
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic effect of MOF fillers and succinonitrile in PEO-based electrolytes for long-cycle all-solid-state Li–CO2 batteries
Yilong
Huang
a,
Jinfeng
Ha
b,
Zhixin
Liu
a,
Xingyu
Yu
a,
Yin
Liu
a,
Xiaoli
Fan
c,
Xianli
Huang
a,
Jianping
He
 a,
Shuping
Huang
*b and
Tao
Wang
*a
a,
Shuping
Huang
*b and
Tao
Wang
*a
aCentre for Hydrogenergy, College of Materials Science and Technology, Nanjing University of Aeronautics and Astronautics, Nanjing 210016, P.R. China. E-mail: wangtao0729@nuaa.edu.cn
bFujian Provincial Key Laboratory of Electrochemical Energy Storage Materials, College of Chemistry, Fuzhou University, Fuzhou 350108, P.R. China. E-mail: huangshp@fzu.edu.cn
cSchool of Materials Science and Engineering, Nanjing Institute of Technology, Nanjing 211167, P.R. China
First published on 27th November 2025
Abstract
Li–CO2 batteries have emerged as promising candidates for next-generation energy storage systems due to their ultrahigh theoretical energy density. However, some safety issues primarily arising from the volatility and leakage of liquid electrolytes within semi-open cell configurations remain a critical challenge. Herein, we develop a novel solid polymer electrolyte (SPE) based on a poly(ethylene oxide) (PEO) matrix, incorporated with zeolitic imidazolate framework-8 (ZIF-8) and succinonitrile (SN). ZIF-8 with ample open metal sites (OMSs) can function as a Lewis acidic center to anchor anions effectively, while SN reduces the crystallinity of the polymer. Benefiting from the synergistic effect between ZIF-8 and SN, the SPE exhibits high ionic conductivity (3.6 × 10−4 S cm−1 at 30 °C), an enhanced Li+ transference number (0.36), and an extended electrochemical window (>5.2 V vs. Li/Li+). As a result, the assembled all-solid-state Li–CO2 battery with the SPE delivers a high discharge capacity of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 016 mAh g−1 at 100 mA g−1 and demonstrates exceptional cycling stability exceeding 1600 h at 200 mA g−1. This work provides an innovative strategy for developing safe and high-performance Li–CO2 batteries.
016 mAh g−1 at 100 mA g−1 and demonstrates exceptional cycling stability exceeding 1600 h at 200 mA g−1. This work provides an innovative strategy for developing safe and high-performance Li–CO2 batteries.
Introduction
Since the 21st century, with the increasing energy consumption in human society, exploring advanced energy storage devices has been an urgent task.1 Li–CO2 batteries have been attracting tremendous attention as next-generation energy storage systems owing to their ultra-high theoretical energy density of 1876 Wh kg−1 based on the four-electron transfer reaction of 4Li+ + 3CO2 + 4e− → 2Li2CO3 + C and their potential for CO2 utilization.2–4 A typical Li–CO2 battery is composed of a Li metal anode, a Li+-conductive electrolyte, and a porous gas cathode.5 Unfortunately, conventional Li–CO2 batteries rely primarily on liquid organic electrolytes, which face the risks of liquid electrolyte volatilization and leakage in semi-open cell configurations.6,7 Additionally, the inevitable growth of Li dendrites during charge–discharge cycles and uncontrollable reactions arising from the interaction of Li metal with CO2 or H2O dissolved in the electrolyte significantly impair the mechanical stability of the electrode structure, leading to structural collapse and failure.8–11 Therefore, it is crucial to explore alternatives to liquid organic electrolytes.Solid-state electrolytes (SSEs) have emerged as promising candidates in this context. A key advantage of SSEs lies in their superior mechanical properties, which enable effective suppression of Li dendrite growth, thereby markedly enhancing battery safety and cycle life.12,13 Up to now, the reported SSEs can be broadly categorized into two types: inorganic solid electrolytes (ISEs) and solid polymer electrolytes (SPEs).14 Compared with ISEs, SPEs offer numerous advantages, including excellent processability, chemical stability and superior interface compatibility, making them ideal candidates for Li–CO2 batteries.15–17 Among various SPEs, poly(ethylene oxide) (PEO)-based electrolytes are particularly notable for their ability to effectively dissolve lithium salts. However, these electrolytes still face significant challenges, such as low ionic conductivity (10−6 to 10−5 S cm−1 at room temperature) and inadequate mechanical strength, impeding their broader application.18,19
To alleviate these critical issues and achieve excellent performance, metal–organic frameworks (MOFs) have been employed as fillers for PEO-based SPEs due to their unique structures and properties. MOFs are an emerging class of porous crystalline organic–inorganic hybrid materials assembled through the linkage of metal centers (clusters) and organic ligands.20,21 In particular, these ample open metal sites (OMSs) can function as Lewis acidic centers to anchor anions effectively. Besides, the tunable pore structure of MOFs can restrict the movement of anionic groups and thus preferentially promote cation transfer.22–24 It must also be mentioned that PEO-based SPEs transport Li ions via complexation and decomplexation with ether oxygen in amorphous polymer chain segments.25,26 Consequently, increasing the number of active segments is recognized as an effective approach to improving ionic conductivity. Plastic crystal succinonitrile (SN) can reduce the crystallinity of the polymer and be a carrier for the migration of Li ions.27–29 On the other hand, since SN has a tendency to degrade mechanical properties and can even react with lithium, producing harmful by-products, its content must be strictly controlled.30–32
In this work, we synthesized zeolitic imidazolate framework (ZIF-8) nanoparticles as a filler via a one-pot solvothermal reaction, and moderate proportions of SN were selected to prepare PEO-based composite SPEs by a simple casting method. The composite electrolyte consisting of PEO/ZIF-8/15 wt% SN (named PZS15) exhibited a high ionic conductivity of 3.6 × 10−4 S cm−1 at 30 °C. This remarkable improvement can be attributed to the SN plasticizer effectively reducing crystallinity and interfacial resistance. Furthermore, ZIF-8 could reduce the side reactions between SN and the Li anode, regulating the deposition of Li metal. Benefiting from the homogeneous structure, the PZS15 SPEs exhibited a wide electrochemical window of more than 5.2 V. As a result, the assembled all-solid-state Li–CO2 batteries with PZS15 exhibited a high discharge capacity of 15016 mAh g−1 at 100 mA g−1 and demonstrated an impressive cycle life exceeding 1600 h at 200 mA g−1. Thus, we believe that our research puts forward an innovative strategy for the development of high-performance and safe Li–CO2 batteries.
Results and discussion
Fig. 1 illustrates the structural model of the PEO/ZIF-8 (PZ) composite electrolyte and the schematic diagram of the Li–CO2 battery. As a typical MOF, ZIF-8 possesses a highly Lewis acidic surface, which induces strong adsorption of TFSI− anions. This facilitates lithium salt dissociation and enhances Li+ transfer kinetics.33 The fabrication procedure of the PEO/ZIF-8/SN composite electrolyte is schematically depicted in Fig. 2a. Initially, ZIF-8 nanoparticles were synthesized through coordination between zinc ions and 2-methylimidazole ligands. These synthesized nanoparticles were subsequently incorporated as nanofillers into a homogeneous slurry containing a PEO matrix, lithium salt (LiTFSI) and SN plasticizer. The resultant mixture was then cast onto a polytetrafluoroethylene (PTFE) mold to form a freestanding electrolyte membrane with uniform dispersion. The X-ray diffraction (XRD) pattern of the as-synthesized ZIF-8 powder confirms its well-developed crystal structure, aligning well with the simulated pattern (Fig. S1a).34,35 Fourier transform infrared (FT-IR) spectra further analyzed the chemical bond structures of ZIF-8 (Fig. S1b). The absorption peaks at 3134 cm−1 and 2928 cm−1 are attributed to the stretching vibrations absorption of the aromatic C–H bonds in the imidazole ring and the aliphatic C–H bonds in the methyl group, respectively. The peaks observed at 1146 cm−1 and 995 cm−1 correspond to C–N stretching vibrations, while the peak at 420 cm−1 is assigned to the Zn–N stretching.36 In addition, the synthesized ZIF-8 exhibits a high specific surface area of 1724.9 m2 g−1 and a dominant pore size of less than 2 nm, which are characteristic of its microporous structure (Fig. S1c and d). The corresponding scanning electron microscopy (SEM) image shown in Fig. 2b revealed a uniform dodecahedral morphology, with dimensions of approximately 200 nm. Fig. 2c presents the optical image of the prepared PZ (without SN) composite polymer electrolyte. The electrolyte film exhibits a smooth and homogeneous surface without obvious agglomeration. This uniform dispersion can be attributed to the hybrid inorganic–organic nature of the MOF material, which facilitates its even distribution within the PEO polymer electrolyte matrix. Fig. 2d displays a representative cross-sectional SEM image of the PZ electrolyte, indicating a thickness of approximately 400 µm. | ||
| Fig. 1 Schematic illustration showing ZIF-8 facilitating the dissociation of LiTFSI into Li+ and TFSI−. | ||
 | ||
| Fig. 2 (a) Schematic illustration of the preparation of the PEO/ZIF-8/SN composite electrolyte. (b) SEM images of ZIF-8. (c) Digital photograph of PZ electrolyte. (d) The cross-section SEM image of PZ electrolyte. (e) Top-view SEM image and (f) corresponding EDS mapping of PZ. | ||
Furthermore, elemental distribution mapping of Zn, S, and F across the composite polymer electrolyte membrane surface reveals uniform dispersion of these elements. This observation demonstrates the homogeneous distribution of both the MOF filler and LiTFSI salt within the PEO matrix, thereby facilitating the construction of continuous pathways for ionic conduction (Fig. 2e and f). In addition, comparative SEM images (Fig. S2a–h) show that the pristine PEO electrolyte exhibits a coarse morphology characterized by large polymer spherulites and substantial intergranular porosity. In contrast, incorporation of the SN plasticizer progressively transforms the surface morphology into a smoother, more homogeneous, and densely packed structure. This plasticizer-induced morphological refinement offers critical advantages for SSEs:37,38 elimination of macroscopic pores drastically reduces interfacial impedance through ensured intimate electrode contact, while the enhanced mechanical robustness from improved physical integrity more effectively suppresses Li dendrite penetration relative to the porous pristine structure.
The crystallinity of the SPEs was characterized by XRD (Fig. 3a). The pristine PEO pattern shows two distinct peaks at 2θ = 19° and 23.3°, corresponding to its crystalline phase. With the introduction of SN, these peaks diminish alongside the appearance of a broad diffraction peak, indicating reduced crystallinity. This confirms that SN effectively suppresses the crystallinity of PEO, which enhances polymer chain mobility and leads to higher ionic conductivity.39 FT-IR spectroscopy was employed to identify the structure of the samples. As shown in Fig. 3b, the peaks observed at 2884 cm−1 and 1466 cm−1 are attributed to the stretching and bending vibrations of CH2 groups in PEO, respectively. The other prominent peaks at 1054 cm−1 and 1185 cm−1 correspond to the asymmetric and symmetric stretching vibrations of the –CF3 group in LiTFSI.40 Notably, the CH2 wagging vibrations at 1341 cm−1 and 1353 cm−1 gradually merged into a single peak with increasing SN content, indicating the absence of a crystalline PEO phase (Fig. S3a).41,42 Detailed spectra in the 1300–700 cm−1 region (Fig. S3b) reveal peaks at 1281, 1096, and 961 cm−1, assigned to the asymmetric stretching, symmetric stretching, and deformation vibrations of the C–O–C group, respectively, while the peak at 842 cm−1 is ascribed to the deformation vibration of CH2CH2O.43–46
 | ||
| Fig. 3 (a) XRD profiles of various SPEs. (b) FT-IR spectra of the corresponding SPEs. (c) Electrochemical impedance spectra of SS|PZS15|SS at 30–70 °C. (d) Arrhenius plots of SPEs at different temperatures. (e) Chronoamperometric curve of the Li|PZS15|Li symmetric cell after a perturbation with a 10 mV direct current pulse. Inset: impedance spectra of the cell before and after the DC polarization. (f) Linear-sweep voltammetry of SPEs. (g) Stress–strain curves of PEO, PZ and PZS15. (h) TGA curves of PEO, PZ and PZS15 from room temperature to 600 °C. (i) DSC curves of PEO, PZ and PZS15. | ||
Electrochemical impedance spectroscopy (EIS) was performed to determine the ionic conductivities of the electrolytes over a temperature range of 30–70 °C (Fig. 3c and S4). It can be seen that the PZS15 electrolyte exhibits a high ionic conductivity of 3.6 × 10−4 S cm−1 at 30 °C. Additionally, all electrolytes exhibited temperature-dependent ionic conductivity, as confirmed by Arrhenius plots (Fig. 3d) used to calculate the activation energies (Ea) for the SPEs. Critically, a distinct inflection point observed in the Arrhenius plot corresponds to the melting point temperature (Tm) of SN (50–54 °C). This correspondence demonstrates that SN effectively reduces polymer crystallinity and increases the population of active chain segments, thereby significantly lowering the migration barrier for Li+. Fig. 3e presents the Li+ transference number (tLi+) for PZS15 (0.36), measured using chronoamperometry combined with EIS. As shown in Fig. S5, the pristine PEO electrolyte exhibits a low tLi+ of 0.15. Incorporation of ZIF-8 (forming PZ) increases tLi+ to 0.28, attributed to enhanced LiTFSI dissociation and partial anion immobilization by the MOF.33 As the SN content was further increased, the tLi+ continued to rise. This can be attributed to the dual role of SN: it effectively suppresses the crystallinity of PEO and introduces polar nitrile groups that actively participate in the solvation and transport of Li+.47 However, when the SN content reaches 20 wt% (PZS20), there is an observed reduction in tLi+, which is ascribed to the aggregation of SN at higher concentrations, leading to the formation of insulating domains that obstruct Li+ conduction pathways. The electrochemical stability of the SPEs was investigated via linear sweep voltammetry (LSV) measurements. The results in Fig. 3f reveal that the PZS15 electrolyte possesses a wide stability window of 5.2 V, indicating its superior oxidative stability. Consequently, based on the comprehensive evaluation presented in Fig. S6, PZS15 was identified as the optimal composition and selected for subsequent investigations.
The mechanical properties of the SPEs were characterized via stress–strain testing. PZS15 exhibits a fracture elongation of 1638.5%, significantly higher than those of PEO (871.2%) and PZ (278.3%), demonstrating excellent flexibility (Fig. 3g and S7). This enhanced flexibility promotes intimate interfacial adhesion to the electrodes. Thermogravimetric analysis (TGA) was performed to assess the thermal stability of the SPEs. As revealed in Fig. 3h, an initial negligible weight loss occurs due to residual solvent evaporation. Subsequently, the second weight-loss stage observed for PZS15 corresponds to the decomposition of SN. Derivative thermogravimetry (DTG) analysis (Fig. S8) further shows a sharp decrease between 350 °C and 450 °C, attributed to the irreversible decomposition of the PEO matrix. Collectively, these results demonstrate that the SPEs possess excellent thermal stability within typical application temperature ranges, ensuring safe operation under elevated temperatures. Differential scanning calorimetry (DSC) was employed to analyze the crystallinity behavior of the SPEs. As shown in Fig. 3i, the Tm of pure PEO was the highest at 56.2 °C, indicating strong crystallinity. In contrast, PZS15 exhibited a reduced Tm of 49.5 °C and the smallest melting enthalpy peak area, demonstrating a marked decrease in crystallinity. Furthermore, the glass transition temperatures (Tg) of PEO and PZ were −44.1 and −46.8 °C, respectively. The observed disappearance of Tg for PZS15 suggests that the addition of SN enhances the mobility of PEO chain segments, thereby promoting the formation of an amorphous phase. As summarized in Table S1 (SI), the calculated crystallinities of PEO, PZ, and PZS15 were 23%, 19.2%, and 14.7%, respectively. In addition, the DSC cooling curves (from 100 to 10 °C) revealed that PZS15 had the lowest crystallization temperature (Tc) at 23.1 °C, consistent with the aforementioned results (Fig. S9).
The cycling performance of Li|SPE|Li symmetric cells was further evaluated at 0.1 mA cm−2 and 0.1 mAh cm−2 at room temperature (Fig. 4a). It is evident that the Li|PEO|Li cell exhibits a sharp increase in polarization voltage, followed by a short circuit at 120 hours. This failure is primarily attributed to Li dendrite formation during the plating/stripping processes. In contrast, the Li|PZS15|Li cell demonstrates stable cycling for over 500 hours, while the Li|PZ|Li cell exhibits increasing polarization voltage after only 300 hours of cycling. In addition, density functional theory (DFT) calculations were conducted to assess the redox properties of SPEs. The orbital energy levels of the key constituents in SPEs are illustrated in Fig. 4b and S10. The highest occupied molecular orbital (HOMO) energy level is −7.13 eV for PEO and −8.75 eV for SN. A lower HOMO energy level indicates a higher resistance to oxidation, as it signifies a lower tendency to lose electrons. These results account for the superior oxidative stability of SN. The incorporation of SN effectively enhances the antioxidant properties and broadens the electrochemical window of PZS15. Furthermore, the lowest unoccupied molecular orbital (LUMO) energy level of LiTFSI was calculated to be −2.29 eV; the lower value suggests a higher electron affinity, rendering LiTFSI more susceptible to reduction at the Li anode. This process facilitates SEI formation and prevents side reactions between SN and the lithium. To confirm the superior electrochemical stability of the PZS15 electrolyte with the Li metal anode, we disassembled the cycled Li|SPE|Li cells and analyzed the morphology and composition of the Li anode surface. As shown in Fig. 4c, the surface of the Li anode cycled with PEO exhibits severe erosion and a honeycomb-like structure, attributed to continuous Li dendrite growth and accumulation of side reaction products. Similarly, the Li anode surface from the Li|PZ|Li cell shows uneven dendrites and the presence of dead lithium after cycling. In contrast, the Li anode cycled with PZS15 demonstrates a smooth surface morphology largely free of large aggregates of dead lithium, consistent with the SEM images of the electrolyte surface in Fig. S11. These observations demonstrate that the PZS15 electrolyte effectively inhibits Li metal corrosion.
 | ||
| Fig. 4 (a) Galvanostatic cycling curves of Li|SPE|Li symmetric cells at 0.1 mA cm−2 and 0.1 mAh cm−2 at room temperature. (b) Calculated HOMO and LUMO energy levels of PEO, LiTFSI and SN. (c) SEM images of Li anodes in PEO, PZ and PZS15 electrolytes from Li|SPE|Li symmetric cells after cycling. XPS spectra of the lithium metal surface after cycling: (d) C 1s, (e) N 1s, (f) O 1s and (g) F 1s. | ||
To further understand the difference in the electrochemical properties of SPEs, the surface composition of the Li anode was investigated using X-ray photoelectron spectroscopy (XPS) (Fig. 4d–g). In the C 1s spectrum, the peaks at 284.8 and 286.8 eV are assigned to C–C and C–O bonds, respectively, originating from residual PEO polymer. The peaks at 288.6 and 292.8 eV correspond to –CO32− and C–F (from the Li salt LiTFSI), respectively, indicating the presence of both organic and inorganic components within the solid electrolyte interface (SEI) formed on the lithium metal surface.48,49 The N 1s spectrum reveals four peaks corresponding to –NO3 (407.5 eV), –NOx (403.8 eV), TFSI− (400.2 eV) and Li3N (398.2 eV).50,51 Notably, the SEI for PZS15 exhibits a significantly higher concentration of Li3N, a superionic Li+ conductor known for its high ionic conductivity and low energy barrier, which facilitates rapid Li+ migration and promotes uniform Li deposition.52 Furthermore, the N 1s spectrum for PZS15 shows a significant reduction in the intensities of the –NO3 and –NOx peaks, indicating effective suppression of associated side reactions. Meanwhile, the O 1s spectrum of Li metal cycled with PZS15 electrolyte displays the lowest proportion of Li2CO3 (531.2 eV) compared to samples cycled with PEO and PZ electrolytes, consistent with the Li 1s spectrum results (Fig. S12). Li2CO3 was reported to cause interfacial passivation due to its low Li+ conductivity (≈10−8 S cm−1), which manifests as a large overpotential during lithium plating/stripping.53 The F 1s spectrum exhibits three peaks at 684.8, 686.6 and 688.6 eV, attributed to LiF, 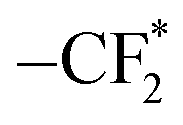 and –CF3, respectively. The highest intensity of the
and –CF3, respectively. The highest intensity of the  peak in PEO originates from the intermediate reduction of the TFSI− anion.54,55 In contrast, PZS15 shows the strongest –CF3 peak intensity among the electrolytes (PEO, PZ, and PZS15), suggesting continuous decomposition of the TFSI− anion at the Li anode surface, leading to the sustained formation of LiF and Li3N.
peak in PEO originates from the intermediate reduction of the TFSI− anion.54,55 In contrast, PZS15 shows the strongest –CF3 peak intensity among the electrolytes (PEO, PZ, and PZS15), suggesting continuous decomposition of the TFSI− anion at the Li anode surface, leading to the sustained formation of LiF and Li3N.
Further DFT calculation was performed to evaluate the binding energies between Li+ and each electrolyte component. As shown in Fig. 5a, the binding energy of SN–Li+ was calculated to be −4.34 eV, exceeding those of PEO–Li+ (−3.63 eV) and TFSI−–Li+ (−1.04 eV), indicating a stronger coordination tendency of SN toward Li+. The enhanced interaction promotes the dissociation of LiTFSI and facilitates the rapid release of more Li+, which is beneficial for efficient Li+ transport at room temperature.56 The interactions between Li+ and polymer fragments were further elucidated through electrostatic potential (ESP) calculations (Fig. 5b). The blue regions on the ESP surfaces represent electrophilic regions, while the red regions correspond to nucleophilic sites. The working mechanism of SN is illustrated in Fig. 5c. Briefly, in amorphous PEO-based solid electrolytes, lithium ions migrate through a complexation–decomplexation process with the PEO chains. However, the relatively high crystallinity of PEO at room temperature hinders the formation of fast ion-conduction pathways. In contrast, incorporation of SN effectively suppresses PEO crystallization and promotes the formation of a polymer⋯[SN⋯Li+] complex. The synergistic interaction between polymer chain segments and SN molecules not only facilitates rapid Li+ migration but also effectively inhibits side reactions triggered by free SN molecules. As shown in Fig. 5d, in addition to the interaction between Li+ and the electrolyte component, the binding energies of TFSI− with ZIF-8 (−2.52 eV) clusters and PEO (−0.83 eV) fragments are further compared. The results indicate a stronger interaction between TFSI− and ZIF-8, suggesting that TFSI− is preferentially adsorbed by ZIF-8 and thus tends to dissociate from LiTFSI. Besides, the charge density difference for the PEO and ZIF-8 systems is analyzed in Fig. 5e. According to our theoretical calculation, the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group in the TFSI− anion exhibits an electron-rich state. For the PEO system, the oxygen atoms in TFSI− are close to the hydrogen atoms of the PEO chain. In contrast, a higher charge density accumulates around the oxygen atoms in the ZIF-8 system, indicating that they are strongly attracted through Lewis acid–base interactions. Raman spectroscopy was conducted to analyze the TFSI− coordination state. As summarized in Fig. S13, the contents of free TFSI−, contact ion pairs (CIPs) and aggregate clusters (AGGs) of the PEO electrolyte are 13.57%, 58.71% and 27.72%, respectively. Obviously, upon incorporation of ZIF-8, the proportion of free TFSI− significantly increased to 29.14% in the PZ electrolyte, similar to that of the PZS15 electrolyte (30.83%). However, the PZS15 electrolyte shows a lower AGG content (20.55%) than that of the PZ electrolyte (26.09%). These results confirm that the synergistic effect of ZIF-8 fillers and SN effectively facilitates lithium salt dissociation to form more mobile Li+.
O group in the TFSI− anion exhibits an electron-rich state. For the PEO system, the oxygen atoms in TFSI− are close to the hydrogen atoms of the PEO chain. In contrast, a higher charge density accumulates around the oxygen atoms in the ZIF-8 system, indicating that they are strongly attracted through Lewis acid–base interactions. Raman spectroscopy was conducted to analyze the TFSI− coordination state. As summarized in Fig. S13, the contents of free TFSI−, contact ion pairs (CIPs) and aggregate clusters (AGGs) of the PEO electrolyte are 13.57%, 58.71% and 27.72%, respectively. Obviously, upon incorporation of ZIF-8, the proportion of free TFSI− significantly increased to 29.14% in the PZ electrolyte, similar to that of the PZS15 electrolyte (30.83%). However, the PZS15 electrolyte shows a lower AGG content (20.55%) than that of the PZ electrolyte (26.09%). These results confirm that the synergistic effect of ZIF-8 fillers and SN effectively facilitates lithium salt dissociation to form more mobile Li+.
 | ||
| Fig. 5 (a) Binding energies between Li+ and each electrolyte component. (b) Electrostatic potential of PEO and SN. (c) Schematic illustration of the working mechanism of SN in PEO. (d) Binding energy of TFSI− anions with ZIF-8 and PEO. (e) Electron density difference of TFSI− with PEO and ZIF-8. | ||
To elucidate the coordination environment of Li+ across different electrolyte systems, we performed molecular dynamics (MD) simulations (Fig. 6a–c). Fig. 6d presents the corresponding radial distribution functions (RDFs) and coordination numbers (CNs) for Li–O interactions involving both polymer chains and anions. In the PEO-based system, the RDF between Li+ and oxygen atoms from TFSI− shows an average CN of 1.92. With the incorporation of ZIF-8, the Li–O coordination number for TFSI− decreases to 1.63. This indicates an increased population of free Li+ and a weaker binding affinity between Li+ and TFSI− in the PZ electrolyte, aligning with the former DFT calculations. Moreover, a new peak emerges at 2.1 Å in the RDF for Li–N (SN) in the PZS15 system, indicating an ion–dipole interaction between the Li+ and nitrogen atom of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N group. We also analyzed the interactions of Li+ with polymer chains; the Li–O coordination number of EO segments decreased from 3.40 (in PEO and PZ systems) to 3.19 in the PZS15 system. The mean square displacement (MSD) was calculated at 298 K for all systems to evaluate the diffusion dynamics of Li+ (Fig. S14). As shown in Fig. 6e, the Li+ diffusion coefficient in PEO is 5.61 × 10−8 cm2 s−1, while that in PZ reaches 2.16 × 10−7 cm2 s−1. Notably, the PZS15 system exhibits the highest Li+ diffusion coefficient, which is 3.92 times that of PEO (2.20 × 10−7 cm2 s−1). The computational results are in unambiguous agreement with experimental observations, corroborating the accuracy of the proposed theoretical framework.
N group. We also analyzed the interactions of Li+ with polymer chains; the Li–O coordination number of EO segments decreased from 3.40 (in PEO and PZ systems) to 3.19 in the PZS15 system. The mean square displacement (MSD) was calculated at 298 K for all systems to evaluate the diffusion dynamics of Li+ (Fig. S14). As shown in Fig. 6e, the Li+ diffusion coefficient in PEO is 5.61 × 10−8 cm2 s−1, while that in PZ reaches 2.16 × 10−7 cm2 s−1. Notably, the PZS15 system exhibits the highest Li+ diffusion coefficient, which is 3.92 times that of PEO (2.20 × 10−7 cm2 s−1). The computational results are in unambiguous agreement with experimental observations, corroborating the accuracy of the proposed theoretical framework.
 | ||
| Fig. 6 Snapshots of MD simulations of (a) PEO, (b) PZ and (c) PZS15. (d) Radial distribution functions (RDFs) and coordination numbers (CNs) of PEO, PZ and PZS15 systems. (e) Calculated MSDs of Li+ for PEO, PZ and PZS15 with simulation time at 25 °C. | ||
Subsequently, given the high ionic conductivity, favorable electrochemical stability, and Li dendritic suppression capability of the PZS15 electrolyte, all-solid-state Li–CO2 batteries were assembled to demonstrate its potential for practical applications. The cathode was initially fabricated by utilizing Co/NC single-atom catalysts (SACs) as functional electrocatalysts. As shown in Fig. S15, the obtained Co/NC exhibits a folded lamellar structure, which offers a superior specific surface area conducive to CO2 adsorption. The XRD patterns suggest that no characteristic peak associated with cobalt is observed in Co/NC, due to the highly dispersed state of cobalt (Fig. S16). Further XPS spectra verify the successful introduction of Co single atoms (Fig. S17). First, the deep discharge capability of Li–CO2 batteries employing PEO, PZ, and PZS15 electrolytes was evaluated through full discharge tests. As shown in Fig. 7a, the initial discharge capacities of the Li–CO2 batteries using PEO and PZ electrolytes are 2126 mAh g−1 and 4413 mAh g−1, respectively. In contrast, the battery employing PZS15 delivers a significantly higher initial discharge capacity of 15016 mAh g−1. Besides, the discharge plateau of the PZS15-based battery is closer to the theoretical potential of 2.8 V. Fig. 7b compares the cycling performance of Li–CO2 batteries with different electrolytes at a current density of 200 mA g−1 and a fixed capacity of 1000 mAh g−1. Remarkably, the Li–CO2 battery with PZS15 electrolyte achieves a cycle life of over 1600 hours, which is several times longer than those based on PEO and PZ. As illustrated in Fig. 7c, the PZS15-based battery maintains a round-trip energy efficiency of 65.01% after 15 cycles, outperforming those with PEO (50.14%) and PZ (52.36%), thereby demonstrating superior cycling stability. The discharge/charge voltage profiles further reveal that the PZS15-based Li–CO2 battery exhibits a first-cycle overpotential of only 1.34 V, significantly lower than those of PEO (1.81 V) and PZ (1.79 V). In addition, it displays highly reversible and stable cycling behavior, with only a minor increase in overpotential observed after 150 cycles (Fig. S18). Due to the poor room-temperature performance of the PEO electrolyte, its cycling stability was evaluated at 60 °C. The results show a rapid decline in the discharge plateau and pronounced degradation with increasing discharge time. The embedded digital image indicates that this failure is due to pore clogging, a consequence of polymer melting at the elevated temperature (Fig. S19).
 | ||
| Fig. 7 (a) The full discharge curves of various Li–CO2 batteries at 100 mA g−1. (b) The comparison of cycling performance at 200 mA g−1 under a specific capacity limit of 1000 mAh g−1. (c) The energy efficiency curves of various Li–CO2 batteries. (d) Electrochemical impedance spectroscopy of the three samples for Li–CO2 batteries. (e) Electrochemical performance comparison of the three electrolytes. (f) Rate performance at current densities from 100 to 1000 mA g−1. (g) The electrochemical performance comparisons of different Li–CO2 batteries reported in the literature. | ||
The deactivation mechanism of the all-solid-state Li–CO2 battery was further investigated and attributed to the formation of Li2CO3. SEM characterization of the cathode surface after discharge revealed uniformly distributed aggregates composed of small particles, which contrasted with the pristine electrode's morphology (Fig. S20). However, the formed Li2CO3 exhibited sluggish decomposition kinetics during the subsequent charging process. Consequently, the residual Li2CO3 passivated the cathode surface and blocked the active sites, ultimately leading to the gradual performance degradation and eventual failure of the battery.57,58 The charge transfer kinetics of the Li–CO2 batteries were also investigated by using EIS. The equivalent circuit fitting results in Fig. 7d reveal that the PZS15 electrolyte possesses the lowest charge-transfer resistance (Rct). This result is attributed to the synergistic effect of ZIF-8 and SN, which enhances Li+ ion transport kinetics and improves electrochemical performance. Furthermore, the comprehensive evaluation in Fig. 7e demonstrates that PZS15 delivers the most superior overall performance among the various electrolytes.
The rate performance of the batteries was evaluated with a cut-off capacity of 1000 mAh g−1 (Fig. 7f). The PZS15-based battery displayed stable plateaus across current densities ranging from 100 to 1000 mA g−1, exemplifying the robust electrochemical performance and superior charge transport capabilities of PZS15 under high-rate operating conditions. In contrast, the PEO and PZ-based batteries exhibit larger polarization, indicating poorer ion transfer kinetics. Notably, the PEO-based battery failed to recover its initial discharge plateau following high-current cycling. As shown in Fig. S21, the PZS15-based battery consistently exhibited the lowest overpotential over the entire current density range (100–1000 mA g−1). Remarkably, even at a current density of 1000 mA g−1, the PZS15-based battery maintains polarization below 1.5 V during cycling (Fig. S22). Fig. 7g and Table S2 summarize a performance comparison between this work and other reported studies.59–69 The PZS15-based battery demonstrates a significant improvement in cycling stability, achieving over 1600 hours of stable operation, which compares favorably with previously reported Li–CO2 systems. Moreover, the developed electrolyte exhibits a high discharge capacity of 15016 mAh g−1, outperforming many existing solid-state and liquid-based configurations. While earlier studies have laid important groundwork in cathode catalysis and electrolyte formulation, our strategy integrates a MOF with plastic crystal enhancement to simultaneously tackle the long-standing challenges of interfacial instability and limited ionic conductivity in conventional designs. These advances highlight a meaningful step forward in the development of high-performance and safe Li–CO2 batteries.
Conclusions
In conclusion, we developed a novel composite solid polymer electrolyte based on a PEO matrix, incorporated with zeolitic imidazolate framework-8 and succinonitrile, to address the critical safety and performance issues associated with liquid electrolytes in Li–CO2 batteries. The synergistic effect between ZIF-8 and SN significantly enhances the ionic conductivity (3.6 × 10−4 S cm−1 at 30 °C) and broadens the electrochemical stability window (>5.2 V vs. Li/Li+) of the optimized PZS15 electrolyte. These improvements are attributed to the effective suppression of PEO crystallinity by SN, combined with the ability of ZIF-8 to facilitate LiTFSI dissociation and anchor anions, thereby promoting Li+ transport and deposition. The assembled all-solid-state Li–CO2 battery with the PZS15 electrolyte delivers a high discharge capacity of 15016 mAh g−1 at 100 mA g−1 and exhibits exceptional cycling stability exceeding 1600 hours at 200 mA g−1. DFT calculations and MD simulations further elucidate the mechanisms underlying the enhanced Li+ conduction and interfacial stability. This study provides an innovative and effective strategy for designing high-performance, safe, and durable solid-state Li–CO2 batteries, highlighting the potential of composite electrolytes in next-generation energy storage systems.
Author contributions
Y. Huang and T. Wang designed the experiment and wrote the manuscript. J. Ha completed the simulation calculation. Z. Liu, X. Yu and Y. Liu conducted the experiments. X. Fan, X. Huang, J. He and S. Huang analyzed the data. All authors discussed the results at all stages and participated in the development of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Data availability
The data supporting this study are available within the article and the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc07513k.Acknowledgements
The authors acknowledge the financial support for this work from the National Defense Technology Innovation Special Zone Spark Project (2016300TS00911901) and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). Postgraduate Research & Practice Innovation Program of NUAA (xcxjh20240619). We acknowledge the Center for Microscopy and Analysis at Nanjing University of Aeronautics and Astronautics.References
- Z. Peng, S. A. Freunberger, Y. Chen and P. G. Bruce, Science, 2012, 337, 563–566 CrossRef CAS
.
- Y. Qiao, J. Yi, S. Wu, Y. Liu, S. Yang, P. He and H. Zhou, Joule, 2017, 1, 359–370 CrossRef CAS
- S. Yang, Y. Qiao, P. He, Y. Liu, Z. Cheng, J. Zhu and H. Zhou, Energy Environ. Sci., 2017, 10, 972–978 RSC
- Q. Pan, X. Ma, H. Wang, Y. Shu, H. Liu, L. Yang, W. Li, J. Liu, Y. Wu, Y. Mao, J. Xie, G. Zou, H. Hou, W. Deng and X. Ji, Adv. Mater., 2024, 36, 2406905 CrossRef CAS
- B. Liu, Y. Sun, L. Liu, J. Chen, B. Yang, S. Xu and X. Yan, Energy Environ. Sci., 2019, 12, 887–922 RSC
- J. Zhou, X. Li, C. Yang, Y. Li, K. Guo, J. Cheng, D. Yuan, C. Song, J. Lu and B. Wang, Adv. Mater., 2019, 31, 1804439 CrossRef
- S. Xu, C. Chen, Y. Kuang, J. Song, W. Gan, B. Liu, E. M. Hitz, J. W. Connell, Y. Lin and L. Hu, Energy Environ. Sci., 2018, 11, 3231–3237 RSC
- X. Gao, Y.-N. Zhou, D. Han, J. Zhou, D. Zhou, W. Tang and J. B. Goodenough, Joule, 2020, 4, 1864–1879 CrossRef CAS
- H. Chen, X. Li, Z. Liu, Y. Xu, Y. Yan, P. Li, K. Chang, X. Huang, J. He and T. Wang, Adv. Funct. Mater., 2025, 35, 2412387 CrossRef CAS
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS
- T. Liu, J. P. Vivek, E. W. Zhao, J. Lei, N. Garcia-Araez and C. P. Grey, Chem. Rev., 2020, 120, 6558–6625 CrossRef CAS PubMed
- S. Zhang, L. Sun, Q. Fan, F. Zhang, Z. Wang, J. Zou, S. Zhao, J. Mao and Z. Guo, Nano Res. Energy, 2022, 1, 9120001 CrossRef
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 16141 CrossRef
- X. Zhan, M. Li, S. Li, X. Pang, F. Mao, H. Wang, Z. Sun, X. Han, B. Jiang, Y.-B. He, M. Li, Q. Zhang and L. Zhang, Energy Storage Mater., 2023, 61, 102875 CrossRef
- Q. Zhou, J. Ma, S. Dong, X. Li and G. Cui, Adv. Mater., 2019, 31, 1902029 CrossRef CAS
- S. Li, F. Pei, Y. Ding, X. Guo, X. Zhang, H. Tao, Z. He, H. Hu and L. Zhang, Adv. Funct. Mater., 2025, 35, 2415495 CrossRef CAS
- Q. Zhao, S. Stalin, C.-Z. Zhao and L. A. Archer, Nat. Rev. Mater., 2020, 5, 229–252 CrossRef CAS
- Z. Wan, D. Lei, W. Yang, C. Liu, K. Shi, X. Hao, L. Shen, W. Lv, B. Li, Q.-H. Yang, F. Kang and Y.-B. He, Adv. Funct. Mater., 2019, 29, 1805301 CrossRef
- Z. Song, F. Chen, M. Martinez-Ibañez, W. Feng, M. Forsyth, Z. Zhou, M. Armand and H. Zhang, Nat. Commun., 2023, 14, 4884 CrossRef CAS PubMed
- H. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef
- W. Xue, C. D. Sewell, Q. Zhou and Z. Lin, Angew. Chem., Int. Ed., 2022, 61, e202206512 CrossRef CAS
- R. Zhao, Y. Wu, Z. Liang, L. Gao, W. Xia, Y. Zhao and R. Zou, Energy Environ. Sci., 2020, 13, 2386–2403 RSC
- C. Wang, D. Liu and W. Lin, J. Am. Chem. Soc., 2013, 135, 13222–13234 CrossRef CAS
- L. Wei, Q. Liu, Y. Gao, Y. Yao, B. Hu and Q. Chen, Macromolecules, 2013, 46, 4447–4453 CrossRef CAS
- C. Fang, Y. Huang, Y. Sun, P. Sun, K. Li, S. Yao, M. Zhang, W. Fang and J. Chen, Nat. Commun., 2024, 15, 6781 CrossRef CAS
- L. Z. Fan, Y. S. Hu, A. J. Bhattacharyya and J. Maier, Adv. Funct. Mater., 2007, 17, 2800–2807 CrossRef CAS
- S. Xu, Z. Sun, C. Sun, F. Li, K. Chen, Z. Zhang, G. Hou, H.-M. Cheng and F. Li, Adv. Funct. Mater., 2020, 30, 2007172 CrossRef CAS
- J. Wang, G. Huang, K. Chen and X.-B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 9382–9387 CrossRef CAS PubMed
- C. Wang, K. R. Adair, J. Liang, X. Li, Y. Sun, X. Li, J. Wang, Q. Sun, F. Zhao, X. Lin, R. Li, H. Huang, L. Zhang, R. Yang, S. Lu and X. Sun, Adv. Funct. Mater., 2019, 29, 1900392 CrossRef
- W. Zha, F. Chen, D. Yang, Q. Shen and L. Zhang, J. Power Sources, 2018, 397, 87–94 CrossRef CAS
- C. Fu, Y. Ma, S. Lou, C. Cui, L. Xiang, W. Zhao, P. Zuo, J. Wang, Y. Gao and G. Yin, J. Mater. Chem. A, 2020, 8, 2066–2073 RSC
- Z. Lei, J. Shen, W. Zhang, Q. Wang, J. Wang, Y. Deng and C. Wang, Nano Res., 2020, 13, 2259–2267 CrossRef CAS
- S. A. Moggach, T. D. Bennett and A. K. Cheetham, Angew. Chem., Int. Ed., 2009, 48, 7087–7089 CrossRef CAS PubMed
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS
- Y. Wang, B. Wang, H. Shi, C. Zhang, C. Tao and J. Li, Inorg. Chem. Front., 2018, 5, 2739–2745 RSC
- Y. Xia, Q. Wang, Y. Liu, J. Zhang, X. Xia, H. Huang, Y. Gan, X. He, Z. Xiao and W. Zhang, J. Colloid Interface Sci., 2023, 638, 908–917 CrossRef CAS PubMed
- X. Song, K. Ma, J. Wang, H. Wang, H. Xie, Z. Zheng and J. Zhang, ACS Nano, 2024, 18, 12311–12324 CrossRef CAS
- J. Chen, X. Deng, Y. Gao, Y. Zhao, X. Kong, Q. Rong, J. Xiong, D. Yu and S. Ding, Angew. Chem., Int. Ed., 2023, 62, e202307255 CrossRef CAS
- Y. Lin, X. Wang, J. Liu and J. D. Miller, Nano Energy, 2017, 31, 478–485 CrossRef CAS
- W. Wieczorek, D. Raducha, A. Zalewska and J. R. Stevens, J. Phys. Chem. B, 1998, 102, 8725–8731 CrossRef CAS
- H. Chen, D. Adekoya, L. Hencz, J. Ma, S. Chen, C. Yan, H. Zhao, G. Cui and S. Zhang, Adv. Energy Mater., 2020, 10, 2000049 CrossRef CAS
- J. Hu, K. Chen, Z. Yao and C. Li, Sci. Bull., 2021, 66, 694–707 CrossRef CAS PubMed
- S. Li, Z. Zhang, K. Yang and L. Yang, ChemElectroChem, 2018, 5, 328–334 CrossRef CAS
- S. Yang, Z. Liu, Y. Liu and Y. Jiao, J. Mater. Sci., 2015, 50, 1544–1552 CrossRef CAS
- J. Hu, C. Lai, K. Chen, Q. Wu, Y. Gu, C. Wu and C. Li, Nat. Commun., 2022, 13, 7914 CrossRef CAS
- P.-J. Alarco, Y. Abu-Lebdeh, A. Abouimrane and M. Armand, Nat. Mater., 2004, 3, 476–481 CrossRef CAS
- J. Sun, X. Yao, Y. Li, Q. Zhang, C. Hou, Q. Shi and H. Wang, Adv. Energy Mater., 2020, 10, 2000709 CrossRef CAS
- H. Peng, T. Long, J. Peng, H. Chen, L. Ji, H. Sun, L. Huang and S.-G. Sun, Adv. Energy Mater., 2024, 14, 2400428 CrossRef CAS
- X. Zhang, Q. Su, G. Du, B. Xu, S. Wang, Z. Chen, L. Wang, W. Huang and H. Pang, Angew. Chem., Int. Ed., 2023, 62, e202304947 CrossRef CAS PubMed
- Z. Zhang, J. Wang, S. Zhang, H. Ying, Z. Zhuang, F. Ma, P. Huang, T. Yang, G. Han and W.-Q. Han, Energy Storage Mater., 2021, 43, 229–237 CrossRef
- L. Chen, T. Gu, J. Ma, K. Yang, P. Shi, J. Biao, J. Mi, M. Liu, W. Lv and Y.-B. He, Nano Energy, 2022, 100, 107470 CrossRef CAS
- S. Liu, J. Shen, Z. Wang, W. Tian, X. Han, Z. Chen, H. Pan, L. Wang, D. Bian, C. Yang and S. Zhu, J. Mater. Chem. A, 2024, 12, 256–266 RSC
- Q. Liu, A. Cresce, M. Schroeder, K. Xu, D. Mu, B. Wu, L. Shi and F. Wu, Energy Storage Mater., 2019, 17, 366–373 CrossRef
- Y. Chai, D. Ning, D. Zhou, J. Gao, J. Ni, G. Zhang, R. Gao, W. Wu, J. Wang and Y. Li, Nano Energy, 2024, 130, 110160 CrossRef CAS
- M. Echeverri, N. Kim and T. Kyu, Macromolecules, 2012, 45, 6068–6077 CrossRef CAS
- L. Sun, J. A. Yuwono, S. Zhang, B. Chen, G. Li, H. Jin, B. Johannessen, J. Mao, C. Zhang, M. Zubair, N. Bedford and Z. Guo, Adv. Mater., 2024, 36, 2401288 CrossRef CAS
- L. Sun, Q. Gu, J. A. Yuwono, J. Zhou, B. Johannessen, L. Zhao, C. Zhang, G. Li, Z. Guo and S. Zhang, ACS Nano, 2025, 19, 20051–20062 CrossRef PubMed
- R. Wang, X. Zhang, Y. Cai, Q. Nian, Z. Tao and J. Chen, Nano Res., 2019, 12, 2543–2548 CrossRef CAS
- J. Li, H. Zhao, H. Qi, X. Sun, X. Song, Z. Guo, A. G. Tamirat, J. Liu, L. Wang and S. Feng, Adv. Funct. Mater., 2019, 29, 1806863 CrossRef
- M. Mushtaq, X.-W. Guo, J.-P. Bi, Z.-X. Wang and H.-J. Yu, Rare Met., 2018, 37, 520–526 CrossRef CAS
- J. Zhao, Y. Wang, H. Zhao, L. Liu, S. Li, X. Hu and S. Ding, ACS Nano, 2024, 18, 5132–5140 CrossRef CAS
- C. Li, Z. Guo, B. Yang, Y. Liu, Y. Wang and Y. Xia, Angew. Chem., Int. Ed., 2017, 56, 9126–9130 CrossRef CAS PubMed
- S. Mamidi, D. Na, B. Yoon, H. Sharma, A. D. Pathak, K. K. Sahu, D. Y. Lee, C.-R. Lee and I. Seo, J. Power Sources, 2024, 591, 233867 CrossRef CAS
- Y. Du, Y. Liu, S. Yang, C. Li, Z. Cheng, F. Qiu, P. He and H. Zhou, J. Mater. Chem. A, 2021, 9, 9581–9585 RSC
- Q.-C. Zhu, J. Ma, J.-H. Huang, D.-Y. Mao and K.-X. Wang, Chem. Eng. J., 2024, 482, 148977 CrossRef CAS
- Y. Qiao, S. Xu, Y. Liu, J. Dai, H. Xie, Y. Yao, X. Mu, C. Chen, D. J. Kline, E. M. Hitz, B. Liu, J. Song, P. He, M. R. Zachariah and L. Hu, Energy Environ. Sci., 2019, 12, 1100–1107 RSC
- K. M. Naik, A. K. Chourasia, M. Shavez and C. S. Sharma, ChemSusChem, 2023, 16, e202300734 CrossRef CAS PubMed
- H. Dong, J. Han, Z. Guan, X. Jia, Z. Wang, X. Zhao, X. Li, Z. Cheng, D. Wang, Y. Yin and S.-T. Yang, J. Colloid Interface Sci., 2025, 700, 138315 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2026 |