
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anion-based electrolyte chemistry for sodium-ion batteries: fundamentals, advances and perspectives
Shu-Yu
Li†
a,
Yong-Li
Heng†
b,
Zhen-Yi
Gu
*b,
Xiao-Tong
Wang
b,
Yan
Liu
b,
Xin-Ru
Zhang
b,
Zhong-Hui
Sun
*b,
Dai-Huo
Liu
 c,
Bao
Li
c and
Xing-Long
Wu
*ab
c,
Bao
Li
c and
Xing-Long
Wu
*ab
aDepartment of Chemistry, Northeast Normal University, Changchun, Jilin 130024, P. R. China. E-mail: xinglong@nenu.edu.cn
bState Key Laboratory of Integrated Optoelectronics, MOE Key Laboratory for UV Light-Emitting Materials and Technology, Northeast Normal University, Changchun, Jilin 130024, P. R. China. E-mail: xinglong@nenu.edu.cn; guzy166@nenu.edu.cn; cczhsun@gzhu.edu.cn
cCollaborative Innovation Center of Henan Province for Green Manufacturing of Fine Chemicals, School of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang, Henan 453007, P. R. China
First published on 9th December 2025
Abstract
The scarcity of lithium resources and cost concerns have driven the development of sodium-ion batteries (SIBs), which utilize more abundant and lower-cost resources, as a promising alternative energy storage technology. As a key component of batteries, the electrolyte directly influences their energy density, rate capability, cycle life, and safety. However, conventional electrolytes face limitations such as poor high-voltage compatibility, inferior low-temperature performance, and unstable electrode–electrolyte interfaces, which severely hinder the commercialization of SIBs. In recent years, anion-regulated electrolytes have emerged as a research focus due to their unique anion-dominated solvation structures, demonstrating significant potential in enhancing interfacial stability, facilitating ion transport, improving high- and low-temperature performance, and strengthening safety. This review systematically elucidates the fundamental chemical characteristics and solvation structure design of anion-based electrolytes for SIBs, summarizes the influence of the mechanisms of different anion types on electrochemical performance, and explores the expanded application of anions in new systems. Finally, it outlines future directions and challenges in the field, providing valuable insights for the rational design of high-performance SIB electrolytes.
1 Introduction
The explosive growth of the new energy industry has increasingly highlighted the scarcity and supply–demand imbalance of lithium resources. Global lithium reserves are relatively limited and highly concentrated geographically, which not only drives up raw material costs but also poses potential constraints on large-scale applications, emerging as a critical bottleneck hindering the sustainable development of the industry.1–5 Under such circumstances, the search for novel energy storage systems that combine resource abundance with performance advantages has become an inevitable direction for industrial development. Driven by this demand, sodium-ion batteries (SIBs) have emerged as an ideal alternative. Compared to lithium, sodium resources are vastly more abundant and widely distributed in oceans, salt lakes, and underground rock formations, offering inherent advantages such as broad availability and ease of access. More importantly, SIBs share a highly similar working principle with lithium batteries—both operate based on a “rocking-chair” intercalation/deintercalation mechanism for charge storage and release. This fundamental similarity enables significant technological inheritance in areas such as electrode material design and battery architecture, laying a solid foundation for the rapid iteration and industrial adoption of sodium battery technology.6–11 It is noteworthy that the electrolyte, often regarded as the “blood” of a battery, serves as a central component in the battery system, fulfilling multiple critical functions including ion transport, interface formation, and safety assurance. Its physicochemical properties directly govern key performance metrics of the battery, such as energy density, fast-charging capability, cycling stability, and operational safety.12–15 Given this context, research on electrolytes for SIBs is of great significance for promoting the maturation and application of sodium battery technology.Currently, the development of electrolytes for SIBs still faces several critical challenges that hinder their large-scale commercial application, as illustrated in Fig. 1. Firstly, insufficient high-voltage compatibility restricts the utilization of high-capacity cathode materials, thereby limiting further improvement in energy density and impairing long-term cycling stability.16–18 Secondly, the unstable electrode–electrolyte interphase (EEI) tends to trigger continuous side reactions, consuming active Na+ and reducing both coulombic efficiency (CE) and reversible capacity, while also compromising battery safety and cycle life.19–21 Moreover, the performance across a wide temperature range is poor. At low temperatures, slow ion transport and high desolvation barriers lead to a sharp performance decline,22–25 while at elevated temperatures, severe interfacial side reactions and facile electrolyte decomposition occur.26,27 Conventional electrolyte systems, limited by the intrinsic properties of their solvent–salt combinations, struggle to systematically address these issues. Therefore, designing novel electrolyte systems has become a crucial step in advancing SIB technology.
 | ||
| Fig. 1 Challenges for SIB electrolytes. | ||
In recent years, anion-regulated electrolytes have garnered significant attention from both academia and industry as an emerging strategy. By facilitating an anion-dominated solvation structure, these electrolytes effectively modulate ion transport and interfacial chemistry, directly addressing several bottlenecks in current battery technologies. Specifically, anion-regulated electrolytes demonstrate notable advantages in the following aspects: enhancing EEI stability and suppressing side reactions;28,29 facilitating ion transport and accelerating desolvation, thereby improving low-temperature and fast-charging performance;30,31 increasing oxidation resistance to widen the electrochemical stability window, enabling compatibility with high-voltage cathodes;32–34 and contributing to the formation of a more robust solid electrolyte interphase (SEI), which enhances the overall safety of the battery.35,36 Furthermore, this kind of electrolyte provides the possibility for the development of new battery systems (e.g., dual-ion batteries and anode-free batteries), expanding the application scenarios of SIBs.37,38
Against this background, this paper systematically reviews the research status and development trends of anion-regulated electrolytes. It elucidates their basic structure and solvation chemistry, summarizes key recent research advances in SIBs, and discusses the design principles for different anion types and their coordination environments. This work aims to provide a theoretical foundation and a materials perspective for understanding and designing high-performance anion electrolytes. Finally, the article also outlines future research directions and technical challenges in this field, with the goal of informing the design and application of electrolytes for SIBs.
2 Characteristics of anion solvation structures
Anion-based electrolytes, as an emerging electrolyte system, have become one of the key research directions in the field of SIBs in recent years. By strategically modulating the structure and chemical properties of anions, such electrolytes are expected to address key bottlenecks in the performance and interfacial behavior of conventional SIBs. Unlike traditional solvation structures that rely primarily on “cation–solvent” interactions, anion-dominated solvation structures emphasize the regulation of anion–solvent–cation interactions, thereby constructing an electrolyte environment better aligned with functional requirements.39 In such structures, anions extensively enter the first solvation shell of sodium ions, forming contact ion pairs (CIPs) or even aggregates (AGGs).38,39 In contrast to the solvent-separated ion pair (SSIP) configuration, where anions are largely excluded, the aggregated configurations (CIPs and AGGs) effectively suppress the decomposition of solvent molecules and facilitate the formation of an anion-derived SEI or cathode electrolyte interphase (CEI). These interphases typically exhibit higher stability, compactness, and abundant inorganic components, thereby significantly enhancing electrode interfacial stability and ion transport efficiency.40–42 Moreover, conventional cation-dominated solvation structures are governed by strong cation–dipole interactions, which tend to increase the desolvation energy barrier of Na+ at low temperatures, resulting in sluggish kinetics and promoting the formation of organic-rich, less stable SEI/CEI layers—factors that severely restrict the low-temperature performance of SIBs.43,44 In contrast, anion-involved solvation structures significantly reduce the desolvation energy barrier and improve the interfacial reaction kinetics. They promote the formation of thinner, more uniform, and inorganic-rich SEI/CEI layers. As a result, such structures enable efficient Na+ transport and stable cycling over a wide temperature range, particularly under low-temperature conditions.45–47Hence, the anion plays a crucial regulatory role in several key aspects, including the optimization of the solvation structure, enhancement of electrochemical performance, improvement of interfacial stability and kinetics, and expansion of applications for novel batteries (schematic diagram shown in Fig. 2). The role of anions in solvation structures has evolved from that of a passive charge carrier to a critical regulating factor—a paradigm shift from a “supporting role” to a “leading role”. This transformation provides a novel design strategy for developing next-generation high-performance sodium battery electrolytes.
 | ||
| Fig. 2 Schematic diagram of the anion regulation effect. | ||
3 Involvement of solute anions in the solvation structure
3.1 Single anion
 | ||
| Fig. 3 (a) Schematics of solvation structures and their merits of the three electrolytes. (b) Rate capability of a PB‖HC cell. (c) Long-term cycling performance of a PB‖HC cylindrical cell.50 Copyright 2024 Wiley-VCH GmbH. (d) Ratio of different coordinated structures. (e) Long cycling performance in N-mixTHF at different temperatures (100 mA g−1 beyond −20 °C, 50 mA g−1 at −40 °C, and −60 °C).43 Copyright 2024 Wiley-VCH GmbH. (f) Cycling performance of the HC‖NFPP pouch cell at 1C.53 Copyright 2024 American Chemical Society. (g) Anion-induced uniform and robust CEI for layered metal oxide cathodes of SIBs.54 Copyright 2024 American Chemical Society. (h) Dissociation energy of various Na salts. (i) Tafel curves of TDF and TDF electrolyte. Copyright 2025 Elsevier. | ||
Furthermore, Li et al. employed methyl butyrate, a weakly coordinating carboxylate co-solvent, to modulate ion–dipole interactions within a fluorine-free ester-based electrolyte, thereby promoting an anion-enhanced solvation structure.54 With more ClO4− occupying the Na+ solvation sheath, the Na+ desolvation energy is significantly reduced, accelerating interfacial kinetics. Furthermore, robust anion-rich inorganic EEIs form on both the cathode and anode, effectively suppressing EEI dissolution and enhancing interfacial stability. A PB‖HC full cell achieved stable operation over a wide temperature range from −20 to 100 °C. This study demonstrates the potential of fluorine-free ester-based electrolytes with anion-enhanced solvation chemistry for advanced wide-temperature SIBs.
 | ||
| Fig. 4 HRTEM images of NVPF cathodes after 100 cycles (a) in TDF electrolyte and (b) TF electrolyte. (c) Cycling performance of the Na‖NVPF coin cell using TF and TDF electrolytes cycled at 20C.56 Copyright 2025 Elsevier. (d) Cycling performance of Na‖NVP batteries at 1C.58 Copyright 2025 American Chemical Society. (e) Na‖Na symmetric cells with LHCE-TTE and MIX-CPME at a current density of 0.5 mA cm−2 and an areal capacity of 0.5 mA h cm−2.59 Copyright 2025 Wiley-VCH GmbH. (f) Schematic diagram of representative Na+ solvation structures in NaTFSI-DMS and NaTFSI-DG, respectively. (g) The cycling performance of NFM‖HC full cells at 45 °C and −20 °C.62 Copyright 2025 American Chemical Society. | ||
 | ||
| Fig. 5 (a) Radius comparison of anion and solvated Na+ ions. (b) Binding energy of different anions. (c) In situ EIS at different potentials based on the optimized electrolyte. (d) Charge–discharge curves the of HC‖Na cell from −40 to 60 °C. (e) TOF− SIMS 3D images of the OTF−-derived SEI film.64 Copyright 2025 Wiley-VCH GmbH. (f) Na+ binding energy tabulation of Na salts. (g) Cycling data of Na-LTO‖C–Al foil configuration.65 Copyright 2025 Elsevier. (h) Voltage versus time profile of the Na‖Na symmetric cell at a current rate of 0.5 mA cm−2 (1 mA h cm−2) (inset showing the zoomed potential profiles from 30 to 60 h and 300 to 330 h range). (i) Linear sweep voltammetry curves (scan rate – 0.1 mV s−1 up to 4.5 V).66 Copyright 2024 Wiley-VCH GmbH. | ||
Ban et al. designed a low-cost, fluorine-free sodium salt (sodium tetraphenoxyborate, chemical formula NaBO4C24H20, abbreviated as NaBOPh). This fluorine-free borate-based electrolyte has enabled breakthroughs in low-cost, high-performance sodium batteries, offering a new direction for sustainable energy storage technologies.65 By calculating the salt pairs and ion free energies, Na+ ions exhibit the strongest binding energy with this salt (Fig. 5f), indicating that the NaBOPh–diethylene glycol dimethyl ether (DG) electrolyte exhibits stronger Na+–anion pairing compared to NaPF6–DG or NaBPh–DG electrolytes, resulting in weakened Na+–DG interactions. Furthermore, the tighter binding geometry of cations in NaBOPh restricts the number of DG molecules available for coordination with Na+ ions, while the lack of fluorine in this salt structure contributes to more robust Na+–anion pairs. The distinctive molecular structure of NaBOPh leads to increased anion participation in the solvation sheath and reduced solvent coordination, thereby inducing the anion-rich interfacial chemistry observed in the borate-dominated SEI. These effects promote smoother and more uniform Na electrodeposition morphologies. The fluorine-free interphase demonstrates improved cycling performance across multiple battery configurations, including SIBs, SMBs, and anode-free sodium metal batteries (Fig. 5g).
Mitra et al. proposed a new electrolyte system based on a highly fluorinated borate anion ([B(hfip)4]−), consisting of 1 M Na[B(hfip)4]·3DME/EC:EMC. This design demonstrates remarkable performance enhancement in high-voltage SMBs compared to conventional NaPF6-based electrolytes.66 The Na[B(hfip)4]·3DME electrolyte system exhibited a consistently flat voltage profile for over 350 hours. In contrast, the NaPF6-based electrolyte showed a fluctuating voltage profile, with sodium stripping/plating lasting only 110 hours before a sudden voltage increase due to cell short-circuit (Fig. 5h). This suggests the formation of smooth, uniform, and non-porous Na metal deposition in the designed electrolyte system. Linear sweep voltammetry was conducted for Na‖Al cells to evaluate anodic stability. The 1 M NaPF6 EC:EMC electrolyte exhibited a sharp current increase beyond 4.0 V, indicating poor anodic stability of the [PF6]−-based system. In contrast, the [B(hfip)4]−-based system remained highly stable up to 4.5 V, with no significant current surge even after 4.0 V (Fig. 5i), demonstrating effective passivation of the Al current collector and suppression of further electrolyte oxidation.
To assess practical feasibility, the electrolyte was paired with sodium full-cell configurations. Excellent cycling stability and reversible capacity were achieved with high-voltage cathodes (NVPF@C@CNT and NFMO), along with significantly reduced transition metal dissolution. Analysis of the radial distribution function (RDF) in the ab initio molecular dynamics trajectories for Na–O in NaPF6 and Na[B(hfip)4]·3DME (Fig. 6a) reveals highly similar peak positions at 2.29 Å and 2.39 Å, respectively, representing the Na+–solvent distances within the primary solvation shell. A more pronounced difference appeared in the second solvation shell, with Na–O RDF peaks at 4.39 Å for NaPF6 and 4.57 Å for Na[B(hfip)4]·3DME, suggesting a less dense secondary solvation structure in the latter. This implies that Na+ may undergo easier desolvation and diffuse more readily to the electrode surface in this fluorinated borate-based carbonate electrolyte system. This study highlights the potential of weakly coordinating anions with similar properties for advanced electrolyte design.
 | ||
| Fig. 6 (a) Radial distribution functions of Na–O in Na[PF6] and Na[B(hfip)4]·3DME electrolytes.66 Copyright 2024 Wiley-VCH GmbH. (b) The representative solvation structures. (c) The fitted Arrhenius plots of 1 M TMP, MT and MTD, and the error bars represent the standard deviation of triplicates.73 Copyright 2024 Wiley-VCH GmbH. (d and e) CN of MUL electrolyte and STD electrolyte. (f) GCD profiles over multiple cycles in the voltage range of 1.5–4.15 V.69 Copyright 2025 Elsevier. (g) ESP simulation of SPFO–solvent complexes. (h) SFG signals of DEGDME between NVP and electrolytes.93 Copyright 2024 American Chemical Society. | ||
These novel sodium salts are often designed by modifying the existing anions, with the core advantage of meeting the requirements of specific systems through tailored anions. For instance, introducing conjugated groups like benzene rings may alter the charge distribution of anions and affect frontier orbital energy levels, potentially conferring specific interfacial functions. However, the incorporation of bulky groups might limit solubility and ionic conductivity in conventional solvents, while also complicating synthesis. Consequently, these salts can be utilized to form stable interfacial films through their distinctive products or as sacrificial agents to improve electrolyte tolerance under extreme conditions.
3.2 Synergy of multi-anion systems
Based on our previous discussion of solvation structures governed by single anions, we now extend the scope to multi-anion systems. Compared with single-anion systems, multi-anion cooperation enables a more stable solvation structure by modulating the coordination behavior of different anions67,68 and introducing competitive coordination between them.63,69 Multi-salt formulations inherently facilitate the formation of heterogeneous interphases, enhancing structural and chemical stability, and thereby improving overall battery performance.70–72 The multi-anion strategy better reflects the concept of “proactively designed” solvation structures.Wang et al. designed a unique dual-anion-aggregated Na+ solvation structure by employing cost-effective NaPF6 and NaDFOB(sodium difluoro(oxalato)borate) salts dissolved in trimethyl phosphate (TMP) and 1,1,2,2-tetrafluoroethyl-2,2,3,3-tetrafluoropropyl ether (TTE).73 This unique aggregate preferentially decomposes on the cathode, constructing a gradient CEI rich in phosphides and borides. Based on the trajectory data obtained from molecular dynamics (MD) simulations, the predominant solvation structures in the electrolyte were directly extracted (Fig. 6b). Among these, [Na–TMP6]+, [Na–TMP4]+[PF6]−, and [Na–TMP2]+[PF6]2− represent typical solvation configurations corresponding to the SSIP, CIP, and AGG, respectively. The simulation results reveal the formation of a highly aggregated dual-anion structure, [Na–TMP]+[PF6]2−[DFOB]−, in the target electrolyte (MT–D), demonstrating the participation of DFOB− in the solvation sheath and its coordination with Na+. Variations in Na+ solvation were further corroborated by the kinetic behavior across different electrolyte systems. The activation energy (Ea) for (de)solvation, calculated using the Arrhenius equation (Fig. 6c), indicates that the MT–D electrolyte effectively reduces the energy barrier for the (de)solvation process, thereby facilitating rapid Na+ transport at the interface. This phenomenon is attributed to the anion-rich solvation structure formed in MT–D, which promotes the incorporation of anions into the solvation sheath. The involvement of anions reduces the energy required for complete dissociation of sodium salts, leading to a lower binding energy within the solvation structure. This accelerates desolvation kinetics and, in turn, enables rapid decomposition during cycling.
Zhang et al. proposed a simple strategy of anion competitive coordination for designing weakly solvating electrolytes. By introducing complex salts with high Na+-anion binding energy, abundant anions are retained within the solvation sheath, thereby optimizing both the Na+ solvation structure and the CE.69 In order to construct a more stable inorganic CEI, the anion competitive coordination weakly solvated electrolyte (MUL) containing 0.8 M NaPF6, 0.1 M NaTFSI, 0.05 M NaDFOB and 0.05 M NaDFP (sodium difluorophosphate) was prepared by adding composite salts into PC. For comparison, 1 M NaPF6 solution in PC was also prepared and analyzed, which was designated as STD. The selection of these salts was guided by their calculated Na+–anion binding energies, which follow the trend: NaDFP > NaDFOB > NaTFSI > NaPF6. Classical MD simulations were employed to investigate differences in the solvation structures between the STD and MUL electrolytes. Compared with STD electrolyte, the MUL system exhibits stronger coordination between anions and Na+, accompanied by a reduced coordination number of Na+–PC in the first solvation shell (Fig. 6d and e). The enhanced Na+–anion interaction in MUL electrolyte reduces the binding affinity between Na+ and solvent molecules, enabling more anions (DFOB−, DFP−, and TFSI−) to displace solvent molecules within the solvation sheath. Furthermore, the strong Na+–anion interactions effectively restrict the migration of free solvent molecules to the electrode interface, thereby suppressing solvent decomposition and promoting the formation of an inorganic-rich CEI. Additionally, the weakened Na+–solvent interaction lowers the desolvation energy barrier for Na+, resulting in enhanced reaction kinetics. Pouch cells serve as an ideal platform for comprehensive evaluation of electrolyte performance. In a pouch full cell composed of an HC anode and NFM cathode, the cell employing the MUL electrolyte delivered a reversible capacity exceeding 1550 mA h g−1 over 100 cycles (Fig. 6f). In contrast, the cell using the STD electrolyte exhibited severe instability. This instability originates from the pronounced oxidative decomposition of PC in the NFM‖HC system with the STD electrolyte, which impedes stable cell operation. This strategy, by tailoring the interactions among Na+, anions, and solvent molecules, reinforces anion–solvent competition within the Na+ solvation sheath, offering an innovative pathway to enhance the high-voltage stability of electrolytes for SIBs.
Furthermore, Yu et al. developed a low-concentration dual-salt carbonate electrolyte for SMBs. The combination of NaFSI and NaOTF effectively modulates the solvation structure, reduces the desolvation energy barrier, and enhances the low-temperature kinetics of Na+ ions.74 This dual-salt system promotes the formation of an anion-dominated solvation structure and a robust inorganic-rich SEI layer. Such an innovative electrolyte design significantly improves the ion transport kinetics and extends the cycling lifespan under low-temperature conditions.
Synergistic effects among different sodium salts provide complementary and optimized performance. However, it is crucial to recognize that such complex electrolyte formulations inevitably increase computational demands during simulations. At present, solvation-related investigations primarily involve interactions between cations and anions, yet interactions among anions are also worth consideration, which will be a viable direction for future research.
4 Anion-based additives
Electrolyte additives, despite their low dosage, play a crucial role in determining the overall performance of electrolytes and batteries, representing one of the most cost-effective strategies for enhancing battery performance.75–78 The incorporation of small amounts of functional additives can selectively improve specific battery characteristics, such as protecting high-voltage cathodes,79,80 enabling high energy density,81,82 enhancing cycling performance,83,84 and improving safety.85,86 Reducing the amount of sodium salt and using it as an additive has become a common strategy.87–89 In this section, we will categorize recent studies on sodium salt anions used as additives based on the elements contained in the anions. Drawing from the previous discussion on primary salt anions, it is anticipated that anionic additives can lead to a qualitative leap in battery performance.4.1 Fluorine-containing anionic additives
Fluorine-containing additives have been extensively studied due to their ability to form effective passivation layers on both anodes and cathodes.90,91 Li et al. introduced trifluoroacetate (TFA−), an anion with ultra-high electron density, into a diglyme (G2)-based electrolyte as a co-solvent to enhance anion-derived solvation over a wide temperature range.92 Owing to its high donor number (34.0 kcal mol−1), TFA− exhibits a strong interaction with Na+, enabling it to effectively coordinate within the inner solvation shell and reduce the solvent coordination number. This strong cation–anion interaction facilitates the formation of an anion-dominated solvation structure and promotes the desolvation of Na+.21 The highly coordinated TFA− leads to more PF6− anions and fewer G2 molecules occupying the inner solvation shell of Na+, favoring the establishment of enhanced Na+-anion coordination. This unique solvation configuration induces the formation of an anion-derived, inorganic-rich, and robust SEI layer. As a result, the average mechanical strength of the SEI was significantly enhanced from 0.86 to 5.20 GPa, concurrently with improved stability and reversibility of Na plating/stripping.Li et al. proposed a self-assembled protective layer by applying a perfluorinated anion additive as a dual-chain electrolyte surfactant to enhance the oxidative stability of ether-based electrolytes.93 The perfluorinated anion additive, characterized by a lower HOMO–LUMO energy gap, exhibits high reactivity, indicating a greater propensity for decomposition. The preferential adsorption and oxidation of the additive suppress weak oxidation at low voltage, thereby widening the electrochemical window and potentially forming the CEI rich in C–F components and NaF. Furthermore, molecular electrostatic potential analysis revealed that sodium pentadecafluorooctanoate (SPFO) exhibits high electron density centered on the perfluorinated chain and a uniformly distributed charge profile (Fig. 6g), which is expected to interact with the positive electrostatic potential localized on the –C–H groups of DEGDME. When the electron-donating –C–F groups interact with –C–H groups, the fluorine atoms activate the –C–H bonds, leading to weak –C–F⋯H–C– interactions. These weak interactions, defined by Bergström et al. as pseudo-hydrogen bonds, enable the SPFO–solvent complex to form a well-compatible structure with homogeneous charge distribution. This is anticipated to suppress electrolyte volatility and enhance thermal stability. The evolution of adsorbed species was monitored using electrochemical in situ sum frequency generation (SFG) spectroscopy. Upon applying a 50 mV bias until steady state in two practical systems, the intensities of –CH2 (ν) at 1430 cm−1 and –CH3 (ν) at 1380 cm−1 from DEGDME decreased after the introduction of SPFO (Fig. 6h). This confirms that under an electric field, SPFO preferentially adsorbs at the interface and self-assembles into an ordered layer, effectively shielding the electrode from solvent contact. Moreover, the weak –C–F⋯H–C– pseudo-hydrogen bonds contribute to a significant improvement in the thermal stability of the electrolyte, with an operational tolerance up to 60 °C. In addition, Hagiwara et al. used NaDFP as a strategy for optimizing the performance of sodium-metal ion batteries.94 As previously discussed, NaDFP exhibits strong cation–anion interaction energy. In this study, the incorporation of 1 wt% NaDFP resulted in high-rate capability and long cycling life. The enhanced electrochemical performance of Na‖HC cells further indicates the beneficial role of NaDFP in facilitating the formation of a stable SEI layer, thereby suppressing overpotential and interfacial resistance.
The core function of fluorine-containing additives is to regulate interfacial chemistry and enhance the mechanical strength of interfacial films. However, an overabundance of inorganic fluorinated components may cause excessive interfacial thickness and increased impedance. Therefore, balancing organic and inorganic components to construct gradient interfacial layers with both high modulus and high toughness, represents the key scientific concern for fluorine-containing anionic additives.
4.2 Boron-containing anions as additives
Boron-containing additives represent a highly important and successful category of electrolyte additives. The difluoro(oxalato)borate (DFOB−) anion is the most common boron-containing anion, as it exhibits the lowest unoccupied molecular orbital energy level and the highest occupied molecular orbital energy level when coordinating with either Na+ or Li+. This electronic structure suggests that DFOB− can preferentially undergo reactions at both the cathode and anode, facilitating the formation of a stable electrode–electrolyte interface.95 Leveraging this preferential reactivity of DFOB− (Fig. 7a), Qiu et al. demonstrated that DFOB− rapidly forms a stable passivation layer on the electrode surface.96 A stable SEI rich in NaF and B is formed on the HC surface, which effectively alleviates the excessive reduction and decomposition of electrolyte in the initial charge process and inhibits the repeated dissolution and regeneration of an unstable SEI in the subsequent cycle. On the cathode side, preferential oxidation of NaDFOB enhances the CEI and ensures interfacial stability of the FeMn-based Prussian blue cathode. Furthermore, the pre-formed CEI, composed of sturdy NaF and boron-rich components, prevents undesirable oxidative decomposition of the electrolyte and dissolution of transition metals. The formed uniform and mechanically robust SEI and CEI collaboratively suppress the formation of detrimental sodium dendrites. Even at a low N/P ratio of 0.96, no dendrites were observed throughout the entire charging process (Fig. 7b). | ||
| Fig. 7 (a) HOMO and LUMO energy levels of NaPF6, DEGDME, NaDFOB, and DEGDME–Na+ ions. (b) In situ optical microscope observations of full cells with a N/P ratio of 0.96 at various voltages during the charging process with the NaDFOB additive.96 Copyright 2024 Wiley-VCH GmbH. (c) Comparison of XPS Na 1s spectra of the sodium metal anode in the Na‖Na symmetric cells and (d) B 1s spectra of the sodium metal in the electrolyte with NaDFOB after 10 cycles. (e) Long-term cycling performance of the HC‖NFM111 pouch cell at a charging rate of 0.5C and a discharging rate of 1C.98 Copyright 2025 Elsevier. (f) Comparative electrostatic potential mapping and structural formulae of the predominant solvation structures. (g) Schematic illustration of electrolyte environments and CEI transformations induced by the NaNO3 additive.102 Copyright 2024 Springer Nature. (h) Cycling stability of the NVP‖graphite battery with DCA2 and DCA0 at 0.2C.103 Copyright 2025 Wiley-VCH GmbH. | ||
Similarly, Xia et al. combined theoretical calculations and experimental evidence to show that the DFOB− anion exhibits a lower adsorption energy than other electrolyte components, leading to its preferential adsorption within the inner Helmholtz plane near both electrode surfaces. Introducing a small amount (0.5 wt%) of NaDFOB—a borate additive with electron-rich characteristics—into the electrolyte system enables the construction of a robust inorganic-rich EEI via an anion-preferential adsorption and decomposition mechanism, thereby effectively suppressing side reactions.97 As a result, the NaxNi0.25Fe0.25Mn0.5O2 cathode, HC anode, and the corresponding full cell achieve outstanding cycling performance. Liu et al. used NaDFOB as an additive in the Na+ solvation sheath in SMBs.98 This promotes anion aggregation via van der Waals interactions, leading to the formation of the SEI enriched with NaF and B2O3 (Fig. 7c and d), which significantly enhances the stability of both the SEI and CEI. This modification has been shown to effectively suppress transition metal leaching from the cathode and sodium dendrite growth on the anode. Further evaluation using pouch cells confirms the potential commercial applicability of NaDFOB as a functional electrolyte additive (Fig. 7e).
Beyond its established role in lithium batteries, LiDFOB has found key applications in SIBs. Chen et al. explored its function as an electrolyte additive for improving SIB cathode performance.99 They demonstrated that LiDFOB effectively inhibits electrolyte decomposition and HF-induced corrosion at the electrode surface. By contributing to the formation of a more robust CEI, it shields the active material from damage, thereby inhibiting the dissolution of transition metals.
In addition to the classic boron-containing anion DFOB−, BF4− is also a highly cost-effective boron-containing anion. Even with minimal addition, it can significantly enhance the performance of the entire battery system at a very low cost. Yang et al. introduced a trace amount of NaBF4 into a G2-based electrolyte, enabling stable cycling of the Na3(VOPO4)2F cathode.100 This work provides valuable insights into the application of ultra-low concentration ether-based electrolytes in durable high-voltage SIBs. The improved cycling stability can be attributed to the modulation of the electrolyte solvation structure by NaBF4. Specifically, BF4− anions weaken the interaction between G2 molecules and Na+, while introducing more PF6− into the Na+ solvation sheath. This promotes the enrichment of inorganic species within the CEI, mitigates its dissolution under high voltage, and facilitates stable long-term cycling. Similarly, Yan et al. applied ether-based electrolyte containing NaBF4 as the main salt or additive in the NFM‖HC full cell, which exhibited superior performance compared to an electrolyte with NaPF6.101 This improvement is attributed to the stronger interaction between BF4− and Na+, which reduces the solvation of Na+ with solvent molecules, thereby promoting the formation of a CEI and SEI rich in inorganic species. Furthermore, investigations via SEM, XPS, EDS, and solubility tests revealed that BF4− facilitates the formation of a more stable EEI enriched with boron-containing and inorganic compounds.
Unlike previous fluorinated anions, boron-based anions offer a more pronounced contribution to the cathode CEI. Simultaneously, when the empty orbital remains uncoordinated, the central boron atom acts as a Lewis acid to scavenge trace moisture and HF. Designing boron-containing anions requires integrating organic and inorganic groups, such as coupling boron with fluorinated functional groups to enable simultaneous action at both electrode interfaces.
4.3 Nitrogen-containing anions as additives
Nitrogen-containing additives, such as amines, nitriles, and nitrates, have been widely employed in electrolytes. To address the challenge of ether-based electrolyte oxidation under high voltage, Xia et al. introduced NO3− anions to passivate the reactive terminal hydrogen sites of diglyme molecules.102 The incorporation of this anionic additive induces rearrangement of the primary solvation structure. MD simulations reveal that NO3− ions are incorporated into the inner solvation sheath, and their electron-donating ability weakens Na+–solvent interactions, thereby enhancing ionic conductivity. DFT calculations further demonstrate that the electron-donating NO3− shifts the solvation structures toward the CIP and even promotes the formation of aggregates (Fig. 7f). Moreover, sodium nitrate plays a dual role in the chemistry of CEI formation. The inclusion of NO3− anions in the primary solvation shell releases some OTF− anions into the free ion state. These anions accumulate in the IHP, where they decompose to form a CEI rich in NaF and RSO3Na/RSO2Na species, which act as effective electron insulators and suppress further solvent oxidation (Fig. 7g). Meanwhile, the preferential decomposition of NO3− ion serves as a sacrificial process, incorporating NaNxOy compounds into the CEI and protecting the C–O bonds in G2 molecules from oxidation. This synergy results in a thin, dense CEI layer that selectively permits Na+ transport while blocking electron transfer.Recently, Wu et al. proposed the concept of bridging-donor-ligands, which proved that the NaDCA additive used the dicyandiamide anion (DCA−) as a bridging ligand of the graphite layer, promoting the formation of rich ligand channels and significantly enhancing the structural stability of ternary graphite intercalation compounds.103 During the initial discharge process, DCA− acts as a ligand and driven by its strong electron-withdrawing nature, it actively intercalates into the graphite interlayers. Upon charging, DCA− serves as an electron donor continuously supporting the graphite layers under the influence of the electric field, thereby mitigating anode volume fluctuations during cycling. In NVP‖graphite full cells, this mechanism enables a capacity retention of approximately 80% after 160 cycles (over 1280 hours) (Fig. 7h). This mechanism reveals new potential for achieving high-energy-density graphite anodes in SIBs.
The value of nitrogen-containing anions derives from their unique decomposition products, such as Na3N with high ionic conductivity in the interfacial films. The challenges are the limited solubility of some salts and precise control of their decomposition pathways.
5 Conclusion and outlook
This review systematically summarizes and elaborates on the latest advancements in research concerning anion-based electrolytes for SIBs, focusing primarily on the critical mechanisms by which solvation structures induced by single or composite anions optimize battery electrochemical performance, enhance interfacial ion transport kinetics, and improve EEI stability. It specifically elucidates how diverse types of anions, leveraging their distinct coordination capabilities and modes, effectively regulate the solvation environment of Na+. This regulation facilitates efficient and rapid ion migration at the interface, significantly reduces the desolvation energy barrier for Na+ at the interface, and promotes the formation of denser, more stable interfacial protective layers—particularly the inorganic-rich SEI and CEI. Notably, introducing specific anions as functional additives into the electrolyte system not only directly participates in and optimizes the primary solvation sheath structure of Na+ but also induces the formation of stable, ion-conductive interfacial phases at the electrode surfaces, thereby holistically enhancing the battery's cycle life and safety. Furthermore, this paper critically reviews the remarkable efficacy demonstrated by recently developed novel anions and their corresponding sodium salts in advancing the performance of SIBs.Based on a systematic analysis of recent significant research efforts, constructing anion-centered solvation structures has transcended the conventional strategy employed in HCEs, which solely relies on increasing the anion-to-solvent ratio. Several innovative and highly efficient regulatory pathways have emerged: (i) modulating the Na+ coordination environment via weakly coordinating solvents—for instance, utilizing solvents with inherently weak coordination capabilities such as fluorinated ethers or linear ethers effectively weakens the interactions between solvent molecules and Na+. This creates favorable conditions for anions to enter the inner solvation sheath, thereby enhancing their participation within the coordination structure; (ii) employing strongly associating sodium salts to boost anion coordination competitiveness—sodium salts exhibiting high Na+ binding energy, including NaBF4, NaOTF, and NaTFA, substantially elevate the advantage of anions in the coordination competition. This facilitates their easier entry and stable accommodation within the primary solvation shell of Na+, resulting in anion-enriched coordination architectures. Table 1 summarizes the cation–anion binding energies for typical sodium salts; (iii) introducing functional additives or leveraging solvent synergistic effects—specific additives enable precise tailoring of the composition and configuration of anion-containing solvation sheaths. Meanwhile, synergistic interactions among diverse solvent molecules contribute to forming more loosely packed and flexible solvation layers, promoting greater anion involvement in coordination. These approaches collectively optimize interfacial ion transport and enhance interfacial stability. Furthermore, the distinct effects of anion coordination ability on electrolyte ionic conductivity and viscosity must be comprehensively considered. While strongly coordinating anions can reside more stably within the sodium ion's solvation sheath and effectively participate in interfacial film formation, they often lead to increased viscosity and reduced salt solubility. To mitigate these negative effects stemming from strong coordination, several effective strategies have been developed: one approach involves introducing diluents to construct localized high-concentration electrolytes. This strategy can significantly enhance anion involvement in the solvation structure and interfacial reactions while maintaining low overall viscosity. Another line of strategy focuses on designing weakly coordinating anions (e.g., TFSI−and FSI−) from the outset or utilizing the entropy effect of solvent mixing to induce the formation of weakly solvating structures. These methods have proven effective in markedly improving the ionic conductivity of the system.
| Salt | Anion structure | Binding energy (eV) |
|---|---|---|
| NaPF6 |

|
−5.08 |
| NaClO4 |
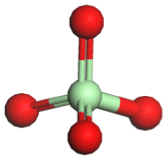
|
−5.23 |
| NaBF4 |

|
−5.55 |
| NaFSI |
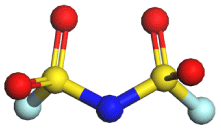
|
−4.85 |
| NaTFSI |

|
−4.98 |
| NaOTF |
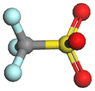
|
−5.24 |
| NaDFOB |

|
−5.32 |
| NaTFA |

|
−5.73 |
| NaNO3 |

|
−5.68 |
In summary, by precisely regulating the solvation behavior of anions and their interfacial chemical reactions, anion-directed electrolyte design strategies play a pivotal role in holistically enhancing the comprehensive electrochemical performance, interfacial compatibility, and long-term cycling stability of SIBs. Building upon current research achievements and remaining challenges, future efforts should focus on exploring novel high-performance anionic sodium salts, deepening studies on multi-anion synergistic mechanisms, developing anion-modulation strategies tailored for high-voltage cathodes/high-capacity anodes, and advancing the integration, validation, and application of relevant electrolyte systems within practical battery configurations. These endeavors will drive research toward higher-performing, more practical anion-dominated SIB electrolytes.
Anion-regulated electrolytes demonstrate pivotal roles in enhancing the overall electrochemical performance, interfacial compatibility, and long-term cycling stability of SIBs by modulating the behavior of anions within solvation structures and their interfacial chemical processes. Building upon current research progress and existing challenges, we systematically propose the following future research directions to drive sustained advancement in this field.
5.1 Sustained innovation in electrolyte systems with systematic optimization of core components
Innovation in electrolyte formulations represents a critical pathway toward high-performance SIBs. Future efforts should prioritize the systematic optimization of three core constituents: sodium salts, solvents, and additives. For sodium salts, emphasis should be placed on developing functional sodium salts featuring novel anion structures to enhance anion participation in solvation architectures, thereby establishing more stable and efficient anion-dominated solvation sheaths. Furthermore, exploration of single-salt and multi-salt synergistic systems is essential to harness collaborative effects among diverse anions for precise control over solvation behavior. In view of the abundance of sodium relative to lithium, the commercial scale production of new sodium salts needs to focus on optimizing the synthesis routes and improving atomic economy. Solvent design requires identifying or engineering environments that promote effective anion coordination, fostering the formation of inorganic-rich, stable interfacial layers (CEI/SEI) to improve safety and cycling lifespan. Additives play a decisive regulatory role; molecules bearing specific functional groups can fine-tune anion behavior within solvation sheaths, reinforcing their dominance and leveraging anion-derived interfaces to enhance interfacial compatibility, thermal stability, and chemical robustness.5.2 Deepening the guiding role of theoretical calculations and simulations in electrolyte research
Theoretical modeling and simulation offer irreplaceable insights into structure–property relationships and underlying mechanisms. DFT is widely applied to evaluate redox stability—for example, by calculating HOMO/LUMO energy levels of solvent molecules to assess electrochemical windows. DFT further enables analysis of Na+–anion binding energies, coordination competition dynamics, and charge distribution patterns, providing a foundational understanding of solvation structures. MD simulations complement this by capturing dynamic evolution of solvation configurations, ion transport kinetics, physicochemical property transitions, and interfacial reaction mechanisms at the molecular level. Integration of multiscale theoretical tools (e.g., DFT + MD) facilitates quantitative dissection of solvation motifs, unraveling complex interactions among anions, cations, and solvent molecules. These approaches not only simulate key processes but also guide rational electrolyte design and predict electrochemical behavior in practical systems.5.3 Accelerating material discovery via machine learning in electrolyte development
Machine learning (ML), as a data-driven accelerator, is increasingly vital for expediting novel electrolyte discovery, predicting critical performance metrics, and optimizing battery systems.104 A central challenge in anion-regulated electrolytes is achieving optimal anion–solvent pairing. ML addresses this through structure–activity relationship models that predict electrochemical performances of emerging anions or solvation configurations, enabling rapid screening of candidate systems. Strategies include high-throughput virtual screening based on physicochemical descriptors and electrochemical databases, allowing evaluation of thousands of anion/solvent/additive combinations. ML integrates multi-source data (experimental + computational) to comprehensively understand electrolyte behavior. Synergy between theoretical calculations and ML will drive rational design and innovation in anion-centric electrolytes, supporting sustainable advancement of sodium battery technology.5.4 Advancing advanced characterization methods to decode solvation structures and interfacial evolution
Solvation structures and their derived interphases are central to optimizing anion-regulated electrolytes. While theoretical simulations provide insights, experimental characterization remains indispensable for directly probing these phenomena. Spectroscopic techniques (e.g., Raman spectroscopy) currently analyze bulk solvation structures but lack spatial/temporal resolution for microscopic details—such as ion rearrangement at electrode/electrolyte interfaces, local coordination evolution, or dynamic solvation sheath changes during cycling.105 To bridge this gap, cutting-edge in situ/operando characterization tools with high spatiotemporal resolution are urgently needed to monitor real-time interfacial formation, structural evolution, and property shifts under operational conditions. Microscopy (SEM/TEM) offers valuable insights into interfacial morphology and composition. Multimodal platforms combining spectroscopy, spectrometry, and imaging will elucidate correlations between solvation structures and interfacial evolution, advancing toward precision-driven, dynamic, and mechanism-guided design of anion-regulated electrolytes.106,107Looking ahead, research on anion-regulated electrolytes will increasingly feature deep multidisciplinary convergence. By establishing a closed-loop “design–simulation–validation” paradigm integrating theoretical calculations, machine learning, and advanced characterization techniques, precise decoding and proactive control of solvation structures and interfacial processes will be progressively realized. This approach will systematically advance electrolyte systems from single-component optimization toward multi-component synergistic design, shifting focus from macroscopic performance enhancement to microscopic mechanism-driven innovation. With deepened understanding of anion behavior, it is anticipated that next-generation electrolyte systems combining high ionic conductivity, wide electrochemical windows, and self-healing interfacial functionality will emerge. Such advancements will not only significantly elevate the energy density, cycle life, and safety performance of SIBs but also offer innovative electrolyte design paradigms for the broader alkali metal battery family. Ultimately, through multiscale, end-to-end collaborative innovation, anion-regulation strategies hold promise to break through current performance bottlenecks in SIB technology, providing critical material foundations for low-cost, high-efficiency, and high-safety sustainable energy storage systems—thereby playing a vital supporting role in global energy transition.
Author contributions
S. Li: investigation, writing – original draft, and writing – review & editing; Y. Heng: investigation, writing – original draft and format analysis; Z. Gu: supervision and writing – review & editing; X. Wang: investigation and conceptualization; Y. Liu: formal analysis; X. Zhang: modification; Z. Sun: writing – review & editing; D. Liu: formal analysis; B. Li: formal analysis; X. Wu: resources, writing – review & editing, and supervision.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
This work was supported by the National Key R&D Program of China (No. 2023YFE0202000), National Natural Science Foundation of China (No. 52502221), Natural Science Foundation of Jilin Province (No. 20250101013JJ), Fundamental Research Funds for the Central Universities (No. 2412025QG001), National Postdoctoral Program for Innovative Talents (No. BX20240062), and Guangzhou Basic and Applied Basic Research Project (2023A04J1408).Notes and references
- Y. Li, F. Wu, Y. Li, M. Liu, X. Feng, Y. Bai and C. Wu, Chem. Soc. Rev., 2022, 51, 4484–4536 RSC.
- X. Cai, Y. Yue, Z. Yi, J. Liu, Y. Sheng and Y. Lu, Nano Energy, 2024, 129, 110052 CrossRef CAS.
- F. Zhang, B. He, Y. Xin, T. Zhu, Y. Zhang, S. Wang, W. Li, Y. Yang and H. Tian, Chem. Rev., 2024, 124, 4778–4821 CrossRef CAS.
- J. Y. Hwang, S. T. Myung and Y. K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC.
- T. Zhang, Y. Xiao, S. Zhao, Y. Han, G. He and S. Chen, Energy Environ. Sci., 2025, 18, 7669–7679 RSC.
- Y. Gao, H. Zhang, J. Peng, L. Li, Y. Xiao, L. Li, Y. Liu, Y. Qiao and S. Chou, Carbon Energy, 2024, 6, e464 CrossRef.
- E. Goikolea, V. Palomares, S. Wang, T. Rojo and M. Armand, Adv. Energy Mater., 2020, 10, 2002055 CrossRef CAS.
- H. Che, S. Chen, Y. Xie, H. Wang, K. Amine, X. Z. Liao and Z. F. Ma, Energy Environ. Sci., 2017, 10, 1075–1101 RSC.
- Z. Tian, Y. Zou, G. Liu, Y. Wang, J. Yin, J. Ming and H. N. Alshareef, Adv. Sci., 2022, 9, 2201207 CrossRef CAS PubMed.
- F. Cheng, J. Hu, W. Zhang, B. Guo, P. Yu, X. Sun and J. Peng, Energy Environ. Sci., 2025, 18, 6874–6898 RSC.
- K. Vignarooban, R. Kushagra, A. Elango, P. Badami, B. E. Mellander, X. Xu, T. G. Tucker, C. Nam and A. M. Kannan, Int. J. Hydrogen Energy, 2016, 41, 2829–2846 CrossRef CAS.
- S. Zhou, X. Chen, X. Zhang, W. Kuang, C. Jiao, X. Xu, W. Zhu, G. Fan, Z. Hu, X. Tan, J. Wang and L. Li, Adv. Funct. Mater., 2025, 35, 2418784 CrossRef CAS.
- J. Zhang, J. Li, H. Wang and M. Wang, Front. Chem., 2023, 11, 1253959 CrossRef CAS PubMed.
- K. Deng, Q. Zeng, D. Wang, Z. Liu, G. Wang, Z. Qiu, Y. Zhang, M. Xiao and Y. Meng, Energy Storage Mater., 2020, 32, 425–447 CrossRef.
- S. Wang, J. Shi, Z. Liu and Y. Xia, Adv. Energy Mater., 2024, 14, 2401526 CrossRef.
- Y. Wu, B. Wang, Z. Luo, Z. Hou, B. Xu, L. Zhou and W. Wei, Adv. Funct. Mater., 2025, 35, 2506098 CrossRef.
- Y. Jin, P. M. L. Le, P. Gao, Y. Xu, B. Xiao, M. H. Engelhard, X. Cao, T. D. Vo, J. Hu, L. Zhong, B. E. Matthews, R. Yi, C. Wang, X. Li, J. Liu and J. G. Zhang, Nat. Energy, 2022, 7, 718–725 CrossRef.
- Y. Liu, L. Zhu, E. Wang, Y. An, Y. Liu, K. Shen, M. He, Y. Jia, G. Ye, Z. Xiao, Y. Li and Q. Pang, Adv. Mater., 2024, 36, 2310051 CrossRef PubMed.
- X. Chang, J. Chen, W. Xu, M. Li, Q. Zhou, B. Dai, H. Zhou, Y. Liu and Y. Qiao, Energy Storage Mater., 2025, 81, 104526 CrossRef.
- S. Dinda, S. Trivedi, A. Roy, F. D. Pammer and M. Fichtner, Adv. Energy Mater., 2023, 13, 2302176 CrossRef.
- Y. H. Feng, C. Lin, H. Qin, G. X. Wei, C. Yang, Y. Tang, X. Zhu, S. Sun, T. L. Chen, M. Liu, H. Zheng, X. Ji, Y. You and P. F. Wang, J. Am. Chem. Soc., 2025, 147, 16107–16118 CrossRef PubMed.
- P. Li, N. Hu, J. Wang, S. Wang and W. Deng, Nanomaterials, 2022, 12, 3529 CrossRef PubMed.
- Y. Yi, J. Qi, X. Yang, H. Yang and G. Zeng, Mater. Sci. Eng., B, 2025, 320, 118443 CrossRef.
- C. Hu, L. Dai, F. Huang, Y. Yang, S. Liang, G. Fang and Q. Zhang, Angew. Chem., Int. Ed., 2025, 64, e202508584 CrossRef PubMed.
- M. Li, J. Jiang, Y. Chen, S. Huang, X. Liu, J. Yi, Y. Jiang, B. Zhao, W. Li, X. Sun and J. Zhang, Angew. Chem., Int. Ed., 2025, 64, e202413806 CrossRef PubMed.
- X. Zheng, Z. Cao, Z. Gu, L. Huang, Z. Sun, T. Zhao, S. Yu, X. L. Wu, W. Luo and Y. Huang, ACS Energy Lett., 2022, 7, 2032–2042 CrossRef.
- Y. Sun, C. Zheng, Q. Yao, H. Lv, X. Ma and J. Yang, Angew. Chem., Int. Ed., 2025, 64, e202420573 CrossRef PubMed.
- K. Qin, S. Li, D. Yen, W. Zhang, Z. Yang, E. P. Kamphaus, E. Y. Kim, J. Huang, J. J. Shea, E. Hu, L. Cheng, C. Wang and C. Luo, ACS Energy Lett., 2024, 9, 3869–3876 CrossRef.
- Y. Huang, W. Sun, M. Zhi, Y. Wu, K. Liu, S. Zheng, Q. Zhang and S. Dai, J. Energy Chem., 2025, 106, 951–960 CrossRef.
- H. J. Liang, W. Y. Qian, H. H. Liu, X. T. Wang, Z. Y. Gu, F. Dong, Y. Deng, Y. Z. Tang, J. Zhang, J. Zhao and X. L. Wu, J. Am. Chem. Soc., 2025, 147, 17860–17870 CrossRef PubMed.
- H. Lin, F. Liu, E. Yang, X. Shi, A. Zhang, Z. Bi, H. Liu, Y. Xie and Z. S. Wu, Nano Energy, 2025, 142, 111266 CrossRef.
- H. Zhang, Y. Zhao, X. Li, H. Wang, L. Wang, Y. Song, F. Qiao, J. Wang and J. Xu, Adv. Sci., 2025, 12, e03151 CrossRef CAS PubMed.
- X. He, J. Peng, Q. Lin, M. Li, W. Chen, P. Liu, T. Huang, Z. Huang, Y. Liu, J. Deng, S. Ye, X. Yang, X. Ren, X. Ouyang, J. Liu, B. Xiao, J. Hu and Q. Zhang, Nano-Micro Lett., 2025, 17, 45 CrossRef CAS.
- Z. Jiang, J. Mo, C. Li, H. Li, Q. Zhang, Z. Zeng, J. Xie and Y. Li, Energy Environ. Mater., 2023, 6, e12440 CrossRef CAS.
- Z. Ni, C. Wei, Z. Wang, Y. Li, X. Zhang, S. Xiong and J. Feng, Energy Storage Mater., 2024, 71, 103603 CrossRef.
- C. Ma, C. Fu, S. Chang, X. Xu, G. Zhang, Z. Liu, H. Huo, L. Fan, G. Yin and Y. Ma, ACS Energy Lett., 2025, 10, 1700–1711 CrossRef CAS.
- R. Zhou, Y. Xu, C. Han, Y. Li, J. Wang, X. Liang, T. Jing, R. Liu, G. Chen and F. Cao, Adv. Mater., 2025, 37, 2416748 CrossRef CAS.
- Q. Zhu, D. Yu, J. Chen, L. Cheng, M. Tang, Y. Wang, Y. Li, J. Yang and H. Wang, Joule, 2024, 8, 482–495 CrossRef CAS.
- H. Huang, Y. Hu, Y. Hou, X. Wang, Q. Dong, Z. Zhao, M. Ji, W. Zhang, J. Li, J. Xie, H. Guo, X. Han, X. Ouyang and W. Hu, Nature, 2025, 644, 660–667 CrossRef CAS.
- H. Li, H. Chen, X. Shen, X. Liu, Y. Fang, F. Zhong, X. Ai, H. Yang and Y. Cao, ACS Appl. Mater. Interfaces, 2022, 14, 43387–43396 CrossRef CAS.
- L. Liao, Z. Han, X. Feng, P. Luo, J. Song, Y. Shen, X. Luo, X. Li, X. Wen, B. Yu, J. Chen, B. Guo, M. Wang, Y. Huang, H. Zhang, M. Yin, J. Liu, Y. Lin and X. Li, J. Energy Chem., 2024, 97, 156–165 CrossRef CAS.
- D. Zhang, H. Fu, X. Ma, X. Yu, F. Li, J. Zhou and B. Lu, Angew. Chem., Int. Ed., 2024, 63, e202405153 CrossRef CAS.
- C. Zhu, D. Wu, Z. Wang, H. Wang, J. Liu, K. Guo, Q. Liu and J. Ma, Adv. Funct. Mater., 2024, 34, 2214195 CrossRef CAS.
- M. Ma, R. Huang, M. Ling, Y. Hu and H. Pan, Interdiscip. Mater., 2023, 2, 833–854 CAS.
- H. Fang, Y. Huang, W. Hu, Z. Song, X. Wei, J. Geng, Z. Jiang, H. Qu, J. Chen and F. Li, Angew. Chem., Int. Ed., 2024, 63, e202400539 CrossRef CAS.
- F. Huang, Y. Zhong, M. Qin, J. Qiu, C. Hu, P. Xu, H. Zhang, C. Sheng, S. Liang and G. Fang, Chem. Eng. J., 2025, 511, 162150 CrossRef CAS.
- P. Lai, Y. Zhang, J. Liu, Z. Zhang, H. Xie, X. Li, X. Deng, B. Huang, P. Zhang and J. Zhao, J. Energy Chem., 2025, 107, 852–860 CrossRef CAS.
- C. Zheng, Q. Yao, Y. Sun, H. Lv, Z. Bai, G. Zhang, N. Wang and J. Yang, Adv. Funct. Mater., 2025, 2501303 CrossRef.
- P. Huang, J. Lin, Z. Wang, W. Li, Y. Chen, L. Zhou, L. Chen, C. Zhang and W. Wei, Adv. Funct. Mater., 2025, 35, 2401234 Search PubMed.
- F. Cheng, M. Cao, Q. Li, C. Fang, J. Han and Y. Huang, ACS Nano, 2023, 17, 18608–18615 CrossRef CAS PubMed.
- X. Zhou, X. Chen, W. Kuang, W. Zhu, X. Zhang, X. Liu, X. Wu, L. Zhang, C. Zhang, L. Li, J. Wang and S. Chou, Angew. Chem., Int. Ed., 2024, 63, e202410494 CAS.
- W. Zhao, M. Wang, H. Lin, K. Kim, R. He, S. Feng and H. Liu, Prog. Nat. Sci.:Mater. Int., 2024, 34, 263–273 CrossRef CAS.
- X. Zhou, X. Chen, Z. Yang, X. Liu, Z. Hao, S. Jin, L. Zhang, R. Wang, C. Zhang, L. Li, X. Tan and S. Chou, Adv. Funct. Mater., 2024, 34, 2312345 Search PubMed.
- H. Chen, K. Chen, J. Yang, B. Liu, L. Luo, H. Li, L. Chen, A. Zhao, X. Liang, J. Feng, Y. Fang and Y. Cao, J. Am. Chem. Soc., 2024, 146, 15751–15760 CrossRef CAS.
- M. Wu, B. Zhang, Y. Ye, L. Fu, H. Xie, H. Jin, Y. Tang, H. Wang and D. Sun, ACS Appl. Mater. Interfaces, 2024, 16, 15586–15595 CrossRef CAS.
- Y. Wan, X. Shi, Z. Zhou, W. Kuang, X. Xu, X. Zhang, X. Zeng, X. Zhou, J. Liu, S. L. Chou and L. Li, J. Am. Chem. Soc., 2025, 147, 29939–29948 CrossRef CAS PubMed.
- Q. Zhu, J. Wang, L. Wu, H. Lan, J. Chen, L. Cheng, B. Zhou, D. Yang, J. Yang, M. Kurbanov, S. Dong and H. Wang, Natl. Sci. Rev., 2025, 12, nwaf124 CrossRef CAS PubMed.
- S. Zhao, Q. Yu, S. Yang, S. Wan, J. Chen, H. Xu, X. Lou and S. Chen, Nano Energy, 2025, 138, 110913 CrossRef CAS.
- L. Huang, Q. Qiu, M. Yang, H. Li, J. Zhu, W. Zhang, S. Wang, L. Xia and P. Müller-Buschbaum, ACS Appl. Mater. Interfaces, 2024, 16, 46392–46400 CrossRef CAS.
- Z. Cheng, Z. Zhang, F. Qiu, Z. Gao, H. Xie, Z. Xu, M. Jia, X. Zhang and H. Zhou, ACS Energy Lett., 2025, 10, 177–184 CrossRef CAS.
- M. Pai and A. Manthiram, Adv. Energy Mater., 2025, 15, 2500026 CrossRef CAS.
- Y. Gao, H. Tu, J. Xue, Y. Wang, S. Zhang, S. Lu, L. Liu, K. Peng, G. Sun, G. Wu, P. Ding, Y. Yang, Z. Wang, J. Xu and X. Wu, ACS Energy Lett., 2025, 10, 3315–3324 CrossRef CAS.
- X. Liu, X. Zheng, Y. Dai, B. Li, J. Wen, T. Zhao and W. Luo, Adv. Mater., 2023, 35, 2304567 Search PubMed.
- W. Fan, A. Wang, J. Cai, L. Shi, W. Wang and J. Nan, J. Power Sources, 2025, 650, 237519 CrossRef CAS.
- S. Wu, H. Liang, Z. Gu, M. Rong, D. Liu, J. Wang, F. Wang, P. Liu, B. Xie, Q. Yao, J. Deng, Z. Wang and X. Wu, Adv. Funct. Mater., 2025, e08137 CrossRef.
- B. Qiu, N. Sun, X. Li, R. A. Soomro, Y. Guan, H. Mi and B. Xu, Adv. Funct. Mater., 2025, e12543 CrossRef.
- E. S. Flitz, N. R. Singstock, S. B. Son, C. Tezak, M. Lucero, X. Li, C. Musgrave and C. Ban, Joule, 2025, 9, 102045 CrossRef CAS.
- H. Lohani, D. T. Duncan, X. Qin, P. Kumari, M. Kar, A. Sengupta, A. Ahuja, A. Bhowmik and S. Mitra, Small, 2024, 20, 2311157 CrossRef CAS.
- Q. Man, Y. Mu, Z. Qiu, X. Wei, J. Guan, H. Gu, L. Yang, J. Guo, Z. Liao, M. Han, Q. Zhang and L. Zeng, Adv. Energy Mater., 2025, 15, 2501818 CrossRef CAS.
- D. Yu, Z. Wang, J. Yang, Y. Wang, Y. Li, Q. Zhu, X. Tu, D. Chen, J. Liang, U. Khalilov and H. Wang, Small, 2024, 20, 2311810 CrossRef CAS PubMed.
- B. Zhu, J. Gao, S. Li, W. Zhou, Z. Yang, R. Jin, Y. Zhang, Z. Ding, Y. Lai, J. Li and Z. Zhang, Chem. Eng. J., 2025, 519, 165626 CrossRef CAS.
- R. García-Méndez, Joule, 2025, 9, 101875 CrossRef.
- J. Zhang, N. Sawut, H. Fan, Z. Li, X. Ai, Y. Fang and Y. Cao, Nano Energy, 2025, 136, 110744 CrossRef CAS.
- L. Huang, Q. Qiu, M. Yang, H. Li, J. Zhu, W. Zhang, S. Wang, L. Xia and P. Müller-Buschbaum, ACS Appl. Mater. Interfaces, 2024, 16, 46392–46400 CrossRef CAS.
- Y. Feng, M. Liu, W. Qi, H. Liu, Q. Liu, C. Yang, Y. Tang, X. Zhu, S. Sun, Y. Li, T. Chen, B. Xiao, X. Ji, Y. You and P. Wang, Angew. Chem., Int. Ed., 2025, 64, e202415644 CrossRef CAS.
- Y. Yang, Y. Yang, S. Yang, Y. Yao, Y. Yang, L. Liu, X. Rui and Y. Yu, ACS Appl. Mater. Interfaces, 2025, 17, 22624–22632 CrossRef CAS.
- Y. Lin, Q. Peng, L. Chen, Q. Zuo, Q. Long, F. Lu, S. Huang, Y. Chen and Y. Meng, Energy Storage Mater., 2024, 67, 103211 CrossRef.
- Z. X. Huang, X. L. Zhang, X. X. Zhao, Y. Y. Zhao, V. Aravindan, Y. H. Liu, H. Geng and X. L. Wu, Inorg. Chem. Front., 2023, 10, 37–48 RSC.
- X. Zheng, L. Huang, X. Ye, J. Zhang, F. Min, W. Luo and Y. Huang, Chem, 2021, 7, 2312–2346 CAS.
- Y. Liu, C. Zhao, J. Du, X. Zhang, A. Chen and Q. Zhang, Small, 2023, 19, 2205315 CrossRef CAS.
- K. Xie, Y. Ji, L. Yang and F. Pan, Adv. Energy Mater., 2025, 15, 2405301 CrossRef CAS.
- B. Qi, X. Zhang, L. You, X. Yu, Z. Qiao and D. Ruan, J. Energy Storage, 2024, 100, 113523 CrossRef.
- C. Senthil, A. Subramani, R. K. Gupta and Z. Sofer, Small, 2025, 21, 2504276 CrossRef CAS.
- F. Wang, Z. Zheng, Z. Wen, W. Fang, C. Kuang, F. Chen, H. Wan, N. Zhang, X. Liu, R. Ma and G. Chen, Sci. Bull., 2025, 70, 2493–2503 CrossRef CAS PubMed.
- Y. Liao, H. Liu, Y. Zhang, J. Yang, H. Ji, D. Wang, L. Yuan, Y. Huang and Y. Ren, ACS Nano, 2025, 19, 30466–30475 CrossRef CAS PubMed.
- H. Che, X. Yang, H. Wang, X. Z. Liao, S. S. Zhang, C. Wang and Z. F. Ma, J. Power Sources, 2018, 407, 173–179 CrossRef CAS.
- X. Chang, Z. Yang, Y. Liu, J. Chen, M. Wu, L. Li, S. Chou and Y. Qiao, Energy Storage Mater., 2024, 69, 103407 CrossRef.
- C. Zhu, D. Wu, C. Wang and J. Ma, Adv. Funct. Mater., 2024, 34, 2406764 CrossRef CAS.
- S. Lin, Z. Yang, J. Chen, Y. Qiao, L. Li and S. Chou, Adv. Funct. Mater., 2024, 34, 2400731 CrossRef CAS.
- B. Song, X. Xiong, Y. Peng, X. Liu, W. Gao, T. Wang, F. Wang, Y. Ma, Y. Zhong, X. Cheng, Z. Zhu, J. He and Y. Wu, Adv. Energy Mater., 2024, 14, 2401407 CrossRef CAS.
- E. Li, X. Tang, J. Zhou, H. Zhao, J. Teng, J. Huang, B. Dai, T. Lu, Q. Tao, K. Zhang, W. Deng and J. Li, Chem. Eng. J., 2024, 489, 151525 CrossRef CAS.
- S. Bolloju, N. Vangapally, Y. Elias, S. Luski, N. L. Wu and D. Aurbach, Prog. Mater. Sci., 2025, 147, 101349 CrossRef CAS.
- P. Gogoi, X. Ge, M. Janpandit, S. Prakash, N. Kalita and Y. C. Li, ACS Appl. Mater. Interfaces, 2025, 17, 13980–13987 CrossRef CAS PubMed.
- X. Zhou, Y. Huang, B. Wen, Z. Yang, Z. Hao, L. Li, S. L. Chou and F. Li, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2316914121 CrossRef CAS.
- X. Hou, T. Li, Y. Qiu, M. Jiang, H. Lin, Q. Zheng and X. Li, ACS Energy Lett., 2024, 9, 461–467 CrossRef CAS.
- H. Yang, J. Hwang, Y. Tonouchi, K. Matsumoto and R. Hagiwara, J. Mater. Chem. A, 2021, 9, 3637–3647 RSC.
- Q. Yu, Y. Xiao, S. Zhao, Y. Miao, S. Wan, L. Zhou, J. Rong, G. Hou and S. Chen, Adv. Funct. Mater., 2024, 34, 2401234 Search PubMed.
- X. Liu, J. Zhao, H. Dong, L. Zhang, H. Zhang, Y. Gao, X. Zhou, L. Zhang, L. Li, Y. Liu, S. Chou, W. Lai, C. Zhang and S. Chou, Adv. Funct. Mater., 2024, 34, 2402310 CrossRef CAS.
- Q. Qiu, T. Zheng, L. Huang, T. Xu, L. Pan, W. Sun, H. Tian, W. Zhang, Q. Yu, Y. Liang, Y. Yan, J. Yuan, P. Müller-Buschbaum and L. Xia, Energy Storage Mater., 2024, 73, 103858 CrossRef.
- Z. Yang, J. Pan, H. Wu, K. Shi, Z. Chen, J. Li, W. Huang, T. Yang, Y. Hong, R. Zhang, Z. Lu, Y. Min and Q. Liu, Energy Storage Mater., 2024, 65, 103456 Search PubMed.
- S. Li, H. Xu, Y. Zhu, Z. Yang, Y. Bao and Y. Chen, J. Mater. Chem. A, 2024, 12, 31347–31361 RSC.
- S. Li, X. Song, P. Jing, X. Xiao, Y. Chen, Q. Sun, M. Huang, Y. Zhang, G. Li, P. Liu, S. Xu, Q. Dou, J. Zhu and X. Yan, Adv. Funct. Mater., 2025, 35, 2501234 Search PubMed.
- X. Yi, X. Li, J. Zhong, Z. Cui, Z. Wang, H. Guo, J. Wang and G. Yan, ACS Appl. Mater. Interfaces, 2024, 16, 11585–11594 CrossRef CAS PubMed.
- X. Wang, Q. Fan, Z. Liu, X. Zhu, M. Yang, Z. Guo, Y. Chen, L. Wang, Y. Jing and H. Xia, Nat. Commun., 2025, 16, 123456 Search PubMed.
- B. Song, Z. Luo, X. Liu, Y. Peng, J. Wang, F. Jiang, X. Cheng, Y. Wu and J. He, Angew. Chem., Int. Ed., 2025, 64, e202503027 CrossRef CAS.
- N. Shukla, M. Saikia and M. Deka, Mater. Today Phys., 2025, 57, 101799 CrossRef CAS.
- W. Deng, G. Li and X. Wang, Adv. Funct. Mater., 2024, 34, 2405012 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |