares no competing interests.Data availability
Trained models are provided at https://huggingface.co/andrewytp/deeppath.Supplementary information (SI): detailed pseudocodes, additional analysis, and parameter tables. See DOI: https://doi.org/10.1039/d5sc08253f.
Acknowledgements
This project was partially supported by a Texas Advanced Computing Center (TACC) Frontera Fellowship. Frontera is supported by NSF grant OAC-1818253. This work was also supported by the National Institutes of Health (R01-GM148586). Additional computational resources were provided through ACCESS (grant TG-MCB130173), which is supported by National Science Foundation grants 2138259, 2138286, 2138307, 2137603, and 2138296. This work also used the Hive cluster, which is supported by the NSF under grant number 1828187 and is managed by the Partnership for an Advanced Computing Environment (PACE) at the Georgia Institute of Technology.References
- J. Jumper, et al., Highly accurate protein structure prediction with AlphaFold, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- M. Baek, et al., Accurate prediction of protein structures and interactions using a three-track neural network, Science, 2021, 373, 871–876 CrossRef CAS PubMed.
- Z. Lin, H. Akin, R. Rao, B. Hie, Z. Zhu, W. Lu, N. Smetanin, R. Verkuil, O. Kabeli, Y. Shmueli, A. dos Santos Costa, M. Fazel-Zarandi, T. Sercu, S. Candido and A. Rives, Evolutionary-scale prediction of atomic-level protein structure with a language model, Science, 2023, 379, 1123–1130 CrossRef CAS PubMed.
- J. Abramson, et al., Accurate structure prediction of biomolecular interactions with AlphaFold 3, Nature, 2024, 630, 493–500 CrossRef CAS PubMed.
- N. Bordin, C. Dallago, M. Heinzinger, S. Kim, M. Littmann, C. Rauer, M. Steinegger, B. Rost and C. Orengo, Novel machine learning approaches revolutionize protein knowledge, Trends Biochem. Sci., 2023, 48, 345–359 CrossRef CAS PubMed.
- R. Wu, F. Ding, R. Wang, R. Shen, X. Zhang, S. Luo, C. Su, Z. Wu, Q. Xie, B. Berger, J. Ma and J. Peng, High-resolution de novo structure prediction from primary sequence, bioRxiv, 2022, preprint, DOI:10.1101/2022.07.21.500999.
- R. Chowdhury, N. Bouatta, S. Biswas, C. Floristean, A. Kharkar, K. Roy, C. Rochereau, G. Ahdritz, J. Zhang, G. M. Church, P. K. Sorger and M. AlQuraishi, Single-sequence protein structure prediction using a language model and deep learning, Nat. Biotechnol., 2022, 40, 1617–1623 CrossRef CAS PubMed.
- K. Weissenow, M. Heinzinger and B. Rost, Protein language-model embeddings for fast, accurate, and alignment-free protein structure prediction, Structure, 2022, 30, 1169–1177.e4 CrossRef CAS PubMed.
- X. Fang, J. Gao, J. Hu, L. Liu, Y. Xue, X. Zhang and K. Zhu, HelixFold-Multimer: Elevating protein complex structure prediction to new heights, arXiv, 2024, preprint, arXiv:2404.10260, DOI:10.48550/arXiv.2404.10260.
- S. Passaro, G. Corso, J. Wohlwend, M. Reveiz, S. Thaler, V. R. Somnath, N. Getz, T. Portnoi, J. Roy, H. Stark, D. Kwabi-Addo, D. Beaini, T. Jaakkola and R. Barzilay, Boltz-2: Towards accurate and efficient binding affinity prediction, bioRxiv, 2025, preprint, DOI:10.1101/2025.06.14.659707.
- N. Bordin, I. Sillitoe, V. Nallapareddy, C. Rauer, S. D. Lam, V. P. Waman, N. Sen, M. Heinzinger, M. Littmann, S. Kim, S. Velankar, M. Steinegger, B. Rost and C. Orengo, AlphaFold2 reveals commonalities and novelties in protein structure space for 21 model organisms, Commun. Biol., 2023, 6, 160 CrossRef CAS PubMed.
- P. Fontana, Y. Dong, X. Pi, A. B. Tong, C. W. Hecksel, L. Wang, T.-M. Fu, C. Bustamante and H. Wu, Structure of cytoplasmic ring of nuclear pore complex by integrative cryo-EM and AlphaFold, Science, 2022, 376, eabm9326 CrossRef CAS PubMed.
- A. Díaz-Holguín, M. Saarinen, D. D. Vo, A. Sturchio, N. Branzell, I. Cabeza de Vaca, H. Hu, N. Mitjavila-Domènech, A. Lindqvist, P. Baranczewski, M. J. Millan, Y. Yang, J. Carlsson and P. Svenningsson, AlphaFold accelerated discovery of psychotropic agonists targeting the trace amine-associated receptor 1, Sci. Adv., 2024, 10, eadn1524 CrossRef.
- M. van Breugel, I. Rosa E Silva and A. Andreeva, Structural validation and assessment of AlphaFold2 predictions for centrosomal and centriolar proteins and their complexes, Commun. Biol., 2022, 5, 312 CrossRef CAS PubMed.
- M. Akdel, et al., A structural biology community assessment of AlphaFold2 applications, Nat. Struct. Mol. Biol., 2022, 29, 1056–1067 CrossRef CAS PubMed.
- K. M. Kuo, J. Liu, A. Pavlova and J. C. Gumbart, Drug binding to BamA targets its lateral gate, J. Phys. Chem. B, 2023, 127, 7509–7517 CrossRef CAS PubMed.
- D. L. Lynch, A. Pavlova, Z. Fan and J. C. Gumbart, Understanding virus structure and dynamics through molecular simulations, J. Chem. Theory Comput., 2023, 19, 3025–3036 CrossRef CAS PubMed.
- Y. T. Pang, A. Acharya, D. L. Lynch, A. Pavlova and J. C. Gumbart, SARS-CoV-2 spike opening dynamics and energetics reveal the individual roles of glycans and their collective impact, Commun. Biol., 2022, 5, 1170 CrossRef CAS PubMed.
- B. Russell Lewis, M. R. Uddin, M. Moniruzzaman, K. M. Kuo, A. J. Higgins, L. M. N. Shah, F. Sobott, J. M. Parks, D. Hammerschmid, J. C. Gumbart, H. I. Zgurskaya and E. Reading, Conformational restriction shapes the inhibition of a multidrug efflux adaptor protein, Nat. Commun., 2023, 14, 3900 CrossRef CAS PubMed.
- D. D. Boehr, R. Nussinov and P. E. Wright, The role of dynamic conformational ensembles in biomolecular recognition, Nat. Chem. Biol., 2009, 5, 789–796 CrossRef CAS PubMed.
- M. Amaral, D. B. Kokh, J. Bomke, A. Wegener, H. P. Buchstaller, H. M. Eggenweiler, P. Matias, C. Sirrenberg, R. C. Wade and M. Frech, Protein conformational flexibility modulates kinetics and thermodynamics of drug binding, Nat. Commun., 2017, 8, 2276 CrossRef CAS.
- M. Karplus and J. A. McCammon, Molecular dynamics simulations of biomolecules, Nat. Struct. Biol., 2002, 9, 646–652 CrossRef CAS PubMed.
- J. C. Phillips, et al., Scalable molecular dynamics on CPU and GPU architectures with NAMD, J. Chem. Phys., 2020, 153, 044130 CrossRef CAS PubMed.
- M. I. Andersson, N. A. Murugan, A. Podobas and S. Markidis, Lecture Notes in Computer Science; Lecture notes in computer science, Springer International Publishing, Cham, 2023, pp 333–345 Search PubMed.
- T.-S. Lee, D. S. Cerutti, D. Mermelstein, C. Lin, S. LeGrand, T. J. Giese, A. Roitberg, D. A. Case, R. C. Walker and D. M. York, GPU-Accelerated Molecular Dynamics and Free Energy Methods in Amber18: Performance Enhancements and New Features, J. Chem. Inf. Model., 2018, 58, 2043–2050 CrossRef CAS PubMed.
- G. M. Torrie and J. P. Valleau, Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling, J. Comput. Phys., 1977, 23, 187–199 CrossRef.
- Y. Sugita and Y. Okamoto, Replica-exchange molecular dynamics method for protein folding, Chem. Phys. Lett., 1999, 314, 141–151 CrossRef CAS.
- A. Barducci, M. Bonomi and M. Parrinello, Metadynamics, Wiley Interdiscip. Rev.:Comput. Mol. Sci., 2011, 1, 826–843 CAS.
- J. Comer, J. C. Gumbart, J. Hénin, T. Lelièvre, A. Pohorille and C. Chipot, The adaptive biasing force method: everything you always wanted to know but were afraid to ask, J. Phys. Chem. B, 2015, 119, 1129–1151 CrossRef CAS PubMed.
- Y. T. Pang, Y. Miao, Y. Wang and J. A. McCammon, Gaussian accelerated molecular dynamics in NAMD, J. Chem. Theory Comput., 2017, 13, 9–19 CrossRef CAS PubMed.
- K. Kuczera, G. S. Jas and R. Elber, Kinetics of helix unfolding: molecular dynamics simulations with milestoning, J. Phys. Chem. A, 2009, 113, 7461–7473 CrossRef CAS PubMed.
- F. Hooft, A. Pérez de Alba Ortíz and B. Ensing, Discovering collective variables of molecular transitions via genetic algorithms and neural networks, J. Chem. Theory Comput., 2021, 17, 2294–2306 CrossRef CAS PubMed.
- L. Bonati, G. Piccini and M. Parrinello, Deep learning the slow modes for rare events sampling, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2113533118 CrossRef CAS PubMed.
- W. Chen and A. L. Ferguson, Molecular enhanced sampling with autoencoders: On-the-fly collective variable discovery and accelerated free energy landscape exploration, J. Comput. Chem., 2018, 39, 2079–2102 CrossRef CAS PubMed.
- J. M. L. Ribeiro, P. Bravo, Y. Wang and P. Tiwary, Reweighted autoencoded variational Bayes for enhanced sampling (RAVE), J. Chem. Phys., 2018, 149, 072301 CrossRef PubMed.
- A. Mardt, L. Pasquali, H. Wu and F. Noé, VAMPnets for deep learning of molecular kinetics, Nat. Commun., 2018, 9, 5 CrossRef PubMed.
- R. A. Stein and H. S. Mchaourab, SPEACH_AF: Sampling protein ensembles and conformational heterogeneity with Alphafold2, PLoS Comput. Biol., 2022, 18, e1010483 CrossRef CAS PubMed.
- H. K. Wayment-Steele, A. Ojoawo, R. Otten, J. M. Apitz, W. Pitsawong, M. Hšmberger, S. Ovchinnikov, L. Colwell and D. Kern, Predicting multiple conformations via sequence clustering and AlphaFold2, Nature, 2023, 625, 832–839 CrossRef.
- Y. Kalakoti and B. Wallner, AFsample2 predicts multiple conformations and ensembles with AlphaFold2, Commun. Biol., 2025, 8, 373 CrossRef CAS PubMed.
- D. del Alamo, D. Sala, H. S. Mchaourab and J. Meiler, Sampling alternative conformational states of transporters and receptors with AlphaFold2, eLife, 2022, 11, e75751 CrossRef CAS.
- B. Jing, B. Berger and T. Jaakkola, AlphaFold meets flow matching for generating protein ensembles, Proceedings of the 41st International Conference on Machine Learning, 2024 Search PubMed.
- N. Raisinghani, M. Alshahrani, G. Gupta, H. Tian, S. Xiao, P. Tao and G. Verkhivker, Probing Functional Allosteric States and Conformational Ensembles of the Allosteric Protein Kinase States and Mutants: Atomistic Modeling and Comparative Analysis of AlphaFold2, OmegaFold, and AlphaFlow Approaches and Adaptations, J. Phys. Chem. B, 2024, 128, 11088–11107 CrossRef CAS PubMed.
- P. Bryant and F. Noé, Structure prediction of alternative protein conformations, Nat. Commun., 2024, 15, 7328 CrossRef CAS.
- K. Cheng, C. Liu, Q. Su, J. Wang, L. Zhang, Y. Tang, Y. Yao, S. Zhu and Y. Qi, 4D diffusion for dynamic protein structure prediction with reference and motion guidance, Proceedings of the Thirty-Ninth AAAI Conference on Artificial Intelligence and Thirty-Seventh Conference on Innovative Applications of Artificial Intelligence and Fifteenth Symposium on Educational Advances in Artificial Intelligence, 2025 Search PubMed.
- G. Janson, G. Valdes-Garcia, L. Heo and M. Feig, Direct generation of protein conformational ensembles via machine learning, Nat. Commun., 2023, 14, 774 CrossRef CAS PubMed.
- J. Kacher, O. S. Sokolova and M. Tarek, A Deep Learning Approach to Uncover Voltage-Gated Ion ChannelsÕ Intermediate States, J. Phys. Chem. B, 2024, 128, 8724–8736 CrossRef CAS PubMed.
- V. Satalkar, G. D. Degaga, W. Li, Y. T. Pang, A. C. McShan, J. C. Gumbart, J. C. Mitchell and M. P. Torres, Generative β-hairpin design using a residue-based physicochemical property landscape, Biophys. J., 2024, 123, 2790–2806 CrossRef CAS PubMed.
- S. Lewis, et al., Scalable emulation of protein equilibrium ensembles with generative deep learning, Science, 2025, 389, eadv9817 CrossRef CAS PubMed.
- H. H. Loeffler, S. Wan, M. KlŠhn, A. P. Bhati and P. V. Coveney, Optimal Molecular Design: Generative Active Learning Combining REINVENT with Precise Binding Free Energy Ranking Simulations, J. Chem. Theory Comput., 2024, 20, 8308–8328 CAS.
- P. Ren, Y. Xiao, X. Chang, P.-Y. Huang, Z. Li, B. B. Gupta, X. Chen and X. Wang, A survey of deep active learning, ACM Comput. Surv., 2022, 54, 180 Search PubMed.
- A. Hernández-García, N. Saxena, M. Jain, C.-H. Liu and Y. Bengio, Multi-Fidelity Active Learning with GFlowNets, arXiv, 2024, preprint, arXiv:2306.11715, DOI:10.48550/arXiv.2306.11715.
- I. Filella-Merce, A. Molina, L. D’az, M. Orzechowski, Y. A. Berchiche, Y. M. Zhu, J. Vilalta-Mor, L. Malo, A. S. Yekkirala, S. S. Ray and V. Guallar, Optimizing drug design by merging generative AI with a physics-based active learning framework, Commun. Chem., 2025, 8, 238 CrossRef PubMed.
- S. Lee, Z. Zhang and G. X. Gu, Generative machine learning algorithm for lattice structures with superior mechanical properties, Mater. Horiz., 2022, 9, 952–960 RSC.
- T. Karras, T. Aila, S. Laine and J. Lehtinen, Progressive Growing of GANs for Improved Quality, Stability, and Variation, International Conference on Learning Representations, 2018 Search PubMed.
- T. Karras, S. Laine, M. Aittala, J. Hellsten, J. Lehtinen and T. Aila, Analyzing and Improving the Image Quality of StyleGAN, 2020 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR), 2020, pp 8107–8116 Search PubMed.
- D. Xu, S. Yuan, L. Zhang and X. Wu, FairGAN: Fairness-aware Generative Adversarial Networks, 2018 IEEE International Conference on Big Data (Big Data), 2018, pp 570–575 Search PubMed.
- X. Wei, Z. Liu, L. Wang and B. Gong, Improving the Improved Training of Wasserstein GANs. International Conference on Learning Representations, 2018 Search PubMed.
- Y. Zhao, C. Xu and Y. Cao, Advanced Data Mining and Applications; Lecture notes in computer science, Springer, Berlin, Heidelberg, 2006, pp 985–991 Search PubMed.
- Y. Matsunaga, H. Fujisaki, T. Terada, T. Furuta, K. Moritsugu and A. Kidera, Minimum free energy path of ligand-induced transition in adenylate kinase, PLoS Comput. Biol., 2012, 8, e1002555 CrossRef CAS PubMed.
- H. D. Song and F. Zhu, Conformational dynamics of a ligand-free adenylate kinase, PLoS One, 2013, 8, e68023 CrossRef CAS PubMed.
- D. Li, M. S. Liu and B. Ji, Mapping the dynamics landscape of conformational transitions in enzyme: the adenylate kinase case, Biophys. J., 2015, 109, 647–660 CrossRef CAS PubMed.
- Q. Shao, Enhanced conformational sampling technique provides an energy landscape view of large-scale protein conformational transitions, Phys. Chem. Chem. Phys., 2016, 18, 29170–29182 RSC.
- Z.-Y. Zhang, Protein tyrosine phosphatases: prospects for therapeutics, Curr. Opin. Chem. Biol., 2001, 5, 416–423 CrossRef CAS PubMed.
- J.-J. Ventura and Á. R. Nebreda, Protein kinases and phosphatases as therapeutic targets in cancer, Clin. Transl. Oncol., 2006, 8, 153–160 CrossRef CAS PubMed.
- S. Bobone, et al., Targeting oncogenic Src homology 2 domain-containing phosphatase 2 (SHP2) by inhibiting its protein-protein interactions, J. Med. Chem., 2021, 64, 15973–15990 CrossRef CAS PubMed.
- Y. Song, M. Zhao, H. Zhang and B. Yu, Double-edged roles of protein tyrosine phosphatase SHP2 in cancer and its inhibitors in clinical trials, Pharmacol. Ther., 2022, 230, 107966 CrossRef CAS PubMed.
- Y. Tao, et al., A novel partially open state of SHP2 points to a “multiple gear” regulation mechanism, J. Biol. Chem., 2021, 296, 100538 Search PubMed.
- Y. Liu, W. Zhang, H. Jang and R. Nussinov, SHP2 clinical phenotype, cancer, or RASopathies, can be predicted by mutant conformational propensities, Cell. Mol. Life Sci., 2023, 81, 5 CrossRef PubMed.
- M. D. Asmamaw, X.-J. Shi, L.-R. Zhang and H.-M. Liu, A comprehensive review of SHP2 and its role in cancer, Cell. Oncol., 2022, 45, 729–753 CrossRef CAS PubMed.
- N. Wang, S. Zhu, D. Lv, Y. Wang, M. B. Khawar and H. Sun, Allosteric modulation of SHP2: Quest from known to unknown, Drug Dev. Res., 2023, 84, 1395–1410 CrossRef CAS PubMed.
- A. M. Taylor, et al., Identification of GDC-1971 (RLY-1971), a SHP2 inhibitor designed for the treatment of solid tumors, J. Med. Chem., 2023, 66, 13384–13399 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- L. A. Abriata and M. Dal Peraro, Assessment of transferable forcefields for protein simulations attests improved description of disordered states and secondary structure propensities, and hints at multi-protein systems as the next challenge for optimization, Comput. Struct. Biotechnol. J., 2021, 19, 2626–2636 CrossRef CAS PubMed.
- S. K. Aoki, R. Pamma, A. D. Hernday, J. E. Bickham, B. A. Braaten and D. A. Low, Contact-dependent inhibition of growth in Escherichia coli, Science, 2005, 309, 1245–1248 CrossRef CAS PubMed.
- C. Baud, J. Guérin, E. Petit, E. Lesne, E. Dupré, C. Locht and F. Jacob-Dubuisson, Translocation path of a substrate protein through its Omp85 transporter, Nat. Commun., 2014, 5, 5271 CrossRef CAS PubMed.
- B. Clantin, H. Hodak, E. Willery, C. Locht, F. Jacob-Dubuisson and V. Villeret, The crystal structure of filamentous hemagglutinin secretion domain and its implications for the two-partner secretion pathway, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 6194–6199 CrossRef CAS PubMed.
- A.-S. Delattre, N. Saint, B. Clantin, E. Willery, G. Lippens, C. Locht, V. Villeret and F. Jacob-Dubuisson, Substrate recognition by the POTRA domains of TpsB transporter FhaC, Mol. Microbiol., 2011, 81, 99–112 CrossRef CAS PubMed.
- Z. C. Ruhe, P. Subramanian, K. Song, J. Y. Nguyen, T. A. Stevens, D. A. Low, G. J. Jensen and C. S. Hayes, Programmed secretion arrest and receptor-triggered toxin export during antibacterial contact-dependent growth inhibition, Cell, 2018, 175, 921–933.e14 CrossRef CAS PubMed.
- J. Guérin, C. Baud, N. Touati, N. Saint, E. Willery, C. Locht, H. Vezin and F. Jacob-Dubuisson, Conformational dynamics of protein transporter FhaC: large-scale motions of plug helix, Mol. Microbiol., 2014, 92, 1164–1176 CrossRef PubMed.
- T. Maier, B. Clantin, F. Gruss, F. Dewitte, A.-S. Delattre, F. Jacob-Dubuisson, S. Hiller and V. Villeret, Conserved Omp85 lid-lock structure and substrate recognition in FhaC, Nat. Commun., 2015, 6, 7452 CrossRef PubMed.
- J. Guerin, I. Botos, Z. Zhang, K. Lundquist, J. C. Gumbart and S. K. Buchanan, Structural insight into toxin secretion by contact-dependent growth inhibition transporters, eLife, 2020, 9, e58100 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD – Visual Molecular Dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- S. L. Seyler, A. Kumar, M. F. Thorpe and O. Beckstein, Path similarity analysis: a method for quantifying macromolecular pathways, PLoS Comput. Biol., 2015, 11, e1004568 CrossRef PubMed.
- J. Tommassen, Assembly of outer-membrane proteins in bacteria and mitochondria, Microbiology, 2010, 156, 2587–2596 CrossRef CAS PubMed.
- R. Misra, Assembly of the β-barrel outer membrane proteins in Gram-negative bacteria, mitochondria, and chloroplasts, ISRN Mol. Biol., 2012, 2012, 708203 CrossRef PubMed.
- S. E. Rollauer, M. A. Sooreshjani, N. Noinaj and S. K. Buchanan, Outer membrane protein biogenesis in Gram-negative bacteria, Philos. Trans. R. Soc., B, 2015, 370, 20150023 CrossRef PubMed.
- N. Noinaj, J. C. Gumbart and S. K. Buchanan, The β-barrel assembly machinery in motion, Nat. Rev. Microbiol., 2017, 15, 197–204 CrossRef CAS PubMed.
- J. Bakelar, S. K. Buchanan and N. Noinaj, The structure of the β-barrel assembly machinery complex, Science, 2016, 351, 180–186 CrossRef CAS PubMed.
- Y. Gu, H. Li, H. Dong, Y. Zeng, Z. Zhang, N. G. Paterson, P. J. Stansfeld, Z. Wang, Y. Zhang, W. Wang and C. Dong, Structural basis of outer membrane protein insertion by the BAM complex, Nature, 2016, 531, 64–69 CrossRef CAS PubMed.
- M. G. Iadanza, A. J. Higgins, B. Schiffrin, A. N. Calabrese, D. J. Brockwell, A. E. Ashcroft, S. E. Radford and N. A. Ranson, Lateral opening in the intact β-barrel assembly machinery captured by cryo-EM, Nat. Commun., 2016, 7, 12865 CrossRef CAS PubMed.
- L. Han, J. Zheng, Y. Wang, X. Yang, Y. Liu, C. Sun, B. Cao, H. Zhou, D. Ni, J. Lou, Y. Zhao and Y. Huang, Structure of the BAM complex and its implications for biogenesis of outer-membrane proteins, Nat. Struct. Mol. Biol., 2016, 23, 192–196 CrossRef CAS PubMed.
- R. Wu, J. W. Bakelar, K. Lundquist, Z. Zhang, K. M. Kuo, D. Ryoo, Y. T. Pang, C. Sun, T. White, T. Klose, W. Jiang, J. C. Gumbart and N. Noinaj, Plasticity within the barrel domain of BamA mediates a hybrid-barrel mechanism by BAM, Nat. Commun., 2021, 12, 7131 CrossRef CAS PubMed.
- D. Tomasek, S. Rawson, J. Lee, J. S. Wzorek, S. C. Harrison, Z. Li and D. Kahne, Structure of a nascent membrane protein as it folds on the BAM complex, Nature, 2020, 583, 473–478 CrossRef CAS PubMed.
- H. Takeda, et al., A multipoint guidance mechanism for β-barrel folding on the SAM complex, Nat. Struct. Mol. Biol., 2023, 30, 176–187 CrossRef CAS PubMed.
- M. T. Doyle, J. R. Jimah, T. Dowdy, S. I. Ohlemacher, M. Larion, J. E. Hinshaw and H. D. Bernstein, Cryo-EM structures reveal multiple stages of bacterial outer membrane protein folding, Cell, 2022, 185, 1143–1156.e13 CrossRef CAS PubMed.
- C. Shen, S. Chang, Q. Luo, K. C. Chan, Z. Zhang, B. Luo, T. Xie, G. Lu, X. Zhu, X. Wei, C. Dong, R. Zhou, X. Zhang, X. Tang and H. Dong, Structural basis of BAM-mediated outer membrane β-barrel protein assembly, Nature, 2023, 617, 185–193 CrossRef CAS PubMed.
- A. C. Pan, D. Sezer and B. Roux, Finding transition pathways using the string method with swarms of trajectories, J. Phys. Chem. B, 2008, 112, 3432–3440 CrossRef CAS PubMed.
- V. K. Ramaswamy, S. C. Musson, C. G. Willcocks and M. T. Degiacomi, Deep learning protein conformational space with convolutions and latent interpolations, Phys. Rev. X, 2021, 11, 011052 CAS.
- A. Das, M. Gur, M. H. Cheng, S. Jo, I. Bahar and B. Roux, Exploring the conformational transitions of biomolecular systems using a simple two-state anisotropic network model, PLoS Comput. Biol., 2014, 10, e1003521 CrossRef PubMed.
- A. Dashti, G. Mashayekhi, M. Shekhar, D. Ben Hail, S. Salah, P. Schwander, A. des Georges, A. Singharoy, J. Frank and A. Ourmazd, Retrieving functional pathways of biomolecules from single-particle snapshots, Nat. Commun., 2020, 11, 4734 CrossRef CAS PubMed.
- F. H. Schopf, M. M. Biebl and J. Buchner, The HSP90 chaperone machinery, Nat. Rev. Mol. Cell Biol., 2017, 18, 345–360 CrossRef CAS PubMed.
- L. Neckers and P. Workman, Hsp90 molecular chaperone inhibitors: Are we there yet?, Clin. Cancer Res., 2012, 18, 64–76 CrossRef CAS PubMed.
- K. S. Bhullar, N. O. Lagarón, E. M. McGowan, I. Parmar, A. Jha, B. P. Hubbard and H. P. V. Rupasinghe, Kinase-targeted cancer therapies: progress, challenges and future directions, Mol. Cancer, 2018, 17, 48 CrossRef PubMed.
- C. Zhang and G. Bollag, Scaffold-based design of kinase inhibitors for cancer therapy, Curr. Opin. Genet. Dev., 2010, 20, 79–86 CrossRef CAS PubMed.
- N. Michaud-Agrawal, E. J. Denning, T. B. Woolf and O. Beckstein, MDAnalysis: a toolkit for the analysis of molecular dynamics simulations, J. Comput. Chem., 2011, 32, 2319–2327 CrossRef CAS PubMed.
- R. Gowers, M. Linke, J. Barnoud, T. Reddy, M. Melo, S. Seyler, J. Domański, D. Dotson, S. Buchoux, I. Kenney and O. Beckstein, MDAnalysis: A python package for the rapid analysis of molecular dynamics simulations, Proceedings of the Python in Science Conference, 2016, pp 98–105 Search PubMed.
- P. Eastman, J. Swails, J. D. Chodera, R. T. McGibbon, Y. Zhao, K. A. Beauchamp, L.-P. Wang, A. C. Simmonett, M. P. Harrigan, C. D. Stern, R. P. Wiewiora, B. R. Brooks and V. S. Pande, OpenMM 7: Rapid development of high performance algorithms for molecular dynamics, PLoS Comput. Biol., 2017, 13, e1005659 CrossRef PubMed.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. de Groot, H. Grubmüller and A. D. MacKerell Jr, CHARMM36m: an improved force field for folded and intrinsically disordered proteins, Nat. Methods, 2017, 14, 71–73 CrossRef CAS PubMed.
- R. B. Best, X. Zhu, J. Shim, P. E. M. Lopes, J. Mittal, M. Feig and A. D. Mackerell Jr, Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles, J. Chem. Theory Comput., 2012, 8, 3257–3273 CrossRef CAS PubMed.
- A. Onufriev, D. Bashford and D. A. Case, Exploring protein native states and large-scale conformational changes with a modified generalized born model, Proteins, 2004, 55, 383–394 CrossRef CAS PubMed.
- D. P. Kingma and J. Ba, Adam: A Method for Stochastic Optimization, International Conference on Learning Representations (ICLR), 2015 Search PubMed.
- M. Abadi et al., TensorFlow: Large-Scale Machine Learning on Heterogeneous Systems, 2015, https://www.tensorflow.org/, Software available from tensorflow.org Search PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, Scalable molecular dynamics with NAMD, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- J. B. Klauda, R. M. Venable, J. A. Freites, J. W. O'Connor, D. J. Tobias, C. Mondragon-Ramirez, I. Vorobyov, A. D. MacKerell Jr and R. W. Pastor, Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types, J. Phys. Chem. B, 2010, 114, 7830–7843 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of simple potential functions for simulating liquid water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- C. W. Hopkins, S. Le Grand, R. C. Walker and A. E. Roitberg, Long-time-step molecular dynamics through hydrogen mass repartitioning, J. Chem. Theory Comput., 2015, 11, 1864–1874 CrossRef CAS PubMed.
- C. Balusek, H. Hwang, C. H. Lau, K. Lundquist, A. Hazel, A. Pavlova, D. L. Lynch, P. H. Reggio, Y. Wang and J. C. Gumbart, Accelerating membrane simulations with hydrogen mass repartitioning, J. Chem. Theory Comput., 2019, 15, 4673–4686 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- S. E. Feller, Y. Zhang, R. W. Pastor and B. R. Brooks, Constant pressure molecular dynamics simulation: The Langevin piston method, J. Chem. Phys., 1995, 103, 4613–4621 CrossRef CAS.
- W. Ma and K. Schulten, Mechanism of substrate translocation by a ring-shaped ATPase motor at millisecond resolution, J. Am. Chem. Soc., 2015, 137, 3031–3040 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |