
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpatially confined single-molecule folding achieves multicolor phosphorescence
Xiaolu
Zhou†
ab,
Xin-Kun
Ma†
a,
Xiaoye
Zhang
a,
Shuihuan
Yu
a,
Zhaoyuan
Zhang
a and
Yu
Liu
 *a
*a
aCollege of Chemistry, State Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, P. R. China. E-mail: yuliu@nankai.edu.cn
bResearch Centre for Information Technology, Shenzhen University of Information Technology, Shenzhen 518172, P. R. China
First published on 24th November 2025
Abstract
Possessing two ethylene units, guest triphenylamine derivatives are encapsulated by the cucurbit[8]uril (CB[8]) cavity to induce molecule folding to form a biaxial pseudorotaxane. Crucially, this structure facilitates the oriented polymerization of ethylene units exposed outside the cavity with acrylamide, producing a pure organic supramolecular polymer (p-TAPCB) that exhibits near-infrared (NIR) room-temperature phosphorescence (RTP) with a high quantum yield of 47.03% through the synergistic effect of macrocyclic confinement and polymerization. By adjusting the ratio of CB[8], the resulting copolymers show tunable phosphorescence color that changes depending on the excitation wavelength and concentration. Furthermore, phosphorescence resonance energy transfer (PRET) is employed in triphenylenylborate (TPE) donor-doped polymeric films to flexibly manipulate the NIR emission lifetime from 9.87 ms up to 1490 ms with a large Stokes shift (390 nm). With the advantages of variable emission color and on-demand lifetime, these materials have potential applications in dynamic information encryption.
Introduction
Supramolecular macrocyclic assemblies has garnered widespread attention due to their extensive applications in the fields of catalysis,1 adsorption, and separation,2 luminescent materials,3 particularly in room-temperature phosphorescence (RTP) materials4 and supramolecular catalysis.5 These macrocyclic host molecules can induce guest chromophores' phosphorescence by restricting molecular motion and suppressing non-radiative transitions.6 Macrocyclic cascade assemblies further allow tuning of luminescence and enable phosphorescence resonance energy transfer (PRET).7 Additionally, the excellent molecular recognition capabilities of macrocyclic compounds and reversible guest encapsulation make them ideal for nanocatalysis. They can act as nanoreactors to catalyze cycloaddition,8 photodimerization,9 photolysis,10 oxidation,11 and hydrolysis,12 or as “reverse catalysts” by controlling reaction pathways through selective guest protection or suppression of radical group activity.13 However, macromolecular-confined single-molecule folding-driven selective oriented polymerization with controllable phosphorescence behavior has rarely been reported to the best of our knowledge.Developing efficient organic RTP materials typically involves crystallization,14 doping,15 polymerization,16 and host–guest interaction.17 Notably, most of the reported RTP systems emit visible light such as blue, green and yellow. Owing to the energy gap law, few pure organic NIR-RTP materials have been reported, with limitations of millisecond scale lifetimes and low quantum yields (Table S1).18 Furthermore, stimulus-responsive multicolor afterglow materials have received extensive attention for their potential applications in bioimaging agents, multicolor displays, and information encryption.19 For instance, Zhao et al. reported color-tunable ultralong organic RTP from bicomponent polymers of benzene and naphthalene.20 Qi et al. reported an excitation-dependent, water stimulus-responsive, and color-tunable RTP cellulose by introducing extended aromatic groups.21 Liu et al. reported a reversible thermally responsive multicolor-tunable supramolecule film based on phosphorescence resonance energy transfer and a thermally controlled host–guest binding mode.22 However, achieving efficient NIR luminescence in RTP materials, especially coupled with tunable afterglow emission, remains a great challenge.
Herein, we constructed a chain supramolecular biaxial polyrotaxane via photoinitiated polymerization of cucurbit[8]uril (CB[8]) encapsulated ethylene-bridged bis(triphenylamine) fold-dimers (TAP) with acrylamide (Scheme 1). Results from 2D rotating frame Overhauser effect spectroscopy (ROESY) and photoluminescence spectra indicated that CB[8] induces TAP folding into a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex, driving the oriented polymerization with acrylamide to form a “lantern-like” biaxial ring rotaxane polymer (p-TAPCB). Benefitting from the non-covalent host–guest interactions with CB[8] and the rigid covalent polymeric environment provided by the polymer network, the resultant supramolecular polymer p-TAPCB displayed robust NIR phosphorescence emission at 680 nm with a photoluminescence quantum yield of 47.03%. Crucially, by adjusting the CB[8] ratio during polymerization from 0 to 0.75 equivalent, an excitation-dependent, concentration-dependent, and color-tunable phosphorescence behavior ranging from blue, yellow, and red was achieved. To extend the NIR afterglow, we leveraged PRET by doping with long-lived phosphorescence donor triphenylenylborate (TPE), which prolonged the NIR emission lifetime of the energy acceptor p-TAPCB from 9.87 ms to 1490 ms with a large Stokes shift (390 nm). Significantly, these water-soluble NIR phosphorescence supramolecular copolymers displayed imaging ability to penetrate tissue and served as security inks applied in multilevel information encryption.
:1 complex, driving the oriented polymerization with acrylamide to form a “lantern-like” biaxial ring rotaxane polymer (p-TAPCB). Benefitting from the non-covalent host–guest interactions with CB[8] and the rigid covalent polymeric environment provided by the polymer network, the resultant supramolecular polymer p-TAPCB displayed robust NIR phosphorescence emission at 680 nm with a photoluminescence quantum yield of 47.03%. Crucially, by adjusting the CB[8] ratio during polymerization from 0 to 0.75 equivalent, an excitation-dependent, concentration-dependent, and color-tunable phosphorescence behavior ranging from blue, yellow, and red was achieved. To extend the NIR afterglow, we leveraged PRET by doping with long-lived phosphorescence donor triphenylenylborate (TPE), which prolonged the NIR emission lifetime of the energy acceptor p-TAPCB from 9.87 ms to 1490 ms with a large Stokes shift (390 nm). Significantly, these water-soluble NIR phosphorescence supramolecular copolymers displayed imaging ability to penetrate tissue and served as security inks applied in multilevel information encryption.
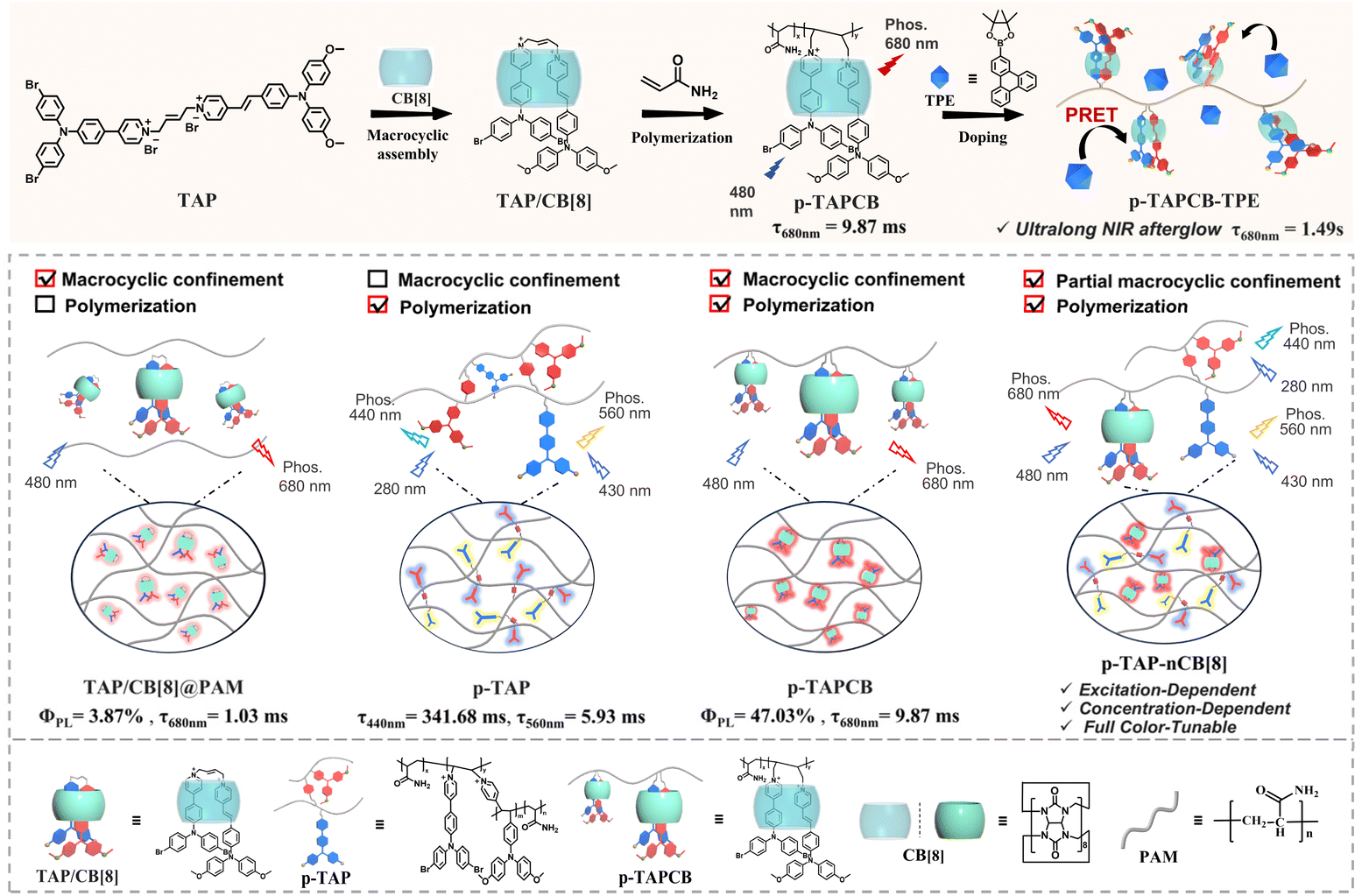 | ||
| Scheme 1 Schematic diagram of the synergistic assembly of the supramolecular polymer p-TAPCB, and concentration-dependent, excitation-dependent, multicolor luminescence, and PRET formation process. | ||
Results and discussion
A series of triphenylamine pyridine derivatives (G1, G2, TAP) were successfully synthesized and characterized by nuclear magnetic resonance spectroscopy (NMR) and high-resolution mass spectrometry (HR-MS) (Fig. S1–S4). The target molecule monomer TAP was assembled with CB[8] in a stoichiometric ratio of 1:1 and further utilized for binary radical copolymerization of acrylamide with a mass ratio of 1:200 (Fig. 1a), and the polymers p-TAPCB were produced and characterized by 1H NMR spectroscopy and gel permeation chromatography (GPC) (Fig. S5 and Table S2). Impressively, p-TAPCB emitted NIR phosphorescence at 680 nm in the solid state with a lifetime of 9.87 ms (Fig. 1b and S6). The temperature-dependent PL spectra further revealed the RTP emission characteristics, meanwhile excluding the possibility of thermally activated delayed fluorescence, in which the PL intensity greatly increased as the temperature decreased from 293 K to 80 K (Fig. S7). In addition, supramolecular polymers containing different mass ratios of phosphors were synthesized and characterized to explore the effect of the phosphor ratio on the photoluminescence of polymers. The X-ray powder diffraction spectrum showed no presence of sharp peaks, indicating that the obtained polymer p-TAPCB was amorphous (Fig. S8). As the chromophore ratio decreased, the luminous intensity increased around 2-fold, and the lifetime extended from 6.18 ms (1.5 wt%) to 9.87 ms (0.5 wt%) (Fig. 1c and S9). This improvement can be attributed to the low concentration of phosphorescent species that increased the molecular spacing, suppressed aggregation, and maintained the rigidity of the matrix, which significantly reduced the non-radiative transition rate. Therefore, we chose p-TAPCB (0.5 wt%) with a high photoluminescence quantum yield (47.03%) for further study.
 | ||
| Fig. 1 (a) Chemical structural scheme of supramolecular polymers p-TAPCB; (b) normalized excitation, PL, and phosphorescence spectra of copolymer p-TAPCB (delay time = 1 ms); (c) the photoluminescence quantum yield and lifetime at 680 nm of supramolecular polymers containing different mass ratios of TAP/CB[8] phosphors (p-TAPCB-1.5%, p-TAPCB-1.0% and p-TAPCB-0.5%) in the solid state at room temperature. | ||
Notably, the formation of a self-folding assembly via the binding of triphenylamine derivatives with CB[8] is a critical step in constructing the supramolecular polymer p-TAPCB, which depends on strong host–guest interaction. To explore the binding behavior of TAP/CB[8], we first investigated the binding affinity between guest molecule TAP and CB[8] through UV-vis spectroscopy. The absorption spectrum of TAP showed a strong absorption peak around 420 nm, and a weak absorption peak around 500 nm ascribed to the intramolecular charge transfer (ICT) (Fig. 2a). With the addition of CB[8], the absorption of TAP was redshifted from 440 to 480 nm with the solution color changing from yellow to red (Fig. 2a, inset), and the fluorescence was red-shifted from 680 nm to 690 nm accompanied by the enhancement of intensity, revealing the formation of assembly TAP/CB[8] (Fig. S10). The association constant (Ks) of TAP/CB[8] was determined to be 1.53 × 107 M−1 in aqueous solution by nonlinear least-squares analysis based on UV-vis spectroscopy titration (Fig. 2b). A Job plot indicated that the host–guest binding stoichiometry was 1:1 with a maximum molar fraction of 0.5 (Fig. 2b, inset). Moreover, the binding stoichiometry and binding constant were also investigated by the ITC method. The ITC curves fit well to one set of binding site models with n = 1 and binding constant K = 2.12 × 106 M−1 in the aqueous solution (Fig. S11). Then, the results of the 1H NMR titration experiment (Fig. 2c and S12), 2D correlation spectroscopy (COSY) and 2D ROESY (Fig. S13 and S14) provided more adequate details for the host–guest binding mode. As shown in Fig. 2c and S12, significant up-field shift of protons on pyridine salts in the aromatic region (H1−4, H1’−4’) occurred as a result of the shielding effect of the CB[8] cavity, while the hydrogen proton on the triphenylamine unit (H5, H5’, H6’) and 2-butylene moiety (Ha, Ha’) outside the cavity shifted downfield. 2D ROESY of TAP/CB[8] showed obvious correlation signals between the pyridinium protons and vinyl protons (H(1’, 2), H(3’, 4)) in the two arms, indicating that the guest TAP folded into the cavity of CB[8] in ‘head-to-head’ to form a 1:1 host–guest complex (Fig. S14).
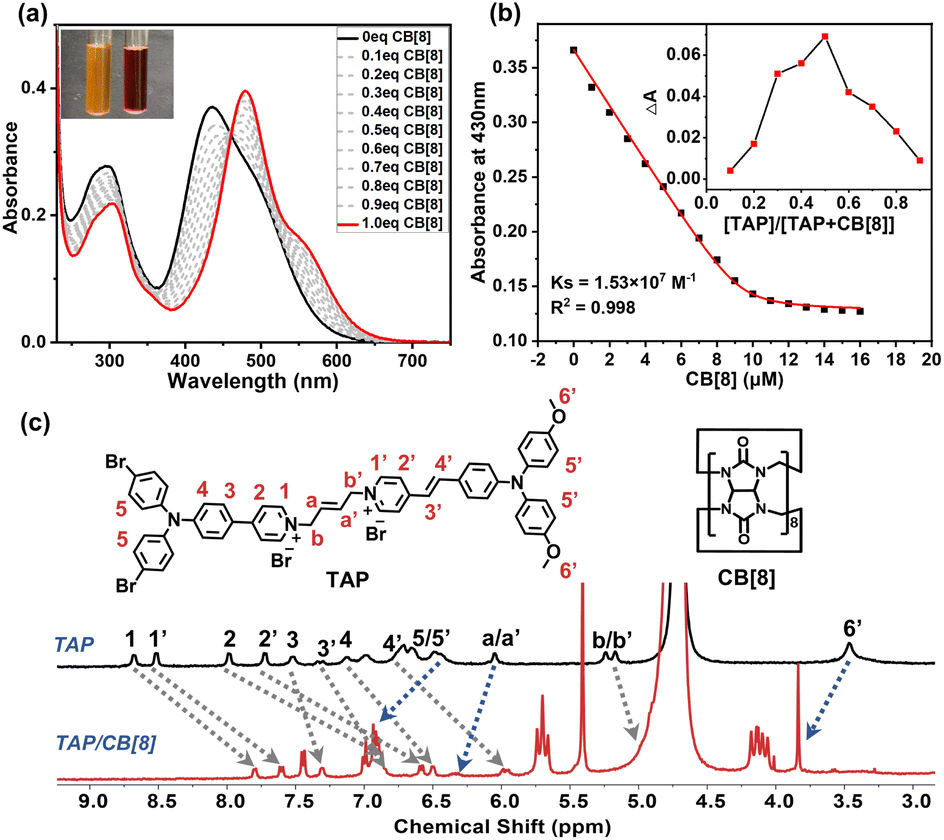 | ||
| Fig. 2 (a) UV-vis absorption spectra of aqueous solutions of TAP (10 µM), and CB[8] at concentrations ranging from 0 to 10 µM under ambient conditions. (b) The nonlinear least-squares analyses of the UV absorbance changes with addition 0–1.6 eq CB[8] to calculate the association constant between TAP and CB[8] (absorbance recorded at 430 nm). Inset: Job plots of the complex between TAP and CB[8] ([TAP] + [CB[8]] = 10 µM). (c) 1H NMR spectral changes of TAP and TAP/CB[8] ([TAP] = [CB[8]] = 0.5 mM) (400 MHz, D2O with 5%DMSO-d6, 298 K). | ||
The synergistic enhancement strategy of polymerization and host–guest complexation was proved effective to achieve ultralong and efficient RTP by suppressing molecular vibrations and isolating quenchers.23 To verify the effect of macrocyclic confinement interaction and polymerization on the luminescence properties, a series of TAP/CB[8]-doping systems based on polyvinyl pyrrolidone (TAP/CB[8]@PVP), polyacrylamide (TAP/CB[8]@PAM) and polyvinyl alcohol (TAP/CB[8]@PVA), and a macrocyclic-free polymer (p-TAP) were prepared and characterized. Photoluminescence and phosphorescence spectra showed that the doping system of TAP/CB[8]@PVP and TAP/CB[8]@PAM exhibited weak phosphorescent peaks at 680 nm with a relatively short lifetime of 0.68 ms and 1.03 ms, and low photoluminescence quantum yield of 3.31% and 3.87%, respectively (Table S3 and Fig. S15). For TAP/CB[8]@PVA, due to the confinement and cavity shielding effect of CB[8] and the stronger hydrogen-bonding abundancy provided by PVA, the phosphorescence emission lifetime increased to 5.19 ms, and the quantum yield increased to 14.59% (Table S3 and Fig. S15). Impressively, compared with macrocyclic supramolecule polymer p-TAPCB, the PL spectrum of macrocyclic-free polymer p-TAP without CB[8] showed phosphorescence emission at 560 nm with a short lifetime of 5.93 ms and a low photoluminescence quantum yield of 10.53% (Table S3 and Fig. S16). The above experimental results confirmed that the encapsulation of CB[8] confined self-folding of TAP is the prerequisites for achieving NIR phosphorescence, and the weaker luminescence performance of the doped polymer TAP/CB[8]@PAM and TAP/CB[8]@PVA compared to the copolymerized one p-TAPCB, revealed the important role of copolymerization in enhancing the phosphorescence properties.
Interestingly, by altering the CB[8] ratio, the polymer p-TAP-nCB[8] demonstrated excitation-dependent color-tunable phosphorescence behavior. As shown in Fig. 3a and S16, with an excitation wavelength increase from 270 nm to 500 nm, the maximum phosphorescence peak of the phosphorescence spectrum for p-TAP shifted from 440 nm to 560 nm accompanied by tunable color from blue to green to yellow (Fig. 3c). For p-TAP-0.2CB[8], the phosphorescent emission peak red-shifted from 440 nm to 630 nm upon the increase of excitation wavelength, and the afterglow color changed from blue to orange and then to red (Fig. 3b and c and S17). Similarly, the CIE diagram of the afterglow of p-TAP-0.1CB[8] moved from the blue region to the orange region with the red-shifted excitation wavelength, while p-TAP-0.75CB[8] ranged from blue to pink and red (Fig. 3c and S18). Accordingly, under the same excitation, by adjusting the concentration of CB[8] from 0 to 0.75 equivalent, the resulting polymer also exhibited concentration-dependent phosphorescent luminescence. As shown in Fig. 3d and e, with the increase of the molar ratio of CB[8], the phosphorescence spectra showed an obvious redshift at the excitation wavelength of 330–440 nm. The afterglow images showed that p-TAP, p-TAP-0.1CB[8], p-TAP-0.2CB[8] and p-TAP-0.75CB[8] exhibited distinct afterglow colors when excited at various wavelengths of 254 nm, 300 nm, 365 nm and 405 nm. Moreover, under identical ultraviolet excitation, the increasing concentration of CB[8] drove the afterglow of p-TAP-nCB[8] transformation from the blue-green to the yellow-red region (Fig. 3f).
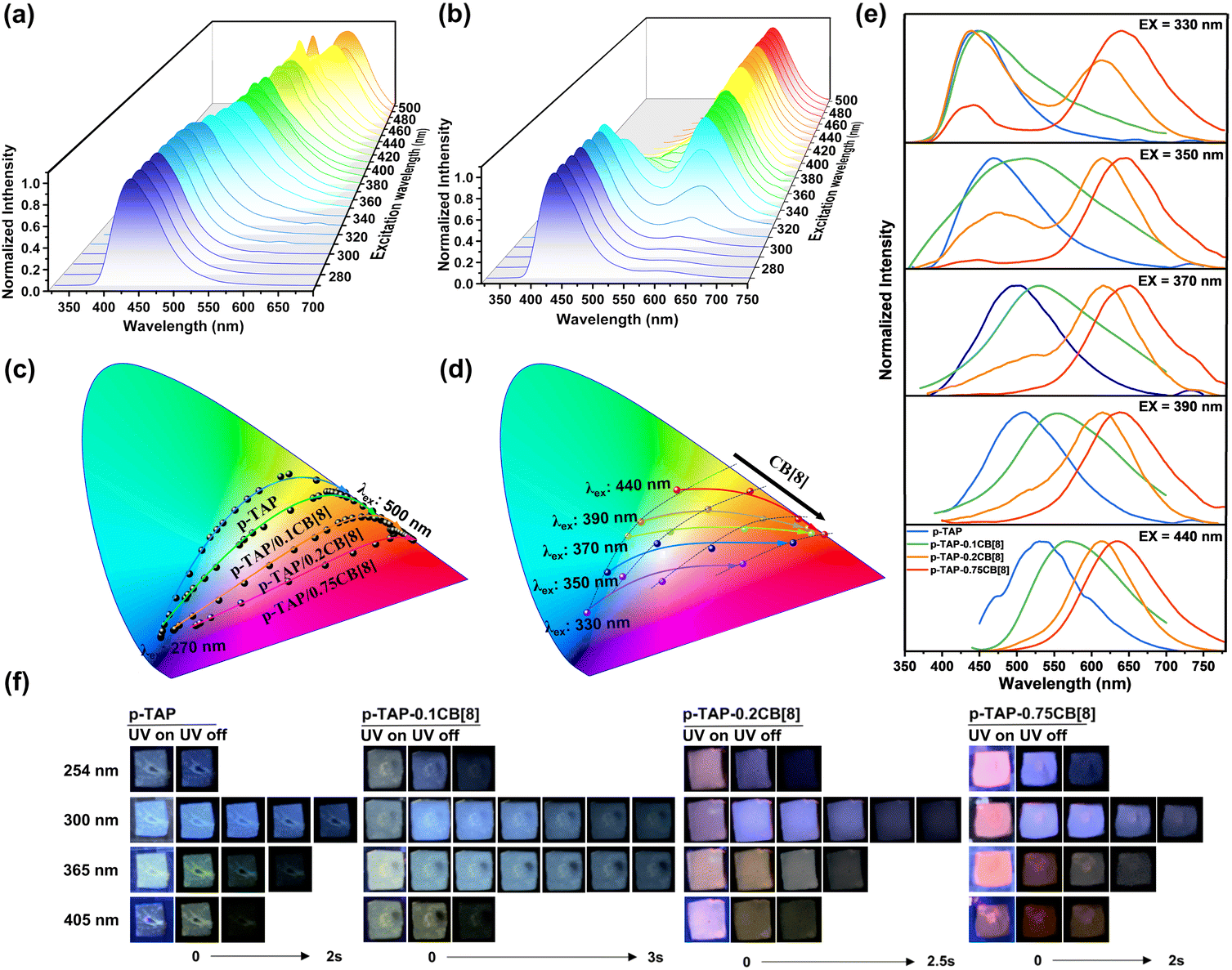 | ||
| Fig. 3 (a and b) Phosphorescence spectra of the polymers p-TAP (a) and p-TAP-0.2CB[8] (b) at different excitation wavelengths. (c) CIE coordinate diagrams of the excitation-dependent color-tunable phosphorescence emission. (d) CIE coordinate diagrams of the CB[8] concentration-dependent color-tunable phosphorescence emission. (e) Phosphorescence spectra of the polymers p-TAP, p-TAP-0.1CB[8], p-TAP-0.2CB[8], and p-TAP-0.75CB[8] at excitation wavelengths of 330 nm, 350 nm, 370 nm, 390 nm and 440 nm. (f) Under different UV excitations, the afterglow images of supramolecular polymers at different CB[8] polymerization concentrations. | ||
To study the principle of excitation-dependence and concentration-dependent behavior, the luminescence characteristics of reference compounds G1 and G2 were investigated. The experimental results demonstrated that the doped system G1@PAM emitted 720 nm fluorescence with a lifetime of 0.96 ns, but no phosphorescence signal was captured (Fig. S19). However, when G1 reacted with acrylamide through photoinitiated polymerization, the resultant copolymer G1-PAM exhibited a blue phosphorescence emission at 440 nm with a lifetime of up to 337.03 ms (Fig. S20). Fourier transform infrared (FTIR) spectra revealed that the ethylenic bond functional group in G1 vanished following polymerization, and the source of the emission was identified as the methoxytriphenylamine unit24 (Fig. S21). The doping system G2@PAM and copolymer G2-PAM both displayed phosphorescent emission peaks at 560 nm under the excitation of 440 nm and lifetimes were recorded as 0.29 ms and 4.95 ms, respectively, indicating that polymerization promoted the phosphorescence properties of the chromophore (Fig. S22). Correspondingly, the polymer p-TAP exhibited a blue phosphorescence emission at 440 nm (λex = 280 nm) with a lifetime of 341.68 ms and yellow phosphorescence emission at 560 nm (λex = 430 nm) with a lifetime of 5.93 ms (Fig. S16). Polymer p-TAP-0.2CB[8] displayed three phosphorescence emission peaks at 440 nm (τ = 211.01 ms), 560 nm (τ = 12.05 ms) and 640 nm (τ = 11.73 ms) under excitation of 280 nm, 430 nm and 480 nm, respectively (Fig. S17). In addition, compared with p-TAP-CB[8], the control experiments with CB[7]-based analogous polymers (p-TAP-CB[7]) displayed a similar luminescence behavior to p-TAP, including phosphorescence peaks at 450 nm with a lifetime of 183.47 ms and 560 nm with a lifetime of 6.02 ms (Fig. S23). The above experimental results confirmed that the excitation dependence was caused by the covalent connection of different components, and the assembly of CB[8] could effectively protect the vinyl group in the aromatic region from polymerization with acrylamide to achieve NIR phosphorescence emission. By adjusting the concentration of CB[8] for polymerization, the proportion of three luminous components with different phosphorescence emission centers in the covalent system was changed, including the methoxytriphenylamine unit with a phosphorescence peak at 440 nm, the bromotrianiline-pyridine unit with a phosphorescence peak at 560 nm, and CB[8]-biaxial pseudorotaxane with a phosphorescence peak at 680 nm, thereby achieving distinct excitation-dependent and concentration-dependent phosphorescence effects (Scheme 1).
It is well known that PRET has been proven to be a concise approach to achieve persistent tunable-color afterglow.25 Accordingly, we selected ultralong phosphor TPE as an energy donor to construct donor-doped polymeric films, expecting to prolong the NIR emission lifetime of p-TAPCB via PRET. The afterglow emission spectrum of TPE is well overlapped with the excitation spectrum of p-TAPCB (Fig. 4a and S24), providing the probability of realizing highly efficient PRET. Experimentally, to illustrate the detailed energy transfer process, we investigated the luminescence properties of TPE-doped films (p-TAPCB-TPE) with doping weight concentrations ranging from 0.1 to 10 weight percent (wt%). As shown in Fig. 4b, the delayed spectra of the p-TAPCB-TPE films exhibited an obvious additional red emission peak at 680 nm under a short-wavelength excitation of 290 nm, the optimum excitation wavelength on the TPE part inside. With the increase of the donor (TPE) content (0.1% to 10%), the peak intensity at 680 nm in delayed spectra exhibited an obvious rise, thus resulting in a gradual red-shift of the CIE coordinates (Fig. S25). In the meantime, the luminescence lifetime at 445 nm of p-TAPCB-TPE films correspondingly reduced from 3.39 s to 2.28 s (Fig. 4c), while, in the control group, the phosphorescence lifetime of TPE@PAM films exhibited a slight change (3.48 s – 3.10 s) in different contents (0.1% to 10%) (Fig. S26). The decreased lifetime indicated the energy transfer process occurred at the excited triplet states, which was further confirmed by the increased emission lifetime at 680 nm from 9.87 ms of p-TAPCB to 1.49 s of p-TAPCB-TPE (10%) (Fig. 4d). In contrast, the optimal excitation wavelength (480 nm) of p-TAPCB cannot excite the doped film to produce long-lifetime emission. Additionally, the p-TAPCB-TPE film with a TPE doping ratio of 10% emitted an orange afterglow for 5 seconds; meanwhile, a naked-eye invisible red afterglow pattern could be captured by a mobile phone camera covered with a filter (630 nm cut-off) (Fig. 4e). This macroscopic afterglow color change hinted at the occurrence of an energy transfer process. The above results demonstrated that the long-lived triplet excitons of the TPE donor and efficient PRET process are the key factors to realize the ultra-long lifetime characteristic at 680 nm of p-TAPCB-TPE.
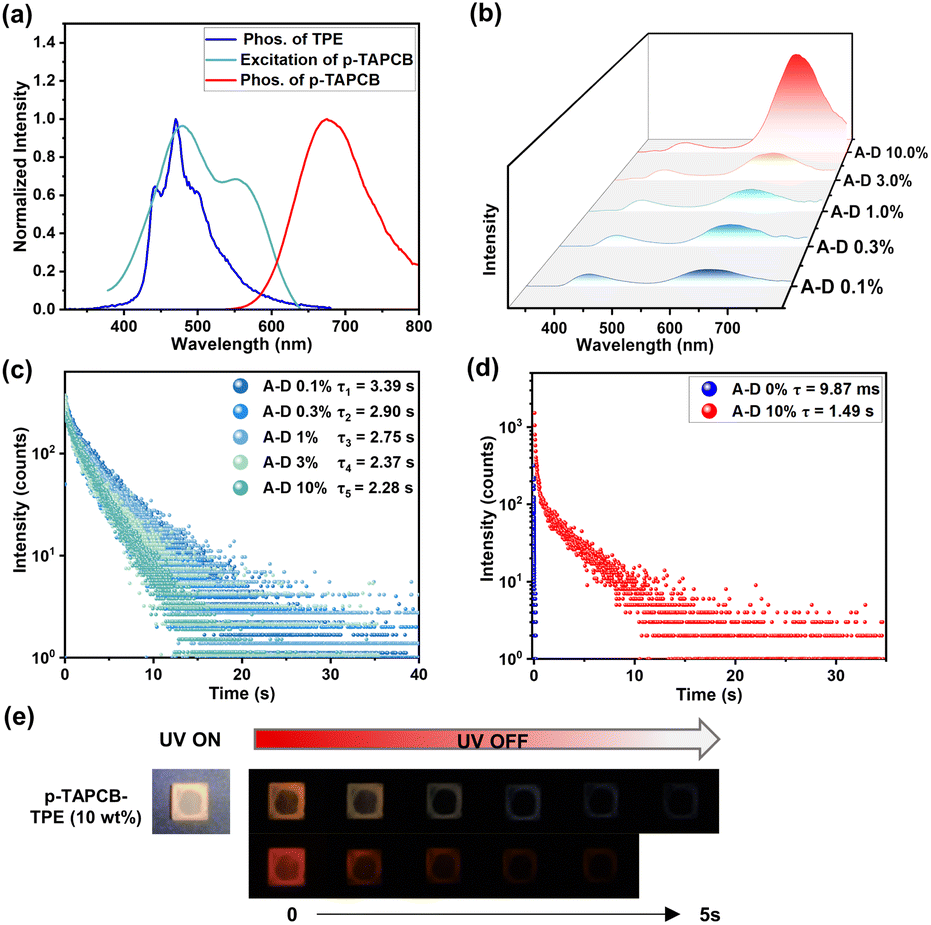 | ||
| Fig. 4 (a) Normalized phosphorescence emission spectrum of TPE@PAM, and the excitation and phosphorescence spectra of p-TAPCB. (b) Delayed spectra of TPE-doped systems (p-TAPCB-TPE) with different doping weight concentrations (A: p-TAPCB; D: TPE; λex = 290 nm, delay time = 1 ms, normalized at 450 nm). (c) The time-correlated decay curves of TPE-doped systems (p-TAPCB-TPE) with different doping weight concentrations (recorded at 450 nm). (d) The time-correlated decay curves of p-TAPCB and p-TAPCB-TPE (10%) (recorded at 680 nm). (e) Photographs of p-TAPCB-TPE (10%) under 300 nm UV light, the visible afterglow and the NIR afterglow with a filter (630 nm cut-off) after turning off the UV light. | ||
Another ultralong phosphor PAMCz, with 450 nm afterglow emission, was selected as the energy donor for the repeated experiments (Fig. S27 and S28). The experimental results indicated that PAMCz doped systems also exhibited long-lived emission at 680 nm (τ = 2.27 s with 2 wt% PAMCz), implying the energy transfer from PAMCz to p-TAPCB (Fig. S29–32). Moreover, the construction of TPE and PAMCz doped systems with changing the monomer content of p-TAPCB-1.5% also obtained similar phenomena (Fig. S33, S34 and Table S4). These results demonstrate that the NIR emission of the p-TAPCB can be extended by an efficient PRET process, regardless of single-molecule or polymer doping systems, providing an alternative general strategy for developing NIR afterglow materials with tunable lifetime and high quantum yield. Impressively, these donor-doped polymeric films displayed a large Stokes shift (390 nm) with one single-step energy transfer.
To study the application of macromolecule-induced single-molecule folding and polymerization synergism color-tunable afterglow luminescence, we systematically evaluated its performance in multi-level optical anti-counterfeiting and dynamic information encryption. For the encryption experiments, we designed a UV-triggered hierarchical encryption system using the aqueous solution of p-TAPCB and its doped PRET system as ink. Under ambient daylight, the pre-written numbers “8, 8, 8, 8” created by p-TAPCB and its doped system exhibited no discernible difference between the interference and confidential information (Fig. 5a). While, under 450 nm, the UV lamp which can only excite the p-TAPCB part, the first level encrypted information ‘838U’ in red color was captured. Then, the second level encrypted information ‘LIFE’ in a different color was captured after the 300 nm UV lamp was turned off and finally exhibited the word ‘IE’ with change over time. Impressively, the NIR letters “LFL” can be captured by a mobile phone camera covered with a filter (630 nm cut-off) with the bright red afterglow emission and finally exhibited the word ‘FL’ with change over time, which gives prominence to the third level encrypted information. Due to the good penetration to the tissue of NIR light, this persistent NIR light can be captured behind a piece of 3 mm thick pig skin (Fig. 5b). Furthermore, the aqueous solution of p-TAP-nCB[8] with different polymerization concentrations of CB[8] exhibiting multicolor afterglow also can be used as a commercial colorful anti-counterfeiting ink. This ink was deposited on a pre-printed image, forming a pattern of one moon and two stars, and then dried at 80 °C. As shown in Fig. 5c, after stopping the ultraviolet excitation, a logo of “Moon and Stars” appeared, and different afterglow colors ranging from blue, green, yellow and orange appeared depending on different excitation wavelengths of 254 nm, 300 nm, 365 nm, 405 nm, thus achieving the encryption process. These experiments establish the material's versatility in advanced encryption. The combination of time-resolved, excitation-dependent, and concentration-dependent afterglow provides a robust platform for multi-modal security.
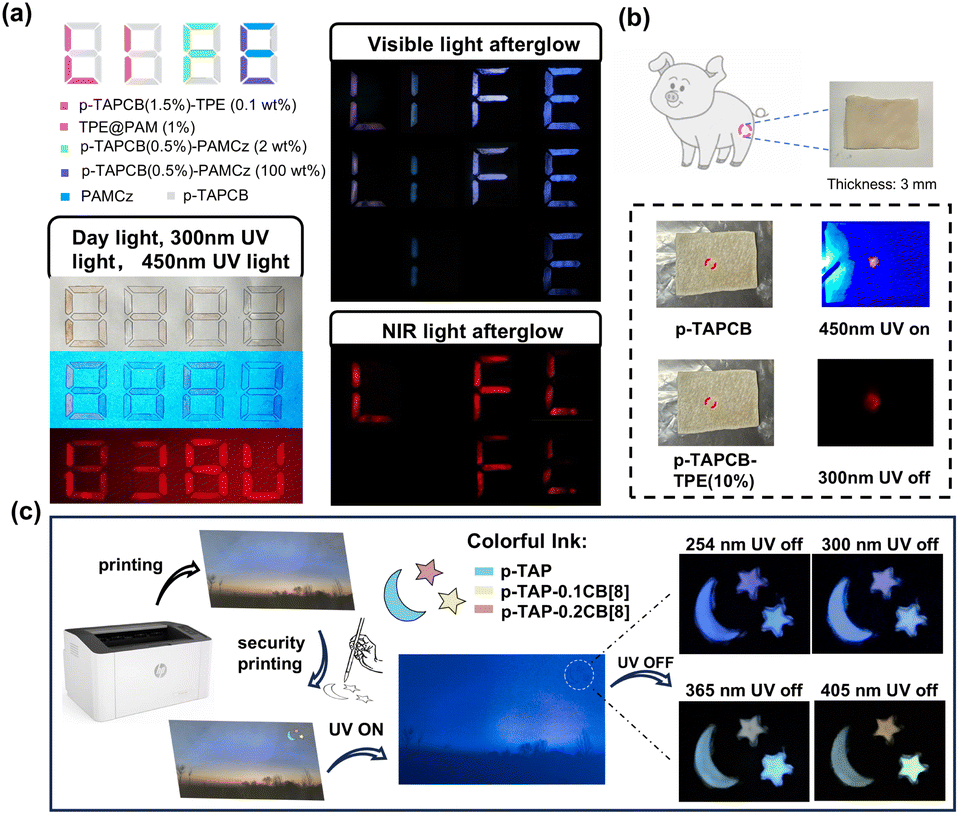 | ||
| Fig. 5 (a) Schematic of multi-mode numeric encryption based on p-TAPCB doped with different donors: the encrypted information image under daylight, 300 nm, and 450 nm UV light; the encrypted information afterglow image after 300 nm UV light was turned off; the encrypted information NIR afterglow image recorded by the cellphone camera covered with an optical filter (630 nm cutoff). (b) The photos were taken behind a piece of pig skin with a 3 mm thickness. (c) Using p-TAP, p-TAP-0.1CB[8], and p-TAP-0.2CB[8] as ink to make anti-counterfeiting marks and locally enlarged views of anti-counterfeiting afterglow marks applied to multicolor anti-counterfeiting. | ||
Conclusions
In summary, a NIR phosphorescent supramolecular polymer with biaxial polyrotaxane side chains was constructed based on configuration-confined covalent polymerization between CB[8] encapsulated ethylene-linked triphenylamine fold-dimers and acrylamide. The non-covalent host–guest assembly and the rigid covalent polymeric framework provided by CB[8]-driven molecular folding-oriented polymerization endowed the supramolecular polymer with an ultra-high NIR photoluminescence quantum yield of 47.03% and a lifetime of 9.87 ms at room temperature. Further studies demonstrated that the NIR emission lifetime could be significantly extended from milliseconds to seconds (1490 ms) by incorporating a long-lived phosphor as a donor to construct a PRET system. More interestingly, by altering the concentration of CB[8], sustainable afterglows with color-tunable, excitation-dependence, and concentration-dependence can be achieved. This study not only enriches high-performance NIR phosphorescent materials but also provides a versatile platform to construct multicolor smart luminescent materials with long-lived afterglow.Author contributions
X. Z. and X.-K. M. designed and performed all experiments. X. Z. wrote the manuscript. X. Z., S. Y. and Z. Z. gave valuable advice on data analysis and revision. Y. L. supervised the work and edited the manuscript. All authors analyzed and discussed the results and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: details of the characterization of target compounds, NMR data, photoluminescence/delayed spectra and time-resolved spectra. See DOI: https://doi.org/10.1039/d5sc08027d.Acknowledgements
The authors thank the National Nature Science Foundation of China (NNSFC, Grant No. 22131008) for financial support.Notes and references
- (a) U. S. Kanchana, E. J. Diana, T. V. Mathew and G. Anilkumar, Carbohydr. Res., 2020, 489, 107954 CrossRef CAS PubMed; (b) K. I. Assaf and W. M. Nau, Chem. Soc. Rev., 2015, 44, 394 RSC.
- (a) J.-R. Wu, G. Wu, D. Li, D. Dai and Y.-W. Yang, Sci. Adv., 2022, 8, eabo2255 CrossRef CAS; (b) R.-H. Gao, Y. Huang, K. Chen and Z. Tao, Coord. Chem. Rev., 2021, 437, 213741 CrossRef CAS; (c) Q. Liu, Y. Zhou, J. Lu and Y. Zhou, Chemosphere, 2020, 241, 125043 CrossRef CAS.
- (a) X. Ma and H. Tian, Acc. Chem. Res., 2014, 47, 1971 CrossRef CAS PubMed; (b) K. Huang, Q. Fang, W. Sun, S. He, Q. Yao, J. Xie, W. Chen and H. Deng, J. Phys. Chem. Lett., 2022, 13, 419 CrossRef CAS; (c) X. Zhou, X. Bai, F. Shang, H.-Y. Zhang, L.-H. Wang, X. Xu and Y. Liu, Nat. Commun., 2024, 15, 4787 CrossRef CAS PubMed.
- (a) X.-K. Ma and Y. Liu, Acc. Chem. Res., 2021, 54, 3403 CrossRef CAS; (b) X.-K. Ma, X. Zhou, J. Wu, F.-F. Shen and Y. Liu, Adv. Sci., 2022, 9, 2201182 CrossRef CAS PubMed; (c) T. Zhang, X. Ma, H. Wu, L. Zhu, Y. Zhao and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 11206 CrossRef CAS PubMed.
- (a) N. Dai, R. Qi, H. Zhao, L. Liu, F. Lv and S. Wang, Chem.–Eur. J., 2021, 27, 11567–11573 CrossRef CAS; (b) S. Cao, W. Wu, C. Liu, L. Song, Q. Xu, H. Zhang and Y. Zhao, Energy Environ. Mater, 2024, 7, e12702 CrossRef CAS.
- (a) Z.-Y. Zhang, Y. Chen and Y. Liu, Angew. Chem., Int. Ed., 2019, 58, 6028 CrossRef CAS PubMed; (b) D. Li, F. Lu, J. Wang, W. Hu, X.-M. Cao, X. Ma and H. Tian, J. Am. Chem. Soc., 2018, 140, 1916 CrossRef CAS PubMed; (c) X.-K. Ma, W. Zhang, Z. Liu, H. Zhang, B. Zhang and Y. Liu, Adv. Mater., 2021, 33, 2007476 CrossRef CAS PubMed.
- (a) X.-Y. Dai, M. Huo and Y. Liu, Nat. Rev. Chem., 2023, 7, 854 CrossRef CAS PubMed; (b) D. Li, Z. Liu, M. Fang, J. Yang, B. Z. Tang and Z. Li, ACS Nano, 2023, 17, 12895 CrossRef CAS; (c) M. Huo, X.-Y. Dai and Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 27171 CrossRef CAS.
- (a) D. Tuncel, Ö. Özsar, H. B. Tiftik and B. Salih, Chem. Commun., 2007, 13, 1369 RSC; (b) Y. C. Tse and H. Y. Au-Yeung, Chem.–Asian J., 2023, 18, e202300290 CrossRef CAS.
- (a) J.-s. Geng, L. Mei, Y.-y. Liang, L.-y. Yuan, J.-p. Yu, K.-q. Hu, L.-h. Yuan, W. Feng, Z.-f. Chai and W.-q. Shi, Nat. Commun., 2022, 13, 2030 CrossRef CAS PubMed; (b) C. Yang, T. Mori, Y. Origane, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, J. Am. Chem. Soc., 2008, 130, 8574 CrossRef CAS PubMed.
- A. L. Koner, C. Márquez, M. H. Dickman and W. M. Nau, Angew. Chem., Int. Ed., 2011, 50, 545 CrossRef CAS.
- (a) Y. Jiao, B. Tang, Y. Zhang, J.-F. Xu, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2018, 57, 6077 CrossRef CAS; (b) H. Cong, T. Yamato and Z. Tao, New J. Chem., 2013, 37, 3778 RSC.
- P. Ruz, S. Banerjee, R. Khurana, N. Barooah, V. Sudarsan, A. C. Bhasikuttan and J. Mohanty, ACS Appl. Mater. Interfaces, 2021, 13, 16218 CrossRef CAS.
- (a) L. S. Berbeci, W. Wang and A. E. Kaifer, Org. Lett., 2008, 10, 3721 CrossRef PubMed; (b) S. Mommer, K. Sokołowski, M. Olesińska, Z. Huang and O. A. Scherman, Chem. Sci., 2022, 13, 8791 RSC.
- (a) H. Zhu, I. Badía-Domínguez, B. Shi, Q. Li, P. Wei, H. Xing, M. C. Ruiz Delgado and F. Huang, J. Am. Chem. Soc., 2021, 143, 2164 CrossRef CAS; (b) X. Zhang, X. Li, Z. Wang, L. Bai, H. Qu and S. Xu, Chem. Res. Chinese U., 2023, 39, 960 CrossRef CAS.
- K. C. Chong, C. Chen, C. Zhou, X. Chen, D. Ma, G. C. Bazan, Z. Chi and B. Liu, Adv. Mater., 2022, 34, 2201569 CrossRef CAS.
- N. Gan, X. Zou, M. Dong, Y. Wang, X. Wang, A. Lv, Z. Song, Y. Zhang, W. Gong, Z. Zhao, Z. Wang, Z. Zhou, H. Ma, X. Liu, Q. Chen, H. Shi, H. Yang, L. Gu, Z. An and W. Huang, Nat. Commun., 2022, 13, 3995 CrossRef CAS PubMed.
- (a) Z.-Y. Zhang and Y. Liu, Chem. Sci., 2019, 10, 7773 RSC; (b) X. Zhou, H. Zhang and Y. Liu, Chem. Sci., 2024, 15, 18259 RSC.
- F. Xiao, H. Gao, Y. Lei, W. Dai, M. Liu, X. Zheng, Z. Cai, X. Huang, H. Wu and D. Ding, Nat. Commun., 2022, 13, 186 CrossRef CAS.
- (a) D. Li, J. Yang, M. Fang, B. Z. Tang and Z. Li, Sci. Adv., 2022, 8, eabl8392 CrossRef CAS PubMed; (b) S. Xiong, Y. Xiong, D. Wang, Y. Pan, K. Chen, Z. Zhao, D. Wang and B. Z. Tang, Adv. Mater., 2023, 35, 2301874 CrossRef CAS PubMed.
- L. Gu, H. Wu, H. Ma, W. Ye, W. Jia, H. Wang, H. Chen, N. Zhang, D. Wang, C. Qian, Z. An, W. Huang and Y. Zhao, Nat. Commun., 2020, 11, 944 CrossRef CAS.
- F. Peng, Y. Chen, H. Liu, P. Chen, F. Peng and H. Qi, Adv. Mater., 2023, 35, 2304032 CrossRef CAS PubMed.
- X. Zhou, X. Zhao, X. Bai, Q. Cheng and Y. Liu, Adv. Funct. Mater., 2024, 34, 2400898 CrossRef CAS.
- Z.-Y. Zhang, W.-W. Xu, W.-S. Xu, J. Niu, X.-H. Sun and Y. Liu, Angew. Chem., Int. Ed., 2020, 59, 18748 CrossRef CAS PubMed.
- A. Cheng, Y. Jiang, H. Su, B. Zhang, J. Jiang, T. Wang, Y. Luo and G. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202206366 CrossRef CAS PubMed.
- S. Kuila and S. J. George, Angew. Chem., Int. Ed., 2020, 59, 9393 CrossRef CAS PubMed.
Footnote |
| † Authors with equal contribution to the work. |
| This journal is © The Royal Society of Chemistry 2026 |