
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-centered, yet persistent: isolation of N2O-based radicals through FLP-type stabilization
Andrea
Orellana Ben Amor
 a,
Laure
Vendier
a,
Vincent
César
a,
Vincent
Maurel
*b,
Julien
Panetier
*c and
Nicolas
Queyriaux
*a
a,
Laure
Vendier
a,
Vincent
César
a,
Vincent
Maurel
*b,
Julien
Panetier
*c and
Nicolas
Queyriaux
*a
aUniv. Toulouse, CNRS, LCC, Toulouse, France. E-mail: nicolas.queyriaux@lcc-toulouse.fr
bUniv. Grenoble Alpes, CEA, CNRS, IRIG, SyMMES, 38000 Grenoble, France. E-mail: vincent.maurel@cea.fr
cDepartment of Chemistry, State University of New York at Binghamton, Binghamton, NY 13902, USA. E-mail: panetier@binghamton.edu
First published on 27th November 2025
Abstract
The electrochemical one-electron reduction of IDipp·N2O (IDipp = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene) is inherently irreversible, highlighting the short-lived nature of the corresponding radical anion. However, stabilization by Lewis acidic boranes enables electrochemically reversible behavior, unlocking access to persistent radical species. These radicals are synthesized via chemical reduction and fully characterized, revealing a highly N-centered nature alongside remarkable persistence at room temperature under an inert atmosphere. A comprehensive investigation of their electronic structure is presented, integrating electron paramagnetic resonance techniques (both continuous-wave and pulsed) with theoretical analysis to provide deeper insight into their properties.
Introduction
N-heterocyclic carbenes (NHCs) have recently emerged as powerful molecular tools capable of stabilizing N-centered radicals.1–4 Through a combination of both steric protection and π-accepting properties, NHCs greatly limit the reactivity of the radicals, typically resulting in persistent derivatives. In this context, a series of NHC-stabilized aminyl radicals has been isolated by the Severin group (Scheme 1A).5 Those radicals – stable under an inert atmosphere – are characterized by a significant localization of the spin density of the unpaired electron (ca. 41%) on one of the nitrogen atoms of the diazenyl group. Open-shell diradicals, bridged by a permethylated biphenyl unit, were similarly isolated (Scheme 1B). When exposed to air, these diradicaloid derivatives were found to be significantly less stable than their monoradical analogues. This was attributed to increased steric congestion, which limits the coplanarity of the diazenyl units with the adjacent arene ring, thereby accounting for low electron delocalization. Related triazenyl radicals were also synthesized by the Lee group (Scheme 1C).6 Although displaying a greater N-centered character (ca. 55%), the spin density of the radical is, again, computed to be significantly delocalized throughout the molecule.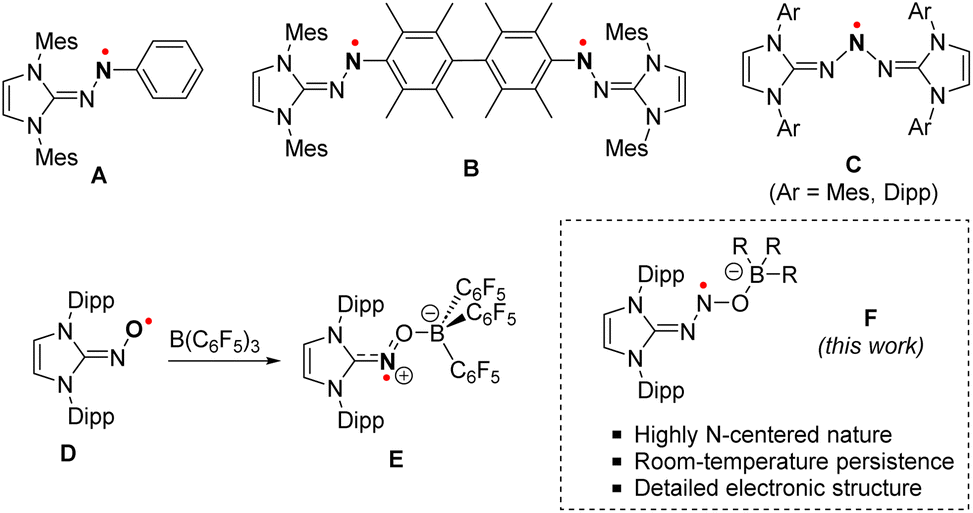 | ||
| Scheme 1 Molecular structures of NHC-stabilized N-centered radicals. | ||
Further stabilization of a radical species may also be achieved through the combined use of NHCs and Lewis acid partners, employing a Frustrated Lewis Pair (FLP) approach, which provides increased steric protection and electronic delocalization. Hence, Lee and co-workers demonstrated that the stabilization of an iminoxyl radical induced by a NHC alone was insufficient, resulting in progressive decomposition in solution.7 Conversely, a redox-active FLP system was recently reported to split NO through single electron oxidation.8 A dramatic increase in stability was achieved upon reaction with tris(pentafluorophenyl)borane (1a), with the resulting zwitterionic radical displaying remarkable stability towards both air and moisture (Scheme 1D and E).9
In γ-radiolysis, nitrous oxide (N2O) has been used for a long time as a chain transfer agent, facilitating the increased generation of hydroxyl radicals in water.10,11 While undoubtedly involved in the process, the N2O˙− radical anion has never been experimentally evidenced. A notable result in this context was the in situ EPR characterization of a silyl-stabilized N2O radical – embedded on a SiO2 surface – by the group of Radtsig.12 From this perspective, it is therefore appealing to deploy NHC-based strategies that have proven successful in stabilizing related aminyl and iminoxyl radicals to the N2O motif. Interestingly, the group of Severin reported the preparation of NHC·N2O adducts that may serve as convenient entry points in such an approach.13–15 Although they display a rich and diverse chemistry,16–21 their redox behavior remains virtually unexplored to date.
Here, we report the preparation of new FLP-type adducts of N2O varying in the nature of their Lewis acid partners. Examination of their redox behavior specifically highlights the remarkable persistence of the corresponding radical anions. Accordingly, such radicals were isolated and fully characterized. A specific focus has been dedicated to elucidating the electronic structures of these species through the use of electron paramagnetic resonance techniques and theoretical investigations. The aminyl nature of the N2O-based radical is evidenced, with a high degree of N-centered character.
Results and discussion
Adducts 3a–c were synthesized through a two-step synthetic route. 1,3-Bis(2,6-diisopropylphenyl)-2H-imidazol-2-ylidene (IDipp) was initially exposed to N2O following a reported procedure to produce the stable IDipp·N2O adduct 2 (Scheme 2).13,14 Subsequent reactions with selected boranes (1a–c) were carried out in dichloromethane at room temperature, resulting in the formation of adducts 3a–c in yields exceeding 80%. The relative Lewis acid strengths of these boranes were assessed using the Gutmann–Beckett method, employing triethylphosphine oxide (Et3PO) as the Lewis base probe (refer to the SI for more details).22 When normalized to tris(pentafluorophenyl)borane (B(C6F5)3, 1a: 100%), the values obtained for 1b–c reveal the selected boranes to cover a significant range of Lewis acidity (1b: 85%, 1c: 57%). | ||
| Scheme 2 Synthesis of the IDipp·N2O·BAr3 compounds 3a–c. | ||
Compounds 3a–c display spectroscopic signatures similar to related species.17,19 More specifically, the formation of adducts diagnostically influences chemical shifts of the imidazolium backbone protons. In the 1H NMR spectra, the corresponding singlet indeed experiences strong deshielding in comparison to the parent IDipp·N2O compound (2: 7.25 ppm, 3a: 7.99 ppm, 3b: 7.78 ppm, 3c: 7.70 ppm). As the Lewis acid becomes stronger, an enhanced weight of the diazotate mesomeric form is expected in the hybrid structure of the corresponding adducts (Scheme 2). As a result, the resonance of the backbone protons is progressively shifted downfield, in line with an increased aromatic character. For comparison purposes, the 2-phenyl imidazolium analogue, 4·BF4 (1,3-bis(2,6-diisopropylphenyl)-2-phenylimidazolium tetrafluoroborate), whose aromaticity is unambiguous, exhibits signals for these same protons at a value of 8.52 ppm. In the 13C{1H} NMR spectra, the resonances for the carbene carbon atom of 3a–c are in the region of δ = 144–148 ppm, in good agreement with previous findings.17,1911B{1H} NMR spectra display sharp resonances at close-to-zero δB values, confirming the expected tetrahedral geometry around the boron atom. When present, signals of the fluorine atoms lying on the phenyl groups of the boranes are observed at chemical shifts consistent with adduct formation.
X-ray quality crystals were grown from THF/pentane solutions, allowing for X-ray diffraction analysis of all three compounds 3a–c. In all cases, the resulting structures confirm the coordination of the borane moiety to the IDipp·N2O adduct 2 (Fig. 1). The relevant bond lengths and angles for compounds 3a–c are summarized in Table 1, together with a comparison with the parent compound 2.13
 | ||
| Fig. 1 Molecular structures of 3a (left), 3b (centre) and 3c (right), as determined by X-ray diffraction. Thermal ellipsoids shown at the 50% probability level. Hydrogen atoms and solvent molecules were omitted for sake of clarity. | ||
| 2 13 | 3a | 3b | 3c | |
|---|---|---|---|---|
| N1–N2 (Å) | 1.352(4) | 1.283(2) | 1.284(2) | 1.277(2) |
| N1–O1 (Å) | 1.250(4) | 1.317(2) | 1.313(2) | 1.310(1) |
| B1–O1 (Å) | — | 1.537(3) | 1.551(2) | 1.593(2) |
| C1–N2 (Å) | 1.358(4) | 1.374(3) | 1.374(2) | 1.376(2) |
| N2–N1–O1 (°) | 112.9(3) | 108.0(2) | 108.4(1) | 109.1(1) |
In detail, the N2O motif adopts a bent geometry with its proximal nitrogen atom connected to the NHC carbon atom and its oxygen atom linked to the borane group. The N–N–O angles are in the range of 108–109°, thus showing increased bending compared to 2 (112.9°). Interestingly, the nature of the borane slightly – but coherently – influences the N–N–O angle: the stronger the Lewis acid is, the more bent the motifs are. As a general trend, the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds tend to contract in 3a–c (1.27–1.28 Å) compared to 2 (1.35 Å), while the N–O bonds are elongated (1.30–1.32 Å in 3a–cvs. 1.25 Å in 1). Such a bond length inversion aligns well with the results obtained other FLP-type adducts of N2O.17,19,23,24 Upon increased acidity of the borane partner, enhanced coplanarity of the N2O motif and the heterocycle is accessed: the solid angle formed by the mean planes associated with these two motifs is indeed shifting from 39.08° in 2 to 19.80, 23.55 and 35.35° for 3a–c, respectively.
N bonds tend to contract in 3a–c (1.27–1.28 Å) compared to 2 (1.35 Å), while the N–O bonds are elongated (1.30–1.32 Å in 3a–cvs. 1.25 Å in 1). Such a bond length inversion aligns well with the results obtained other FLP-type adducts of N2O.17,19,23,24 Upon increased acidity of the borane partner, enhanced coplanarity of the N2O motif and the heterocycle is accessed: the solid angle formed by the mean planes associated with these two motifs is indeed shifting from 39.08° in 2 to 19.80, 23.55 and 35.35° for 3a–c, respectively.
To gain further insight into the electronic structures of compounds 3a–c, we conducted DFT calculations. Initial geometry optimizations were performed using the ωB97X-D25 functional in solution (tetrahydrofuran, ε = 7.4257) with the SMD approach (see the SI for more details).26 A good agreement was typically found between the X-ray and DFT geometries, with the most significant error being an underestimation of the N1–O1 bond distances by an average of 2.8% (Table S2). We note that, as observed experimentally, the computed N–N–O bond angle increases across the series in going from 3a (110.7°) to 3c (111.0°), consistent with 3a featuring the strongest Lewis acid borane moieties. Inspection of the frontier molecular orbitals in this series of compounds reveals that the lowest unoccupied molecular orbitals are primarily localized on the N2O motif (see SI), suggesting that the one-electron reduced species will yield a N2O-centered radical.
We then investigated the electrochemical behaviour of compounds 3a–c, along with the parent compound 2, using cyclic voltammetry (CV). In dry THF (containing 0.1 M nBu4PF6 as the supporting electrolyte), 2 exhibits two irreversible one-electron processes at Ep,c = −2.86 V vs. Fc+/Fc and Ep,a = +0.68 V vs. Fc+/Fc (Fig. 2, green trace). Despite attempts to increase the scan rate (up to 1000 mV s−1) or decrease the temperature (down to 5 °C), redox reversibility could not be achieved, even to a limited extent. Such a result suggests that the in situ electrogenerated radical is highly unstable and further evolves in solution. Interestingly, dramatic differences were observed when recording the CVs of compounds 3a and 3b (Fig. 2, blue and red traces, respectively). In both cases, a single quasi-reversible process was observed to occur at strongly anodically-shifted potentials of −1.70 and −1.93 V vs. Fc+/Fc, for 3a and 3b, respectively. The borane moiety thus appears to have a dual effect: (i) by providing an increased steric hindrance and a partial electronic density delocalization, it firstly results in increased kinetic stability of the radical, ultimately leading to full reversibility recovery, and (ii), the decreased electronic density of the NHC·N2O moiety simultaneously lowers the energy required to initiate electron transfer from the electrode, resulting in the observed anodic shift. As a result, the potential required can be tuned by the nature of the Lewis acid partner, as evidenced by the 230 mV gain in potential when switching from the partially fluorinated borane 1b to the perfluorinated one 1a. Noteworthy, a similar beneficial effect of the addition of a Lewis-acidic borane was reported very recently in the one-electron reduction of NHC·CO2 adducts.27 Linear trends were observed when plotting the variation of the anodic and cathodic peak currents (ip,a and ip,c) of the electrochemical process versus the square root of the scan rate (from 100 mV s−1 to 1000 mV s−1, see SI), confirming the compounds to freely diffuse in solution. Meanwhile, the CV of 3c suggests that the Lewis acidity of the borane 1c is insufficient to achieve efficient stabilization of the associated radical species. An irreversible process was indeed observed, similar to parent compound 2 (see SI). Interestingly, the 2-phenyl imidazolium 4+ displays a reversible one-electron process at a similar potential of −2.18 V vs. Fc+/Fc (Fig. 2, dark grey trace). As previously investigated by the Ghadwal group,28 the unpaired electron density of the resulting neutral radical was found to be partially located on the carbene carbon atom, with significant contributions from the carbon atoms of the aryl rings. This prior work thus questions the spin distribution in Lewis-acid stabilized radical anions [3a–b˙]−, and more specifically, the extent of their delocalization.
 | ||
| Fig. 2 Cyclic voltammograms of compounds 2 (green trace), 3a (blue trace), 3b (red trace) and 4·BF4 (dark grey trace), recorded in THF (0.1 M nBu4NPF6) at a glassy carbon electrode (analyte concentration: 1 mM, scan rate: 100 mV s−1). | ||
To specifically address this point, we decided to synthesize the radical anions corresponding to the reduction of 3a and 3b. Encouraged by the reversible electrochemical behaviour of those compounds on the CV timescale, we thus turned our attention to their preparative chemical reduction. Notably, one-electron reduction using potassium graphite (KC8) in dry tetrahydrofuran enabled the isolation of the desired species in reasonable yields (>50%), as dark red solids. The structures of [K][3a˙] and [K][3b˙] were confirmed by single-crystal X-ray diffraction (Fig. 3, together with relevant bond lengths and angles summarized in Table 2). Upon comparison with the parent neutral compounds, notable modifications are identified. Increased bending of the N–N–O angles is observed, with values as low as 103.2° in [3a˙]−. To the best of our knowledge, it is one of the most acute angles reported for this structural motif so far, excluding cyclic derivatives.29–32 Meanwhile, the elongation of both the N–N and N–O bonds in the N2O motif is observed. The N–N bonds are observed in the range 1.34–1.35 Å in [3a˙]− and [3b˙]−, which are typically halfway between single and double bonds and thus suggest an overall bond order of 1.5. As for the N–O bonds, they range from 1.40 to 1.41 Å, consistent with single bond character. Conversely, contraction of the B–O and CNHC–N bonds is observed, suggesting increased interactions within the FLP-type adducts that may account for the observed persistence of the radicals. A significant change in the structural features of these radicals lies in the increased coplanarity of the N2O motif and the imidazolium ring: the dihedral angles associated with these two moieties shift from 19.80° and 23.55° in 3a–b to 5.6° and 2.5° for [3a˙]− and [3b˙]−, respectively. Similarly observed in radicals based on NHC–CO2–borane adducts,27 this overall flattening probably arises from increased conjugation within the radical.
 | ||
| Fig. 3 Molecular structures of [K][3a˙] (left), [K][3b˙] (centre) and [K][3c˙] (right), as determined by X-ray diffraction. Thermal ellipsoids shown at the 50% probability level. Hydrogen atoms were omitted for sake of clarity. | ||
| [K][3a˙] | [K][3b˙] | [K][3c˙] | |
|---|---|---|---|
| N1–N2 (Å) | 1.354(3) | 1.340(4) | 1.359(3) |
| N1–O1 (Å) | 1.412(4) | 1.400(3) | 1.392(3) |
| B1–O1 (Å) | 1.488(3) | 1.493(4) | 1.525(4) |
| C1–N2 (Å) | 1.316(4) | 1.312(4) | 1.315(4) |
| N2–N1–O1 (°) | 103.2 | 103.7 | 102.9 |
Interestingly, preparative one-electron reduction of 3c under identical conditions allowed the isolation of a few dark red crystals. Suitable for X-ray diffraction, they allowed the characterization of the molecular structure of [K][3c˙] (Fig. 3). No particular structural differences were noticed when compared with [K][3a˙] and [K][3b˙]. At first glance, this result may seem somewhat puzzling, as it is inconsistent with the electrochemical data that support a low-stability radical. We suspect, however, that the potassium cation derived from the reduction using KC8 plays a significant role in the stabilization of the radical. Mougel's group recently demonstrated that the presence of Lewis acidic alkali cations (K+, Rb+, Cs+) is an effective strategy for stabilizing low-valent iron-sulfur cubanes.33 We believe that a similar scenario is at play here, with K+ supplementing the limited Lewis acidity of BPh3.
Electron Paramagnetic Resonance (EPR) techniques were then used to gain further experimental insights into the electronic structure of the radicals [3a˙]− and [3b˙]− (Fig. 4). The continuous wave (cw) EPR spectrum of [3b˙]−, recorded at room temperature, exhibits three main features separated by 37.1 MHz (1.32 mT), consistent with the strong isotropic hyperfine coupling computed by DFT (Aiso,DFT = 29.1 MHz) for the N1 nucleus. Each main feature of the cw-EPR exhibits 7 lines separated by 3–5 MHz and additional shoulders. To better identify such smaller couplings, the HYSCORE spectrum of [3b˙]− was recorded at 70 K. In the left quadrant, two pairs of features are observed at [±5.00; ±8.71] and [±4.70; ±2.45] MHz. Their position and shape correspond to those expected for the double quantum/double quantum and double quantum/single quantum transitions of a nitrogen nucleus having a mainly isotropic hyperfine interaction (weak values of T tensor components compared to Aiso). In this case, the hyperfine Aiso and quadrupolar K constants can be computed using the analysis developed by Dikanov and the relationship:34
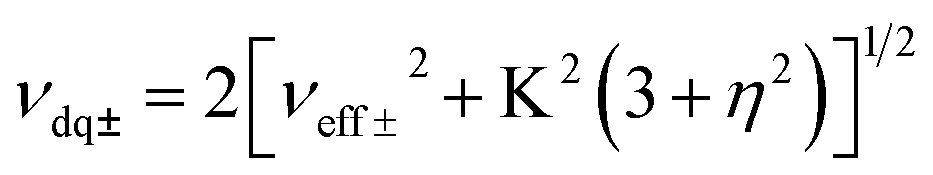

 | ||
| Fig. 4 EPR spectra of [3a˙]− and [3b˙]−, in solution at room temperature, together with their corresponding numerical simulations (left) and HYSCORE spectrum of [3b˙]− recorded at T = 70 K (right). | ||
This analysis provides hyperfine coupling of Aiso,exp = 5.9 MHz and a quadrupolar interaction in the range 0.81 < K < 0.93 (in MHz), in very good agreement with the values computed by DFT (Aiso,DFT = −5.4 MHz, KDFT = 0.87 MHz) for N2 (Table 3). In the right quadrant two very intense features are observed at [±4.71; ±2.44] MHz. They are typical of nitrogen nuclei with hyperfine coupling constants close to the so-called cancellation condition: A/2 ∼ ν(14N) (nuclear Larmor frequency for 14N, 1.07 MHz at 3460 G). These signals can be attributed to the N3 and N4 nuclei, as determined by DFT computations, since it is clear that, for some orientations of [3b˙]−vs. magnetic field, the hyperfine couplings of these nuclei fulfill the cancellation condition. However, the pronounced anisotropic character of the hyperfine tensors of N3 and N4 predicted by DFT computations (see the higher values of T tensors for N3 and N4 compared with N2 in Table 3), does not allow the use of the same quantitative analysis as for the N2 nucleus. The last two features are observed centered on the diagonal at 1.6 MHz and 4.8 MHz, which can be attributed, respectively, to the 10B and 11B isotopes of the boron nucleus. The signal centered at 4.8 MHz extends over a range of ∼3–4 MHz in the antidiagonal direction, in good agreement with the small and anisotropic hyperfine interaction computed by DFT for this 11B nucleus (Table 3). Noteworthy, no significant signal was observed that could be attributed to 1H or 19F nuclei, beyond a weak signal close to the diagonal at 14.7 MHz, corresponding to very small hyperfine couplings (<2 MHz; see full scale spectrum in the SI).
| N1 | N2 | N3 | N4 | B | Linewidth | ||
|---|---|---|---|---|---|---|---|
| [3a˙]− | A iso,exp (Aiso,DFT) | 36.8 (28.7) | EPR 4.3 (−4.4) | 3.6 (4.0) | 4.1 (4.3) | 1.4 (−3.7) | 4.7 |
| HYSCORE 5.9 | |||||||
| T DFT | [−29.3 −30.5 59.8] | [2.8 −0.7 −2.1] | [−4.4 −4.6 9.0] | [−4.1 −3.8 7.9] | [2.7 −1.2 −1.5] | ||
| K exp (KDFT) | n.m. (1.13) | 0.81 < K < 0.93 (0.86) | n.m. (−1.13) | n.m. (−1.14) | n.m. (0.04) | ||
| [3b˙]− | A iso,exp (Aiso,DFT) | 37.1 (28.7) | EPR 4.6 (−5.3) | 3.8 (3.8) | 4.5 (4.4) | 1.7 (4.1) | 3.4 |
| HYSCORE 5.9 | |||||||
| T DFT | [−29.3 −30.4 59.7] | [1.2 0.1 −1.3] | [−4.2 −4.4 8.6] | [−4.0 −3.6 7.6] | [2.8 −1.3 −1.5] | ||
| K exp (KDFT) | n.m. (1.11) | 0.81 < K < 0.93 (0.86) | n.m. (−1.15) | n.m. (−1.15) | n.m. (0.06) |
Moreover, the experimental room-temperature cw-EPR spectrum can be numerically simulated (Easyspin software package), considering only the isotropic hyperfine coupling of N1, N2, N3, N4 and B nuclei. The agreement between the numerically fitted and the experimental spectrum is excellent. The Aiso values extracted from this simulation match reasonably well those computed by DFT, which, in addition to the HYSCORE experiment, further validates experimentally the electronic structure of [3b˙]− calculated by DFT.
EPR and HYSCORE investigations of the radical [3a˙]− yielded very similar results. The HYSCORE spectrum is similar to that of [3b˙]− (see SI). The EPR spectrum of [3a˙]− recorded at room temperature is less resolved than the spectrum of [3b˙]−, but careful numerical simulation leads to very similar hyperfine coupling constants in good agreement with DFT computations. As expected, the plot of the Mulliken spin density for the one-electron reduced species indicates that most of the electron density is localized on the N2O moiety (e.g., ρ(N2O) = 0.71, Fig. 5A), in agreement with the EPR properties. We also note that the frontier molecular orbitals for [3a˙]− indicate that the highest occupied molecular orbital, which is located in the α-space, involves a π* orbital on the N2O moiety, consistent with the experimentally observed elongation of both the N–N and N–O bonds. Taken together, structural information, spectroscopic data and DFT calculations all point towards the reduced species being best described as aminyl radicals (Fig. 5B).
 | ||
| Fig. 5 (A) Isosurface (0.007 au) plot of the Mulliken spin population of the one-electron reduced species [3a˙]−. Hydrogen atoms have been omitted for clarity. (B) Corresponding Lewis representation of the aminyl radical [3a˙]−. | ||
Interestingly, we found that the radical anion [3a˙]− exhibits remarkable weeks-long persistence in solution at room temperature under an inert atmosphere (as determined by cw-EPR measurements compared to a TEMPO control). In contrast, exposure to oxygen leads to immediate re-oxidation to the neutral adduct 3a, likely via an outer-sphere electron transfer mechanism. While extended stability has been reported for structurally related radicals, it remains surprising in this case due to the strongly N-centered character of the radical, which is typically associated with short-lived, highly reactive species.
These results, therefore, point to an important kinetic stabilization capable of balancing the thermodynamic drivers typically associated with strongly localized organic radicals.35 The effective steric protection of the spin density provided by the highly congested molecular environment of our radicals is most probably responsible for this stabilization.
Conclusions
In summary, we report the synthesis, isolation and full characterization of the first molecular examples of N2O-based radical anions. Access to these species, best described as aminyl-type radicals, was enabled by a FLP-type strategy combining N-heterocyclic carbenes with boron-based Lewis acids. Electrochemical studies demonstrated that the nature and strength of the Lewis acid partner directly influence both the redox properties and the stability of the resulting radicals. A combination of cw-EPR spectroscopy, HYSCORE experiments and DFT calculations, confirmed that the spin density is primarily localized on the central nitrogen atom, yielding a highly N-centered radical. Remarkably, despite this localization, the radicals display significant persistence in solution under inert atmosphere – a feature attributed to the substantial steric protection afforded by the surrounding molecular environment.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
CCDC 2440410 to 2440415 contain the supplementary crystallographic data for this paper.60a–fThe authors have also cited additional references within the supporting information (SI).36–59 The data supporting this article (detailed synthetic procedures, NMR/IR/Raman spectra, additional EPR measurements, computational details and radical persistence assessment) have been included as part of the SI. Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc08152a.
Acknowledgements
Dr Dmitry Valyaev is kindly acknowledged for providing compound 4·BF4. NMR Spectroscopy Service (A. Bonnet) from the Laboratoire de Chimie de Coordination is acknowledged for technical assistance with NMR experiments. Dr Baptiste Martin is acknowledged for preliminary EPR experiments. Marine Tassé is acknowledged for Raman spectroscopy measurements. Dr Alix Sournia-Saquet is gratefully acknowledged for granting access to the electrochemistry facility at LCC. Dr Sébastien Bontemps is thanked for fruitful discussions. N. Q. would like to thank the CNRS and the ANR (grant ANR-24-CE07-5771-01) for financial support. A. O. B. acknowledges funding from the University of Toulouse for her PhD fellowship. Financial support from the IR INFRANALYTICS FR2054 for conducting the research is similarly gratefully acknowledged. This material is also based upon work supported by the National Science Foundation under Grant No. CHE-2400072.References
- Y. Kim and E. Lee, Chem.–Eur. J., 2018, 24, 19110–19121 CrossRef CAS.
- H. Song, E. Pietrasiak and E. Lee, Acc. Chem. Res., 2022, 55, 2213–2223 CrossRef CAS.
- S. Gao and F. Li, Adv. Funct. Mater., 2023, 33, 2304291 CrossRef CAS.
- S. Choe, C. Lim, S. Kang and E. Lee, Chem, 2025, 102714 Search PubMed.
- L. Y. M. Eymann, A. G. Tskhovrebov, A. Sienkiewicz, J. L. Bila, I. Živković, H. M. Rønnow, M. D. Wodrich, L. Vannay, C. Corminboeuf, P. Pattison, E. Solari, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2016, 138, 15126–15129 CrossRef CAS.
- J. Back, J. Park, Y. Kim, H. Kang, Y. Kim, M. J. Park, K. Kim and E. Lee, J. Am. Chem. Soc., 2017, 139, 15300–15303 CrossRef CAS PubMed.
- J. Park, H. Song, Y. Kim, B. Eun, Y. Kim, D. Y. Bae, S. Park, Y. M. Rhee, W. J. Kim, K. Kim and E. Lee, J. Am. Chem. Soc., 2015, 137, 4642–4645 CrossRef CAS PubMed.
- C. A. Lenart, A. Bhattacharjee, T. P. L. Cosby, G. Bélanger-Chabot and C. B. Caputo, J. Am. Chem. Soc., 2025, 147, 27043–27048 CrossRef CAS.
- Y. Kim and E. Lee, Chem. Commun., 2018, 54, 6824–6827 RSC.
- M. I. Al-Sheikhly, H.-P. Schuchmann and C. von Sonntag, Int. J. Radiat. Biol. Relat. Stud. Phys., Chem. Med., 1985, 47(4), 457–462 CAS.
- P. Popov, N. Getoff, J. Grodkowski, Z. Zimek and A. G. Chmielewski, Radiat. Phys. Chem., 2004, 69, 39–44 CrossRef CAS.
- V. A. Radtsig, Kinet. Catal., 2001, 42, 631–652 CrossRef CAS.
- A. G. Tskhovrebov, E. Solari, M. D. Wodrich, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2012, 51, 232–234 CrossRef CAS PubMed.
- A. G. Tskhovrebov, B. Vuichoud, E. Solari, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2013, 135, 9486–9492 CrossRef CAS PubMed.
- L. Y. M. Eymann, R. Scopelliti, F. T. Fadaei, G. Cecot, E. Solari and K. Severin, Chem. Commun., 2017, 53, 4331–4334 RSC.
- A. G. Tskhovrebov, E. Solari, M. D. Wodrich, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2012, 134, 1471–1473 CrossRef CAS.
- E. Theuergarten, T. Bannenberg, M. D. Walter, D. Holschumacher, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2014, 43, 1651–1662 RSC.
- A. G. Tskhovrebov, L. C. E. Naested, E. Solari, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2015, 54, 1289–1292 CrossRef CAS.
- A. Thakur, P. K. Vardhanapu, G. Vijaykumar, P. Kumar Hota and S. K. Mandal, Eur. J. Inorg. Chem., 2016, 2016, 913–920 CrossRef CAS.
- L. Y. M. Eymann, R. Scopelliti, F. F. Tirani and K. Severin, Chem.–Eur. J., 2018, 24, 7957–7963 CrossRef CAS PubMed.
- L. Y. M. Eymann, P. Varava, A. M. Shved, B. F. E. Curchod, Y. Liu, O. M. Planes, A. Sienkiewicz, R. Scopelliti, F. Fadaei Tirani and K. Severin, J. Am. Chem. Soc., 2019, 141, 17112–17116 CrossRef CAS.
- S. Keess, A. Simonneau and M. Oestreich, Organometallics, 2015, 34, 790–799 CrossRef CAS.
- E. Otten, R. C. Neu and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 9918–9919 CrossRef CAS PubMed.
- R. C. Neu, E. Otten, A. Lough and D. W. Stephan, Chem. Sci., 2011, 2, 170–176 RSC.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- A. Morales, C. Gonçalves, A. Sournia-Saquet, L. Vendier, A. Lledós, O. Baslé and S. Bontemps, Chem. Sci., 2024, 15, 3165–3173 RSC.
- D. Rottschäfer, B. Neumann, H.-G. Stammler, M. van Gastel, D. M. Andrada and R. S. Ghadwal, Angew. Chem., Int. Ed., 2018, 57, 4765–4768 CrossRef.
- T. J. King, P. N. Preston, J. S. Suffolk and K. Turnbull, J. Chem. Soc., Perkin Trans. 2, 1979, 1751–1757 RSC.
- G. Fischer, D. Hunkler, H. Prinzbach, G. Rihs and H. Fritz, Tetrahedron Lett., 1984, 25, 2459–2462 CrossRef CAS.
- D. Grossie, L. Harrison and K. Turnbull, Acta Crystallogr., Sect. E, 2013, 69, o1196 CrossRef CAS PubMed.
- D. Dumitrescu, S. Shova, C. Draghici, M. M. Popa and F. Dumitrascu, Molecules, 2021, 26, 3693 CrossRef CAS.
- L. Grunwald, M. Inoue, P. C. Carril, M. Wörle and V. Mougel, Chem, 2024, 10(1), 365–387 CAS.
- Q. Lam, C. Van Stappen, Y. Lu and S. A. Dikanov, J. Phys. Chem. B, 2024, 128, 3350–3359 CrossRef CAS PubMed.
- S. S. S. V., P. C. S. John and R. S. Paton, Chem. Sci., 2021, 12, 13158–13166 RSC.
- C. Wang, G. Erker, G. Kehr, K. Wedeking and R. Fröhlich, Organometallics, 2005, 24, 4760–4773 CrossRef CAS.
- J. A. Nicasio, S. Steinberg, B. Inés and M. Alcarazo, Chem.–Eur. J., 2013, 19, 11016–11020 CrossRef CAS.
- J. E. Borger, A. W. Ehlers, M. Lutz, J. C. Slootweg and K. Lammertsma, Angew. Chem., Int. Ed., 2016, 55, 613–617 CrossRef CAS.
- R. S. Ghadwal, S. O. Reichmann and R. Herbst-Irmer, Chem.–Eur. J., 2015, 21, 4247–4251 CrossRef CAS.
- R. K. Harris, E. D. Becker, S. M. C. De Menezes, P. Granger, R. E. Hoffman and K. W. Zilm, Magn. Reson. Chem., 2008, 46, 582–598 CrossRef CAS.
- R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow and P. Granger, Pure Appl. Chem., 2001, 73, 1795–1818 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian Inc., Wallingford CT Search PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, Chem.–Eur. J., 2012, 18, 9955–9964 CrossRef CAS PubMed.
- Y.-P. Li, J. Gomes, S. Mallikarjun Sharada, A. T. Bell and M. Head-Gordon, J. Phys. Chem. C, 2015, 119, 1840–1850 CrossRef CAS.
- A. V. Marenich, J. Ho, M. L. Coote, C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2014, 16, 15068–15106 RSC.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- V. Barone, in Recent Advances in Density Functional Methods, World Scientific, 1995, vol. 1, pp. 287–334 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A, 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C, 2015, 71, 3–8 CrossRef PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- International Tables for X-Ray Crystallography, ed. J. A. Ibers and W. C. Hamilton, Kynoch Press, Birmingham, England, 1974, vol. IV Search PubMed.
- P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A, 1990, 46, 194–201 CrossRef.
- F. Zimmermann, Th. Lippert, Ch. Beyer, J. Stebani, O. Nuyken and A. Wokaun, Appl. Spectrosc., 1993, 47, 986–993 CrossRef CAS.
- (a) CCDC 2440410: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2mxfxx; (b) CCDC 2440411: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2mxfyy; (c) CCDC 2440412: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2mxfzz; (d) CCDC 2440413: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2mxg01; (e) CCDC 2440414: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2mxg12; (f) CCDC 2440415: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2mxg23.
| This journal is © The Royal Society of Chemistry 2026 |