
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpatiotemporally regulated mitochondrial genome editing via enzyme and NIR-activated CRISPR/Cas9 nanoplatform
Fei
Yang†
a,
Qianqin
Ran†
a,
Jiahui
Chen
a,
Guochen
Bao
b,
Yuezhong
Xian
 *a and
Cuiling
Zhang
*a
*a and
Cuiling
Zhang
*a
aShanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, Department of Chemistry, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200241, China. E-mail: yzxian@chem.ecnu.edu.cn; clzhang@chem.ecnu.edu.cn
bSchool of Mathematical and Physical Sciences, Faculty of Science, University of Technology Sydney, Sydney, New South Wales 2007, Australia
First published on 13th January 2026
Abstract
Mitochondrial DNA (mtDNA) mutations play critical roles in tumor progression and metabolic reprogramming. Controllable gene editing within tumor cell mitochondria remains a challenge due to the double-membrane barrier and the lack of tumor-selective activation. Herein, we report a dual-responsive CRISPR/Cas delivery platform (UCRP-TPP) that enables spatiotemporally regulated mtDNA editing for targeted tumor therapy. This nanoplatform integrates near infrared light-responsive upconversion nanoparticle (UCNP), an apurinic endonuclease 1 (APE-1)-responsive DNA complex, and a mitochondrial-targeting ligand (TPP), ensuring selective activation and mitochondrial release of Cas9/sgRNA complexes. Upon activation by endogenous APE-1 enzyme and exogenous NIR light, UCRP-TPP induces mtDNA editing by CRISPR/Cas, which leads to mtDNA copy number reduction, mitochondrial membrane depolarization, reactive oxygen species generation, and tumor cell apoptosis. In vivo studies further confirm the robust antitumor efficacy of the UCRP-TPP-based nanoplatform. This work presents a versatile and controllable mitochondrial gene-editing strategy.
Introduction
The CRISPR/Cas9 system, composed of a single guide RNA (sgRNA) and the Cas9 protein, enables site-specific genome editing through double-strand breaks (DSBs) at protospacer adjacent motif (PAM, 5′–NGG–3′) sites.1–5 This powerful tool has been widely applied in biomedical research, particularly gene therapy and diagnostics.6–8 However, intracellular delivery remains a major challenge due to the system's instability (e.g., nuclease-mediated degradation of sgRNA and proteolysis of Cas9) and poor membrane permeability.9,10 Current delivery strategies11 include using plasmid DNA,12,13 RNA (Cas9 mRNA and sgRNA),14,15 and ribonucleoprotein (RNP) complexes.16,17 Among them, direct RNP delivery can bypass transcription and translation, offering fast and efficient editing.17 To improve delivery efficiency, various nanocarriers have been explored, such as metal–organic framework materials,18,19 liposomes,20,21 gold nanoparticles,22–24 DNA nanostructures,16 and upconversion nanoparticles (UCNPs).25,26 However, there still remain challenges (e.g. off-target effects), and most strategies are limited to nuclear genome editing. Thus, there is an urgent need to develop spatiotemporally controlled delivery platforms and mitochondrial gene editing strategies.The conventional CRISPR/Cas9 system suffers from “always-on” activity, resulting in off-target effects and reduced editing precision.27 To address this, spatiotemporal control of the CRISPR/Cas9 activity is highly desirable.28,29 Stimulus-responsive CRISPR/Cas9 platforms have thus attracted growing interest, as they enable controlled editing by restricting Cas9 activity to specific types of cells. These systems can be activated by endogenous triggers (e.g., miRNA,30 pH,13,27 redox reactions,31,32 and enzymes16,33) and exogenous stimuli (e.g., ultrasound,33 light23,26,34), offering enhanced precision and minimized side effects. The system's integration with internal and external stimuli represents a promising direction for refined and programmable genome editing.
Compared to nuclear gene editing, mitochondrial DNA (mtDNA) editing based on the CRISPR system is challenging35 due to the difficulty of transporting sgRNA and Cas proteins across the mitochondrial double membrane.36 Unlike nuclear DNA, mtDNA lacks introns and histone protection, and exhibits a limited repair capacity, leading to a ten times higher damage rate than nuclear DNA.37–39 As a result, mtDNA represents a potentially vulnerable and effective therapeutic target. Current strategies for mtDNA-targeted therapy through direct mitochondrial disruption include DNA-damaging agents such as doxorubicin and cisplatin 40,41, ultra-thin nanosheets,42 and small molecule probes.43 Doxorubicin and cisplatin can directly cause mtDNA damage and interfere with mitochondrial function to initiate apoptotic cascades.40,41 Ultra-thin nanosheets can selectively target mitochondrial compartments and disrupt mitochondrial integrity to trigger cell death signaling.42 Small molecule probes, by virtue of their ability to accumulate in mitochondria via membrane potential gradients, can directly induce structural impairment, such as mitochondrial membrane potential dissipation or oxidative stress elevation.43 Zhang's group reported a different therapy approach based on mtDNA mutation-induced photosensitizer release. It leverages the characteristics of mtDNA mutations to design a responsive drug release system, thereby expanding the application of mtDNA-targeted therapy.44
Herein, we developed a dual-responsive CRISPR/Cas9 delivery nanoplatform (UCRP-TPP, UCNP@SiO2-sgAPC/Cas9-TPP) for targeted mtDNA editing with high spatiotemporal resolution. This system is activated by apurinic endonuclease 1 (APE1, an endogenous stimulus) and near-infrared (NIR, 980 nm) light-driven UV emission (an exogenous stimulus), inducing mtDNA double-strand breaks (DSBs) and triggering tumor cell apoptosis. APE1 is a base excision repair enzyme that is expressed in the cytoplasm of many cancer cells, whereas it is minimally present in the nucleus of normal cells.45–48 Therefore, it enables tumor-specific activation of the CRISPR/Cas9 system and minimization of off-target effects. UCNPs serve as carriers of CRISPR/Cas9 RNPs as well as transducers for converting NIR light into UV emission to trigger mitochondrial release of the editing machinery.49 As illustrated in Fig. 1A, UCRP-TPP is formed by loading dual-responsive DNA, CRISPR/Cas9 RNPs, and triphenylphosphine (TPP) onto UCNPs. The assembly process involves three steps: (i) nucleic acid complex (sg-APC) formation for dual-stimuli responsiveness, (ii) conjugation of sgRNA/Cas9 onto UCNPs (UCRP), and (iii) TPP modification for mitochondrial localization (UCRP-TPP). As depicted in Fig. 1B, uptaken by tumor cells, UCRP-TPP is first activated by cytoplasmic APE1, followed by NIR light-triggered UV emission. The combination of endogenous activation (APE-1) and exogenous stimulation (NIR light) induces RNP release into mitochondria. The released CRISPR/Cas9 then cleaves mtDNA, generating DSBs and inducing apoptosis. It can be confirmed by the reduction of mtDNA copy number in cancer cells (HeLa and MCF-7 cells) (Fig. 1C). As for normal cells, this process cannot occur due to the rare expression of APE-1. Thus, the UCRP-TPP-based dual-responsive nanoplatform enables highly selective, spatiotemporally controlled mitochondrial gene editing and effective tumor suppression.
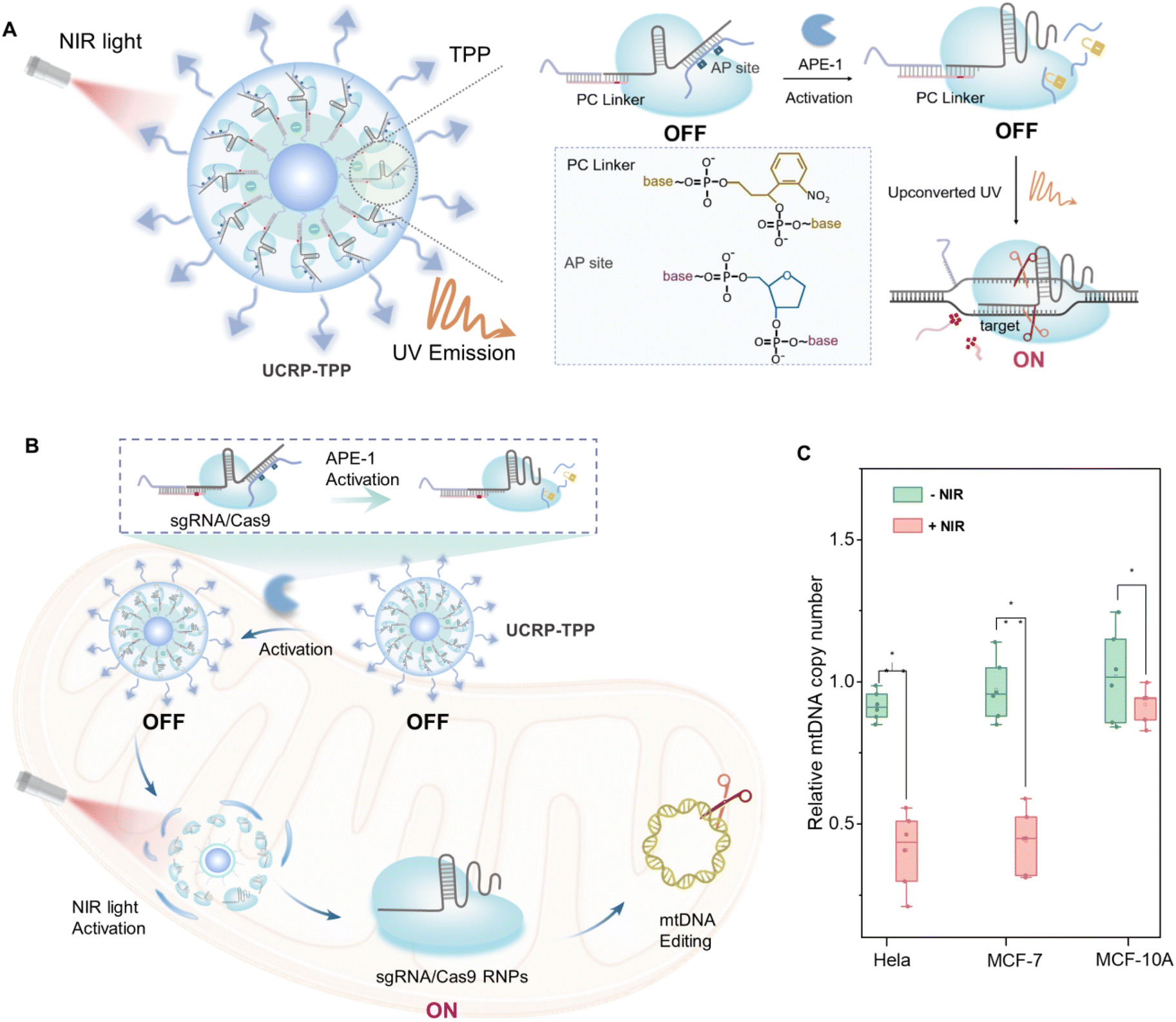 | ||
| Fig. 1 Schematic illustration of a dual-responsive CRISPR/Cas9 delivery system (UCRP-TPP) for mitochondrial gene editing in tumor cells. (A) Scheme of UCRP-TPP (UCNP@SiO2-sgAPC/Cas9-TPP; sgAPC: sgRNA-APC complex composed of sgRNA, AP strand, PC strand, and SH-DNA). (B) APE-1-triggered activation of the DNA complex and NIR light-induced UV emission (NIR: 980 nm) initiate mitochondrial release of CRISPR/Cas9 RNPs, leading to mtDNA cleavage and apoptosis. (C) Quantification of mtDNA copy number in HeLa, MCF-7, and MCF-10A cells after NIR-triggered UCRP-TPP. | ||
Results and discussion
We synthesized the UCRP in a stepwise manner based on Tm-doped UCNPs (Fig. 2A). The TEM image in Fig. S1 shows the monodisperse UCNPs with uniform particle size (27.0 ± 2.2 nm). These particles can convert 980 nm NIR light into UV emission, enabling photo-triggered release of CRISPR/Cas9 components. To optimize luminescence performance, the modification of the silicon shell on UCNPs with different thicknesses was performed (Fig. S2A–E). The maximum upconversion emission was observed with a shell thickness of ∼4.5 nm (particle size ∼38.5 ± 1.9 nm) (Fig. S2F and 2B). Fig. S3 indicates the scheme of the conjugation of thiol-modified DNA complexes (sgAPC) with UC@SiO2-NH2-sulfo-SMCC, followed by Cas9 loading to yield UC@SiO2-sgAPC/Cas9 (UCRP). The process was confirmed via UV-vis absorption spectra (Fig. S4).50 TEM images of UCRP (Fig. 2C) showed a clear increase in shell thickness to ∼14.7 nm, confirming the successful loading of sgRNA/Cas9.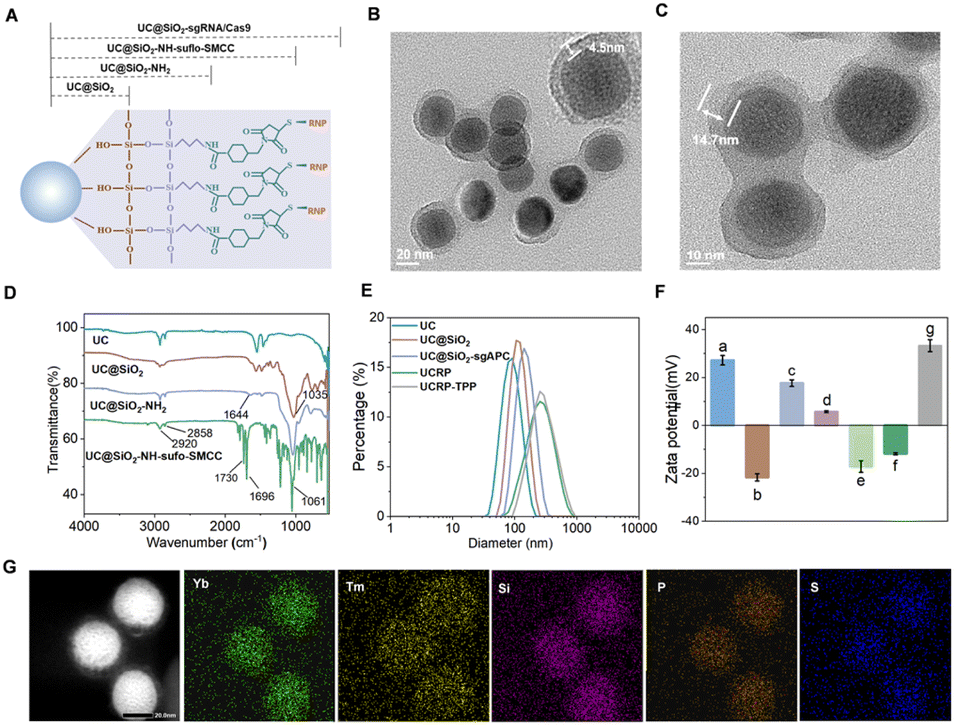 | ||
| Fig. 2 Characterization of the CRISPR/Cas9-based UCRP delivery platform. (A) Schematic representation of UCRP. TEM images of (B) core–shell UC@SiO2 and (C) UC@SiO2-RNP (UCRP). (D) FTIR characterization of surface modification of UCNPs. (E) DLS analysis of the hydrodynamic diameter of UCNPs for surface modification. (F) Zeta potential analysis of (a) UCNPs (+27.2 mV), (b) UC@SiO2 (−21.7 mV), (c) UC@SiO2-NH2 (+17.6 mV), (d) UC@SiO2-SMCC (+5.5 mV), (e) UC@SiO2-sgAPC (−17.2 mV), (f) UC@SiO2-RNP (UCRP, −11.9 mV), (g) UCRP-TPP (+33.3 mV). (G) Elemental mapping of UCRP. | ||
The surface of as-prepared UCNPs was coated with oleic acid, as evidenced by distinct C–H stretching vibrations in the FTIR spectrum (Fig. 2D). UCNPs were coated with a silica layer, resulting in a Si–O–Si stretching band at 1035 cm−1. Subsequent amination generated a peak at 1644 cm−1, confirming the successful synthesis of UC@SiO2-NH2. Further modification with sulfo-SMCC yielded characteristic peaks associated with UC@SiO2-NH2-sulfo-SMCC.49 To quantify the encapsulation efficiency of sgRNA, carboxyfluorescein (FAM)-labeled sgRNA was used and the fluorescence intensity of the supernatant was measured before and after the synthesis of UC@SiO2-sgRNA. The encapsulation efficiency was calculated as approximately 82.3% (Fig. S5). DLS analysis revealed a moderate increase in hydrated particle size upon UCRP formation (Fig. 2E). To improve cellular uptake and escape, UCRP was coated with positively charged poly-L-lysine (PLL), then conjugated with TPP via EDC/NHS chemistry.51 The as-obtained UCRP-TPP showed a further size increase. Zeta potential measurements confirmed the successful modification of each step (Fig. 2F). Elemental mapping further verified the presence of Yb and Tm (from UCNPs), phosphorus (from sgRNA), and sulfur (from Cas9) (Fig. 2G).
Enzyme- and light-activated CRISPR/Cas9
To enable spatiotemporally controlled genome editing, we designed an sgRNA structure with the APE-1 enzyme and light-responsive sites. The sgRNA comprises a target recognition region and a hairpin structure.52 To enable the regulation of CRISPR/Cas9 activity through two keys, we engineered the recognition and hairpin domains with distinct responsive elements. As shown in Fig. 3A, for APE-1 responsiveness, an AP strand complementary to the hairpin was introduced to generate AP sites for enzymatic cleavage. UV light-induced regulation was obtained by hybridizing the recognition region with SH chains and PC strands containing photolysis groups. Gel electrophoresis analysis in Fig. S6 confirmed the successful formation of the APC-sgRNA complex. CRISPR/Cas9 cleavage activity involved amplifying a segment of mtDNA to produce dsDNA (852 bp) containing cleavage sites. As shown in Fig. 3B, APE-1 could recover the cleavage ability of CRISPR/Cas9 by removing the AP strand. Similarly, upon UV light exposure (Fig. 3C), 594 bp and 258 bp fragments were obtained due to the restoration of CRISPR/Cas9 cleavage activity. After individually validating the CRISPR/Cas9 cleavage activity in response to UV or APE1 enzyme alone, we proceeded to assess the dual-responsivity (APE-1 enzyme and UV irradiation) simultaneously for cleavage activity. As shown in Fig. 3D, the coexistence of APE-1 and UV light can fully recover CRISPR/Cas9 cleavage activity, validating the successful dual-lock design.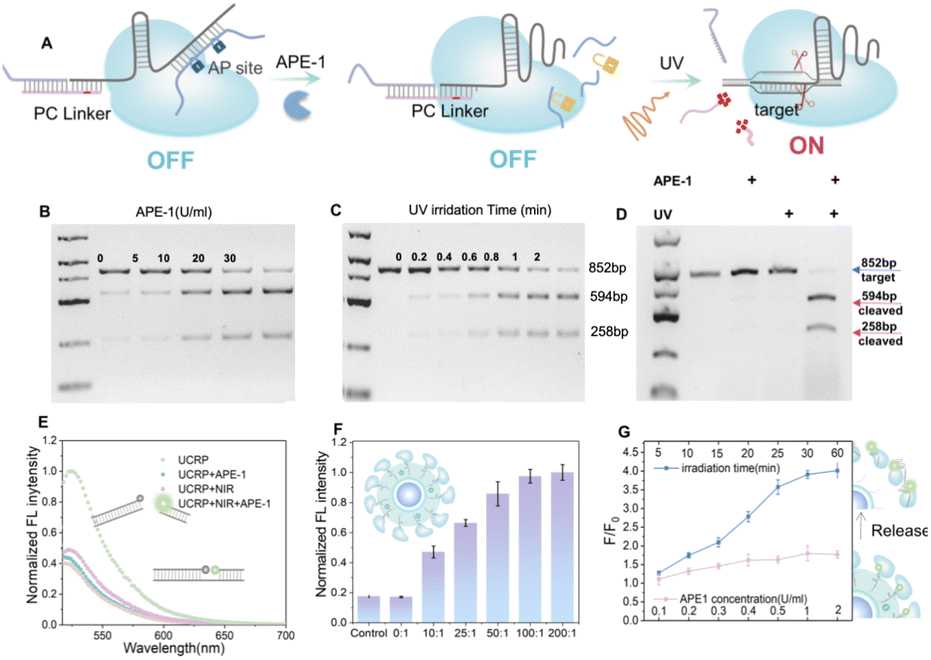 | ||
| Fig. 3 UV light- and APE-1-triggered activation of the CRISPR/Cas9 system. (A) Schematic illustration of the activation of sgRNA/Cas9 via APE1-mediated strand cleavage and UV-induced photolysis. (B–D) Gel electrophoresis analysis of Cas9/sgRNA-mediated cleavage of 852 bp mtDNA upon (B) APE1 interaction, (C) UV light irradiation, respectively and (D) the simultaneous interaction with APE1 and UV light stimulation. Fragments of 594 bp and 258 bp were detected. (E) Fluorescence intensity of FAM-BHQ1-labeled target DNA after treatment with UCRP under different conditions (±APE1, ±NIR). (F) Optimizing the activity of UCRP by varying the molar ratio of Cas9/UCNP. (G) Fluorescence intensity ratios of released FAM-labeled sgRNA in the supernatant as a function of APE1 concentration and NIR irradiation time. F represents the fluorescence intensity of FAM-labeled sgRNA released into the supernatant after treatment with APE1 and NIR irradiation, while F0 refers to the fluorescence intensity of FAM-labeled sgRNA in the supernatant under initial conditions (without APE1 exposure or NIR irradiation). n = 3; data are presented as mean ± SD. | ||
To realize spatiotemporal control in biological systems, we replaced UV light with NIR irradiation for downstream activation of the CRISPR system. As shown in Fig. 3E, the APE-1 and NIR light dual-responsive UCRP was evaluated with a FAM-BHQ1-labeled DNA substrate. The simultaneous treatment with APE-1 and NIR light could restore the cleavage activity, evidenced by a marked increase in fluorescence intensity. The fluorescence intensity gradually increased with prolonged NIR light exposure, and approximately 2.7-fold enhancement was obtained after 30 min illumination (Fig. S7). To optimize the editing efficiency, UCRP was prepared by varying the molar ratio of Cas9 to UCNP (Fig. 3F). The maximum cleavage efficiency was achieved at a ratio of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Furthermore, Cas9/sgRNA release was monitored using fluorescent dye-labeled sgRNA. As shown in Fig. 3G, the fluorescence intensity in the supernatant did not show any significant increase only with APE-1 enzyme addition. In contrast, the fluorescence intensity increased markedly with prolonged NIR irradiation in the presence of APE-1. This indicates that APE1 alone is insufficient to promote the release of Cas9/sgRNA, and the UCRP structure remains stable. The simultaneous existence of APE1 and NIR light can trigger the release of Cas9/sgRNA. These findings indicate that the dual-lock design of UCRP can be efficient in controlling the activity of CRISPR/Cas9 through the combination of APE1 modulation and NIR light exposure.
:1. Furthermore, Cas9/sgRNA release was monitored using fluorescent dye-labeled sgRNA. As shown in Fig. 3G, the fluorescence intensity in the supernatant did not show any significant increase only with APE-1 enzyme addition. In contrast, the fluorescence intensity increased markedly with prolonged NIR irradiation in the presence of APE-1. This indicates that APE1 alone is insufficient to promote the release of Cas9/sgRNA, and the UCRP structure remains stable. The simultaneous existence of APE1 and NIR light can trigger the release of Cas9/sgRNA. These findings indicate that the dual-lock design of UCRP can be efficient in controlling the activity of CRISPR/Cas9 through the combination of APE1 modulation and NIR light exposure.
Gene editing in mitochondria with UCRP-TPP targeting
After confirming the activity, we next evaluated the intracellular performance of the UCRP–TPP system. TPP, a mitochondrial-targeting ligand, was covalently conjugated with PLL using the EDC/NHS crosslinking method.53 Then, the as-obtained TPP-PLL was assembled onto UCRP to form UCRP-TPP for mitochondrial targeting. Subcellular localization was tracked by FAM-labeled sgRNA. Compared with UCRP-PLL, UCRP-TPP showed superior colocalization ability (Fig. 4A), which was supported by fluorescence intensity profiling (Fig. 4B and C).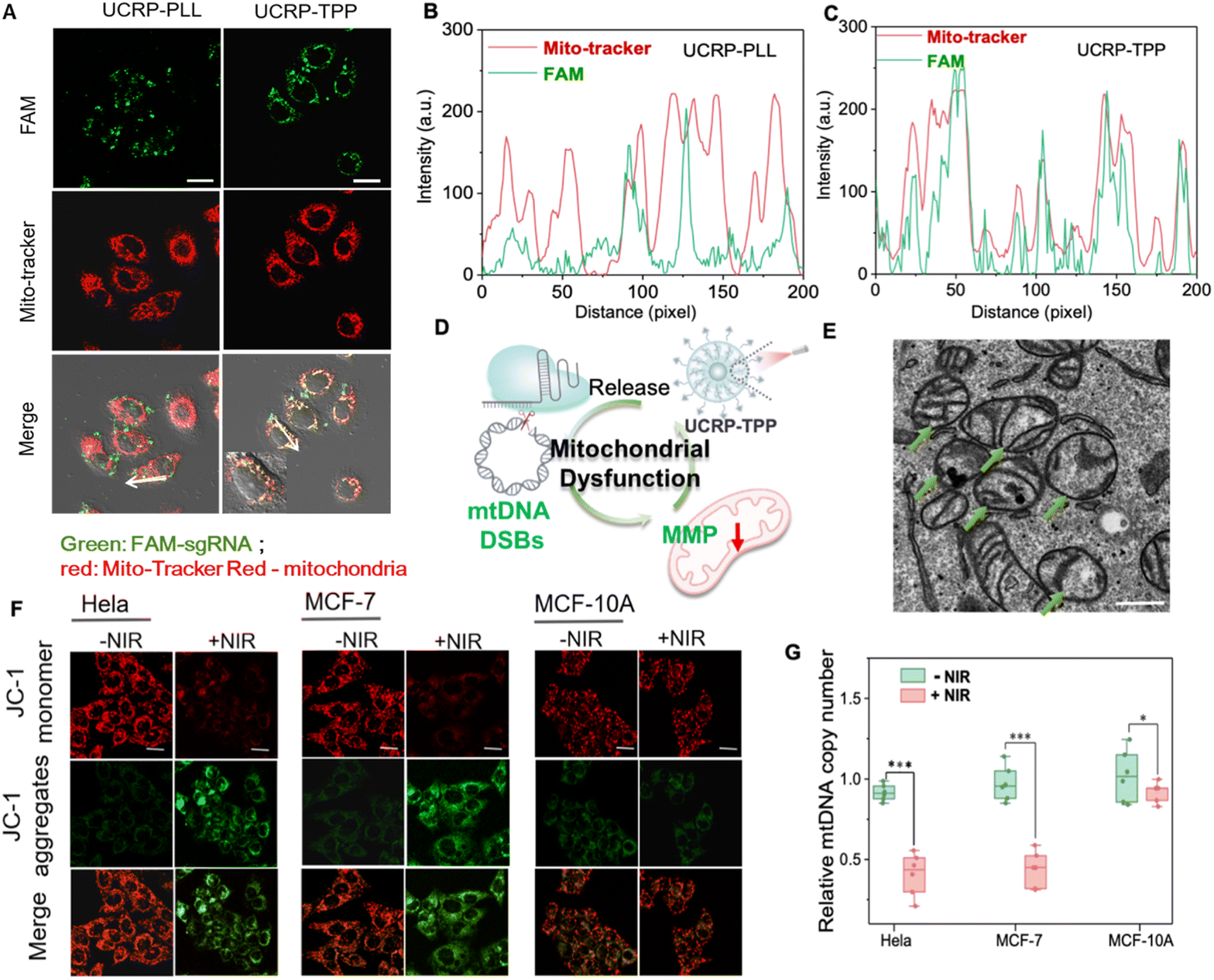 | ||
| Fig. 4 Mitochondrial dysfunction induced by APE-1 and NIR light-triggered UCRP-TPP. (A) CLSM images of HeLa cells treated with UCRP-PLL and UCRP-TPP (FAM-labeled sgRNA), colocalized with MitoTracker (red); scale bar: 20 µm. (B and C) Fluorescence intensity profiles of MitoTracker with (B) UCRP-PLL and (C) UCRP-TPP along the white arrow in (A). (D) Schematic illustration of mitochondrial damage induced by UCRP-TPP. (E) Bio-TEM image of mitochondria in MCF-7 cells after treatment with UCRP-TPP under NIR light irradiation; scale bar: 500 nm. (F) CLSM images of JC-1 staining in HeLa, MCF-7, and MCF-10A cells before and after NIR light irradiation (1.2 w cm−2, 30 min, 980 nm); scale bar: 20 µm. (G) Quantification of mtDNA copy number in HeLa, MCF-7, and MCF-10A cells by qPCR. *P < 0.1, ***P < 0.001. Data represent mean ± SD (n = 3). | ||
Given that mtDNA can be easily damaged due to lack of introns and histone protection, double-strand breaks (DSBs) may irreversibly impair mitochondrial function.54,55 To validate CRISPR-induced mtDNA editing, we designed a 20-bp guide RNA (sgND4, Table S1) targeting the MT-ND4 gene. NIR light irradiation triggered the release of sgRNA/Cas9 from UCRP-TPP after targeting delivery, and then induced mtDNA DSBs in mitochondria. MCF-7, HeLa, and MCF-10A with the target gene via sequencing (Fig. S8) were selected for evaluation. The copy number analysis of mtDNA revealed a marked reduction in MCF-7 and HeLa cells after UCRP-TPP treatment, while minimal change was observed in normal MCF-10A cells (Fig. 4G). This selectivity is attributed to APE1, an enzyme overexpressed in the cytoplasm of tumor cells, that activates CRISPR/Cas9 specifically.56 Upon activation and mitochondrial localization, UCRP-TPP initiated genome editing and mtDNA cleavage, triggering mitochondrial quality control pathways and accelerating mtDNA degradation57,58 (Fig. S9). The structural integrity of mitochondria was examined by TEM. Cells without NIR light illumination showed normal cristae and morphology. In contrast, UCRP-TPP-treated cancer cells under NIR light exposure displayed cristae fragmentation and mitochondrial swelling, indicating structural damage (Fig. 4E and S10).59–61 The observed changes may be attributed to mtDNA disruption, impairing mitochondrial membrane proteins and respiratory function, leading to morphological collapse.62
To further assess mitochondrial dysfunction, JC-1 staining was employed to evaluate changes in mitochondrial membrane potential (MMP). All three cell types exhibited red fluorescence without NIR light illumination, indicating high MMP. After NIR irradiation, a pronounced red-to-green shift occurred in MCF-7 and HeLa cells (Fig. 4F). Compared to MCF-10A, the green/red fluorescence ratio increased by ∼5.4-fold and ∼6.5-fold, respectively (Fig. S11). These results confirm that NIR light-activated UCRP-TPP can induce mitochondrial depolarization and MMP reduction in tumor cells due to mitochondrial function disruption (Fig. 4D).54 Thus, APE1- and NIR light-activated UCRP-TPP enables mtDNA editing and subsequent disruption of mitochondrial function in tumor cells, providing a promising therapeutic strategy.
Induction of cancer cell apoptosis through NIR light-triggered UCRP-TPP disruption of mitochondrial function
To evaluate the therapeutic potential of UCRP-TPP, we examined its impact on mitochondrial damage-induced apoptosis at the cellular level. CCK-8 assays confirmed that UCRP-TPP exhibited good biocompatibility (Fig. 5A). Under NIR light irradiation, a marked increase in intracellular reactive oxygen species (ROS) was observed for the UCRP-TPP-treated MCF-7 cells (Fig. 5B). It was likely a result of the mitochondrial dysfunction-induced disruption of the respiratory chain and oxidative phosphorylation processes.63 Elevated ROS levels can induce oxidative stress and trigger apoptosis.51 Flow cytometry revealed that NIR-activated UCRP-TPP significantly increased apoptotic cell populations compared to control groups (Fig. 5C). Furthermore, the cell viability of UCRP-TPP decreased notably with increasing NIR light power and duration (Fig. S12 and S13). Comparative analysis for different cell lines showed that UCRP-TPP generated substantially higher ROS in tumor cells (MCF-7 and HeLa) than in normal cells (MCF-10A) (Fig. S14).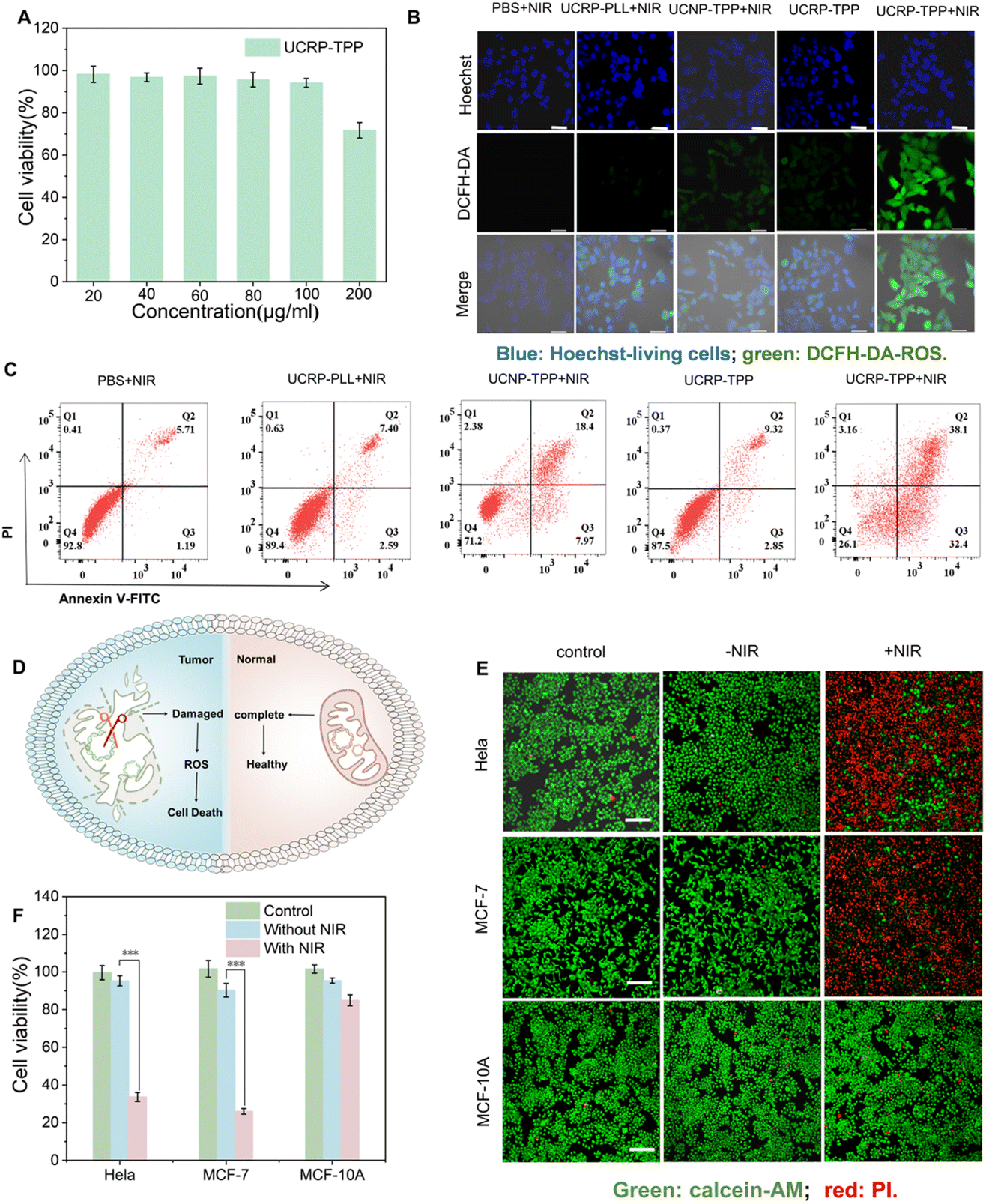 | ||
| Fig. 5 APE-1 and NIR light-triggered disruption of mitochondrial function and apoptosis of cancer cells. (A) CCK-8 analysis of the biocompatibility of UCRP-TPP in MCF-7 cells. (B) CLSM images of MCF-7 cells under various conditions; scale bar: 50 µm. (C) Flow cytometry assay of MCF-7 cells treated with UCRP-TPP with/without NIR light irradiation. (D) Schematic illustration of cancer cell elimination triggered by mitochondrial damage via UCRP-TPP. (E) CLMS images of HeLa, MCF-7, and MCF-10A cells with/without NIR light irradiation; scale bar: 200 µm. (F) CCK-8 viability assay of HeLa, MCF-7, and MCF-10A cells before and after NIR light irradiation. | ||
We further assessed the selective killing effect of UCRP-TPP (Fig. 5D). Without NIR light irradiation, all cell lines maintained high viability. Upon 980 nm light exposure, significant viability loss occurred in tumor cells, while normal cells remained almost unaffected (Fig. 5E). These findings were further validated by CCK-8 analysis (Fig. 5F), confirming the selective killing effect of UCRP-TPP in cancer cells under NIR light activation. These results demonstrate that UCRP-TPP, as a mitochondria-targeted CRISPR/Cas9 delivery system, can effectively induce mitochondrial dysfunction and apoptosis in tumor cells via APE-1 and NIR light activated gene editing.
In vivo antitumor efficacy of UCRP-TPP
To further evaluate the therapeutic efficacy of UCRP-TPP, in vivo experiments were performed using MCF-7 tumor-bearing nude mice. Mice were randomly divided into five groups and administered different treatments by intratumoral injection: PBS, UCNP-TPP, UCRP-PLL, UCRP-TPP, and UCRP-TPP with NIR light irradiation. As shown in Fig. 6A, tumors were locally irradiated with NIR light once daily on days 2 and 3 after intratumoral injection. Each irradiation session lasted 30 minutes, and an intermittent irradiation mode was used to avoid excessive local heating, as described in the experimental section. During the 20-day treatment period, both tumor volume and body weight were monitored. As shown in Fig. 6B, no significant changes in body weight were observed, indicating good biocompatibility of all treatments. Tumor growth curves revealed a marked suppression of tumor progression in the UCRP-TPP + NIR light group, while only a marginal inhibition of tumor growth was observed in the UCNP-TPP + NIR light group (Fig. 6C). The other groups exhibited rapid tumor growth comparable to the PBS control. Upon termination of the study, tumors were harvested and weighed. Both tumor size and weight were significantly reduced in the UCRP-TPP + NIR group (Fig. 6D and E). Histological analyses further validated the therapeutic effect (Fig. 6F). H&E staining revealed decreased tumor cell density, while TUNEL staining showed abundant apoptotic cells in the UCRP-TPP + NIR light group. Moreover, Ki-67 immunostaining confirmed suppressed tumor cell proliferation. These results demonstrate that UCRP-TPP can effectively inhibit tumor growth by inducing apoptosis in the overexpressed APE-1 tumor microenvironment and upon NIR light activation.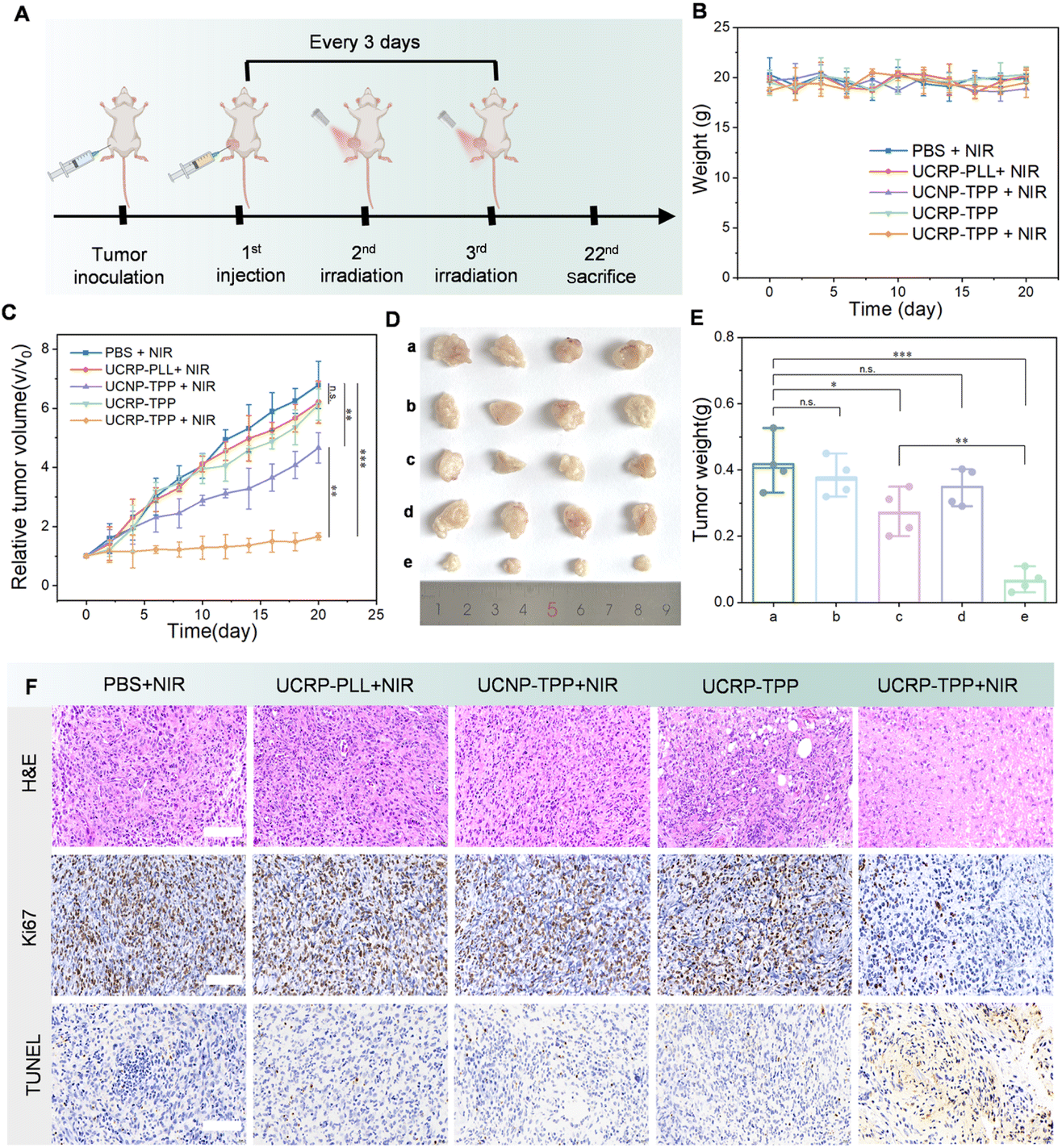 | ||
| Fig. 6 In vivo antitumor efficacy of UCRP-TPP. (A) Schematic illustration of the treatment protocol for MCF-7 tumor-bearing nude mice. (B) Body weight profiles and (C) tumor growth curves of mice in different treatment groups over 20 days. (D) Representative tumor images from each group: (a) PBS + NIR, (b) UCRP-PLL + NIR, (c) UCNP-TPP + NIR, (d) UCRP-TPP, and (e) UCRP-TPP + NIR. (E) Tumor weight data for each group at the end of the treatment. (F) Histological analyses of tumor tissues by H&E staining, Ki67 immunohistochemistry, and TUNEL staining under different treatment conditions; scale bar: 50 µm. | ||
Conclusions
We developed a dual-responsive CRISPR/Cas9 delivery platform (UCRP-TPP) that enables precise mitochondrial gene editing for tumor therapy. By integrating endogenous APE-1 enzyme responsiveness and exogenous NIR light control, the system achieves spatiotemporal activation of CRISPR/Cas9 in tumor cells. The nanoplatform ensures efficient intracellular transport, mitochondrial localization, and minimizes off-target effects. Upon APE-1 and NIR light activation, UCRP-TPP induces mtDNA editing by CRISPR/Cas9, disrupts mitochondrial membrane potential, and triggers ROS generation and apoptosis, resulting in selective killing of cancer cells in vitro and in vivo. This strategy provides a powerful tool for stimulus-responsive gene-editing systems in cancer therapeutics.Author contributions
Fei Yang: conceptualization, investigation, writing – original draft. Qianqin Ran: investigation, visualization. Jiahui Chen: data, investigation. Guochen Bao: visualization. Yuezhong Xian: supervision, writing – review & editing, funding acquisition. Cuiling Zhang: supervision, conceptualization, writing – review & editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Data availability
Additional experimental data supporting this article are included in the supplementary information (SI). Supplementary information: Chemicals and materials, instruments, experimental section, TEM images, UV-visible absorption spectra, fluorescence spectra, PAGE, Western blot images, CCK-8 analysis, CLSM images, Table S1. See DOI: https://doi.org/10.1039/d5sc07976d.Reasonable requests for additional information can be made to the corresponding authors.
Acknowledgements
This research was supported by the Natural Science Foundation of Shanghai (25ZR1401107), National Natural Science Foundation of China (22274054), and Shanghai Magnolia Talent Plan Pujiang Project (24PJD027).References
- H. Li, Y. Xie, F. Chen, H. Bai, L. Xiu, X. Zhou, X. Guo, Q. Hu and K. Yin, Chem. Soc. Rev., 2023, 52, 361–382 Search PubMed.
- J. Y. Wang and J. A. Doudna, Science, 2023, 379, eadd8643 CrossRef CAS PubMed.
- K. Yi, H. Kong, Y. H. Lao, D. Li, R. L. Mintz, T. Fang, G. Chen, Y. Tao, M. Li and J. Ding, Adv. Mater., 2024, 36, 2300665 CrossRef CAS PubMed.
- Z. Weng, Z. You, J. Yang, N. Mohammad, M. Lin, Q. Wei, X. Gao and Y. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202214987 CrossRef CAS PubMed.
- H. Yang, R. Ledesma-Amaro, H. Gao, Y. Ren and R. Deng, Biosens. Bioelectron., 2023, 228, 115189 CrossRef CAS PubMed.
- A. Katti, B. J. Diaz, C. M. Caragine, N. E. Sanjana and L. E. Dow, Nat. Rev. Cancer, 2022, 22, 259–279 CrossRef CAS PubMed.
- E. Tan, T. Wan, Q. Pan, J. Duan, S. Zhang, R. Wang, P. Gao, J. Lv, H. Wang and D. Li, Sci. Adv., 2024, 10, eadl4336 Search PubMed.
- D. Tang, Y. Yan, Y. Li, Y. Li, J. Tian, L. Yang, H. Ding, G. Bashir, H. Zhou and Q. Ding, Theranostics, 2024, 14, 203 Search PubMed.
- S. K. Alsaiari, B. Eshaghi, B. Du, M. Kanelli, G. Li, X. Wu, L. Zhang, M. Chaddah, A. Lau and X. Yang, Nat. Rev. Mater., 2025, 10, 44–61 Search PubMed.
- J. Yan, D. D. Kang and Y. Dong, Biomater. Sci., 2021, 9, 6001–6011 RSC.
- X. Tang, Z. Wang, Y. Zhang, W. Mu and X. Han, Chem. Eng. J., 2022, 435, 135584 Search PubMed.
- Y. J. Kim, D. Yun, J. K. Lee, C. Jung and A. J. Chung, Nat. Commun., 2024, 15, 8099 CrossRef CAS PubMed.
- Y. Xing, J. Yang, Y. Wang, C. Wang, Z. Pan, F. L. Liu, Y. Liu and Q. Liu, ACS Nano, 2023, 17, 5713–5726 Search PubMed.
- L. Zhang, L. Wang, Y. Xie, P. Wang, S. Deng, A. Qin, J. Zhang, X. Yu, W. Zheng and X. Jiang, Angew. Chem., Int. Ed., 2019, 58, 12404–12408 Search PubMed.
- R. Masarwy, D. Breier, L. Stotsky-Oterin, N. Ad-El, S. Qassem, G. S. Naidu, A. Aitha, A. Ezra, M. Goldsmith and I. Hazan-Halevy, Adv. Sci., 2025, 12, 2411032 Search PubMed.
- J. W. Liu, N. Song, Y. Chu, S. Li and X. Fan, et al. , Adv. Funct. Mater., 2023, 33, 2306634 Search PubMed.
- K. Chen, H. Han, S. Zhao, B. Xu, B. Yin, A. Lawanprasert, M. Trinidad, B. W. Burgstone, N. Murthy and J. A. Doudna, Nat. Biotechnol., 2024, 1–13 Search PubMed.
- S. K. Alsaiari, S. Patil, M. Alyami, K. O. Alamoudi, F. A. Aleisa, J. S. Merzaban, M. Li and N. M. Khashab, J. Am. Chem. Soc., 2018, 140, 143–146 Search PubMed.
- Y. Huang, G. Qin, T. Cui, C. Zhao, J. Ren and X. Qu, Nat. Commun., 2023, 14, 4647 Search PubMed.
- W. Deng, W. Chen, S. Clement, A. Guller, Z. Zhao, A. Engel and E. M. Goldys, Nat. Commun., 2018, 9, 2713 Search PubMed.
- Y. Zhao, J. Qin, D. Yu, Y. Liu, D. Song, K. Tian, H. Chen, Q. Ye, X. Wang and T. Xu, Nat. Nanotechnol., 2024, 19, 1869–1879 Search PubMed.
- K. Lee, M. Conboy, H. M. Park, F. Jiang, H. J. Kim, M. A. Dewitt, V. A. Mackley, K. Chang, A. Rao and C. Skinner, et al. , Nat. Biomed. Eng., 2017, 1, 889–901 Search PubMed.
- J. Wu, H. Peng, X. Lu, M. Lai, H. Zhang and X. C. Le, Angew. Chem., Int. Ed., 2021, 60, 11104–11109 Search PubMed.
- X. Zhao, N. Na and J. Ouyang, Chem. Sci., 2022, 13, 11433–11441 Search PubMed.
- N. Song, X. Fan, X. Guo, J. Tang, H. Li, R. Tao, F. Li, J. Li, D. Yang and C. Yao, et al. , Adv. Mater., 2024, 36, 2309534 Search PubMed.
- Y. Pan, J. Yang, X. Luan and X. Liu, et al. , Sci. Adv., 2019, 5, eaav7196 Search PubMed.
- R. Xie, X. Wang, Y. Wang, M. Ye, Y. Zhao, B. S. Yandell and S. Gong, Adv. Mater., 2022, 34, 2110618 Search PubMed.
- H. Shivram, B. F. Cress, G. J. Knott and J. A. Doudna, Nat. Chem. Biol., 2021, 17, 10–19 Search PubMed.
- D. Zhang, G. Wang, X. Yu, T. Wei, L. Farbiak, L. T. Johnson, A. M. Taylor, J. Xu, Y. Hong, H. Zhu and D. J. Siegwart, Nat. Nanotechnol., 2022, 17, 777–787 Search PubMed.
- J. Shi, X. Yang, Y. Li, D. Wang, W. Liu, Z. Zhang, J. Liu and K. Zhang, Biomaterials, 2020, 256, 120221 CrossRef CAS PubMed.
- E. Tan, T. Wan, C. Yu, Q. Fan, W. Liu, H. Chang, J. Lv, H. Wang, D. Li, Y. Ping and Y. Cheng, Nano Today, 2022, 46, 101601 Search PubMed.
- Y. Sun, X. Xu, L. Chen, W. L. Chew, Y. Ping and A. Miserez, ACS Nano, 2023, 17, 16597–16606 Search PubMed.
- Y. Pu, H. Yin, C. Dong, H. Xiang, W. Wu, B. Zhou, D. Du, Y. Chen and H. Xu, Adv. Mater., 2021, 33, 2104641 Search PubMed.
- W. Zhou, W. Brown, A. Bardhan, M. Delaney, A. S. Ilk, R. R. Rauen, S. I. Kahn, M. Tsang and A. Deiters, Angew. Chem., Int. Ed., 2020, 59, 8998–9003 CrossRef CAS PubMed.
- R. Bi, Y. Li, M. Xu, Q. Zheng, D. F. Zhang, X. Li, G. Ma, B. Xiang, X. Zhu and H. Zhao, Innovation, 2022, 3, 100329 CAS.
- Y. Li, Y. Wu, R. Xu, J. Guo, F. Quan, Y. Zhang, D. Huang, Y. Pei, H. Gao and W. Liu, et al. , Nat. Commun., 2023, 14, 7722 CrossRef CAS PubMed.
- K. Zhang, R. Deng, X. Teng, Y. Li, Y. Sun, X. Ren and J. Li, J. Am. Chem. Soc., 2018, 140, 11293–11301 Search PubMed.
- Y. W. Jun, E. Albarran, D. L. Wilson, J. Ding and E. T. Kool, Angew. Chem., Int. Ed., 2022, 61, e202111829 CrossRef CAS PubMed.
- M. Kodavati, H. Wang, W. Guo, J. Mitra, P. M. Hegde, V. Provasek, V. H. M. Rao, I. Vedula, A. Zhang and S. Mitra, Nat. Commun., 2024, 15, 2156 CrossRef CAS PubMed.
- C. Tang, J. Cai, X. M. Yin, J. M. Weinberg, M. A. Venkatachalam and Z. Dong, Nat. Rev. Nephrol., 2021, 17, 299–318 Search PubMed.
- J. Luo, X. Gong, H. Su, Z. Lin, X. Yang, X. Yan and J. Gao, Angew. Chem., Int. Ed., 2021, 60, 1403–1410 Search PubMed.
- J. W. Liu, Y. G. Yang, K. Wang, G. Wang, C. C. Shen, Y. H. Chen, Y. F. Liu, T. D. James, K. Jiang and H. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 3669–3678 Search PubMed.
- K. N. Wang, X. Shao, Z. Tian, L. Y. Liu, C. Zhang, C. P. Tan, J. Zhang, P. Ling, F. Liu and Q. Chen, et al. , Adv. Sci., 2021, 8, 2004566 Search PubMed.
- Y. Li, R. Xu and Y. Wu, et al. , Sci. Adv., 2023, 9, eadi1965 Search PubMed.
- C. Sheng, J. Zhao, Z. Di, Y. Huang, Y. Zhao and L. Li, Nat. Biomed. Eng., 2022, 6, 1074–1084 Search PubMed.
- D. Yi, J. Zhao and L. Li, Angew. Chem., Int. Ed., 2021, 60, 6300–6304 Search PubMed.
- Y. Chen, X. Gong, Y. Gao, Y. Shang, J. Shang, S. Yu, R. Li, S. He, X. Liu and F. Wang, Chem. Sci., 2021, 12, 15710–15718 RSC.
- T. M. Weaver, N. M. Hoitsma, J. J. Spencer, L. Gakhar, N. J. Schnicker and B. D. Freudenthal, Nat. Commun., 2022, 13, 5390 Search PubMed.
- Z. Yang, K. Y. Loh, Y. T. Chu, R. Feng, N. S. R. Satyavolu, M. Xiong, S. M. Nakamata Huynh, K. Hwang, L. Li and H. Xing, J. Am. Chem. Soc., 2018, 140, 17656–17665 Search PubMed.
- J. Zhang, P. Zhao, W. Li, L. Ye, L. Li, Z. Li and M. Li, Angew. Chem., Int. Ed., 2022, 61, e202117562 Search PubMed.
- F. Yu, Y. Shao, X. Chai, Y. Zhao and L. Li, Angew. Chem., Int. Ed., 2022, 61, e202203238 CrossRef CAS PubMed.
- Y. Hao, J. Li, Q. Li, L. Zhang, J. Shi, X. Zhang, A. Aldalbahi, L. Wang, C. Fan and F. Wang, Angew. Chem., Int. Ed., 2020, 59, 20612–20618 Search PubMed.
- D. Yi, H. Zhao, J. Zhao and L. Li, J. Am. Chem. Soc., 2023, 145, 1678–1685 Search PubMed.
- E. Janik-Karpinska, M. Ceremuga, M. Niemcewicz, E. Synowiec, T. Sliwinski and M. Bijak, Molecules, 2023, 28, 2014 CrossRef PubMed.
- D. A. Roubicek and N. C. Souza-Pinto, Toxicology, 2017, 391, 100–108 Search PubMed.
- Y. Shao, J. Zhao, J. Yuan, Y. Zhao and L. Li, Angew. Chem., Int. Ed., 2021, 60, 8923–8931 Search PubMed.
- R. Loutre, A. M. Heckel, A. Smirnova, N. Entelis and I. Tarassov, IUBMB Life, 2018, 70, 1233–1239 Search PubMed.
- W. P. Bian, Y. L. Chen, J. J. Luo, C. Wang, S. L. Xie and D. S. Pei, ACS Synth. Biol., 2019, 8, 621–632 Search PubMed.
- Y. Guo, S. Li, Z. Tong, J. Tang, R. Zhang, Z. Lv, N. Song, D. Yang and C. Yao, J. Am. Chem. Soc., 2023, 145, 23859–23873 Search PubMed.
- K. Lin, Z. Ma, J. Li, M. Tang, A. Lindstrom, M. Ramachandran, S. Zhu, T. Y. Lin, L. Zhang and Y. Li, Adv. Funct. Mater., 2021, 31, 2008423 Search PubMed.
- J. Bao, J. Wang, S. Chen, S. Liu, Z. Wang, W. Zhang, C. Zhao, Y. Sha, X. Yang and Y. Li, et al. , ACS Nano, 2024, 18, 9100–9113 Search PubMed.
- Z. Yi, X. Zhang, W. Tang, Y. Yu, X. Wei, X. Zhang and W. Wei, Nat. Biotechnol., 2024, 42, 498–509 CrossRef CAS PubMed.
- R. Bi, Y. Li, M. Xu, Q. Zheng, D. F. Zhang, X. Li, G. Ma, B. Xiang, X. Zhu and H. Zhao, et al. , Innovation, 2022, 3, 100329 CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |