
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePd-catalyzed stereoretentive synthesis of reversed C-acyl glycosides: access to rare L-sugars and higher-carbon sugars
Guoqiang
Cheng
ab,
Bo
Yang
*ab and
Feng
Zhu
 *abc
*abc
aFrontiers Science Center for Transformative Molecules (FSCTM), Shanghai Key Laboratory for Molecular Engineering of Chiral Drugs, School of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai, 200240, P. R. China. E-mail: yangbo71@sjtu.edu.cn; fzchem@sjtu.edu.cn
bPingyuan Laboratory, State Key Laboratory of Antiviral Drugs, Henan Normal University, Xinxiang, Henan 453007, P.R. China
cState Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, China
First published on 2nd February 2026
Abstract
Reversed C-acyl glycosides represent a versatile class of nonclassical glycosides with potential in complex carbohydrate synthesis, including L-sugars, higher-carbon sugars, and medicinal chemistry. Conventional strategies for L- and higher-carbon sugars are limited by multi-step protection–deprotection sequences and poor stereocontrol. Herein, we report a general Pd-catalyzed reversed acyl C-glycosylation that efficiently couples configurationally stable reversed glycosyl stannanes with C(sp2)- and C(sp3)-derived thioesters under mild conditions. The reaction proceeds with complete stereoretentive transfer, enabling precise access to both D- and L-type glycosides, including higher-carbon sugar derivatives and C-ferrocenecarbonyl glycosides. The broad substrate scope, excellent functional group tolerance, and predictable stereochemical outcome highlight the robustness and synthetic versatility of this approach. Applications of the resulting reversed C-acyl glycosides as chiral synthons enable D-to-L interconversion, construction of L-sugar analogues, and derivatization toward designer carbohydrate frameworks. Importantly, this transformation enables a distinct D-to-L conversion featuring simultaneous C4 and C5 inversion, unlike conventional methods that modify only C5 configuration. Overall, this protocol establishes a general platform for stereocontrolled construction and diversification of structurally defined nonclassical glycosides, providing a foundation for glycodiversification, complex sugar synthesis, and exploration of biologically relevant C-glycosyl scaffolds.
Introduction
Carbohydrates are among the most abundant and structurally diverse biomolecules in nature, serving as essential components in energy storage, cellular architecture, and molecular recognition.1–4 Among them, higher-carbon sugars, defined as carbohydrates containing more than six carbon atoms, are broadly distributed in nature and fulfill diverse biological roles. For instance, L-glycero-D-manno-heptose (L,D-Hep), D-glycero-D-manno-heptose (D,D-Hep), and 3-deoxy-D-manno-oct-2-ulosonic acid (Kdo) are key constituents of Gram-negative bacterial lipopolysaccharides (LPS), predominantly located in the inner core region but also present in the outer core or O-antigen domains (Fig. 1A).5–7 Unlike the more common D-hexoses, their enantiomeric counterparts, L-sugars are less prevalent in nature yet frequently embedded in structurally and functionally significant biomolecules.8 Despite the commercial availability of certain L-sugars and higher-carbon sugars, their high cost and limited supply significantly hinder broader biological and synthetic investigations. To fully explore their bioactivities and potential as functional molecules, the development of efficient synthetic strategies is essential. The synthesis of L-sugars and higher-carbon sugars from readily available carbohydrate feedstocks has long been a central focus in carbohydrate chemistry, and a variety of strategies for accessing L-sugars from their D-counterparts have been developed.9,10 However, due to the inherent structural complexity and diversity of carbohydrates, tedious protecting-group manipulations and consequently lengthy synthetic routes are often inevitable. From the perspective of green synthesis and atom economy, therefore, the development of efficient catalytic methods represents an especially appealing strategy.11,12 | ||
| Fig. 1 Overview and background of reversed C-acyl glycosides. (A) Representative examples of rare L-sugars and higher-carbon sugars found in nature. (B) Significance and synthetic diversification of reversed C-acyl glycosides. (C) Catalytic synthesis of reversed C-acyl glycosides beyond non-classical glycosidic bond formation. (D) Catalytic synthesis of reversed C-acyl glycosides involving reversed anomeric radicals. (E) Stereoretentive synthesis of reversed C-acyl glycosides involving reversed anomeric anions (this work). | ||
Non-anomeric C-acyl glycosides 1, also referred to as reversed C-acyl glycosides,13,14 are characterized by the presence of an acyl substituent at the C-5 position of pyranoses or the C-4 position of furanoses, representing a distinct subclass of C-glycosides. In contrast to classical C-acyl glycosides,15–17 which have been extensively studied, the synthesis of these nonclassical motifs has remained relatively underexplored. Nevertheless, they hold significant potential as versatile building blocks for accessing diverse C-glycomimetics through various downstream transformations (Fig. 1B), including reversed CH2-linked glycosides (L-sugars) 2, glycosyl alcohols 3, reversed C-aryl glycosides 4, and reversed C-vinyl glycosides 5, many of which occur in bioactive compounds or challenging molecular architectures. According to whether the construction of a glycosidic bond is involved, the synthesis of reversed C-acyl glycosides can be divided into two categories. Traditionally, reversed C-acyl glycosides have been prepared through multistep oxidation-coupling sequences that require harsh conditions and highly reactive reagents, often leading to poor chemoselectivity, limited functional group tolerance, and tedious protecting-group manipulations.18–22 Notably, catalytic methods have also been reported. For instance, in independent studies, Ye,16 Hu,23 and Li24 disclosed catalytic variants based on Liebeskind–Srogl cross-coupling reactions of thioesters derived from glycosyl acids with arylboronic acids, thereby enabling the synthesis of reversed C-acyl glycosides. However, the incompatibility of alkylboronic acids limits the structural diversity of accessible reversed C-acyl glycosides. Although such methods bypass the stereochemical challenges associated with reversed glycosidic bond formation, they also restrict access to more diverse sugar scaffolds, such as the conversion of D-sugars to L-forms via C5 stereoinversion during acylation (Fig. 1C). An alternative and elegant approach involves Ni(II)/photoredox-catalyzed radical couplings of sugar-derived carboxylic acids,14 active esters (NHPI),25 and 1,4-dihydropyridines (DHPs).13 Despite these advances, the diastereoselectivity of these radical-mediated reactions is strongly substrate-dependent, and predictable stereocontrol across different sugar scaffolds remains elusive (Fig. 1D).26–28 Consequently, a general, mild, and stereoretentive catalytic platform for constructing reversed C-acyl glycosides, particularly one capable of delivering both D- and L-type configurations with excellent stereoselectivity, has not yet been realized.
In response to the aforementioned synthetic challenges and potential applications of reversed C-acyl glycosides, we herein report the first example of a Pd-catalyzed stereospecific cross-coupling of reversed glycosyl stannanes with aryl, alkenyl, and alkyl carboxylic acid-derived thioesters under mild conditions, affording reversed C-acyl glycosides in good to excellent yields with retention of C5 stereochemistry (Fig. 1E). This new glycosylation strategy has been demonstrated in over 45 examples, showcasing high predictability, modularity, and chemoselectivity. This robust and modular catalytic platform demonstrates remarkable substrate generality and functional group tolerance, accommodating protected and unprotected sugars, diverse aromatic and aliphatic thioesters, and complex drug-derived electrophiles. Importantly, thioesters derived from alkyl carboxylic acids bearing multiple stereocenters participate robustly, delivering higher-carbon sugar precursors in a fully stereoretentive manner, independent of substrate stereochemistry. A further advantage of this method is its applicability to both α- and β-anomers, while uniquely enabling a stereoretentive D-to-L transformation to L-sugar precursors through a revised acylation pathway that achieves formal C5 epimerization.
Results and discussion
Reaction development
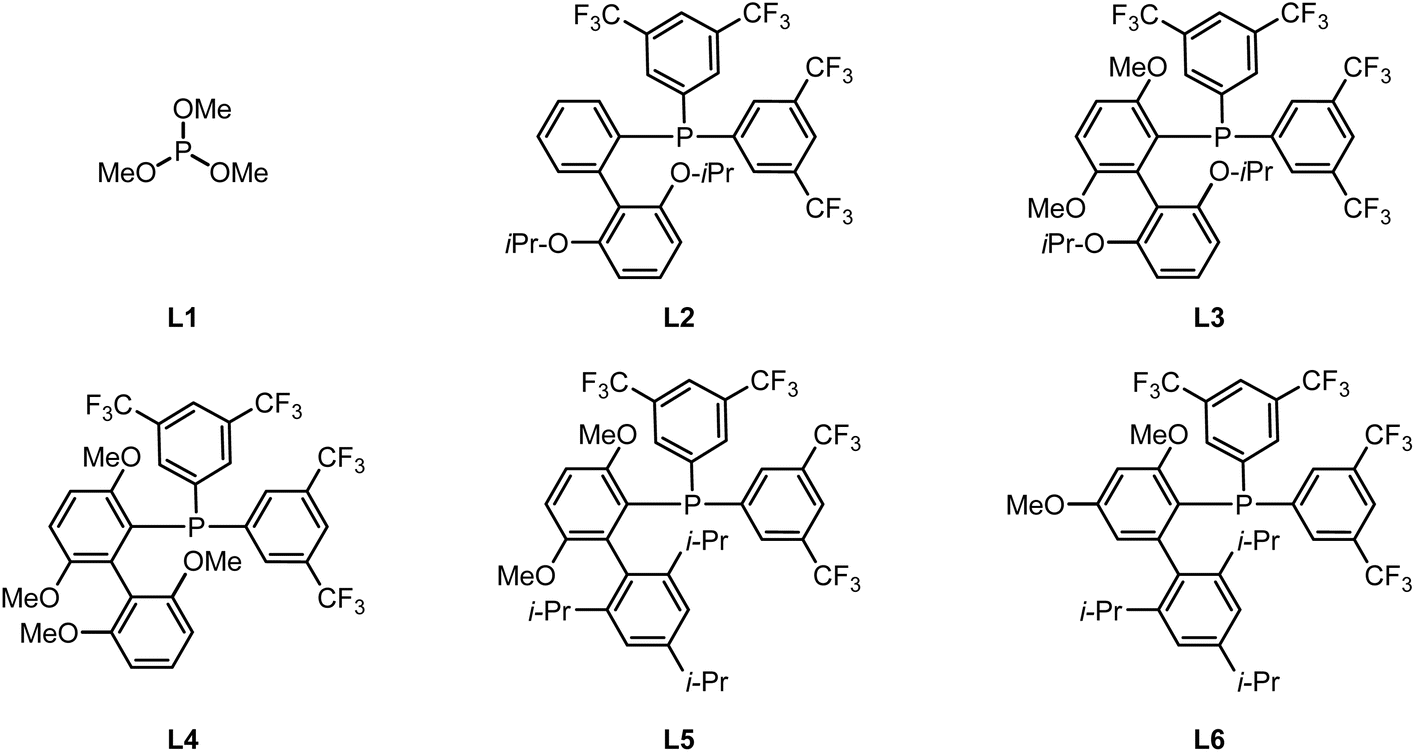
Building on our previous studies on the modular synthesis of reversed C-aryl glycosides,29 we conveniently obtained a series of reversed glycosyl stannanes 9a for further exploration. Our next objective was to identify optimal conditions for their stereoretentive non-classical acylation (Table 1). As a model, we examined the coupling between reversed glycosyl stannane 9a and benzoyl S-phenyl thioester 10a under the catalytic system of Pd2(dba)3/P(OMe)3 (L1).15 Although the desired reversed C-acyl glycoside 12a was obtained, the NMR yield was modest (32%, entry 1). To enhance the reaction efficiency, a series of phosphine ligands were evaluated. Employing a bulky, electron-deficient ligand L2 significantly improved the yield (entry 2). Subsequent fine-tuning of the ligand structure (entries 3–5) revealed that JackiePhos (L5), featuring greater steric bulk, afforded the best result (entry 5). Further structural modification of JackiePhos led to the development of ligand L6, which provided an even higher yield (86%, entry 8).The ligand primarily affects reaction efficiency rather than stereochemical outcome. Consistent with prior studies on Pd-catalyzed Stille couplings using electron-deficient phosphine ligands,29,30 the ligand likely facilitates catalyst activation and promotes transmetalation by maintaining an electron-deficient palladium center. In contrast, lowering the reaction temperature or shortening the reaction time both resulted in decreased efficiency (entries 6 and 7). Notably, reducing the Pd loading to 5 mol% or decreasing the amount of copper salt to 3.0 equiv did not compromise the yield (entries 9 and 10). Solvent screening indicated that MeCN or DMF completely suppressed the reaction (entries 11 and 12), while toluene and THF afforded slightly diminished yields (entries 13 and 14). Ultimately, employing 1.0 equiv of CuCl delivered the desired reversed C-acyl glycoside in 85% NMR yield and 80% isolated yield. Replacement of CuCl with CuBr reduced the NMR yield to 60%, whereas Cu I led to a further decrease to 9%. No cross-coupling occurred in the absence of CuCl (see SI for details). These observations are consistent with prior reports that copper salts facilitate transmetalation from organostannanes to palladium by activating the otherwise inert Sn–C bond.31 In addition to thioesters, other commonly used acyl sources were evaluated, including acyl chlorides and acid anhydrides. In these cases, the coupling efficiency decreased significantly, affording the corresponding products in NMR yields of 42% and 67%, respectively (see SI for details). Importantly, no evidence of configurational inversion at the C5 of the nonclassical glycosyl stannane was observed (see SI for details). Across all conditions examined, exclusive formation of the L- and β-configured sugar was observed.|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Solvent | CuCl (equiv) | NMR yield (%) |
| a Genernal reaction conditions: thioester 10a (0.10 mmol), reversed anomeric stannane 9a (2.0 equiv), Pd2(dba)3 (7.5 mol%), ligand (30.0 mol%), CuCl (4.0 equiv), and anhydrous 1,4-dioxane (2.0 mL) under Ar, 90 °C, 72 h; NMR yields are reported, and values in parentheses refer to isolated yields. b 70 °C was used. c 24 h was used. d Pd2(dba)3 (2.5 mol%) and ligand (10.0 mol%) were used. The stereochemical outcome was determined by 1H NMR analysis of crude reaction mixture. | ||||
| 1 | L1 | 1,4-Dioxane | 4.0 | 32% |
| 2 | L2 | 1,4-Dioxane | 4.0 | 63% |
| 3 | L3 | 1,4-Dioxane | 4.0 | 71% |
| 4 | L4 | 1,4-Dioxane | 4.0 | 72% |
| 5 | L5 | 1,4-Dioxane | 4.0 | 82% |
| 6b | L5 | 1,4-Dioxane | 4.0 | 63% |
| 7b,c | L5 | 1,4-Dioxane | 4.0 | 53% |
| 8 | L6 | 1,4-Dioxane | 4.0 | 86% |
| 9d | L6 | 1,4-Dioxane | 4.0 | 86% |
| 10d | L6 | 1,4-Dioxane | 3.0 | 86% |
| 11d | L6 | MeCN | 3.0 | 2% |
| 12d | L6 | DMF | 3.0 | 2% |
| 13d | L6 | Toluene | 3.0 | 81% |
| 14d | L6 | THF | 3.0 | 81% |
| 15d | L6 | 1,4-Dioxane | 1.0 | 85% (80%) |
|
||||

Scope and application
 | ||
| Scheme 1 Scope of thioesters derived from both C(sp2)- and C(sp3)-carboxylic acidsa. aStandard reaction conditions: thioesters 14 (0.10 mmol), reversed anomeric stannane 9a (2.0 equiv), Pd2(dba)3 (2.5 mol%), ligand L6 (10.0 mol%), CuCl (1.0 equiv), anhydrous 1,4-dioxane (2.0 mL), 90 °C, 72 h, Ar, isolated yields. | ||
 | ||
| Scheme 2 Scope of reversed glycosyl stannanesa. aStandard reaction conditions: thioesters 10b (0.10 mmol), reversed anomeric stannane 9 (2.0 equiv), Pd2(dba)3 (2.5 mol%), ligand L6 (10.0 mol%), CuCl (1.0 equiv), and anhydrous 1,4-dioxane (2.0 mL), 90 °C, 72 h, Ar, isolated yields. | ||
 | ||
| Scheme 3 Synthesis of higher-carbon sugar derivatives and downstream transformation of reversed C-acyl alycosidesa. aStandard reaction conditions: thioesters (0.10 mmol), reversed anomeric stannane (2.0 equiv), Pd2(dba)3 (2.5 mol%), ligand L6 (10.0 mol%), CuCl (1.0 equiv), and anhydrous 1,4-dioxane (2.0 mL), 90 °C, 72 h, Ar, isolated yields. | ||
 | ||
| Scheme 4 Sythesis of L-Sugars via a D to L strategy and access to C-ferrocenecarbonyl glycosides. | ||
C-Ferrocenyl glycosides are known to display a broad spectrum of biological activities, including anticancer, antifucosidase, and antimicrobial effects, as exemplified by compound 38.36 However, C-ferrocenecarbonyl glycosides have not yet been explored, despite their potential as versatile precursors for the synthesis of other C-linked C-ferrocenyl glycosides. To address this gap, we envisioned that the reversed acyl C-glycosylation platform could be employed to construct these novel ferrocenyl glycosides in a stereocontrolled manner. As illustrated in Scheme 4B, L- and D-type reversed anomeric stannanes were reacted with ferrocenecarbonyl thioester 36 under the optimized conditions, efficiently delivering the corresponding C-ferrocenecarbonyl glycosides 39–42 in good yields and with complete stereochemical fidelity. This strategy not only provides direct access to previously unexplored C-ferrocenecarbonyl glycosides but also establishes a general route for the preparation of diverse C-linked ferrocenyl sugar derivatives.
Conclusions
In summary, we have developed a general and stereoretentive reversed acyl C-glycosylation of nonclassical anomeric nucleophiles with thioesters. This method enables a stereoretentive transfer of configuration from configurationally stable reversed glycosyl stannanes, allowing efficient access to both D- and L-type reversed C-acyl glycosides with complete stereochemical fidelity. The broad substrate scope encompasses C(sp2)- and C(sp3)-derived thioesters as well as a variety of protected and unprotected glycosyl stannanes, highlighting the robustness and functional group tolerance of the transformation. Furthermore, the resulting reversed C-acyl glycosides serve as versatile chiral synthons for the synthesis of higher-carbon sugars, L-sugar analogues viaD-to-L interconversion, and C-ferrocenecarbonyl glycosides, providing a practical route to both natural and designer sugar frameworks. Taken together, the reversed acyl C-glycosylation offers an unprecedented combination of stereocontrol, efficiency, and synthetic flexibility, establishing a general platform for the preparation and diversification of structurally defined nonclassical glycosides. Applications in glycodiversification, medicinal chemistry, and the synthesis of complex architectures are currently underway.Author contributions
F. Z. conceived and directed the project. G. C. performed the experiments and analyzed the data. F. Z. and B. Y. wrote the manuscript with input from all the authors. All authors have read and approved the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc08224b.Acknowledgements
This work was sponsored by the National Key Research & Development Program of China (Grant No. 2023YFA1508800), the National Natural Science Foundation of China (Grant No. 22301178, 22301180, and 22577072), the Fundamental Research Funds for the Central Universities (Grant No. 25X010202131), Shanghai Municipal Science and Technology Major Project, the Open Grant from the Pingyuan Laboratory (2023PY-OP-0102), the Open Fund from the State Key Laboratory of Antiviral Drugs (Grant No. SKLAD-2024-0103 to B. Y.), the Foundation of National Facility for Translational Medicine (Shanghai).Notes and references
- C. R. Bertozzi and L. L. Kiessling, Chemical Glycobiology, Science, 2001, 291, 2357–2364 Search PubMed.
- K. Thaçi and R. M. Anthony, The importance of IgG N-glycosylation in Health, Disease, and Neonatal hemochromatosis, Glycosci. Ther., 2025, 1, 100002 Search PubMed.
- T. J. Boltje, T. Buskas and G.-J. Boons, Opportunities and Challenges in Synthetic Oligosaccharide and Glycoconjugate Research, Nat. Chem., 2009, 1, 611–622 CrossRef CAS PubMed.
- A. Varki, Biological roles of glycans, Glycobiology, 2017, 27, 3–49 CrossRef CAS PubMed.
- L. Cipolla, L. Gabrielli, D. Bini, L. Russo and N. Shaikh, Kdo: a Critical Monosaccharide for Bacteria Viability, Nat. Prod. Rep., 2010, 27, 1618–1629 RSC.
- T. Angata and A. Varki, Chemical Diversity in the Sialic Acids and Related α-Keto Acids: An Evolutionary Perspective, Chem. Rev., 2002, 102, 439–470 CrossRef CAS PubMed.
- A. Zamyatina, S. Gronow, M. Puchberger, A. Graziani, A. Hofinger and P. Kosma, Efficient Chemical Synthesis of both Anomers of ADP L-glycero- and D-glycero-D-manno-heptopyranose, Carbohydr. Res., 2003, 338, 2571–2589 CrossRef CAS PubMed.
- R. M. de Leder Kremer and C. Gallo-Rodriguez, Naturally Occurring Monosaccharides: Properties and Synthesis, Adv. Carbohydr. Chem. Biochem., 2004, 59, 9–67 CrossRef CAS PubMed.
- T. G. Frihed, M. Bols and C. M. Pedersen, Synthesis of l-Hexoses, Chem. Rev., 2015, 115, 3615–3676 CrossRef CAS PubMed.
- M. Wenninger, M. L. Poulsen, M. E. Larsen and N. M. Hauser, Recent Progress in the Synthesis and Glycosylation of Rare Sugars, Synthesis, 2024, 57, 1265–1279 Search PubMed.
- T. G. Frihed, C. M. Pedersen and M. Bols, Synthesis of All Eight L-Glycopyranosyl Donors Using C-H Activation, Angew. Chem., Int. Ed., 2014, 53, 13889–13893 Search PubMed.
- Y. Wang, H. M. Carder and A. E. Wendlandt, Synthesis of Rare Sugar Isomers through Site-Selective Epimerization, Nature, 2020, 578, 403–408 CrossRef CAS PubMed.
- S. O. Badir, A. Dumoulin, J. K. Matsui and G. A. Molander, Synthesis of Reversed C-Acyl Glycosides through Ni/Photoredox Dual Catalysis, Angew. Chem., Int. Ed., 2018, 57, 6610–6613 CrossRef CAS PubMed.
- M. Escolano, M. J. Cabrera-Afonso, M. Ribagorda, S. O. Badir and G. A. Molander, Nickel-Mediated Synthesis of Non-Anomeric C-Acyl Glycosides through Electron Donor-Acceptor Complex Photoactivation, J. Org. Chem., 2022, 87, 4981–4990 CrossRef CAS PubMed.
- F. Zhu, J. Rodriguez, S. O'Neill and M. A. Walczak, Acyl Glycosides through Stereospecific Glycosyl Cross-Coupling: Rapid Access to C(sp3)-Linked Glycomimetics, ACS Cent. Sci., 2018, 4, 1652–1662 CrossRef CAS PubMed.
- L.-N. Wang, Y.-H. Niu, Q.-H. Cai, Q. Li and X.-S. Ye, A General Approach to C-Acyl Glycosides via Palladium/Copper Co-catalyzed Coupling Reaction of Glycosyl Carbothioates and Arylboronic acids, Tetrahedron, 2021, 82, 131955 CrossRef CAS.
- L.-J. Zou, Q. Pan, C.-Y. Li, Z.-T. Zhang, X.-W. Zhang and X.-G. Hu, Cyanide-Free Synthesis of Glycosyl Carboxylic Acids and Application for the Synthesis of Scleropentaside A, Org. Lett., 2020, 22, 8302–8306 CrossRef CAS PubMed.
- E. Lindbäck, Y. Zhou, L. Marinescu, C. M. Pedersen and M. Bols, The Grignard Reaction of Cyclodextrin-6-aldehydes Revisited: A Study of the Stereoselectivity Upon Addition of Organometallic Reagents to Aldehydes and Ketones, Eur. J. Org Chem., 2010, 2010, 3883–3896 CrossRef.
- S. Chakraborty and D. Mal, A Representative Synthetic Route for C5 Angucycline Glycosides: Studies Directed toward the Total Synthesis of Mayamycin, J. Org. Chem., 2018, 83, 1328–1339 CrossRef CAS PubMed.
- S. Jarosz, S. Skóra, A. Stefanowicz, M. Mach and J. Frelek, Application of Sugar Phosphonates for the Preparation of Higher Carbon Monosaccharides, J. Carbohydr. Chem., 1999, 18, 961–974 CrossRef CAS.
- M. A. Potopnyk, P. Cmoch, M. Cieplak, A. Gajewska and S. Jarosz, The Synthesis of Higher Carbon Sugars: a Study on the Rearrangement of Higher Sugar Allylic Alcohols, Tetrahedron: Asymmetry, 2011, 22, 780–786 Search PubMed.
- M. G. Kulkarni, Y. B. Shaikh, A. S. Borhade, S. W. Chavhan, A. P. Dhondge, D. D. Gaikwad, M. P. Desai, D. R. Birhade and N. R. Dhatrak, Greening the Wacker process, Tetrahedron Lett., 2013, 54, 2293–2295 Search PubMed.
- Z.-T. Zhang, Y. Ma, N.-L. Fan and X.-G. Hu, Synthesis of (non-classical) C-Acyl-Glycosides via Liebeskind–Srogl Coupling: Scope, Limitation, Improved Synthesis and Antioxidant Activity of Scleropentaside A, Tetrahedron, 2021, 92, 132255 CrossRef CAS.
- L. Xu, Z. Hou, X. Ma, Y. Ma, J. Wang, X. Zhang, P. Wang and M. Li, Synthesis of C4-Acyl-tetrofuranosides and C5-Acyl-pentopyranosides Enabled by the Liebeskind-Srogl Cross-Coupling Reaction, Asian J. Org. Chem., 2022, 11, e202100665 CrossRef CAS.
- J.-H. Liu, Z.-Y. Tian, Z.-Y. Wu, T.-L. Huang, Z. Lin, L. Zhang, J. Chen, L. Hai, L. Guo and Y. Wu, Access to Ketones via Nickel-Catalyzed Coupling between S-2-Pyridyl Thioesters and Redox-Active Esters Using an Organic Reductant, J. Org. Chem., 2024, 89, 17059–17068 CrossRef CAS PubMed.
- L.-Y. Xu, N.-L. Fan and X.-G. Hu, Recent Development in the Synthesis of C-Glycosides involving Glycosyl Radicals, Org. Biomol. Chem., 2020, 18, 5095–5109 Search PubMed.
- A. Chen, B. Yang, Z. Zhou and F. Zhu, Recent Advances in Transition-Metal-Catalyzed Glycosyl Cross-Coupling Reactions, Chem Catal., 2022, 2, 3430–3470 CAS.
- A. Chen, L. Xu, Z. Zhou, S. Zhao, T. Yang and F. Zhu, Recent Advances in Glycosylation involving Novel Anomeric Radical Precursors, J. Carbohydr. Chem., 2021, 40, 361–400 Search PubMed.
- G. Cheng, B. Yang, Y. Han, W. Lin, S. Tao, Y. Nian, Y. Li, M. A. Walczak and F. Zhu, Pd-Catalyzed Stereospecific Glycosyl Cross-Coupling of Reversed Anomeric Stannanes for Modular Synthesis of Nonclassical C-Glycosides, Precis. Chem., 2024, 2, 587–599 CrossRef CAS PubMed.
- F. Zhu, J. Rodriguez, T. Y. Yang, I. Kevlishvili, E. Miller, D. Yi, S. O'Neill, M. J. Rourke, P. Liu and M. A. Walczak, Glycosyl Cross-Coupling of Anomeric Nucleophiles: Scope, Mechanism, and Applications in the Synthesis of Aryl C-glycosides, J. Am. Chem. Soc., 2017, 139, 17908–17922 Search PubMed.
- S. P. H. Mee, V. Lee and J. E. Baldwin, Stille Coupling Made Easier—The Synergic Effect of Copper(I) Salts and the Fluoride Ion, Angew. Chem., Int. Ed., 2004, 43, 1132–1136 Search PubMed.
- D. Li, L. Wang, J. Wang, P. Wang, N. Song, P. Chen and M. Li, Synthesis of Branched-Chain Sugars and Higher-Carbon Sugars Enabled by Site-selective C–H Alkylation Relying on 1,5-Hydrogen Atom Transfer of Ethylenoxy Radicals, Org. Chem. Front., 2024, 11, 1332–1340 Search PubMed.
- S. J. Danishefsky and M. P. DeNinno, Totally Synthetic Routes to the Higher Monosaccharides, Angew Chem. Int. Ed. Engl., 1987, 26, 15–23 CrossRef.
- O.-K. Anna, C. Maciej and J. Slawomir, A Review on the Stereoselective Synthesis of Higher Carbon Sugars with an Eye to Making Higher Alditols, Curr. Org. Chem., 2014, 18, 327–340 Search PubMed.
- L. Alberch, G. Cheng, S. K. Seo, X. Li, F. P. Boulineau and A. Wei, Stereoelectronic Factors in the Stereoselective Epoxidation of Glycals and 4-Deoxypentenosides, J. Org. Chem., 2011, 76, 2532–2547 Search PubMed.
- M. M. Abd-Elzaher and I. A. I. Ali, Preparation, Characterization and Biological Studies of some Novel Ferrocenyl Compounds, Appl. Organomet. Chem., 2006, 20, 107–111 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |