
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-innocent behaviour of aromatic isocyanides under visible light: a pathway to thioformimidates and dehydroalanine
Minghui
Wu
 a,
Jay
Hanssens
a,
Camilla
Russo
b,
Rajat
Walia
c,
Mariateresa
Giustiniano
*b,
Ludovic
Troian-Gautier
de,
Jordy M.
Saya
a,
Romano
Orru
*a and
Prabhat
Ranjan
*ad
a,
Jay
Hanssens
a,
Camilla
Russo
b,
Rajat
Walia
c,
Mariateresa
Giustiniano
*b,
Ludovic
Troian-Gautier
de,
Jordy M.
Saya
a,
Romano
Orru
*a and
Prabhat
Ranjan
*ad
aBiobased Organic Chemistry, Aachen-Maastricht Institute for Biobased Materials (AMIBM), Maastricht University, Urmonderbaan 22, 6167RD Geleen, The Netherlands
bDepartment of Pharmacy, University of Naples Federico II, Via D. Montesano 49, 80131 Napoli, Italy
cInstitute of Functional Nano and Soft Materials (FUNSOM), Joint International Research Laboratory of Carbon-Based Functional Materials and Devices, Soochow University, Suzhou, People's Republic of China
dUCLouvain, Institut de la Matière Condensée et des Nanosciences (IMCN), Molecular Chemistry, Materials and Catalysis (MOST), Place Louis Pasteur 1/L4.01.02, B-1348 Louvain-la-Neuve, Belgium. E-mail: prabhat.ranjan@uclouvain.be; pranjan057@gmail.com
eWel Research Institute, Avenue Pasteur 6, 1300 Wavre, Belgium
First published on 24th February 2026
Abstract
Despite the significant potential of isocyanides to engage in reactions with thiyl radicals to produce nitrogen-containing organic molecules, their widespread use has been limited by the need for high-energy UV light activation. Herein, we disclose the synthesis of thioformimidates under mild reaction conditions employing visible light without additives. This study demonstrates the ability of aromatic isocyanides to generate thiyl radicals under visible light, functioning as radical intitators. Furthermore, the developed methodology reveals its utility in the selective activation of the thiol group in cysteine, which is subsequently converted to dehydroalanine, opening the door to late-stage peptide modification.
Introduction
Isocyanides represent important and versatile building blocks in organic synthetic chemistry due to their unique chameleonic character.1 Their partial carbenoid nature makes them iso-electronic to carbon monoxide (CO), while their ionic character makes them akin to alkynes. The carbene character of isocyanides has been extensively explored in coordination chemistry, whereas their ionic character enabled the development of well-known multi-component reactions such as the Ugi and Passerini reactions.2–4 In addition, isocyanides are excellent radical acceptors in one-electron chemistry (known as radical sinks), enabling the efficient generation of diverse heterocyclic and complex molecular scaffolds through cascade reactions.5 Initially, this chemistry heavily relied on radical initiators like AIBN or peroxides.6,7 However, recent advances in visible-light photoredox catalysis have revitalized the field, offering a mild and versatile approach to achieve these transformations.8–10In particular, the addition of heteroatom-based radicals to isocyanides holds great potential in organic synthesis to generate heterocyclic scaffolds. Among these, thioformimidate formation following the addition of sulfur-based radicals on isocyanides has been widely investigated.7 The most common method for the synthesis of thioformimidates involves S-alkylation of thioformamides with aryl or alkyl halides (Scheme 1A).11,12 However, these processes often suffer from several limitations and purification problems due to the instability of thioformimidates and consequently suffer from limited substrate scope. Conversely, thioformimidates can also be synthesized through the reaction of isocyanides and thiols using metal catalysts, UV-light or in the presence of radical initiators such as AIBN (Scheme 1A).13,14 Generally, the reaction proceeds with the formation of a thiyl radical followed by thiyl radical addition to the isocyanide to generate an α-thioimidoyl radical.13 This radical can then undergo H-atom abstraction from thiols to form the final thioformimidates product. Despite the convenience of this method, less attention has been directed to this pathway due to the following reasons: (a) the requirement of UV-light; (b) poor reaction efficiency and (c) the formation of unwanted side products such as 1,1-bisthiolation adducts.
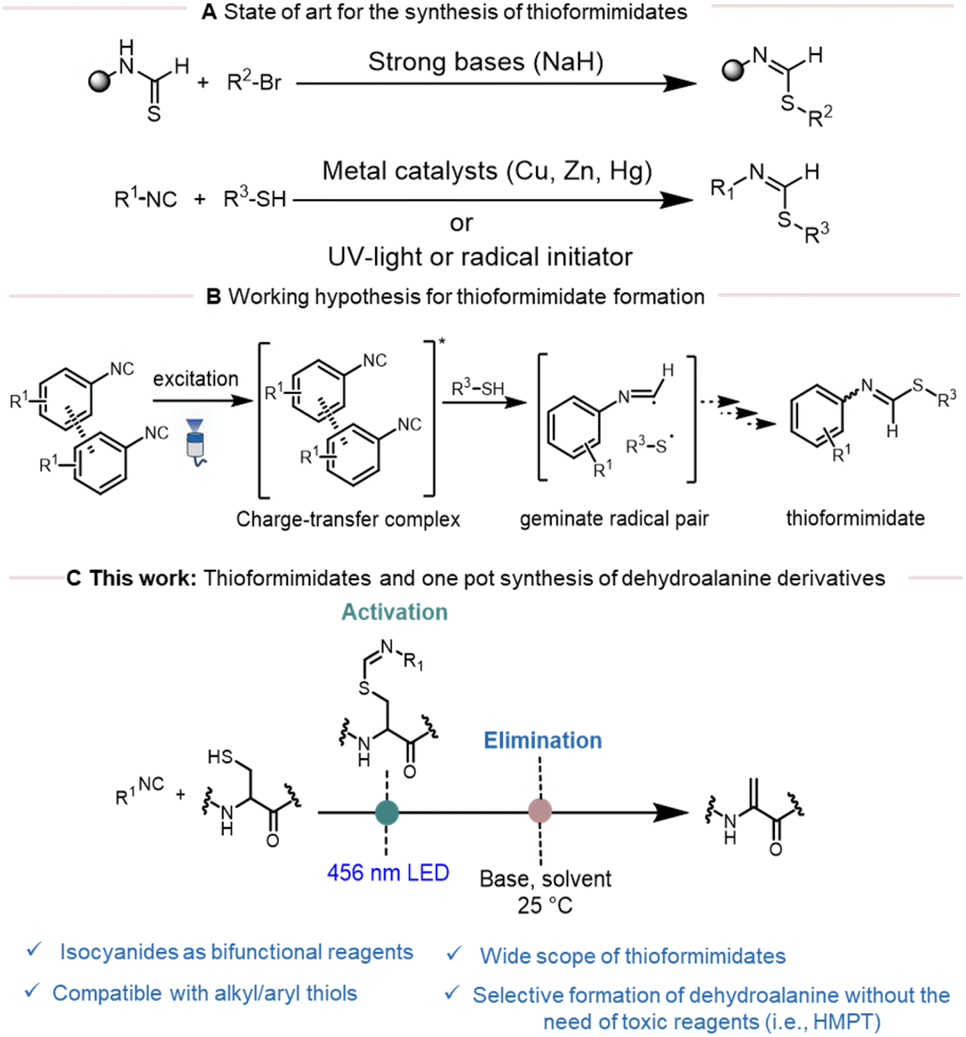 | ||
| Scheme 1 (A) previous work on the synthesis of thioformimidates; (B) our working hypothesis for the formation of thioformimidates; (C) this work: thioformimidates and one pot synthesis of dehydroalanine derivatives. | ||
Recently, our research group reported the ability of tryptamine isocyanide to undergo aggregation-based charge transfer. Building on this concept and previous literature,15,16 we hypothesized that this phenomenon could also be applied to other aromatic isocyanides to trigger the generation of thiyl radicals under visible light followed by the formation of thioformimidates (Scheme 1B). In light of their chemical properties and to broaden the synthetic utility of thioformimidates, we envisioned their use as an activating group for thiol derivatives. In the context of peptide chemistry, we further explored whether this strategy could enable the chemo- and regioselective activation of cysteine residues as a precursor of dehydroalanine (Scheme 1C).
Adhering to this hypothesis and given both the significance of thioformimidates as synthetic intermediates and the limitations of current methods, we sought to develop an innovative, exogeneous photocatalyst-free, visible-light-driven approach for their synthesis. During this study, we also investigated a previously camouflaged role of isocyanides in the generation of thiyl radicals using steady-state and time-resolved fluorescence spectroscopy. Moreover, we discovered the involvement of thioformimidates in the chain initiation and propagation step, a role that had been overlooked until now. Beyond reaction development, we report an application of this strategy to the selective activation of cysteine residues, to be able to further convert them into dehydroalanine derivatives, thereby introducing a sustainable and selective alternative to traditional methods of synthesizing dehydroalanine from cysteine residues in peptides.
Results and discussion
Optimization studies
Our optimization studies began with isocyanide 1a and thiophenol 1b in 1,2-dichloroethane (DCE) under visible light irradiation (λ = 456 nm). To our delight, the desired product 2 was obtained in 91% yield after 12 h (Table 1, entry 1). Being aware of the role of the solvent in charge transfer complexes, we evaluated a variety of polar and non-polar solvents (for more details see SI).17 We observed good to moderate yields in polar as well as moderately polar solvents (Table 1, entries 2 and 3). Interestingly, water also proved to be an effective solvent, providing 87% of the desired product (Table 1, entry 4). However, the addition of an additive like meglumine, which can increase the solubility of isocyanide 1a and thiol 1b in water, had an inverse effect on the product yield (11%, see SI).18 This result could be rationalized by the better chance of forming aggregates of 1a in pure water due to the hydrophobic effect. Following the solvent screening, the effect of a base was evaluated, as in a basic medium, thiol 1b (pKa = 10.2 in DMSO)19 is deprotonated.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
||
|---|---|---|
| Entry | Deviations from the optimized conditions | Yieldb (%) |
| a All reactions were performed using 1a (0.24 mmol, 1.2 equiv.) and 1b (0.2 mmol, 1 equiv., 0.1 M). b Yields were determined by 1H NMR using CH2Br2 as an internal standard. | ||
| 1 | None | 91 |
| 2 | MeCN | 81 |
| 3 | 2-MeTHF | 60 |
| 4 | Water | 87 |
| 5 | Cs2CO3 | 8 |
| 6 | DIPEA | 10 |
| 7 | 0.5 M instead of 0.1 M | 83 |
| 8 | 0.05 M instead of 0.1 M | 67 |
| 9 | No light, 80 °C | 0 |
| 10 | Air instead of N2 | <10 |

The thiolate formed under basic conditions could either potentially form an intermolecular electron donor–acceptor (EDA) complex with isocyanide 1a or be excited under visible light to induce a single electron transfer (SET).20,21 However, the addition of a base had a detrimental effect on the product yield, possibly due to the inhibition of the hydrogen atom transfer (HAT) step to the intermediate imidoyl radical (Table 1, entries 5 and 6).13 Finally, we performed some control experiments to substantiate the need for light and to provide evidence of the radical nature of this transformation (Table 1, entries 9 and 10). Quantum yield measurements further indicated a radical chain mechanism, in accordance with the reported literature (see SI).22
Reaction scope
Having established the optimal conditions, the scope and limitations of thioformimidate formation were investigated. Aromatic thiols bearing electron-withdrawing or donating substituents delivered the desired products in good to moderate yields (47–91%, Scheme 2). However, ortho-Me and ortho-CO2Me substituted aromatic thiols delivered the corresponding products in lower yields (Scheme 2, 4 and 9). Pleasingly, an aromatic thiol having a carboxylic acid group at the benzylic position (Scheme 2, 10) was also compatible under these conditions. This transformation is typically challenging to be achieved in the presence of photocatalysts due to the tendency of the benzylic carboxylic group to undergo decarboxylation.23 Based on our initial hypothesis, we anticipated that the generation of a thiyl radical should also be feasible from aliphatic thiols employing aromatic isocyanides. This expectation materialized as aliphatic thiols like N-acetyl-L-cysteine methyl ester and cyclohexyl thiol delivered the desired products in good yields (Scheme 2, 11–16). In particular, thioformimidate formation from N-acetyl-L-cysteine methyl ester paves the way to harness isocyanides as thiol-activating groups in peptides and proteins. It is worth emphasizing that the reaction described herein is compatible with aqueous conditions and other additives (Table S3).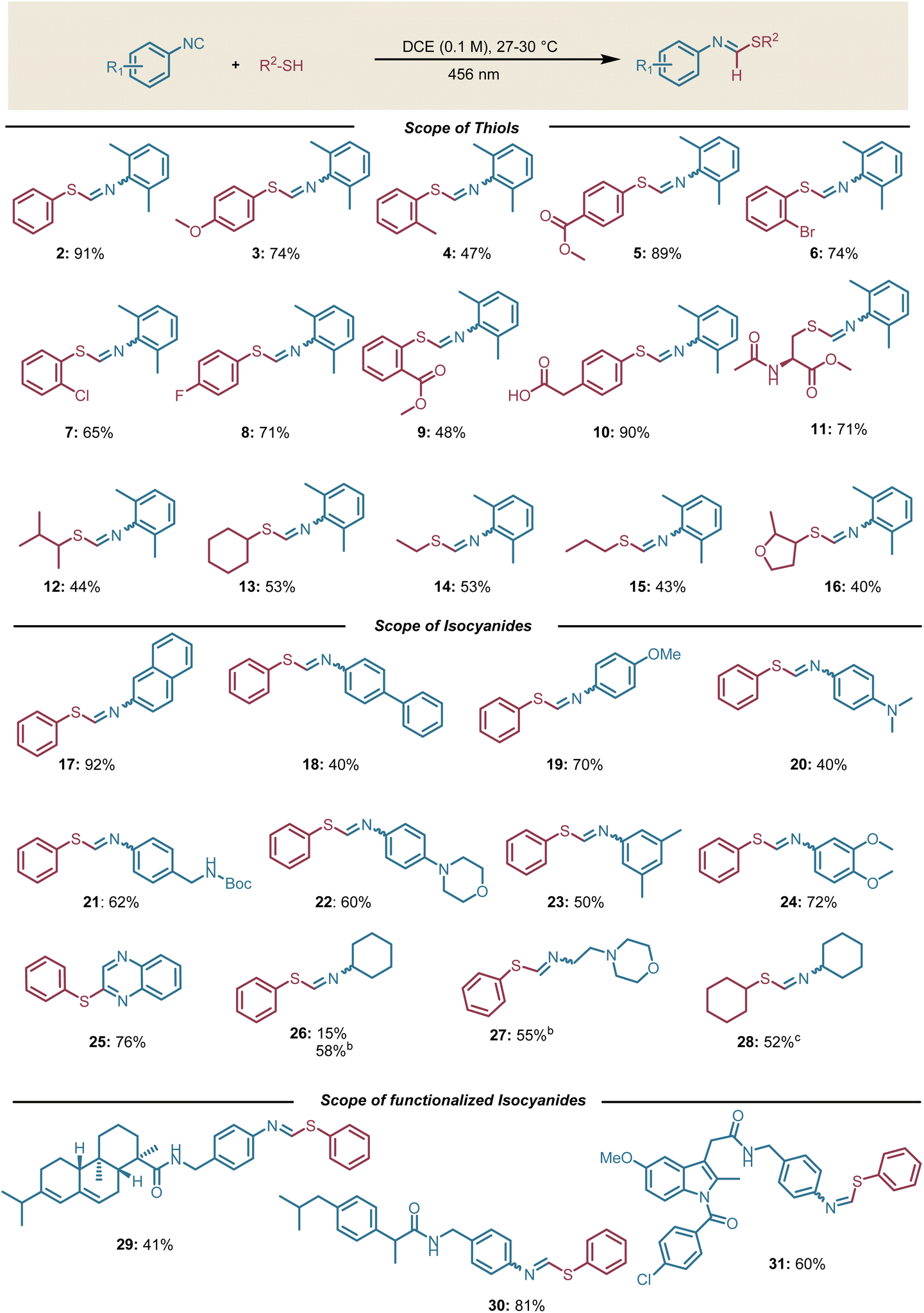 | ||
| Scheme 2 Scope of thioformimidates: aconditions unless otherwise noted: all reactions were performed using isocyanide (0.24 mmol, 1.2 equiv.), thiol (0.2 mmol, 1 equiv.), dry DCE (0.1 M), 12 h, irradiating with a Kessil lamp 456 nm (40 W). bConcentration (1 M). cPhSSPh (10–20 mol%) used as an additive, concentration (1 M). | ||
After screening thiol compatibility, we evaluated various aromatic isocyanides under the optimized conditions. We obtained good yields in all cases (Scheme 2, 17–24). Furthermore, 1,2-diisocyanobenzene selectively afforded 2-(phenylthio)quinoxaline without any detectable formation of 2,3-bis(phenylthio)quinoxaline (Scheme 2, 25).7 Based on our initial hypothesis, we assumed that aliphatic isocyanides would not be suitable substrates under our optimal conditions. Nevertheless, we obtained 15% yield of the desired thioformimidate when cyclohexyl isocyanide was employed (Scheme 2, 26). Surprised by this result, we performed additional control experiments and we found that the diphenyl disulfide is responsible for the desired product formation.13
The in situ formation of diphenyl disulfide can be explained by the tendency of thiols to self-oxidize in the presence of residual oxygen in the solvent (see SI, Section 4.8). As the only absorbing species in the reaction mixture would have been the diphenyl disulfide, we increased its concentration in an attempt to prove our assumption and promote the reactivity. As expected, we obtained a higher yield at 1 M reaction concentration compared to the reaction with 0.1 M concentration. Similarly, with cyclohexyl isocyanide and cyclohexyl thiol, we employed a catalytic amount of diphenyl disulfide to start the radical chain reaction to deliver the desired product (Scheme 2, 28). It is important to note that we did not observe any 1,1-bisthiolation side product in any case. To further probe the functional group compatibility, we employed isocyanides functionalized with complex molecular scaffolds such as the benzoamide derivatives of the drugs abietic acid, ibuprofen, and indomethacine that gave the desired products in moderate to good yields (29, 30, and 31, respectively, Scheme 2).
Application of thioformimidates
After establishing the scope of the thioformimidate formation, we focused our attention on the synthesis of dehydroalanine (DHA) derivatives, an amino acid residue important in both biological and synthetic chemistry.24,25 There is substantial research focused on the site-selective incorporation of dehydroalanine into peptides and proteins. Among these, the conversion of cysteine residues to dehydroalanine holds great potential for protein modification because of the strong nucleophilicity of sulfur and the ease of incorporating cysteine into proteins using standard biochemical techniques. Over the past decade, many methods have been documented, but none of them enables a general, chemo-and site-selective incorporation of dehydroalanine.26 For instance, the synthesis of dehydroalanine from cysteine using the O-mesitylenesulfonylhydroxylamine (MSH) is incompatible with peptides containing serine, asparagine, tyrosine, or tryptophane derivatives.26We were therefore pleased to achieve the formation of DHA using N-acetyl cysteine methyl ester and eight different dipeptides (32–40, Scheme 3A). We first carried out the photochemical transformation on the cysteine residues for the formation of the thioformimidate intermediates. After confirming the formation of the latter through LC-MS, we redissolved the crude reaction products in N,N-dimethylformamide (DMF), in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as a base, to yield the desired dehydroalanine derivatives. Dipeptides containing amino acids such as alanine, phenylalanine, methionine, and proline delivered the desired products in good yields within 1 h (Scheme 3A, 33–36). Next, we employed proteinogenic amino acids such as serine, asparagine, tyrosine and tryptophan (Scheme 3A, 37–40) that also led to the selective formation of dehydroalanine, showcasing good functional group tolerance.
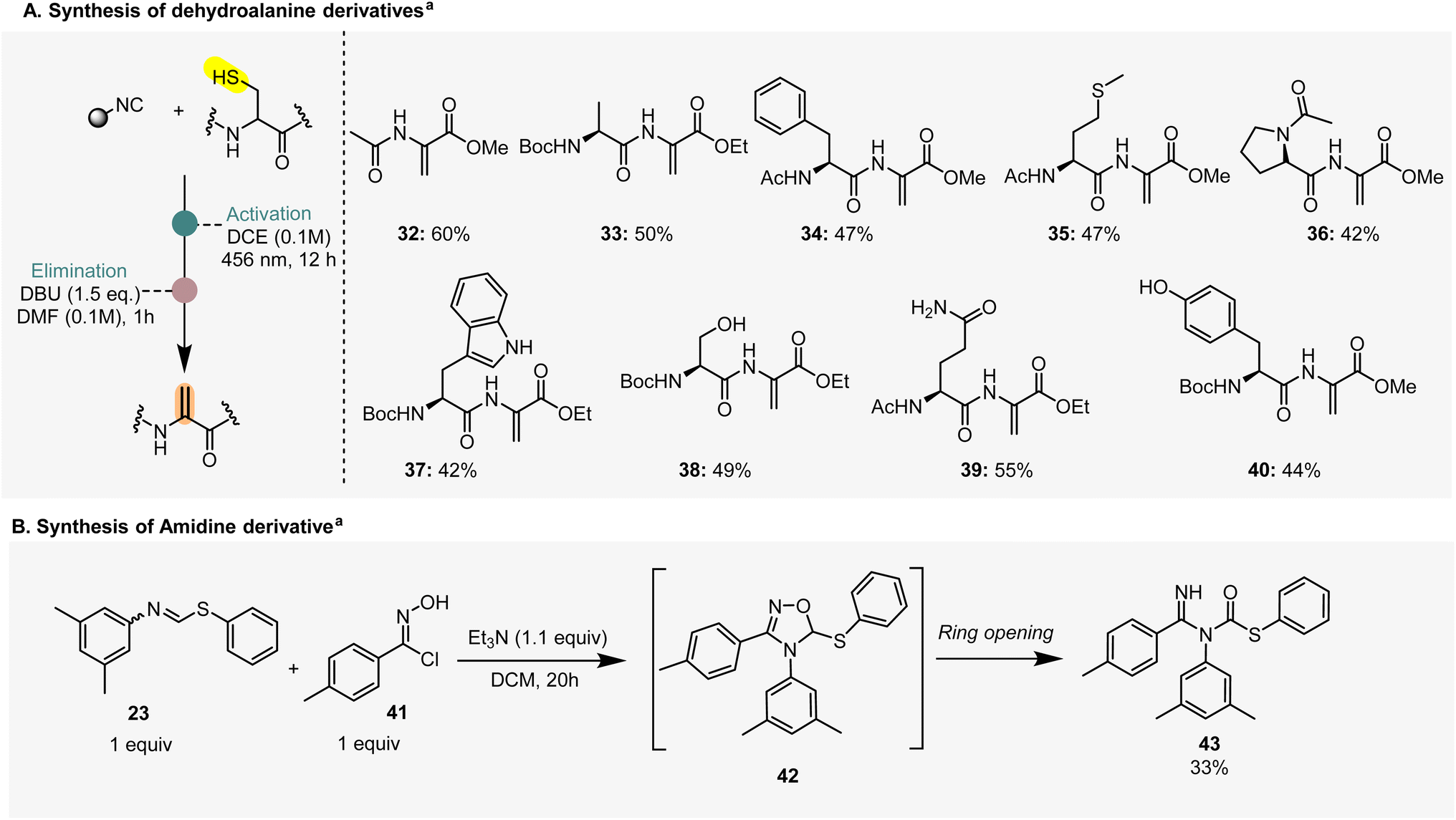 | ||
| Scheme 3 Application of thioformimidates to the synthesis of dehydroalanine and amidine derivatives. aYields refer to a two-step process. | ||
We also explored the possibility to involve thioformimidates in [3 + 2]-cycloaddition reactions with 1,3-dipoles such as chlorooxime 41 (Scheme 3B) to get amidine derivatives such as 43, likely formed upon a based catalyzed ring-opening of intermediate 42 (Scheme 3B). A similar product was previously reported by Yamamoto and co-workers in 1971, using benzyl cyanide and n-butyl di-n-butylthioboronite.27 Our approach offers direct access to generate this complex molecular scaffold in a two-step one-pot reaction.
Mechanistic investigations
Based on both the initial hypothesis and the previous findings, we anticipated that aromatic isocyanides could undergo aggregation-based charge transfer. Yu, Ma, and co-workers previously reported a similar behaviour in 1,2-diisocyanoarenes.16 To investigate if a similar mechanism operates in the presence of compound 1a, we first recorded its UV-Vis absorption spectra in DCE at varying concentrations (Fig. 1a). A pronounced bathochromic shift was observed at higher concentrations, indicating potential aggregation effects. It is important to note that the purity of 1a was crucial for this observation as samples exhibiting a yellow colour showed an absorption peak around 390 nm at higher concentrations, likely due to impurities or gradual decomposition/polymerization over time. Unlike the well-defined absorption peak in the visible region reported by Yu, Ma and coworkers, we observed a broader absorption band.16 This difference may derive from the distinct packing structure of 1a in solution. Indeed, the reported crystal structure of 1a shows intermolecular distances ranging from 3.47 Å to 4.65 Å, which could influence the observed absorption features.28 We then recorded the steady-state fluorescence spectra of 1a at different excitation wavelengths. Excitation at 355 nm led to a vibronically-resolved photoluminescence centered around 400 nm. Interestingly, exciting at 420 nm, i.e. within the novel absorption features apparent at high concentrations, a steady-state photoluminescence spectra with a maximum at 500 nm was observed. This observation suggests relaxation from different electronic states, a common characteristic of aggregates (Fig. 1b).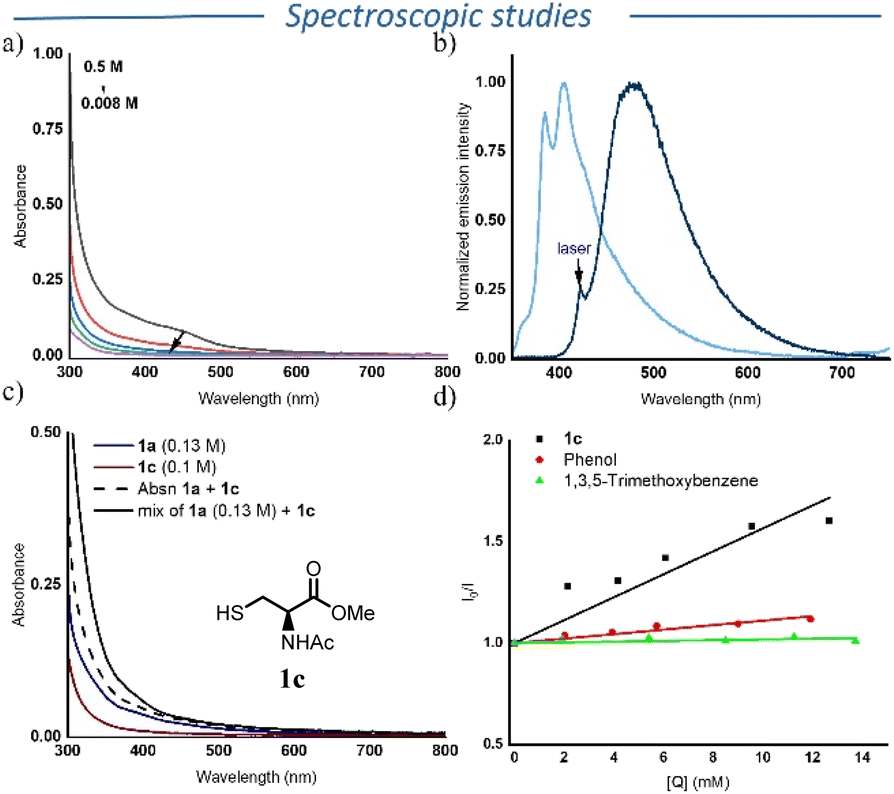 | ||
| Fig. 1 (a) absorption spectra of 1a in DCE at different concentrations; (b) emission spectra of 1a (0.1 M) in DCE, sky blue excitation at 355 nm and navy blue excitation at 420 nm; (c) absorption spectra of individual components and reaction mixture, recorded in 40% MeCN in DCE: [1c] = 0.13 M; [1a] = 0.1 M; [1a + 1c] = solution of 0.13 M of 1c and 0.1 M of 1a, dotted black line represent the sum of absorption of 1a and 1c; (d) Stern–Volmer quenching of 4-isocyanoanisole with 1c, phenol and 1,3,5-trimethoxybenzene. | ||
Following the confirmation of aggregate formation in 1a, we next recorded the UV-Vis spectra of mixtures of 1a and 1c (N-acetyl-L-cysteine methyl ester) to check the possibility of a ground-state donor–acceptor (EDA) complex formation. We selected 1c because it is a solid, non-volatile compound devoided of the strong pungent odor typical of more volatile thiols. However, the study in DCE was inconclusive due to the poor solubility of 1c, which led to significant scattering. Therefore, we recorded the UV-vis spectrum in a mixture of DCE and MeCN, where the solubility of both 1a and 1c was better. We did not observe any appreciable bathochromic shift above 420 nm (Fig. 1c and Fig. S6–S8). However, the absorption spectrum of 1a and 1c exhibited a hyperchromic shift compared to the sum of the individual absorption spectra below 420 nm (dashed and solid lines in Fig. 1c). Although not conclusive, this hyperchromic shift could point towards a ground-state association between 1a and 1c. To further probe a ground-state interaction, we measured the UV-vis spectrum of a mixture of 1a, 1b and cyclohexyl thiol, which revealed again no significant bathochromic shift (Fig. S9 and S12). In addition, the formation of an excimer between 1a and 1c was also ruled out as we did not observe any appreciable shift in the steady-state photoluminescence spectrum of 1a and in the mixture of 1a and 1c.
After having identified the main photo-absorbing species under our reaction conditions, we turned our attention to delineate the mechanism of radical generation from thiol. To do that, we performed Stern–Volmer fluorescence quenching experiments using either 1a or 4-isocyanoanisole (Fig. S21–S25). In case of 1a, a very low Stern–Volmer constant (Ksv: 8 M−1) was obtained. In the case of 4-isocyanoanisole, the Stern–Volmer constant rose to 56 M−1. Based on this result, we envision two possible pathways for the generation of thiyl radical from 1c. The first involves the electron transfer from 1c to the excited isocyanide, followed by protonation, while the second possibility involves a proton coupled electron transfer (PCET) process.
To further discriminate between these two pathways, we performed Stern–Volmer quenching experiments with phenol and 1,3,5-trimethoxybenzene. Whereas 1c efficiently quenched the excited state of 4-isocyanoanisole isocyanide (Ksv = 56 M−1), only moderate quenching was observed using phenol (Ksv = 11 M−1) and trimethoxybenzene showed no measurable quenching (Fig. 1d, S24 and S25). This indicates the possibility of proton coupled electron transfer, since the oxidation potential of phenol and 1,3,5-trimethoxybenzene are comparable. Note however that the corresponding Stern–Volmer plot for the quenching of 4-isocyanoanisole by 1c exhibits some deviation from linearity, especially at low concentration of 1c (Fig. 1d). We believe that this deviation occurs from the degradation of 4-isocyanoanisole that has been observed in solution, leading to an additional emission peak around 650 nm that would in fine influence the corresponding Stern–Volmer plot. Furthermore, TD-DFT calculations reveal that the first excited state (S1) possesses a 16.6% charge-transfer (CT) character from the phenyl ring to the isocyanide (–NC) unit, with the remaining contribution arising from locally excited (LE) transitions within the phenyl framework (Fig. S30). This partial CT character indicates a redistribution of electron density over the phenyl ring accompanied by electron migration toward the NC bond. This shift of electron density can enhance hydrogen bonding in the excited state, which can lead to higher quenching, as observed in the case of 1c and phenol in comparison with 1,3,5-trimethoxybenzene, supporting the PCET mechanism.
To complete the mechanistic investigation, we further analysed the role of the reaction product. We wondered whether there could be the possibility of an autocatalytic reaction, where product 11 acts as a radical initiator, or participates in the radical propagation step. We therefore carried out reactions using 11 (that absorbs in the visible region) in varying amounts, in the presence of an aliphatic thiol and an aliphatic isocyanide (Fig. 2b), that do not absorb in the visible region. To our surprise, we observed the formation of the desired product, though in low yields, even when higher loadings of product 11 were added (for details see SI). Based on these results, the product can act as a radical initiator (though not efficiently). Nevertheless, we cannot generalize this behaviour because not all the products acted as initiators.
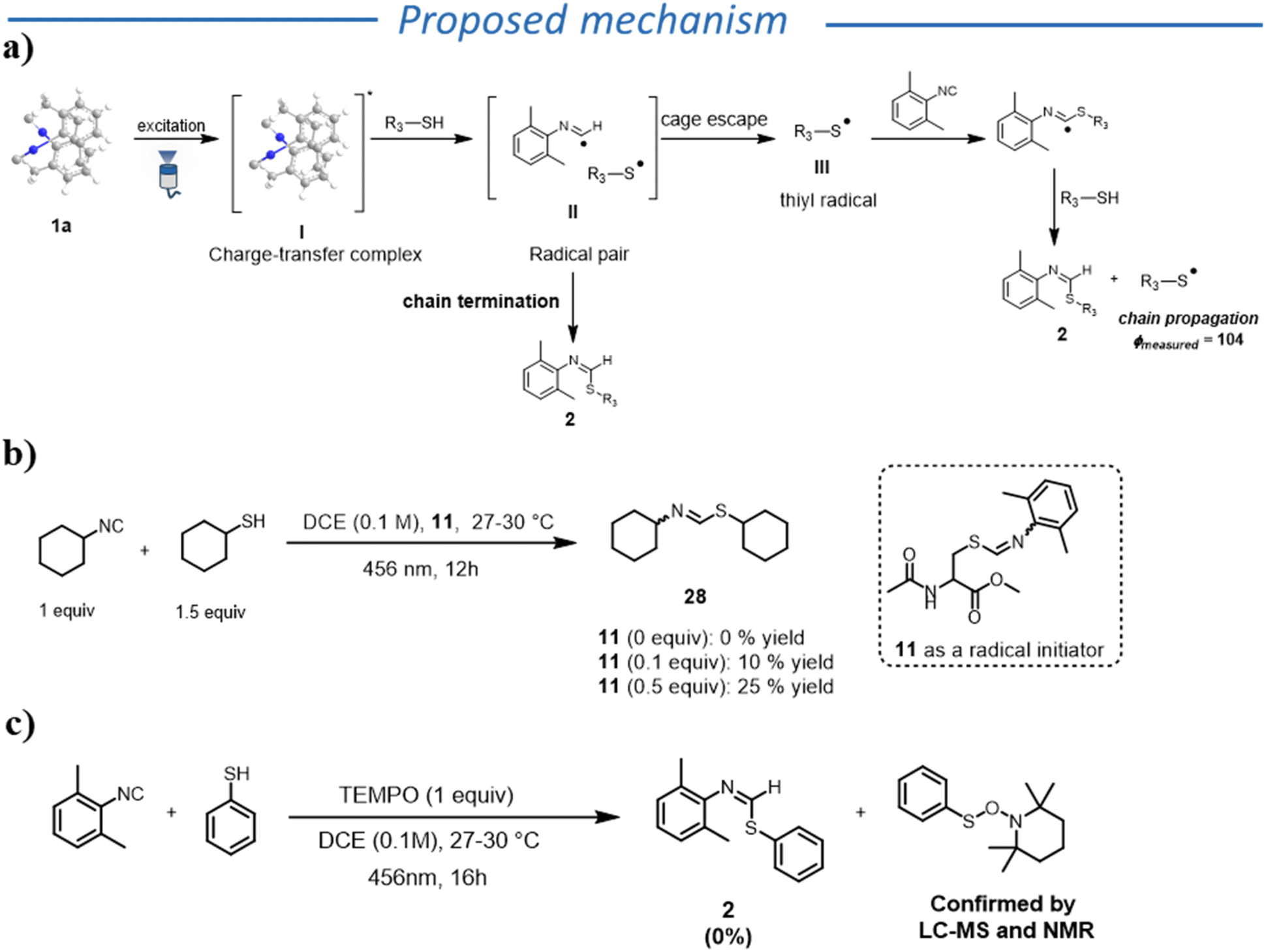 | ||
| Fig. 2 (a) proposed mechanism; (b and c) control experiments. | ||
Taking into account all these observations, we propose the following mechanism for the formation of thioformimidates (Fig. 2a). Upon excitation of 1a, a PCET transfer between 1a* and thiol happens, forming radical pair II. At this stage, the radical pair can undergo in-cage radical coupling to generate the desired product (termination step). Alternatively, the thiyl radical escapes the solvent cage, initiating a radical chain by reacting with another isocyanide molecule. The formation of a TEMPO adduct with the thiyl derivative supports the formation of thiyl radical species (Fig. 2c). We would also like to emphasize that, in case of aromatic thiols, other mechanistic scenarios, such as the involvement of diphenyl disulfide or EDA-complex formation, could play a relevant role in the radical initiation step. Compared to previously reported UV-light-induced reactions between thiols and isocyanides, we hypothesize that the high selectivity for the thioformamide formation in our reaction is due to the unique ability of aromatic isocyanides to be involved in the generation of a thiyl radical and the subsequent fast HAT step (see Scheme S3).
Conclusions
In conclusion, we demonstrated the potential of aromatic isocyanides to serve as radical initiators in the formation of thioformimidates. Beyond the thioformimidate formation, we developed a one-pot method for synthesizing dehydroalanine in dipeptides with good yields and excellent chemo-selectivity. We also explored [3 + 2] cycloaddition reactions with 1,3-dipoles, such as Z-chlorooximes, to generate amidine derivatives. Overall, this new methodology paves the way for sustainable and cost-effective routes to thioformimidates and catalyst free generation of thiyl radicals.Author contributions
The authors confirm their contribution to the paper as follows: study conception: P. R.; design and methodology: P. R., M. W., M. G. and J. M. S.; experimentation: P. R., M. W., C. R. and J. H.; spectroscopy: P. R. and L. T. G.; theoretical studies: R. W.; writing, review and editing: P. R. with input from M. W., M. G., L. T. G., J. M. S., and R. V. A. O.; acquiring funding: P. R. and R. V. A. O.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: optimization details, NMR spectra, spectroscopic data and DFT data. See DOI: https://doi.org/10.1039/d5sc07984e.Acknowledgements
This project has received funding from the European Union's Horizon 2021 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 101063688 (project name: SusIsocyanide). This publication reflects only the author's view, exempting the Community from any liability. Neither the European Union nor the granting authority can be held responsible for them. P. R. is thankful to the Marie Skłodowska-Curie Postdoctoral Fellowship (Grant Agreement No. 101063688). M. W. is thankful to China Scholarship Council (No. 202207720033) for providing a doctoral scholarship. L. T.-G. is a chercheur qualifié of the Fonds de la Recherche Scientifique – F.R.S-FNRS. P. R. is thankful to Dr Serena Pillitteri and Dr Felix Glaser for their valuable suggestions.Notes and references
- G. dos P. Gomes, Y. Loginova, S. Z. Vatsadze and I. V. Alabugin, J. Am. Chem. Soc., 2018, 140, 14272–14288 CrossRef CAS PubMed.
- M. Knorn, E. Lutsker and O. Reiser, Chem. Soc. Rev., 2020, 49, 7730–7752 RSC.
- L. Banfi, A. Basso, C. Lambruschini, L. Moni and R. Riva, Chem. Sci., 2021, 12, 15445–15472 RSC.
- E. Ruijter, R. Scheffelaar and R. V. A. Orru, Angew. Chem., Int. Ed., 2011, 50, 6234–6246 CrossRef CAS PubMed.
- B. Zhang and A. Studer, Chem. Soc. Rev., 2015, 44, 3505–3521 RSC.
- L. Benati, R. Leardini, M. Minozzi, D. Nanni, R. Scialpi, P. Spagnolo, S. Strazzari and G. Zanardi, Angew. Chem., Int. Ed., 2004, 43, 3598–3601 CrossRef CAS.
- A. Ogawa and Y. Yamamoto, Beil. J. Org. Chem., 2024, 20, 2114–2128 CrossRef CAS PubMed.
- G. A. Coppola, S. Pillitteri, E. V. Van der Eycken, S.-L. You and U. K. Sharma, Chem. Soc. Rev., 2022, 51, 2313–2382 RSC.
- M. Martín, R. M. Romero, C. Portolani and M. Tortosa, ACS Catal., 2024, 14, 17286–17292 CrossRef PubMed.
- I. Quirós, M. Martín, M. Gomez-Mendoza, M. J. Cabrera-Afonso, M. Liras, I. Fernández, L. Nóvoa and M. Tortosa, Angew. Chem., Int. Ed., 2024, 63, e202317683 CrossRef.
- K. de la Vega-Hernández, R. Senatore, M. Miele, E. Urban, W. Holzer and V. Pace, Org. Biomol. Chem., 2019, 17, 1970–1978 RSC.
- T. S. Jagodziński, Chem. Rev., 2003, 103, 197–228 CrossRef PubMed.
- F. Dénès, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev., 2014, 114, 2587–2693 CrossRef PubMed.
- T. Saegusa, S. Kobayashi, K. Hirota, Y. Okumura and Y. Ito, Bull. Chem. Soc. Jpn., 1968, 41, 1638–1642 CrossRef CAS.
- M. Wu, J. M. Saya, P. Han, R. Walia, B. Pradhan, M. Honing, P. Ranjan and R. V. A. Orru, Chem. Sci., 2024, 15, 6867–6873 RSC.
- W. Wang, X. Sun, J. Qu, X. Xie, Z.-H. Qi, D. Hong, S. Jing, D. Zheng, Y. Tian, H. Ma, S. Yu and J. Ma, Phy, Phys. Chem. Chem. Phys., 2017, 19, 31443–31451 RSC.
- C. G. S. Lima, T. de M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389–1407 CrossRef CAS.
- Y. Tian, E. Hofmann, W. Silva, X. Pu, D. Touraud, R. M. Gschwind, W. Kunz and B. König, Angew. Chem., Int. Ed., 2023, 62, e202218775 CrossRef CAS PubMed.
- F. G. Bordwell and D. L. Hughes, J. Org. Chem., 1982, 47, 3224–3232 CrossRef CAS.
- B. Liu, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 13616–13619 CrossRef CAS.
- S. Wang, H. Wang and B. König, Chem, 2021, 7, 1653–1665 CAS.
- T. Saegusa, S. Kobayashi and Y. Ito, J. Org. Chem., 1970, 35, 2118–2121 CrossRef CAS.
- J. Xuan, Z. Zhang and W. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632–15641 CrossRef CAS PubMed.
- D. E. Palmer, C. Pattaroni, K. Nunami, R. K. Chadha, M. Goodman, T. Wakamiya, K. Fukase, S. Horimoto and M. Kitazawa, J. Am. Chem. Soc., 1992, 114, 5634–5642 CrossRef CAS.
- G. Jung, Angew. Chem., Int. Ed., 1991, 30, 1051–1068 CrossRef.
- J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernández-González, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Chem. Sci., 2011, 2, 1666 RSC.
- T. Mukaiyama, S. Yamamoto and K. Inomata, Bull. Chem. Soc. Jpn., 1971, 44, 2807–2810 CrossRef CAS.
- W. W. Brennessel, B. E. Kucera, V. G. Young and J. E. Ellis, Acta Crystallogr., 2019, 75, 1118–1127 CAS.
| This journal is © The Royal Society of Chemistry 2026 |