
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel-catalyzed decarbonylative addition of acylsilanes across alkynes via the cleavage of a carbon–silicon bond
Yui
Morimoto
a,
Takahiro
Ando
a,
Tetsuya
Inagaki
a,
Hayato
Fujimoto
 ab and
Mamoru
Tobisu
*ab
ab and
Mamoru
Tobisu
*ab
aDepartment of Applied Chemistry, Graduate School of Engineering, The University of Osaka, Suita, Osaka 565-0871, Japan. E-mail: tobisu@chem.eng.osaka-u.ac.jp
bInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (ICS-OTRI), The University of Osaka, Suita, Osaka 565-0871, Japan
First published on 19th November 2025
Abstract
While hydrosilylation, the formal insertion of alkenes or alkynes into an H–Si bond, has been widely developed, analogous transformations involving inert C–Si bonds are rare due to their high bond dissociation energies. Herein, we report a nickel-catalyzed decarbonylative insertion of alkynes into the C–Si bond of acylsilanes, representing a formal insertion into an Ar–Si bond, which has not been previously achieved. Using a bulky N-heterocyclic carbene ligand (IPr*), this reaction enables the stereoselective formation of (Z)-alkenylsilanes from a range of acylsilanes and internal alkynes. This transformation not only expands the synthetic utility of acylsilanes but also illustrates a strategy for structural reprogramming through carbonyl group replacement.
Introduction
Given the broad utility of organosilicon compounds across diverse fields, including synthetic,1 materials,2 and medicinal3 chemistry, the development of efficient methods for their synthesis remains an active area of research. Hydrosilylation of alkenes or alkynes is one of the most valuable methods for the synthesis of organosilicon compounds, both in academic and industrial settings, owing to its intrinsically high atom economical nature via formal insertion into an H–Si bond (Fig. 1A, top).4 If analogous insertion into a C–Si bond of readily available organosilicon compounds could be achieved, it would offer a powerful strategy to access structurally complex organosilicon molecules from simple precursors with perfect atom economy (hereafter referred to as C–Si insertion; Fig. 1A, bottom). However, in contrast to the relatively weak H–Si bonds in hydrosilanes, C–Si bonds are generally much stronger, posing a significant challenge to the development of such insertion reactions. To date, successful examples of C–Si insertion have been limited to highly reactive organosilicon species, such as strained silacycles,5 allylsilanes,6 silyl cyanides,7 and acylsilanes8 (Fig. 1B). Only a few examples of insertion into unactivated C–Si bonds have been reported, but they are restricted to intramolecular processes.9 A key transformation that remains elusive is the intermolecular C–Si insertion of arylsilanes, which would enable the simultaneous introduction of both an aryl and a silyl group into a C2 unit.10 While silicon substitution reactions of arylsilanes, wherein the silyl group acts as a leaving group, are well documented,1c,11 insertion into an Ar–Si bond has not been previously achieved. Herein, we report that this transformation can be formally realized using acylsilanes as arylsilane surrogates via a nickel-catalyzed decarbonylative insertion of alkynes (Fig. 1C). | ||
| Fig. 1 C–Si insertion: background and this work. | ||
Results and discussion
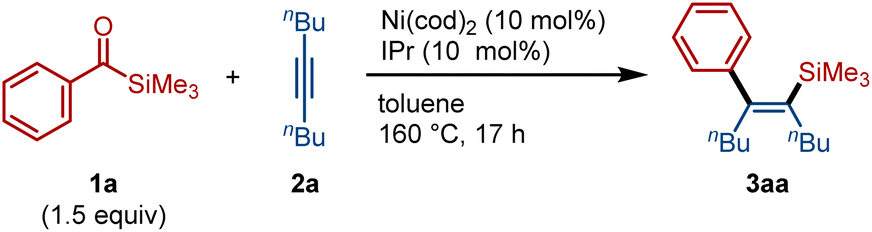
We previously demonstrated that acylsilanes undergo oxidative addition under palladium catalysis, enabling subsequent insertion of allenes8b and alkynes,8c and that nickel catalysts can promote decarbonylation of acylsilanes.12 Building on these findings, we hypothesized that nickel catalysis could facilitate decarbonylative C–Si bond insertion upon reaction of acylsilanes with alkenes or alkynes. To evaluate this hypothesis, we first examined the reaction of benzoylsilane (1a, 1.5 equiv.) with 5-decyne (2a) in the presence of Ni(cod)2 (10 mol%) and IPr (10 mol%)—a ligand previously identified as effective for catalytic decarbonylation of 1a (ref. 12a)—in toluene at 160 °C (Table 1). While the decarbonylated product 4 was obtained in 40% yield, the desired C–Si insertion product 3aa was not detected (entry 1). After extensive ligand screening, we found that the use of the more sterically demanding IPr* enabled the formation of 3aa in 26% yield with excellent Z-selectivity (entry 2). In this reaction, byproducts including 4 (11%) and the indene derivative 5aa (ca. 5%)8c were also observed. Gratifyingly, the yield of 3aa increased significantly to 69% when the reaction was carried out with 2 equiv. of 2a using IPr* as the ligand (entry 3). These conditions were subsequently employed for exploration of the substrate scope. Notably, the Pd/IPr* catalytic system, which is effective for C–Si insertion without decarbonylation, proved completely ineffective for the formation of 3aa (entry 4).Having established effective catalyst systems, we next explored the scope of the decarbonylative C–Si insertion of acylsilanes (Fig. 2). A variety of electron-donating and electron-withdrawing groups on the aryl ring of benzoylsilanes were well tolerated, affording the corresponding insertion products with excellent Z-selectivity (3aa–3fa). In addition to aryl groups, a naphthyl substituent could also be employed, enabling the rapid synthesis of π-extended alkenylsilane 3ga. The silicon substituent was not limited to trimethylsilyl (TMS); the more derivatizable dimethylphenylsilyl group was also compatible (3fa, 3ha). While diphenylacetylene (2b) exhibited lower reactivity toward this C–Si insertion reaction, the corresponding product 3ab was obtained in 42% isolated yield by employing an excess amount (5 equiv.) of 1a. Various substituted alkynes, including cyclic (2c), phenyl-substituted (2d), and oxygen-containing aliphatic alkynes (2e), successfully underwent the C–Si insertion to furnish the corresponding trisubstituted alkenylsilanes 3ac–3ae. In the case of the unsymmetrical alkyne 2f, bearing phenyl and butyl substituents at the termini, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of regioisomers 3af and 3af′ was obtained, indicating that aryl and alkyl groups are not distinguished in this nickel-mediated insertion process. Terminal alkynes such as phenylacetylene and 1-octyne did not furnish the desired C–Si insertion products under the current conditions.
:1 mixture of regioisomers 3af and 3af′ was obtained, indicating that aryl and alkyl groups are not distinguished in this nickel-mediated insertion process. Terminal alkynes such as phenylacetylene and 1-octyne did not furnish the desired C–Si insertion products under the current conditions.
 | ||
| Fig. 2 Scope of Ni-catalyzed decarbonylative addition of acylsilanes across alkynes. aIPr# was used instead of IPr*. b1a (3 equiv.) was used. cFor 24 h. d2a (3 equiv.) was used. e1a (5 equiv.) was used. | ||
The alkenylsilane derivatives synthesized through this C–Si insertion reaction serve as versatile intermediates for further structural elaboration. For instance, alkenylsilane 3fb, obtained from the nickel-catalyzed reaction of acylsilane 1f with alkyne 2b, could be transformed into benzosilole 6,10b a scaffold of interest in organic optoelectronic materials (Fig. 3, top).2c,d Beyond enhancing the reactivity of the C–Si bond, the use of acylsilanes as arylsilane surrogates offers notable synthetic advantages. Specifically, the carbonyl group in benzoylsilanes is known to act as an effective directing group for ortho C–H functionalization.13 For example, simple benzoylsilane 1a can be converted into the ortho-substituted derivative 7via acylsilane-directed C–H functionalization (Fig. 3, bottom).14 This intermediate 7 can then undergo the C–Si insertion reaction, enabling the rapid and atom-economical construction of a densely functionalized alkenylsilane 8.
 | ||
| Fig. 3 Synthetic applications. | ||
Several mechanistic experiments were conducted. First, the nickel-catalyzed reaction of phenylsilane 4 with alkyne 2a under the standard conditions was examined to assess the possible involvement of 4 as an intermediate, since it was observed as a byproduct in the reaction with acylsilane 1a (Table 1). However, no C–Si insertion product was detected, confirming that 4 cannot act as a substrate in this transformation. Second, a crossover experiment was carried out using two acylsilane derivatives bearing different aryl and silyl groups. As a result, only the two expected C–Si insertion products were obtained, with no evidence of aryl or silyl group scrambling (see the SI for details). These findings indicate that each acylsilane reacts specifically with one alkyne molecule and ligand (aryl or silyl) exchange between nickel intermediates is slow under the present conditions.
|
|
|||
|---|---|---|---|
| Entry | Deviation from the above conditions | NMR yielda (%) | |
| 3aa | 4 | ||
| a The yield was determined by 1H NMR analysis using 1,1,2,2-tetrachloroethane as an internal standard. b The ratio in parentheses refers to the Z/E ratio determined by 1H NMR. c 5aa was also formed in 3–5% yield. d Isolated yield. | |||
| 1 | None | Trace | 40 |
| 2c | IPr* instead of IPr | 26 (>20:1) |
11 |
| 3c | IPr* instead of IPr 1a/2a = 1/2 | 69 (8.8:1)d |
Trace |
| 4c | Pd2(dba)3 instead of Ni(cod)2 IPr* instead of IPr | 0 | 0 |
|
|||
Conclusions
In summary, we have developed a nickel-catalyzed decarbonylative insertion of alkynes into the C–Si bond of acylsilanes, enabling the simultaneous introduction of aryl and silyl groups into unactivated C–C triple bonds. This reaction demonstrates that acylsilanes can serve as surrogates for arylsilanes, thereby formally achieving an otherwise challenging Ar–Si bond insertion. From another perspective, this transformation represents a form of chemical structure reprogramming,15 in which the carbonyl group of an acylsilane is replaced by a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C unit. Further studies aimed at expanding this concept of carbonyl group replacement are currently underway in our laboratory.
C unit. Further studies aimed at expanding this concept of carbonyl group replacement are currently underway in our laboratory.
Author contributions
Y. M., T. A., T. I., H. F. and M. T. conceived the project. Y. M., T. A., and T. I. performed the experiments. H. F. and M. T. wrote the manuscript. All authors discussed the results and reviewed the final manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
Additional data and NMR spectra can be found in the supporting information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc07524f.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number 24H02207 (M. T.) in Transformative Research Areas (A) JP24A202 Integrated Science of Synthesis by Chemical Structure Reprogramming (SReP). T. I. thanks a JSPS Research Fellowship for Young Scientists (23KJ1498) for financial support. We also thank the Instrumental Analysis Center, Faculty of Engineering, The University of Osaka, for assistance with HRMS.Notes and references
- (a) I. Fleming, A. Barbero and D. Walter, Chem. Rev., 1997, 97, 2063–2192 CrossRef CAS; (b) G. R. Jones and Y. Landais, Tetrahedron, 1996, 52, 7599–7662 CrossRef CAS; (c) T. Komiyama, Y. Minami and T. Hiyama, ACS Catal., 2017, 7, 631–651 CrossRef CAS.
- (a) G. Chandra, Organosilicon Materials, Springer Berlin, Heidelberg, 1997 CrossRef; (b) N. Auner and J. Weis, Organosilicon Chemistry V: From Molecules to Materials, John Wiley & Sons, New York, 2008 Search PubMed; (c) M. Shimizu and T. Hiyama, Synlett, 2012, 23, 973–989 CrossRef CAS; (d) Y. Cai, A. Qin and B. Z. Tang, J. Mater. Chem. C, 2017, 5, 7375–7389 RSC.
- (a) E. Rémond, C. Martin, J. Martinez and F. Cavelier, Chem. Rev., 2016, 116, 11654–11684 CrossRef; (b) R. Ramesh and D. S. Reddy, J. Med. Chem., 2018, 61, 3779–3798 CrossRef CAS PubMed; (c) J.-L. Panayides, D. L. Riley, F. Hasenmaile and W. A. L. van Otterlo, RSC Med. Chem., 2024, 15, 3286–3344 RSC.
- L. L. D. de Almeida, H. Wang, K. Junge, X. Cui and M. Beller, Angew. Chem., Int. Ed., 2021, 60, 550–565 CrossRef CAS PubMed.
- A review: (a) L. Li, Y. Zhang, L. Gao and Z. Song, Tetrahedron Lett., 2015, 56, 1466–1473 CrossRef CAS; (b) M. Liu, L. Qi and D. Zhao, Chin. J. Org. Chem., 2023, 43, 3508 CrossRef CAS.
- Selected examples: (a) N. Asao, E. Yoshikawa and Y. Yamamoto, J. Org. Chem., 1996, 61, 4874–4875 CrossRef CAS; (b) E. Yoshikawa, V. Gevorgyan, N. Asao and Y. Yamamoto, J. Am. Chem. Soc., 1997, 119, 6781–6786 CrossRef CAS; (c) N. Asao, T. Shimada and Y. Yamamoto, J. Am. Chem. Soc., 1999, 121, 3797–3798 CrossRef CAS; (d) N. Asao, T. Shimada, T. Shimada and Y. Yamamoto, J. Am. Chem. Soc., 2001, 123, 10899–10902 CrossRef CAS.
- (a) N. Chatani, T. Takeyasu, N. Horiuchi and T. Hanafusa, J. Org. Chem., 1988, 53, 3539–3548 CrossRef CAS; (b) P. Hansjacob, F. R. Leroux, V. Gandon and M. Donnard, Angew. Chem., Int. Ed., 2022, 61, e202200204 CrossRef CAS PubMed.
- (a) H. Sakurai, M. Yamane, M. Iwata, N. Saito and K. Narasaka, Chem. Lett., 1996, 25, 841–842 CrossRef; (b) T. Inagaki, S. Sakurai, M. Yamanaka and M. Tobisu, Angew. Chem., Int. Ed., 2022, 61, e202202387 CrossRef CAS; (c) T. Inagaki, T. Ando, S. Sakurai, M. Yamanaka and M. Tobisu, Chem. Sci., 2023, 14, 2706–2712 RSC; (d) T. Inagaki, Y. Akita and M. Tobisu, Org. Lett., 2024, 26, 2141–2145 CrossRef CAS PubMed; (e) P. Becker, D. L. Priebbenow, H.-J. Zhang, R. Pirwerdjan and C. Bolm, J. Org. Chem., 2014, 79, 814–817 CrossRef CAS PubMed; (f) H.-J. Zhang, P. Becker, H. Huang, R. Pirwerdjan, F.-F. Pan and C. Bolm, Adv. Synth. Catal., 2012, 354, 2157–2161 CrossRef CAS; (g) V. K. Rawat and M. Tobisu, ACS Catal., 2025, 15, 8706–8723 CrossRef CAS; (h) Q. Zhao, Q. Geng, Y. Li, J. Li and Z. Liu, Org. Chem. Front., 2023, 10, 1316–1321 RSC; (i) H.-J. Zhang, D. L. Priebbenow and C. Bolm, Chem. Soc. Rev., 2013, 42, 8540–8571 RSC; (j) X. Shen, Acc. Chem. Res., 2025, 58, 1519–1533 CrossRef CAS PubMed.
- (a) R. Shintani, H. Kurata and K. Nozaki, Chem. Commun., 2015, 51, 11378–11381 RSC; (b) Q. Yang, L. Liu, Y. Chi, W. Hao, W.-X. Zhang and Z. Xi, Org. Chem. Front., 2018, 5, 860–863 RSC; (c) Y. Shi, X. Shi, J. Zhang, Y. Qin, B. Li and D. Zhao, Nat. Commun., 2022, 13, 6697 CrossRef CAS PubMed.
- Introduction of a carbon-based substituent and a silyl group into unsaturated hydrocarbons is also possible by three-component coupling with a silyl nucleophile and a carbon electrophile. Selected examples: (a) T. Fujihara, Y. Tani, K. Semba, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2012, 51, 11487–11490 CrossRef CAS PubMed; (b) R. Shintani, H. Kurata and K. Nozaki, J. Org. Chem., 2016, 81, 3065–3069 CrossRef CAS; (c) T. Iwamoto, T. Nishikori, N. Nakagawa, H. Takaya and M. Nakamura, Angew. Chem., Int. Ed., 2017, 56, 13298–13301 CrossRef CAS; (d) Q. Chen, Z. Li and Y. Nishihara, Org. Lett., 2022, 24, 385–389 CrossRef CAS PubMed; (e) W. Bernhard, I. Fleming and D. Waterson, J. Chem. Soc., Chem. Commun., 1984, 28–29 RSC; (f) Y. Obora, Y. Tsuji and T. Kawamura, J. Am. Chem. Soc., 1995, 117, 9814–9821 CrossRef CAS; (g) D. Ni and M. K. Brown, ACS Catal., 2021, 11, 1858–1862 CrossRef CAS; (h) W. Zheng, Y. Xu, H. Luo, Y. Feng, J. Zhang and L. Lin, Org. Lett., 2022, 24, 7145–7150 CrossRef CAS PubMed; (i) J. Wang, Z. Duan, X. Liu, S. Dong, K. Chen and J. Li, Angew. Chem., Int. Ed., 2022, 61, e202202379 CrossRef CAS; (j) M.-Y. Wu, F.-Y. Yang and C.-H. Cheng, J. Org. Chem., 1999, 64, 2471–2474 CrossRef CAS; (k) F.-Y. Yang, M. Shanmugasundaram, S.-Y. Chuang, P.-J. Ku, M.-Y. Wu and C.-H. Cheng, J. Am. Chem. Soc., 2003, 125, 12576–12583 CrossRef CAS PubMed; (l) Y. Tani, T. Fujihara, J. Terao and Y. Tsuji, J. Am. Chem. Soc., 2014, 136, 17706–17709 CrossRef CAS.
- Our reports on C–Si bond substitution reactions: (a) M. Tobisu, M. Onoe, Y. Kita and N. Chatani, J. Am. Chem. Soc., 2009, 131, 7506–7507 CrossRef CAS PubMed; (b) M. Onoe, K. Baba, Y. Kim, Y. Kita, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2012, 134, 19477–19488 CrossRef CAS PubMed; (c) M. Onoe, T. Morioka, M. Tobisu and N. Chatani, Chem. Lett., 2013, 42, 238–240 CrossRef CAS.
- (a) S. Nakatani, Y. Ito, S. Sakurai, T. Kodama and M. Tobisu, J. Org. Chem., 2020, 85, 7588–7594 CrossRef CAS; (b) W. Srimontree, W. Lakornwong and M. Rueping, Org. Lett., 2019, 21, 9330–9333 CrossRef CAS; (c) T. Yoshida, T. Kodama and M. Tobisu, Asian J. Org. Chem., 2022, 11, e202200610 CrossRef CAS; (d) A. Matsuura, Y. Ito, T. Inagaki, T. Kodama and M. Tobisu, ACS Catal., 2024, 14, 18216–18222 CrossRef CAS; (e) T. Inagaki, T. Kodama and M. Tobisu, Nat. Catal., 2024, 7, 132–138 CrossRef CAS.
- R. L. Pilkington and D. L. Priebbenow, ACS Catal., 2025, 15, 6881–6894 CrossRef CAS.
- X. Lu, C. Shen, K. Meng, L. Zhao, T. Li, Y. Sun, J. Zhang and G. Zhong, Chem. Commun., 2019, 55, 826–829 RSC.
- (a) H. Fujimoto and M. Tobisu, Acc. Chem. Res., 2025, 58, 1168–1180 CrossRef CAS; (b) R. Shimazumi and M. Tobisu, JACS Au, 2024, 4, 1676–1695 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |