
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRoom temperature synthesis of α,β-unsaturated nitriles by manganese-catalysed base-free coupling of α-saturated nitriles with aldehydes
Subramanian
Thiyagarajan
 ,
Jie
Luo†
,
Yaoyu
Liang‡
,
Irena
Efremenko
,
Michael
Montag
and
David
Milstein
*
,
Jie
Luo†
,
Yaoyu
Liang‡
,
Irena
Efremenko
,
Michael
Montag
and
David
Milstein
*
Department of Molecular Chemistry and Materials Science, Weizmann Institute of Science, Rehovot 7610001, Israel. E-mail: david.milstein@weizmann.ac.il
First published on 27th January 2026
Abstract
α,β-Unsaturated nitriles have broad applications, from polymers to pharmaceuticals, but their synthesis still poses significant challenges, as it often involves multistep procedures, or the use of precious metal catalysts, or the generation of significant amounts of waste. Herein, we report a new approach to the synthesis of such nitriles through single-step coupling of α-saturated nitriles with aldehydes, catalysed by a pincer complex of earth-abundant manganese. This reaction proceeds at room temperature in the absence of base or other additives, does not require substrate pre-activation and generates water as the only byproduct. The catalytic system was shown to tolerate various functional groups, including base-sensitive ones, and afforded a wide range of unsaturated nitriles in generally high yields. α-Deuterated vinyl nitriles were also obtained by coupling CD3CN with different aldehydes in one-step room temperature syntheses. Experimental and computational data demonstrate that the catalytic reaction involves reversible binding of the nitrile substrate across the metal–ligand framework of the pincer complex, leading to ketimido and enamido complexes as intermediates. The enamido species functions as a carbon nucleophile that forms a C–C bond with an aldehyde to generate a β-hydroxynitrile intermediate, which is subsequently dehydrated into the vinyl nitrile product.
Introduction
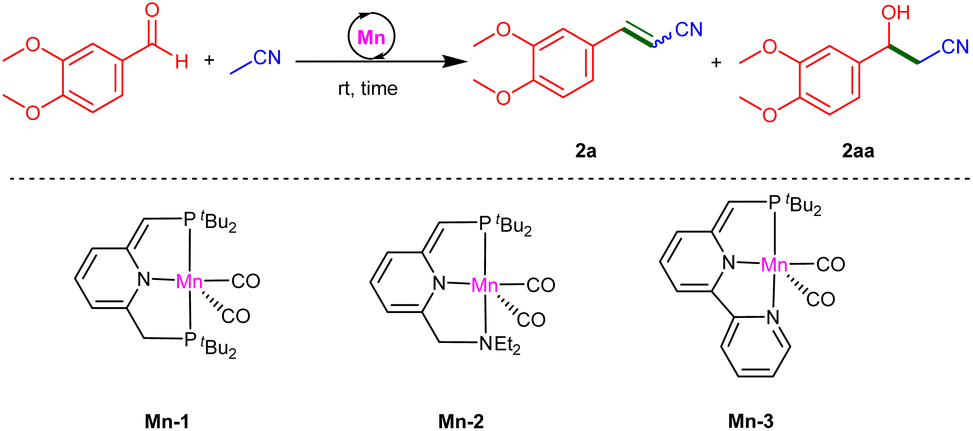
α,β-Unsaturated (vinyl) nitriles are a family of industrially important compounds, the simplest member of which, acrylonitrile, is produced globally on a multimillion tonne scale as a polymer precursor.1a More complex vinyl nitriles can be found among biologically active natural products and man-made pharmaceuticals (Fig. 1),1–6 and are used in various fields of application, e.g., agrochemicals, dyes and functional materials.7 Such nitriles are also valuable building blocks in chemical synthesis, as they are key starting materials in conjugate addition reactions and can be readily transformed into other functional groups.8 | ||
| Fig. 1 Select examples of biologically active vinyl nitrile compounds. | ||
A direct and atom-economical route to the synthesis of α,β-unsaturated nitriles is the condensation of α-saturated nitriles with aldehydes or ketones. Traditionally, this transformation is achieved through the Knoevenagel condensation, which requires a stoichiometric excess of strong bases, such as KOH or NaOH, at elevated temperatures (Scheme 1a).9 However, these conventional base-mediated reactions suffer from significant drawbacks, including side-reactions, such as self-aldol condensation and the Cannizzaro reaction, leading to multiple side-products and reducing the overall atom economy.
 | ||
| Scheme 1 Strategies for the synthesis of α,β-unsaturated nitriles from α-saturated nitriles. (a) Previously reported methods involving the coupling of α-saturated nitriles with aldehydes, promoted by stoichiometric base or heterogeneous catalysis.9 (b) Metal-catalysed synthesis via dehydrogenative coupling of alcohols and α-saturated nitriles.17 (c) Mn-catalysed synthesis from aldehydes and α-saturated nitriles under mild, neutral conditions (this work). | ||
Several alternative approaches have been developed to address these challenges. In 1998, Verkade and coworkers introduced nonionic superbases as catalysts for the synthesis of α,β-unsaturated nitriles directly from aldehydes and acetonitrile (CH3CN) or benzyl nitrile.10 In 2010, Tomioka and coworkers reported a three-step, single-pot (Z)-selective synthesis of acrylonitriles from CH3CN and aldehydes, using stoichiometric amounts of n-BuLi and a boron reagent.11 More recently, Lanari, Vaccaro and coworkers reported a heterogeneous fluoride-catalysed synthesis of vinyl nitriles by coupling α-saturated nitriles with aldehydes and ketones, in the presence of excess silazanes.12 Several groups have also developed protocols for vinyl nitrile synthesis through direct α-olefination of α-saturated nitriles, using alcohols as olefination reagents via dehydrogenative coupling, but such procedures typically require high temperatures, and are mainly limited to benzylic nitriles (Scheme 1b).17 Other notable approaches are Peterson olefination,13 Wittig reactions,14 carbocyanation of alkynes15 and cross-metathesis.16 While the above methods offer advantages, they also have significant drawbacks, including the use of expensive metals, high temperatures, long reaction times, and toxic reagents that are environmentally unfriendly and non-atom-economical, and may also be incompatible with common functional groups.18 Therefore, it is essential to develop new methodologies for the synthesis of α,β-unsaturated nitriles that are both efficient and green.19
A highly useful strategy for C–C and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond formation is transition metal-mediated in situ generation of carbon nucleophiles and their coupling with carbonyl compounds.20,21 In 2013, we showed that benzylic nitriles can be activated by a rhenium pincer complex via metal–ligand cooperation (MLC), wherein the C
C bond formation is transition metal-mediated in situ generation of carbon nucleophiles and their coupling with carbonyl compounds.20,21 In 2013, we showed that benzylic nitriles can be activated by a rhenium pincer complex via metal–ligand cooperation (MLC), wherein the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N moiety adds reversibly across the metal–ligand framework through C–C bonding with the pincer backbone and Re–N coordination.22,23 The nitriles activated in this manner were found to behave like Michael donors that can react with α,β-unsaturated esters and ketones to afford the corresponding C–C coupling products. We subsequently extended this approach to aliphatic nitriles through similar activation by a structurally analogous manganese pincer complex.24 Both complexes exhibited catalytic Michael addition activity, which we termed “template catalysis”, since each complex essentially serves as an anchor for the substrate, while C–C bond formation occurs outside the first coordination sphere. This catalytic strategy facilitates both electrophilic and nucleophilic attack on the metal-bound nitriles, enabling a wide range of organic transformations, including conjugate addition,24 oxa- and aza-Michael addition,25 hydration and α-deuteration,26 and nitrile heterocoupling.27 These processes are atom-economical, waste-free, and proceed under mild, neutral conditions. It should be noted that a similar base-free nitrile activation strategy has also been developed by Otten, de Vries and coworkers, using ruthenium pincer complexes.28
N moiety adds reversibly across the metal–ligand framework through C–C bonding with the pincer backbone and Re–N coordination.22,23 The nitriles activated in this manner were found to behave like Michael donors that can react with α,β-unsaturated esters and ketones to afford the corresponding C–C coupling products. We subsequently extended this approach to aliphatic nitriles through similar activation by a structurally analogous manganese pincer complex.24 Both complexes exhibited catalytic Michael addition activity, which we termed “template catalysis”, since each complex essentially serves as an anchor for the substrate, while C–C bond formation occurs outside the first coordination sphere. This catalytic strategy facilitates both electrophilic and nucleophilic attack on the metal-bound nitriles, enabling a wide range of organic transformations, including conjugate addition,24 oxa- and aza-Michael addition,25 hydration and α-deuteration,26 and nitrile heterocoupling.27 These processes are atom-economical, waste-free, and proceed under mild, neutral conditions. It should be noted that a similar base-free nitrile activation strategy has also been developed by Otten, de Vries and coworkers, using ruthenium pincer complexes.28
As mentioned above, there are various ways to synthesize vinyl nitriles from α-saturated nitriles and aldehydes, including catalytic ones, but they typically involve large amounts of additives or forcing conditions. Coupling of nitriles and aldehydes under mild conditions has been previously achieved using transition metal-based catalysts, but these systems afford β-hydroxynitriles, rather than α,β-unsaturated ones.29 Herein, we present a Mn-catalysed direct condensation of unactivated α-saturated nitriles with aldehydes at room temperature, yielding α,β-unsaturated nitriles in a single synthetic step (Scheme 1c). This mild, base-free approach is highly functional-group tolerant, utilizes the nitrile substrates themselves as solvents, generates water as the sole byproduct, and offers a green, atom-economical pathway for the synthesis of unsaturated nitriles.
Results and discussion
At the outset of our study, we examined the catalytic activity of several Mn(I)-pincer complexes, Mn-1, Mn-2 and Mn-3 (Table 1), which we have previously employed as catalysts for the base-free coupling of nitriles with a variety of substrates.24–27 Using 3,4-dimethoxybenzaldehyde and CH3CN as our model substrates, we found that Mn-1, at a loading of 2 mol% vs. aldehyde, efficiently promotes their conversion into the corresponding α,β-unsaturated nitrile at room temperature, with CH3CN itself being used as solvent (Table 1, entry 1). Thus, after 24 h, gas chromatographic (GC) analysis of the reaction mixture indicated 94% aldehyde conversion, with the vinyl nitrile product 2a having been produced in 80% yield, accompanied by the β-hydroxynitrile 2aa in 12% yield. The presence of this β-hydroxynitrile suggests that it is an intermediate that can undergo catalytic dehydration by Mn-1 to afford the unsaturated product 2a. Nevertheless, after extending the reaction time to 48 h, no significant change was observed in the product composition. Thus, we repeated this reaction with an increased catalyst loading of 3 mol%, and this allowed us to achieve essentially quantitative yield of 2a within 24 h at room temperature (entry 2). Notably, no significant byproducts were observed, including the β-hydroxynitrile. When the neat nitrile was replaced with an equal volume of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of CH3CN and THF, aldehyde conversion and the yield of 2a slightly decreased (95% for both; entry 3). Reducing the amount of nitrile even further, to 1 equiv. per aldehyde, afforded poor results (entry 4), thereby indicating that CH3CN must be used in excess for this transformation to be efficient. Complexes Mn-2 and Mn-3 were also found to be catalytically active, but exhibited lower performance than Mn-1 under otherwise identical conditions, with reduced aldehyde conversions (≤65%) and the generation of product mixtures in lower yields (≤53% for 2a, ≤20% for 2aa; entries 5 and 6). Finally, in the absence of these manganese complexes, no product formation was observed at room temperature, even after 48 h (entry 7), clearly demonstrating the role of these complexes as catalysts in this unsaturated nitrile synthesis.
:1 mixture of CH3CN and THF, aldehyde conversion and the yield of 2a slightly decreased (95% for both; entry 3). Reducing the amount of nitrile even further, to 1 equiv. per aldehyde, afforded poor results (entry 4), thereby indicating that CH3CN must be used in excess for this transformation to be efficient. Complexes Mn-2 and Mn-3 were also found to be catalytically active, but exhibited lower performance than Mn-1 under otherwise identical conditions, with reduced aldehyde conversions (≤65%) and the generation of product mixtures in lower yields (≤53% for 2a, ≤20% for 2aa; entries 5 and 6). Finally, in the absence of these manganese complexes, no product formation was observed at room temperature, even after 48 h (entry 7), clearly demonstrating the role of these complexes as catalysts in this unsaturated nitrile synthesis.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Time (h) | Aldehyde conversion (%)b | Product yields (%)c | |
| 2a | 2aa | ||||
| a General reaction conditions: 3,4-dimethoxybenzaldehyde (0.3 mmol), CH3CN (1 mL) and catalyst (loading as indicated, relative to aldehyde) were stirred at room temperature for the indicated time under an N2 atmosphere. b Conversion of 3,4-dimethoxybenzaldehyde was determined by GC analysis, using mesitylene as an internal standard. c Yields of products 2a and 2aa are for the isolated materials. d Mixture of CH3CN (0.5 mL) and THF (0.5 mL) was used instead of neat CH3CN. e Reaction was performed using 3,4-dimethoxybenzaldehyde (0.3 mmol) and CH3CN (0.3 mmol) in THF (1 mL). | |||||
| 1 | Mn-1 (2 mol%) | 24 | 94 | 80 | 12 |
| 2 | Mn-1 (3 mol%) | 24 | >99 | 99 | — |
| 3d | Mn-1 (3 mol%) | 24 | 95 | 95 | — |
| 4e | Mn-1 (3 mol%) | 24 | 10 | 9 | — |
| 5 | Mn-2 (3 mol%) | 24 | 65 | 53 | 10 |
| 6 | Mn-3 (3 mol%) | 24 | 60 | 34 | 20 |
| 7 | — | 48 | — | ||
After establishing the optimal catalytic conditions (Table 1, entry 2), we applied them in a substrate scope investigation, employing a wide range of aldehydes and unactivated α-saturated nitriles (Fig. 2; data for product 2a from Table 1 are duplicated here for comparative purposes). A gram-scale reaction of 3,4-dimethoxybenzaldehyde in CH3CN gave 2a in 89% yield, demonstrating the synthetic utility of our catalytic system. Benzaldehyde derivatives bearing electroneutral and electron-donating substituents, as well as polycyclic aromatic aldehydes, underwent smooth coupling with CH3CN, affording the respective α,β-unsaturated nitriles, 2b–h, in excellent yields (88–99%). Benzaldehydes featuring para-positioned electron-withdrawing groups, namely, chloride, trifluoromethyl and cyanide, were also effectively coupled with CH3CN, leading to the vinyl nitriles 2i–k in fair to high yields (62–87%). Importantly, base-sensitive functional groups, i.e., ester and carbamate, were tolerated by our catalytic system, and the corresponding benzaldehydes reacted with CH3CN to afford products 2l and 2m in 60 and 90% yield, respectively. Aldehydes with heteroaryl substituents, i.e., 3-pyridyl and 2-furfuryl, were also compatible with our current catalytic protocol and afforded the desired unsaturated nitriles, 2n and 2o, in practically quantitative yields. When we attempted to use an alicyclic aldehyde, cyclopentanecarboxaldehyde, under the optimal reaction conditions, the respective vinyl nitrile product, 2p, was obtained in poor yield. Nevertheless, raising the reaction temperature to 50 °C resulted in formation of this nitrile in 60% yield. A series of other aliphatic and α,β-unsaturated aldehydes were also examined as substrates, to be coupled with acetonitrile or propionitrile under the optimal catalytic conditions, but only marginal vinyl nitrile formation was observed (Fig. S6; see SI). Finally, the regioselectivity of our catalytic system was demonstrated when CH3CN was directly coupled with three conjugated aldehydes, resulting in dienyl nitriles 2q–s in high conversions and yields (84–98%). Conventional base-mediated Michael-type reactions of such carbonyl substrates usually give a mixture of 1,2- and 1,4-addition products, whereas our method exhibits very high 1,2-selectivity.19c,30
 | ||
| Fig. 2 Substrate scope for the coupling of nitriles with aldehydes catalysed by Mn-1. General reaction conditions: aldehyde (0.3 mmol), nitrile (1 mL) and Mn-1 (3 mol% vs. aldehyde) were stirred at room temperature for 24 h [yields correspond to pure isolated products; values in parentheses are aldehyde conversions as determined by GC analysis (mesitylene was used as internal standard); E/Z ratios were determined by GC or 1H NMR spectroscopic analysis of the crude reaction mixtures]. a Gram-scale experiment with increased amounts of reagents: aldehyde (6 mmol), CH3CN (6 mL), Mn-1 (3 mol% vs. aldehyde). b Reaction was conducted at 50 °C. c Increased amount of Mn-1 (5 mol% vs. aldehyde). d The reagents were 2-(4-(tert-butyl)phenyl)acetonitrile (0.3 mmol), 37% aqueous formaldehyde (0.45 mmol, 1.5 equiv.) and Mn-1 (3 mol% vs. nitrile). e Values in parentheses are nitrile conversions as determined by GC analysis (mesitylene was used as internal standard). f The amount of nitrile was 0.3 mmol and the solvent was THF (1 mL). g The reaction was carried out in CD3CN (1 mL). | ||
Having shown that the present catalytic system can couple CH3CN with a host of aldehydes to generate the corresponding α,β-unsaturated nitriles in high efficiency and functional group tolerance, we turned our attention to other unactivated α-saturated nitriles as substrates (Fig. 2). Under the optimal catalytic conditions, albeit with a catalyst loading of 5 mol%, the reactions of propionitrile with 3,4-dimethoxybenzaldehyde and 2-naphthaldehyde proceeded smoothly, giving the vinyl nitriles 2t and 2u in fair to good yield (57–80%). Applying the same procedure to a solution of 4-isopropylbenzaldehyde in neat butyronitrile afforded the desired product, 2v, in 86% yield. We were also able to synthesize a terminal disubstituted olefin by using aqueous formaldehyde as substrate. Thus, coupling it with (4-tert-butyl)phenylacetonitrile, in a THF solution, furnished the unsaturated nitrile 2w in 90% yield.31 2,3-Diphenylacrylonitrile (2x) was obtained quantitatively from benzaldehyde and benzyl nitrile, and the same nitrile was also coupled with 3-phenylpropiolaldehyde to give the product 2y in 90% yield. The chemoselectivity of our catalytic system was demonstrated through the reaction of benzyl nitrile with 4-acetylbenzaldehyde, which features two carbonyl functionalities – aldehyde and ketone – that may undergo condensation. In practice, however, only the aldehyde moiety reacted, furnishing the vinyl nitrile 2z in 97% yield, whereas the ketone group remained intact. The linear aldehydes butyraldehyde and 3,7-dimethylocta-2,6-dienal reacted with benzyl nitrile to afford the products 2ab–ac in very good yields (85–89%), and 2-methylbutyraldehyde was coupled with 4-fluorobenzyl nitrile to give 2ad in a similarly high yield (87%). Notably, no double-bond migration was observed in product 2ac, and both CC bonds of the parent aldehyde remained in their respective positions. The trans isomers of the vinyl nitriles – denoted E for acetonitrile and propionitrile and Z for the heavier nitriles – are preferred across all substrates. Acetonitrile shows high trans selectivity, whereas the moderate stereoselectivity observed for other nitriles may arise from steric or electronic factors. Lastly, the ability to directly couple CH3CN with a range of aldehydes has enabled us to synthesize α-deuterated vinyl nitriles, by using the commercially available isotopolog CD3CN. Thus, various benzaldehyde derivatives, as well as cinnamaldehyde, were allowed to react with CD3CN, selectively affording the corresponding α-deuterated α,β-unsaturated nitriles, 2ae–ai, in generally high yields. Importantly, no deuterium incorporation was observed at other C–H positions in these products.28a
In order to gain insight into the underlying mechanism of our catalytic nitrile–aldehyde coupling system, stoichiometric experiments were performed (Scheme 2). When a THF solution of Mn-1 was treated with 5 equiv. of benzyl nitrile and 5 equiv. of benzaldehyde at room temperature, the only observed product was the nitrile adduct of Mn-1, i.e., the previously-reported rearomatized enamido complex Mn-4 (enamine-containing species; Scheme 2a).24 Notably, no other complexes were observed in solution, including the aldehyde adduct.24,27 This simple competition experiment clearly indicates that Mn-1 reacts preferentially with benzyl nitrile, rather than benzaldehyde. This points to the higher thermodynamic stability of the enamido complex, likely enhanced by conjugation between the CN bond and phenyl group. In an attempt to probe the possible role of Mn-4 as a catalytic intermediate, this complex was independently synthesized and used as catalyst for the coupling of benzyl nitrile with butyraldehyde. Thus, 0.3 mmol of each substrate and 3 mol% of Mn-4 (loading relative to either substrate) were dissolved in THF, and the solution was stirred at room temperature for 24 h, after which the vinyl nitrile product 2ab was isolated in 83% yield (Scheme 2b). This result is nearly identical to the one originally obtained with Mn-1 (Fig. 2). Furthermore, when 4-methylbenzyl nitrile was allowed to react with benzaldehyde under the same catalytic conditions involving Mn-4, the expected product 2aj was obtained in 80% yield (Scheme 2c), but was accompanied by a small amount of compound 2x, which is derived from the enamido ligand of complex Mn-4. Taken together, the above stoichiometric experiments indicate that Mn-4 is indeed an intermediate in the catalytic cycle.
 | ||
| Scheme 2 Mechanistic studies. (a) Competition experiment involving the reaction of Mn-1 with excess benzyl nitrile and benzaldehyde in THF. (b) Reaction of benzyl nitrile with butyraldehyde in THF, catalysed by the independently synthesized complex Mn-4. (c) Reaction of 4-methylbenzyl nitrile with benzaldehyde in THF, catalysed by Mn-4. (d) Competition experiment involving the reaction of Mn-1 with excess CH3CN and benzaldehyde in THF. (e) Stoichiometric reaction of Mn-1 with benzaldehyde in CH3CN. (f) Stoichiometric reaction of 3-hydroxy-3-phenylpropanenitrile with Mn-1 in CH3CN. (g) Reaction of 3-hydroxy-3-phenylpropanenitrile catalysed by Mn-1 in CH3CN. (h) Reaction of 3-hydroxy-3-phenylpropanenitrile with KOtBu (5 mol%) in CH3CN. | ||
As shown above, the present catalytic system can also efficiently couple aldehydes with unactivated aliphatic nitriles, namely, acetonitrile, propionitrile and butyronitrile (Fig. 2). Thus, as was done with the benzylic nitriles, we carried out a stoichiometric competition experiment wherein Mn-1 was mixed with 5 equiv. of CH3CN and 5 equiv. of benzaldehyde in THF, at room temperature (Scheme 2d). Interestingly, only the benzaldehyde adduct of Mn-1, i.e., the previously-reported complex Mn-5 was observed in solution,32 in contrast to the abovementioned competition experiment involving benzyl nitrile, which resulted exclusively in the nitrile adduct. We have previously demonstrated that reaction of Mn-1 with another aliphatic nitrile, propionitrile, is highly reversible at room temperature.24 It generates two nitrile adducts, namely, a ketimido complex (imine-containing species), which is the predominant product, and an enamido complex, which is a minor product, but both complexes were found to be less stable than Mn-1 and the free nitrile. In fact, they could be characterized only in the presence of excess nitrile at sub-ambient temperatures. Hence, in our catalytic system, aliphatic nitriles must be used in large excess relative to the aldehyde, in order to outcompete this substrate for bonding to Mn-1, and to drive the formation of the unstable enamido intermediate. Indeed, when Mn-1 was treated with 1 equiv. of benzaldehyde in CH3CN as solvent, the aforementioned benzaldehyde adduct was not detected. Instead, we observed complexes Mn-6 and Mn-7 (Scheme 2e), which are the cinnamonitrile and water adducts of Mn-1, respectively. The existence of these complexes, which were previously characterized by us,26,27 is consistent with the occurrence of C–C coupling between benzaldehyde and CH3CN, followed by dehydration to generate cinnamonitrile.
During the optimization of the catalytic conditions, we observed β-hydroxynitrile 2aa as a minor product (Table 1, entry 1). In an attempt to examine our assertion that it is an intermediate en route to the α,β-unsaturated nitrile, an acetonitrile solution of Mn-1 was treated, at room temperature, with 1 equiv. of 3-hydroxy-3-phenylpropanenitrile, an analog of 2aa (Scheme 2f). This gave a mixture of the complexes Mn-6 and Mn-7, as was observed for the reaction of Mn-1 with benzaldehyde under the same conditions, thereby indicating that the β-hydroxynitrile is indeed a catalytic intermediate, and that it undergoes facile dehydration by Mn-1 at room temperature. These conclusions were further corroborated when 3-hydroxy-3-phenylpropanenitrile was subjected to the optimal catalytic conditions, using CH3CN as solvent and Mn-1 at 3 mol% loading, resulting in the quantitative formation of cinnamonitrile (Scheme 2g). It should be noted that when this catalytic experiment was repeated, but with Mn-1 having been replaced with the strong base KOtBu (5 mol%), only traces of cinnamonitrile were observed (Scheme 2h). This rules out a general-base dehydration mechanism and highlights the efficiency with which Mn-1 catalyses this reaction at room temperature.
Time-resolved monitoring of the Mn-catalysed reaction of benzaldehyde in CH3CN, using 1H nuclear magnetic resonance (NMR) spectroscopy, enabled us to gain some kinetic insights. The time variation of the reaction mixture composition, under the optimal catalytic conditions, is presented in Fig. 3. As can be deduced from this data, the conversion of benzaldehyde exceeds 50% within the first 2 h at room temperature. During the same time, the β-hydroxynitrile intermediate reaches a yield of nearly 40%, whereas the product, cinnamonitrile, lags behind at 15%. Hydroxynitrile dehydration proceeds slowly, and the amount of cinnamonitrile exceeds that of the hydroxynitrile only after ∼8 h. These data indicate that hydroxynitrile dehydration is the rate-determining step for the overall reaction between the α-saturated nitrile and aldehyde to afford the α,β-unsaturated nitrile.
 | ||
| Fig. 3 Kinetic profile of the reaction between CH3CN and benzaldehyde catalysed by Mn-1. | ||
Based on the above experimental results, as well as our previous work,24–27 we propose a catalytic cycle for the coupling of α-saturated nitriles with aldehydes, promoted by Mn-1, as depicted in Scheme 3. Using CH3CN and benzaldehyde as representative substrates, this cycle was studied by density functional theory (DFT) calculations, with the temperature fixed at 298.15 K and CH3CN set as the solvent. The computed reaction profiles are presented in Fig. 4.
 | ||
| Scheme 3 Proposed reaction mechanism for the direct synthesis of α,β-unsaturated nitriles from α-saturated nitriles and aldehydes catalysed by complex Mn-1. | ||
 | ||
| Fig. 4 Computed reaction profiles for the synthesis of cinnamonitrile from CH3CN and benzaldehyde catalysed by Mn-1 in CH3CN as solvent. (a) C–C coupling between CH3CN and benzaldehyde promoted by complex I (Mn-1). (b) Dehydration of the β-hydroxynitrile intermediate promoted by the in situ-generated complex IV. All energies are in kcal mol−1 and are referenced to complex Mn-1 and the free reactants. | ||
The catalytic reaction begins with addition of a nitrile molecule to species I (complex Mn-1), and follows a template-type mechanism similar to the one reported in our previous work involving conjugate addition of nitriles to α,β-unsaturated carbonyl compounds.24 Initially, the nitrile reacts with complex I to generate the ketimido intermediate IIvia MLC, a process that is endergonic for CH3CN, with ΔG = 7.0 kcal mol−1. It is also endergonic for both propionitrile and benzyl nitrile, with ΔG = 6.0 and 1.9 kcal mol−1, respectively (see SI). Subsequently, II undergoes facile tautomerization into the enamido intermediate III, a reaction that is slightly exergonic for CH3CN, with ΔG = −1.5 kcal mol−1. It is also exergonic for benzyl nitrile, but endergonic for propionitrile, with ΔG = −2.7 and 0.8 kcal mol−1, respectively (see SI). For both aliphatic nitriles, the ketimido and enamido intermediates are less stable than the parent complex I and free nitrile, whereas in the case of benzyl nitrile, the enamido intermediate is more stable. The latter species is apparently stabilized by conjugation between the CN bond and aromatic ring, an effect which is absent for the aliphatic nitriles.24 These computational results are consistent with our experimental observations, which showed that, in contrast to benzyl nitrile, an excess of the aliphatic nitrile is required for shifting the reaction equilibrium toward the enamido complex III. Moreover, they are in line with the results of our competition experiments involving benzaldehyde vs. benzyl nitrile or CH3CN. Nevertheless, regardless of the exact identity of the nitrile and stability of its adduct, once intermediate III is generated, it functions as a carbon nucleophile that reacts with benzaldehyde through C–C bond formation, accompanied by proton transfer, to produce intermediate IV, which is a β-hydroxynitrile adduct. For CH3CN, this is a slightly exergonic step (ΔG = −0.8 kcal mol−1) that passes through a concerted transition state, with an activation energy of 14.9 kcal mol−1. The hydroxynitrile product can then dissociate from IV, with a kinetic barrier of 19.6 kcal mol−1, thereby regenerating complex I and closing the catalytic cycle. Overall, the formation of hydroxynitrile is thermodynamically uphill by 6.0 kcal mol−1, and is driven by the large excess of substrates in the reaction mixture. Moreover, it has an apparent activation barrier of 24.4 kcal mol−1, which is consistent with a facile room temperature process.
As mentioned above, we have shown experimentally that the β-hydroxynitrile compound 3-hydroxy-3-phenylpropanenitrile undergoes facile dehydration into cinnamonitrile in the presence of Mn-1, but is not dehydrated by a strong Brønsted base. However, our computational results indicate that direct dehydration of the hydroxynitrile by Mn-1 is kinetically unattainable at room temperature (see SI). Interestingly, our calculations suggest that dehydration is instead catalysed by the in situ-generated intermediate IV. As can be seen in Fig. 4b, this reaction is exergonic to the tune of 3.7 kcal mol−1, and exhibits an internal kinetic barrier of 22.8 kcal mol−1, giving an overall apparent activation energy of 27.6 kcal mol−1 for cinnamonitrile formation. The latter is 3.2 kcal mol−1 higher than the corresponding barrier for hydroxynitrile generation, in agreement with our experimental data, which indicates that dehydration of the hydroxynitrile is the rate-determining step.
It should be noted that, according to our proposed mechanism, complexes Mn-5 and Mn-7, both of which were observed experimentally, are generated through side equilibria involving the addition of aldehyde or water to Mn-1, and are not directly involved in the catalytic cycle. Our calculations show that in the polar solvent acetonitrile, Mn-5 is less stable than complexes II and III (ΔG = 8.9, 7.0, and 5.6 kcal mol−1, respectively), whereas in the gas phase Mn-5 is the most stable of the three species (ΔG = −0.4, 3.7, and 0.4 kcal mol−1, respectively). Thus, the nitrile adduct III is thermodynamically preferred in the polar nitrile solvent, whereas Mn-5 is preferred in the low-polarity solvent THF, in line with our experimental observations. The formation of Mn-7 can be rationalized by its comparatively high stability (ΔG = −3.9 kcal mol−1 in CH3CN), which enables the effective trapping of water, as it is generated upon hydroxynitrile dehydration.
Our computational study also allows us to rationalize the lower catalytic activity observed for complexes Mn-2 and Mn-3, as compared with Mn-1, through examination of their electronic structures (see SI). Natural bond orbital (NBO) analysis shows that the N-donor arms of the PNN ligands in Mn-2 and Mn-3 are less electron-donating than the corresponding P-donor arm of the PNP ligand in Mn-1. This reduces the overall electron density across the structures of complexes Mn-2 and Mn-3, including that of the bridging C atom of the unsaturated pincer arm, which is crucial for metal–ligand cooperation. These electronic effects render Mn-2 and Mn-3 less efficient in activating the coordinated nitrile substrate and β-hydroxynitrile intermediate.
Conclusions
In this report, we have introduced a catalytic method for the synthesis of α,β-unsaturated nitriles via the coupling of unactivated α-saturated nitriles with aldehydes, promoted by a manganese-based catalyst. This reaction is carried out at room temperature, under base-free conditions and with no other additives, and, in most cases, utilizes the nitrile substrate as solvent. The catalytic protocol was shown to accommodate a broad range of substrates that includes various aliphatic and aromatic nitriles and aldehydes, and exhibit excellent functional group tolerance. Moreover, this method enables the selective synthesis of α-deuterated α,β-unsaturated nitriles, thereby demonstrating further synthetic utility. Based on experimental investigations and DFT calculations, we propose a template-type mechanism for this catalytic system, wherein the manganese pincer complex activates an incoming nitrile via metal–ligand cooperation, thereby forming an enamine-containing intermediate that behaves as a carbon nucleophile. The latter couples with an aldehyde to give a β-hydroxynitrile intermediate that is then dehydrated into the vinyl nitrile product. We believe that this new catalytic method represents a green, efficient alternative to existing techniques for the synthesis of vinyl nitriles.Author contributions
D. M. and S. T. conceived and directed the project and designed the experiments. S. T. performed all of the experiments and analyzed their results. J. L., Y. L. and I. E. carried out the computational studies. M. M. provided insightful discussions. D. M., S. T. and M. M. prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The authors declare that all supporting data are available in the supplementary information (SI) and from the corresponding author upon reasonable request. Supplementary information: detailed experimental procedures, characterization data (1H and 13C NMR spectra), and a detailed description of the computational methods. See DOI: https://doi.org/10.1039/d5sc07492d.Acknowledgements
S.T. is thankful to the Weizmann School of Science for a postdoctoral research fellowship.Notes and references
- (a) J. Zhang, W. Hu, Y. Li, A. Savoy, J. Sun, T. Y. Chi and Y. Wang, Chem Catal., 2024, 4, 100825 CAS; (b) F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902–7917 CrossRef CAS PubMed; (c) F. F. Fleming, Nat. Prod. Rep., 1999, 16, 597–606 Search PubMed.
- P. A. J. Janssen, P. J. Lewi, E. Arnold, F. Daeyaert, M. de Jonge, J. Heeres, L. Koymans, M. Vinkers, J. Guillemont, E. Pasquier, M. Kukla, D. Ludovici, K. Andries, M.-P. de Bé thune, R. Pawels, K. Das, A. D. Clark Jr, Y. V. Frenkel, S. H. Hughes, B. Medaer, F. De Knaep, H. Bohets, F. De Clerck, A. Lampo, P. Williams and P. Stoffels, J. Med. Chem., 2005, 48, 1901–1909 Search PubMed.
- S. Castellino, M. R. Groseclose, J. Sigafoos, D. Wagner, M. de Serres, J. W. Polli, E. Romach, J. Myer and B. Hamilton, Chem. Res. Toxicol., 2013, 26, 241–251 Search PubMed.
- L.-H. Zhang, L. Wu, H. K. Raymon, R. S. Chen, L. Corral, M. A. Shirley, R. K. Narla, J. Gamez, G. W. Muller, D. I. Stirling, J. B. Bartlett, P. H. Schafer and F. Payvandi, Cancer Res., 2006, 66, 951–959 Search PubMed.
- M. Suganuma, H. Fujiki, H. Furuya-Suguri, S. Yoshizawa, S. Yasumoto, Y. Kato, N. Fusetani and T. Sugimura, Cancer Res., 1990, 50, 3521–3525 Search PubMed.
- A. B. Smith III, Z. Liu, A.-M. L. Hogan, D. S. Dalisay and T. F. Molinski, Org. Lett., 2009, 11, 3766–3769 Search PubMed.
- (a) L. Lan and H. Zhang, Angew. Chem., Int. Ed., 2025, 64, e202509140 Search PubMed; (b) R. M. Rodrigues, D. A. Thadathil, K. Ponmudi, A. George and A. Varghese, ChemistrySelect, 2022, 7, e202200081 Search PubMed.
- (a) F. F. Fleming and Q. Wang, Chem. Rev., 2003, 103, 2035–2078 Search PubMed; (b) R. C. Larcok, Comprehensive Organic Transformations: A Guide to Functional Group Preparations, VCH, New York, 2nd edn, 1999 Search PubMed.
- (a) B. Zupančič and M. Kokalj, Synthesis, 1981, 913–915 CrossRef; (b) S. A. DiBiase, B. A. Lipisko, A. Haag, R. A. Wolak and G. W. Gokel, J. Org. Chem., 1979, 44, 4640–4649 Search PubMed.
- B. A. D’Sa, P. Kisanga and J. G. Verkade, J. Org. Chem., 1998, 63, 3961–3967 Search PubMed.
- T. Tomioka, Y. Takahashi, T. G. Vaughan and T. Yanase, Org. Lett., 2010, 12, 2171–2173 Search PubMed.
- D. Lanari, M. Alonzi, F. Ferlin, S. Santoro and L. Vaccaro, Org. Lett., 2016, 18, 2680–2683 CrossRef CAS PubMed.
- (a) S. Kojima, T. Fukuzaki, A. Yamakawa and Y. Murai, Org. Lett., 2004, 6, 3917–3920 CrossRef CAS PubMed; (b) D. J. Ager, Synthesis, 1984, 384–398 Search PubMed.
- B. E. Maryanoff and A. B. Reitz, Chem. Rev., 1989, 89, 863–927 Search PubMed.
- (a) A. F. Palermo, B. S. Y. Chiu, P. Patel and S. A. L. Rousseaux, J. Am. Chem. Soc., 2023, 145, 24981–24989 CAS; (b) Y. Nakao, A. Yada, S. Ebata and T. Hiyama, J. Am. Chem. Soc., 2007, 129, 2428–2429 Search PubMed.
- Y. Mu, T. T. Nguyen, M. J. Koh, R. R. Schrock and A. H. Hoveyda, Nat. Chem., 2019, 11, 478–487 Search PubMed.
- (a) T. K. Roy, R. Babu, G. Sivakumar, V. Gupta and E. Balaraman, Catal. Sci. Technol., 2024, 14, 2064–2089 RSC; (b) D. Song, S. Wang, W. Huang, R. Chen, F. Hu, L. Cheng, X. Zhao, F. Ling and W. Zhong, Org. Chem. Front., 2023, 10, 5908–5915 RSC; (c) R. R. Putta, S. Chun, S. B. Lee, J. Hong, S. H. Choi, D. C. Oh and S. Hong, J. Org. Chem., 2022, 87, 16378–16389 CrossRef CAS PubMed; (d) K. Paudel, S. Xu and K. Ding, Org. Lett., 2021, 23, 5028–5032 Search PubMed; (e) V. Yadav, V. G. Landge, M. Subaramanian and E. Balaraman, ACS Catal., 2020, 10, 947–954 Search PubMed; (f) S. Thiyagarajan and C. Gunanathan, ACS Catal., 2018, 8, 2473–2478 CrossRef CAS; (g) S. Chakraborty, U. K. Das, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2017, 139, 11710–11713 CrossRef CAS PubMed; (h) J. Li, Y. Liu, W. Tang, D. Xue, C. Li, J. Xiao and C. Wang, Chem.--Eur. J., 2017, 23, 14445–14449 CrossRef CAS PubMed.
- (a) G. Wang, R. Zhou, S.-H. Peng, X.-K. Chen and H.-B. Zou, ACS Omega, 2024, 9, 3317–3323 CAS; (b) Z. Gu, Y. Wang, Y. Yao, X. Xia, H. Wang and W. Li, Catal. Lett., 2015, 145, 2046–2054 CrossRef CAS.
- (a) Modern Aldol Reactions, R. Mahrwald, Wiley-VCH, Weinheim, 2004 Search PubMed; (b) Comprehensive Organic Synthesis, B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, vol. 2 Search PubMed; (c) S. Arseniyadis, K. S. Kyler and D. S. Watt, Org. React., 1984, 31, 1–364 Search PubMed.
- (a) R. Ló pez and C. Palomo, Angew. Chem., Int. Ed., 2015, 54, 13170–13184 Search PubMed; (b) B. Alcaide and P. Almendros, Eur. J. Org Chem., 2002, 1595–1601 CrossRef CAS; (c) B. List, Tetrahedron, 2002, 58, 5573–5590 Search PubMed.
- (a) S.-I. Murahashi, T. Naota, H. Taki, M. Mizuno, H. Takaya, S. Komiya, Y. Mizuho, N. Oyasato and M. Hiraoka, J. Am. Chem. Soc., 1995, 117, 12436–12451 Search PubMed; (b) T. Naota, H. Taki, M. Mizuno and S.-I. Murahashi, J. Am. Chem. Soc., 1989, 111, 5954–5955 CrossRef CAS.
- M. Vogt, A. Nerush, M. A. Iron, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2013, 135, 17004–17018 CrossRef CAS PubMed.
- Reviews of metal–ligand cooperation: (a) M. R. Elsby and R. T. Baker, Chem. Soc. Rev., 2020, 49, 8933–8987 Search PubMed; (b) J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed; (c) T. Zell and D. Milstein, Acc. Chem. Res., 2015, 48, 1979–1994 CrossRef CAS PubMed; (d) D. Milstein, Philos. Trans. R. Soc., A, 2015, 373, 20140189 Search PubMed; (e) C. Gunanathan and D. Milstein, Science, 2013, 341, 1229712 Search PubMed; (f) C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 Search PubMed.
- A. Nerush, M. Vogt, U. Gellrich, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2016, 138, 6985–6997 CrossRef CAS PubMed.
- S. Tang and D. Milstein, Chem. Sci., 2019, 10, 8990–8994 RSC.
- Q.-Q. Zhou, Y.-Q. Zou, S. Kar, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2021, 11, 10239–10245 Search PubMed.
- S. Thiyagarajan, Y. Diskin-Posner, M. Montag and D. Milstein, Chem. Sci., 2024, 15, 2571–2577 Search PubMed.
- (a) B. Guo, J. G. de Vries and E. Otten, Adv. Synth. Catal., 2022, 364, 179–186 Search PubMed; (b) B. Guo, J. G. de Vries and E. Otten, Chem. Sci., 2019, 10, 10647–10652 Search PubMed; (c) L. E. Eijsink, S. C. P. Perdriau, J. G. de Vries and E. Otten, Dalton Trans., 2016, 45, 16033–16039 Search PubMed; (d) S. Perdriau, D. S. Zijlstra, H. J. Heeres, J. G. de Vries and E. Otten, Angew. Chem., Int. Ed., 2015, 54, 4236–4240 Search PubMed.
- (a) A. Saito, S. Adachi, N. Kumagai and M. Shibasaki, Angew. Chem., Int. Ed., 2021, 60, 8739–8743 Search PubMed; (b) S. Chakraborty, Y. J. Patel, J. A. Krause and H. Guan, Angew. Chem., Int. Ed., 2013, 52, 7523–7526 Search PubMed; (c) N. Kumagai, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2004, 126, 13632–13633 Search PubMed.
- M. El Bouz, M.-C. Roux-Schmitt and L. Wartski, J. Chem. Soc., Chem. Commun., 1979, 779–780 Search PubMed.
- (a) S. Bindu, S. Mazumder and U. Bandyopadhyay, Biochem. Pharm., 2020, 180, 114147 Search PubMed; (b) E. Brenna, M. Crotti, F. G. Gatti, A. Manfredi, D. Monti, F. Parmeggiani, S. Santangelo and D. Zampieri, ChemCatChem, 2014, 6, 2425–2431 Search PubMed.
- A. Mukherjee, A. Nerush, G. Leitus, L. J. W. Shimon, Y. Ben-David, N. A. Espinosa Jalapa and D. Milstein, J. Am. Chem. Soc., 2016, 138, 4298–4301 Search PubMed.
Footnotes |
| † Current affiliation: Department of Chemistry, Zhejiang University, Hangzhou, Zhejiang, China. |
| ‡ Current affiliation: Advanced Catalysis Research Group, RIKEN, Center for Sustainable Resource Science, Wako, Saitama 351-0198, Japan. |
| This journal is © The Royal Society of Chemistry 2026 |