
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegio-reversed alkenyl–arylation of ynamides via 1,3-olefin shift
Saumya
Verma†
,
Manoj
Sethi†
and
Akhila K.
Sahoo
 *
*
School of Chemistry, University of Hyderabad, 500046, India. E-mail: akhilchemistry12@gmail.com; akssc@uohyd.ac.in
First published on 17th November 2025
Abstract
A cationic Pd-catalysed multicomponent reaction enables the regio-reversed alkenyl–arylation of ynamides through a migratory difunctionalization strategy. A stereoselective 1,3-olefin shift of a keteniminium–alkenyl–Pd intermediate governs the carbopalladation pathway, generating a vinylic Pd species that subsequently couples with aryl boronates. This protocol offers streamlined access to highly substituted branched dienes, trienes, and divinyl ethylene (DVE) derivatives with excellent regio- and stereocontrol. The broad substrate scope, scalability, and mechanistic insights highlight the synthetic potential of this transformation. Additionally, the photophysical properties of these conjugated systems were examined, underscoring their potential as functional materials for optoelectronic applications.
Introduction
Transition metal-catalyzed alkyne difunctionalization has emerged as a powerful strategy for assembling structurally complex molecules from simple and readily available feedstock chemicals.1–7 These transformations offer excellent control over both regio- and stereoselectivity, enabling the modular incorporation of diverse functional groups onto alkyne scaffolds.1–5 Despite these advances, the diastereoselective synthesis of internally substituted conjugated polyenes remains challenging due to difficulties in achieving precise stereochemical control (Fig. 1B). Traditionally, conjugated polyene systems are accessed via cross-coupling strategies that require pre-functionalized alkenyl partners or strained intermediates (Fig. 1A).7–16 In contrast, alkyne difunctionalization allows for the regioselective introduction of two distinct substituents across a triple bond, either through direct addition (Fig. 1A)17,18 or via migratory mechanisms (Fig. 1A), where one group undergoes positional rearrangement before interception by a second coupling partner.19–27 These strategies enable the stereoselective assembly of conjugated polyene frameworks, often surpassing traditional cross-coupling approaches in terms of step-economy, selectivity, and overall synthetic efficiency. | ||
| Fig. 1 Background and this work. | ||
In this context, most established difunctionalization strategies for polarized alkynes (ynamides) have centred on hydro-olefination or stepwise olefin incorporation. Seminal contributions from the Skrydstrup28 and Zhu29 groups have provided access to polarized diene frameworks (Fig. 1C). However, reports of carbo-olefination in such systems are rare. An exception is the study by Lam et al., in which a bimetallic Rh/Zn system for oxazolidinone-derived ynamides forms α-vinylic zinc intermediates that couple with iodoalkenyl reagents, furnishing the corresponding carbo-olefination products (Fig. 1C).30
We recently reported a one-pot aryl-olefination of ynamides, wherein 1,3-aryl shift governed the reaction pathway, enabling the selective synthesis of substituted linear dienes (Fig. 1C).19 Building on these findings, we envisaged that reversing the regioselectivity via alkenyl–arylation could provide access to branched conjugated olefins, which are challenging to synthesize using existing methods (Fig. 1D). To realize this goal, a successful 1,3-olefin shift is essential. In contrast to the facile 1,3-aryl migrations, olefin rearrangements present significant challenges, primarily due to competing migration pathways and the tendency of in situ generated vinylic metal intermediates from alkenyl-electrophile to undergo β-hydride elimination (Fig. 1D).31–34
The present study introduces a general one-pot strategy for the synthesis of internally substituted branched diene and triene frameworks through the three-component coupling of ynamides, alkenyl triflates, and aryl boronates with high selectivity. The reaction proceeds under mild, ligand-free conditions, effectively addressing the challenges associated with olefin migration (Fig. 1D).
Results and discussion
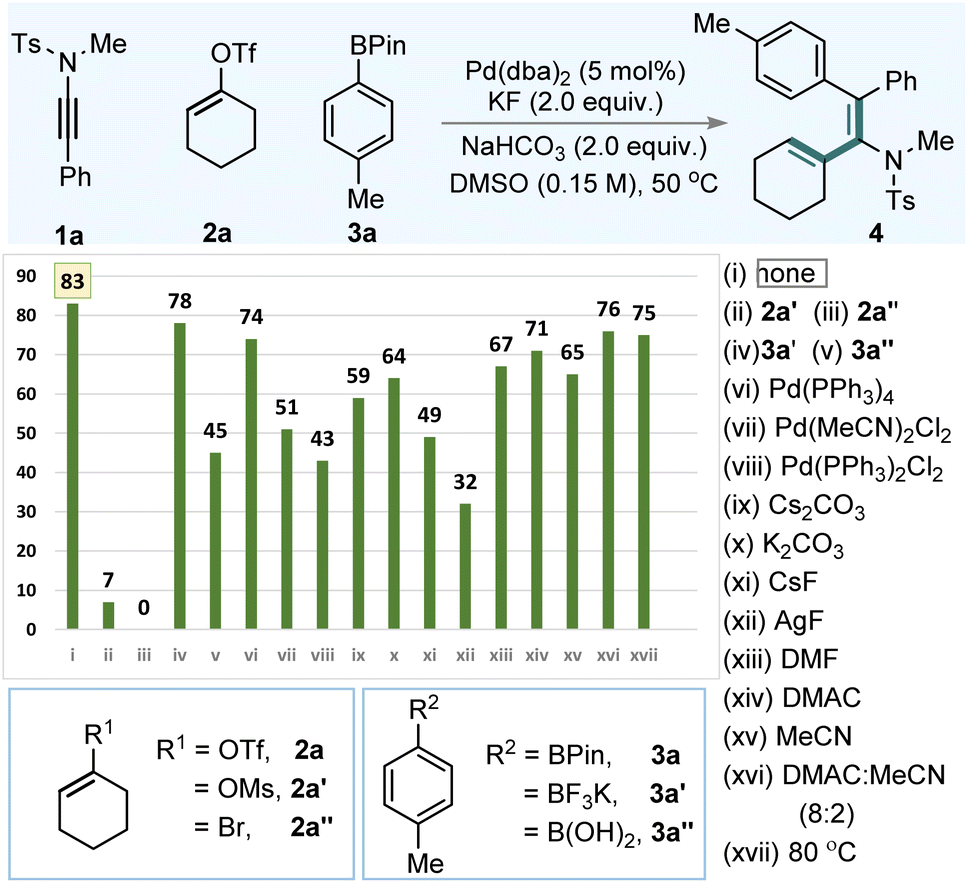
To evaluate our hypothesis, ynamide 1a, cyclohex-1-en-1-yl trifluoromethanesulfonate (2a), and 4,4,5,5-tetramethyl-2-(p-tolyl)-1,3,2-dioxaborolane (3a) were selected as model substrates (Table 1). Reaction optimization revealed that using Pd(dba)2 (5.0 mol%) in combination with KF (2.0 equiv.) and NaHCO3 (2.0 equiv.) in DMSO (0.15 M) at 50 °C delivered the desired alkenyl–arylated product 4 in 83% yield with complete regio- and syn-selectivity (Table 1, entry i). Substituting the alkenyl triflate with cyclohex-1-en-1-yl methanesulfonate (2a′) or 1-bromocyclohex-1-ene (2a″) resulted in significantly reduced yields or complete suppression of product formation (entries ii and iii). Similarly, replacing arylboronate 3a with boronic acid derivatives 3a′ or 3a″ led to only moderate yields (78% and 45%, entries iv and v). Alternative palladium sources, including various Pd(0) and Pd(II) complexes, did not improve the outcome when compared with Pd(dba)2 (entries vi–viii). Base/additive combinations, such as Cs2CO3, K2CO3, CsF, and AgF, led to 4 in 32–64% (entries ix–xii). Finally, alterations of solvent system (entries xiii–xvi) or reaction temperature (entry xvii) did not enhance the product yield.| a Reaction conditions: 1 (0.05 mmol), 2 (0.075 mmol), 3 (0.075 mmol), Pd(dba)2 (0.005 mmol), KF (0.1 mmol), NaHCO3 (0.1 mmol): DMSO (0.15 M), stirred at 50 °C. |
|---|
|
Importantly, the choice of alkenyl electrophile and the solvent system is critical in determining the overall regioselectivity. Labile electrophiles can generate vinylic cationic Pd(II) species that exhibit enhanced electrophilicity stabilized by a coordinating solvent. This promotes the formation of the keteniminium intermediates and the subsequent olefin migration, leading to a regioselective product. Nonetheless, the nature of the ynamide is equally crucial; directing-enabled ynamides (1h) can result in the formation of regioisomeric mixtures.
Under the optimized conditions, the scope of this three-component protocol for synthesizing branched 1,3-dienes was explored (Scheme 1a). The para-substituted arylboronates bearing electron-donating groups p-Me (3a) and p-OMe (3b) reacted independently with 1a and 2a to afford dienes 4 and 5 in 83% and 63% yield, respectively. Despite the potential for Pd deactivation by hydroxyl coordination, the reaction of OH-substituted boronate 3c delivered diene 6 in moderate yield, offering a handle for further derivatization. Electron-deficient and halo-substituted arylboronates, including [p-CF3 (3d), p-Br (3e), and m-Br (3i)] were well tolerated, furnishing dienes 7 (65%), 8 (62%), and 12 (69%) with the bromo group intact. This highlights the potential for downstream cross-coupling or late-stage functionalization. Likewise, the meta-substituted arylboronates [m-CO2Et (3f), m-SO2Me (3g), m-Ac (3h), and m-Br (3i)] also showed broad compatibility delivering 9–12 in 58–80% yield with high regioselectivity. The structure of diene 10 was unambiguously confirmed by single-crystal X-ray diffraction (CCDC 2433173). Notably, the transformation with m-vinyl-substituted arylboronate 3j was smooth affording diene 13 in 62% yield; the vinylic moiety did not affect the reaction outcome. Heteroaryl (3k) and polyaromatic substrates (3l–3o) participated efficiently in the reaction delivering the desired products 14–18 in 57–86% yield.
 | ||
| Scheme 1 Scope of reaction; reaction conditions: 1 (0.3 mmol), 2 (0.45 mmol), 3 (0.45 mmol), Pd(dba)2 (0.015 mmol), KF (0.6 mmol), NaHCO3 (0.6 mmol): DMSO (0.15 M), stirred at 50 °C. The regioisomeric ratio (calculated using 1H NMR) of the products is mentioned in parentheses. | ||
We next examined the reactivity of alkenyl triflates (Scheme 1b). Gratifyingly, both cyclic and acyclic variants of alkenyl triflates coupled efficiently with 1a and 3a, delivering the corresponding 1,3-dienes with excellent stereoselectivity. The sterically demanding cyclic systems, including five-to eight-membered alkenyl triflates (2b–2e) were participated smoothly in the syn-olefinic–arylation of ynamides; the respective products 19–22 were obtained in moderate to good yields (51–73%). Likewise, products 23 (61%), and 24 (57%) were made from the coupling of alkenyl triflates derived from tetralone (2f), and exocyclic framework (2g), respectively.
We then turned our attention to study the ynamides scope (Scheme 1c). Aryl-substituted ynamides bearing both electron-donating and electron-withdrawing groups, such as p-Ph (1b), p-CO2Me (1c), p-CN (1d), p-F (1e), m-NO2 (1f), and m-Cl (1g) reacted efficiently to furnish the corresponding products 25–30 in good yields (69–80%). In contrast, the oxazolidinone ynamide 1h proved less effective, delivering product 31 in 43% yield with poor regioselectivity (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2). This reduced efficiency is likely due to the combined influence of the oxazolidinone directing group and the intrinsic electronic character of the ynamide. Unfortunately, the triflate electrophiles 1-(cyclohex-1-en-1-yl)vinyl triflate (2h) and adamantanyl vinyl triflate (2i) did not undergo the desired transformation with 1a and 3a, resulting in a complex mixture. The outcome with 2h can be attributed to multiple competing migration pathways, whereas for bulky 2i, steric hindrance between the ynamide N-tosyl group and the electrophile likely prevents the intramolecular 1,3-olefin shift. In addition, other alkyne derivatives such as diethyl (phenylethynyl)phosphonate (1i), and phenyl(phenylethynyl)sulfane (1j) failed to deliver the desired diene products under the standard conditions (Scheme 1d).
:2). This reduced efficiency is likely due to the combined influence of the oxazolidinone directing group and the intrinsic electronic character of the ynamide. Unfortunately, the triflate electrophiles 1-(cyclohex-1-en-1-yl)vinyl triflate (2h) and adamantanyl vinyl triflate (2i) did not undergo the desired transformation with 1a and 3a, resulting in a complex mixture. The outcome with 2h can be attributed to multiple competing migration pathways, whereas for bulky 2i, steric hindrance between the ynamide N-tosyl group and the electrophile likely prevents the intramolecular 1,3-olefin shift. In addition, other alkyne derivatives such as diethyl (phenylethynyl)phosphonate (1i), and phenyl(phenylethynyl)sulfane (1j) failed to deliver the desired diene products under the standard conditions (Scheme 1d).
To further demonstrate the versatility of this synthetic method, we extended its application to the synthesis of conjugated trienes using ene-ynamides (Scheme 2). The dual functionality of ene-ynamides, comprising both alkyne and alkene moieties, poses site-selectivity challenges. Specifically, the formation of a π–allyl–Pd intermediate (III″) from the reaction intermediate III′ could complicate the reaction pathway (Scheme 2a). However, the nucleophilic β-carbon of the ynamide strengthens the Pd–C bond, which suppresses π–allyl–Pd formation and consequently directs the reaction toward the selective synthesis of conjugated trienes (Scheme 2a).
 | ||
| Scheme 2 Scope of reaction and scale-up reaction; reaction conditions: 1 (0.3 mmol), 2 (0.45 mmol), 3 (0.45 mmol), Pd(dba)2 (0.015 mmol), KF (0.6 mmol), NaHCO3 (0.6 mmol): DMSO (0.15 M), stirred at 50 °C. The regioisomeric ratio (calculated using 1H NMR) of the products is mentioned in parentheses. | ||
In this context, a variety of aryl boronates including para-(3d) and meta-substituted (3f) derivatives, as well as polyaromatics (3l–3n) were subjected to standard conditions with cyclic ene-ynamide 1k and alkenyl triflate 2a, delivering trienes 32–36 in 67–72% yield. Further, acyclic ene-ynamide (1l and 1m) reacted smoothly furnishing trienes 37–39 in 59–68% yield.
We next examined the reactivity of yne-ynamides (1n) under the standard conditions. Remarkably, despite the coexistence of two potentially reactive sites the ynamide and the terminal alkyne, the transformation proceeded with complete chemoselectivity at the ynamide moiety, affording the highly substituted DVE derivative 40 in 62% yield.
Furthermore, the reaction exhibited excellent tolerance toward a wide range of structurally complex scaffolds incorporated at the periphery of the aryl-boronate unit. Derivatives of gemfibrozil (3p), borneol (3q), naproxen (3r), and ibuprofen (3s) reacted smoothly with 1a and 2a, delivering products 41–44 in 53–70% yield with good selectivity (Scheme 2c).
To our delight, the reaction was successfully performed on a gram scale (Scheme 2d). Specifically, the reaction of the corresponding ynamides (4.0 mmol) with alkenyl triflates and aryl boronates under the standard conditions afforded compounds 6 (0.95 g, 52%), 8 (1.21 g, 58%), 9 (1.67 g, 81%), and 34 (1.29 g, 65%). Notably, these yields are consistent with those obtained in the small-scale experiments, demonstrating the scalability and robustness of the protocol.
To demonstrate the synthetic versatility of the method, further functionalization of selected compounds was carried out (Scheme 3A). Treating compound 6 with ethyl propiolate led to the product 45 in 59% yield. The hydroxyl group in 6 was readily protected providing the complex aryl triflate 46 in 62% yield, thereby offering a handle for further derivatization. The ester hydrolysis of compound 9 provided carboxylic acid, which was then coupled with L-menthol to deliver ester 47 in 64% yield (Scheme 3A).
 | ||
| Scheme 3 Synthetic application and photophysical study; (a) absorption (5 µM, λabs) and (b) emission (50 µM, λem) spectra of the compounds were investigated in MeCN at 298K. The emission spectra were measured upon excitation at 275–320 nm. | ||
Next, the photophysical properties such as absorption and emission spectra of the compounds (36, 34, 38 and 39) were studied in acetonitrile (MeCN) at 298 K (Scheme 3B). The compounds displayed intense high-energy bands at 224–228 nm, assigned to allowed π → π* intra-ligand transitions within the aromatic framework. Less intensity features in the 262–338 nm region are assignable to n → π* transitions from nitrogen lone pairs to π* orbitals, and lower-energy π → π* excitations involving extended aromatic conjugation. The emission spectra of compounds (36, 34, 38 and 39) were recorded in MeCN upon excitation at 290, 275, and 320 nm. All compounds exhibited emission in the near-UV to blue region (365–410 nm) having emission maxima at 365, 385, 410, and 390 nm, respectively. These photophysical features highlight the potential of the synthesized conjugated frameworks as functional materials for optoelectronic applications, and UV-absorbing materials.7
To probe the reaction mechanism and factors controlling regioselectivity, a series of control experiments were conducted, including a competitive reaction between ynamides 1a and 1h with 2a and 3a under standard conditions (Scheme 4A). The reaction afforded products 4 (83%) and 31 (43%) in a 3:2 regioisomeric ratio. The exclusive formation of diene 4 from 1a suggests that N-lone pair delocalization directs regioselectivity, whereas the formation of regioisomer 31 from N-oxazolidinone-substituted ynamide 1h highlights the competing influence of chelation and electronic effects on the reaction outcome.
 | ||
| Scheme 4 Control experiment and plausible mechanism. | ||
Crossover experiments were performed under the optimized conditions to probe chemoselectivity (Scheme 4B and 4C). In the intramolecular case, yne-ynamide 1l reacted exclusively to give product 40, confirming preferential reactivity of the ynamide moiety even when conjugated with an alkyne (Scheme 4B). While in the intermolecular crossover experiment, the reaction independently performed among 1a/1a′ with 2a and 3a, the product 4 was only formed from ynamide 1a (Scheme 4C). These results clearly highlight the preferential reactivity of the ynamide moiety over the alkyne unit (Scheme 4B and 4C).
Based on experimental results and literature precedents, a plausible mechanism is proposed (Scheme 4D). Ligand exchange between Pd(dba)2 and DMSO generates Pd(0)(DMSO)2, which undergoes oxidative addition with alkenyl triflate 2 to form a cationic alkenyl-Pd(II) intermediate I. Nucleophilic attack of ynamide 1 on I, with concomitant triflate displacement, furnishes keteniminium intermediate II. A stereoselective intramolecular 1,3-olefin shift then produces intermediate III. Transmetalation of III with the aryl fluoroborate IV in the presence of KF forms alkenyl-Pd-aryl species V, which undergoes reductive elimination to deliver the final product 4.
Conclusions
In conclusion, we have developed a Pd-catalyzed regio-reversed dicarbofunctionalization strategy of ynamides, enabling highly selective 1,2-alkenylarylation via a stereoselective 1,3-olefin shift. This transformation proceeds through a cationic alkenyl-Pd intermediate, which is efficiently intercepted by aryl boronic esters to furnish α-alkenylated, branched conjugated dienes and trienes with excellent regio- and stereocontrol. The method exhibits broad substrate scope and high tolerance toward various functional groups. This transformation also offers an efficient route for the synthesis of complex π-conjugated systems, including divinyl ethylene (DVE) derivatives. Furthermore, the detailed photophysical studies of the compounds revealed strong π → π* and n → π* absorption and features fluorescence emission in the near-UV to blue region (365–410 nm). These findings suggest that the conjugated frameworks possess strong potential for use as functional materials in optoelectronic devices and UV-absorbing systems. This work not only expands the synthetic utility of ynamides in transition-metal catalysis but also introduces new avenues for the synthesis of divinyl ethylene (DVE) derivatives and their application to showcasing the distinctive photophysical behaviour.Author contributions
All authors have approved the final version of the manuscript. A. K. S., M. S., and S. V. conceived the idea and M. S. and S. V. performed the experiments. Review, editing and supervision done by A. K. S.Conflicts of interest
There are no conflicts to declare.Data availability
CCDC 2433173 (10) and CCDC 2433174 (36) contain the supplementary crystallographic data for this paper.35a,bThe data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc07853a.
Acknowledgements
We thank University of Hyderabad (UoH; UPE-CAS and PURSE-FIST) for the overall facility. M. S. thank UGC for fellowship. We thank Mr Mahesh Srichand Rathod for the help in solving the X-ray crystal data. We also thank Aryan Gautam from Jawaharlal Nehru University, New Delhi for photophysical studies.Notes and references
- J. Corpas, P. Mauleón, R. G. Arrayás and J. C. Carretero, ACS Catal., 2021, 11, 7513–7551 CrossRef CAS.
- F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2004, 104, 3079–3108 CrossRef CAS.
- U. Wille, Chem. Rev., 2013, 113, 813–853 CrossRef CAS PubMed.
- X.-M. Nong, A. Gu, S. Zhai, J. Li, Z.-Y. Yue, M.-Y. Li and Y. Liu, iScience, 2024, 27, 109223 CrossRef CAS PubMed.
- R. Chinchilla and C. Najera, Chem. Rev., 2014, 114, 1783–1826 CrossRef CAS PubMed.
- M. Sethi, S. Dutta and A. K. Sahoo, Org. Lett., 2024, 26, 3224–3229 CrossRef CAS.
- S. Dutta, M. Sethi, A. Maity, A. Sahoo, V. Gandon and A. K. Sahoo, Org. Chem. Front., 2024, 11, 7168–7175 RSC.
- J. Ahmed, G. C. Haug, V. D. Nguyen, A. Porey, R. Trevino and O. V. Larionov, Synthesis, 2023, 55, 1642–1651 CrossRef CAS PubMed.
- J. Corpas, P. Mauleón, R. G. Arrayás and J. C. Carretero, J. Am. Chem. Soc., 2013, 135, 6747–6750 CrossRef.
- M. De Paolis, I. Chataigner and J. Maddaluno, Top. Curr. Chem., 2012, 327, 87 CrossRef CAS PubMed.
- J. Corpas, P. Mauleón, R. G. Arrayás and J. C. Carretero, J. Am. Chem. Soc., 2015, 137, 6747–6750 Search PubMed.
- E. M. Woerly, J. Roy and M. D. Burke, Nat. Chem., 2014, 6, 484–491 CrossRef CAS.
- X.-H. Hu, J. Zhang, X.-F. Yang, Y. H. Xu and T.-P. Loh, J. Am. Chem. Soc., 2015, 137, 3169–3172 CrossRef CAS.
- S. J. Lee, T. M. Anderson and M. D. Burke, Angew. Chem., Int. Ed., 2010, 49, 8860–8863 CrossRef CAS.
- J. Li, S. G. Ballmer, E. P. Gillis, S. Fujii, M. J. Schmidt, A. M. E. Palazzolo, J. W. Lehmann, G. F. Morehouse and M. D. Burke, Science, 2015, 347, 1221–1226 CrossRef CAS.
- J. Li, A. S. Grillo and M. D. Burke, Acc. Chem. Res., 2015, 48, 2297–2307 CrossRef CAS.
- S. Chen, Y.-N. Wang, J. Xie, W. Li, M. Ye, X. Ma, K. Yang, S. Li, Y. Lan and Q. Song, Nat. Commun., 2024, 15, 5479 CrossRef CAS.
- T. Long, C. Zhu, L. Li, L. Shao, S. Zhu, M. Rueping and L. Chu, Nat. Commun., 2023, 14, 55 CrossRef CAS.
- R. Vanjari, S. Dutta, S. Yang, V. Gandon and A. K. Sahoo, Org. Lett., 2022, 24, 1524–1529 CrossRef CAS PubMed.
- M. P. Gogoi, R. Vanjari, B. Prabagar, S. Yang, S. Dutta, R. K. Mallick, V. Gandon and A. K. Sahoo, Chem. Commun., 2021, 57, 7521–7524 RSC.
- S. Dutta, R. K. Mallick and A. K. Sahoo, Angew. Chem., Int. Ed., 2023, 62, e202300816 CrossRef CAS PubMed.
- F. Zhao, D. Zhang, Y. Nian, L. Zhang, W. Yang and H. Liu, Org. Lett., 2014, 16, 5124–5127 CrossRef CAS.
- G. Zhang, B. Feng, Y. Wang, J. Chen, X. Ma and Q. Song, Org. Lett., 2024, 26, 3109–3113 CrossRef CAS PubMed.
- K. Jana, A. Bhunia and A. Studer, Chem, 2020, 6, 512–522 CAS.
- P. Dominguez-Molano, A. Solé-Daura, J. J. Carbó and E. Fernández, Adv. Sci., 2024, 11, 2309779 CrossRef CAS.
- P. Domínguez-Molano, R. Weeks, R. J. Maza, J. J. Carbó and E. Fernández, Angew. Chem., Int. Ed., 2023, 62, e202304791 CrossRef.
- X. Tang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 814–817 CrossRef CAS.
- A. T. Lindhardt, M. L. H. Mantel and T. Skrydstrup, Angew. Chem., Int. Ed., 2008, 47, 2668–2672 CrossRef CAS PubMed.
- Y. Yang, L. Wang, J. Zhang, Y. Jin and G. Zhu, Chem. Commun., 2014, 50, 2347–2349 RSC.
- B. Gourdet, M. E. Rudkin, C. A. Watts and H. W. Lam, J. Org. Chem., 2009, 74, 7849–7858 CrossRef CAS.
- H. Huang, L. Tang, Y. Xi, G. He and H. Zhu, Tetrahedron Lett., 2016, 57, 1873–1876 CrossRef CAS.
- P. Bao, H. Xiao, Q. Li, Y. Wang, Y. Wang and G. He, Org. Biomol. Chem., 2025, 23, 5821–5825 RSC.
- D. Zhang, J. Man, Y. Chen, L. Yin, J. Zhong and Q.-F. Zhang, RSC Adv., 2019, 9, 12567–12571 RSC.
- G. Evano, B. Michelet and C. Zhang, C. R. Chim., 2017, 20, 648–664 CrossRef CAS.
- (a) CCDC 2433173: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2mnxgp; (b) CCDC 2433174: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2mnxhq.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |