
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid preparation of imide-based COF films through electropolymerization integrated with low-temperature annealing for high-performance electrochromic energy storage
Jinming
Zeng
 *a,
Huiling
Hou
a,
Lei
Huang
a,
Zheng
Xie
a,
Qingqing
Qiu
a,
Huan
Li
a,
Dongfa
Liu
ac,
Putrakumar
Balla
d,
Tongxiang
Liang
a and
Ping
Liu
*b
*a,
Huiling
Hou
a,
Lei
Huang
a,
Zheng
Xie
a,
Qingqing
Qiu
a,
Huan
Li
a,
Dongfa
Liu
ac,
Putrakumar
Balla
d,
Tongxiang
Liang
a and
Ping
Liu
*b
aJiangxi Provincial Key Laboratory of Magnetic Metallic Materials and Devices, College of Rare Earths, Jiangxi University of Science and Technology, Ganzhou 341000, PR China. E-mail: zjm1551010758@163.com
bState Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, PR China. E-mail: mcpliu@scut.edu.cn
cNankang No.6 High School, Ganzhou, Jiangxi 341400, PR China
dDepartment of Chemical Engineering and Applied Chemistry, Chungnam National University, Daejeon, 34134, South Korea
First published on 31st December 2025
Abstract
Electrochromic energy-storage materials (EESMs) have received tremendous attention in the field of smart windows and visual energy-storage batteries. Covalent organic frameworks (COFs) are considered as superior candidates for high-performance EESMs owing to the designability of molecular structures, high electron-ion transport rates, and thermodynamically stable networks. In the present study, two triarylamine-based polyimide (TPI) COFs, DFPI and NTPI COF films were first built via a simple and efficient interfacial electropolymerization combined with low-temperature annealing technology. The electropolymerization mechanism of DFPI and NTPI COF films was elucidated in detail. TPPDA first undergoes an oxidative nucleophilic substitution reaction at the anode to produce an electrogenerated acid (EGA), followed by a polycondensation reaction between triphenylamine derivatives and anhydride catalyzed by EGA to form DFPI or NTPI COF films. Additionally, both the DFPI and NTPI COF films exhibited high-performance electrochromic energy-storage, including bipolar four color states (light red, colorless, yellow-green, and blue), high optical contrast (>70%), more than 1000 cycles, and high specific capacitance (80.9 and 72.2 mAh g−1). This work enables the simple and rapid preparation of TPI COF films, which paves the way for the design and fabrication of TPI COF films for high-performance optoelectronics, electrochemical sensing, separation membranes, and EESMs.
Introduction
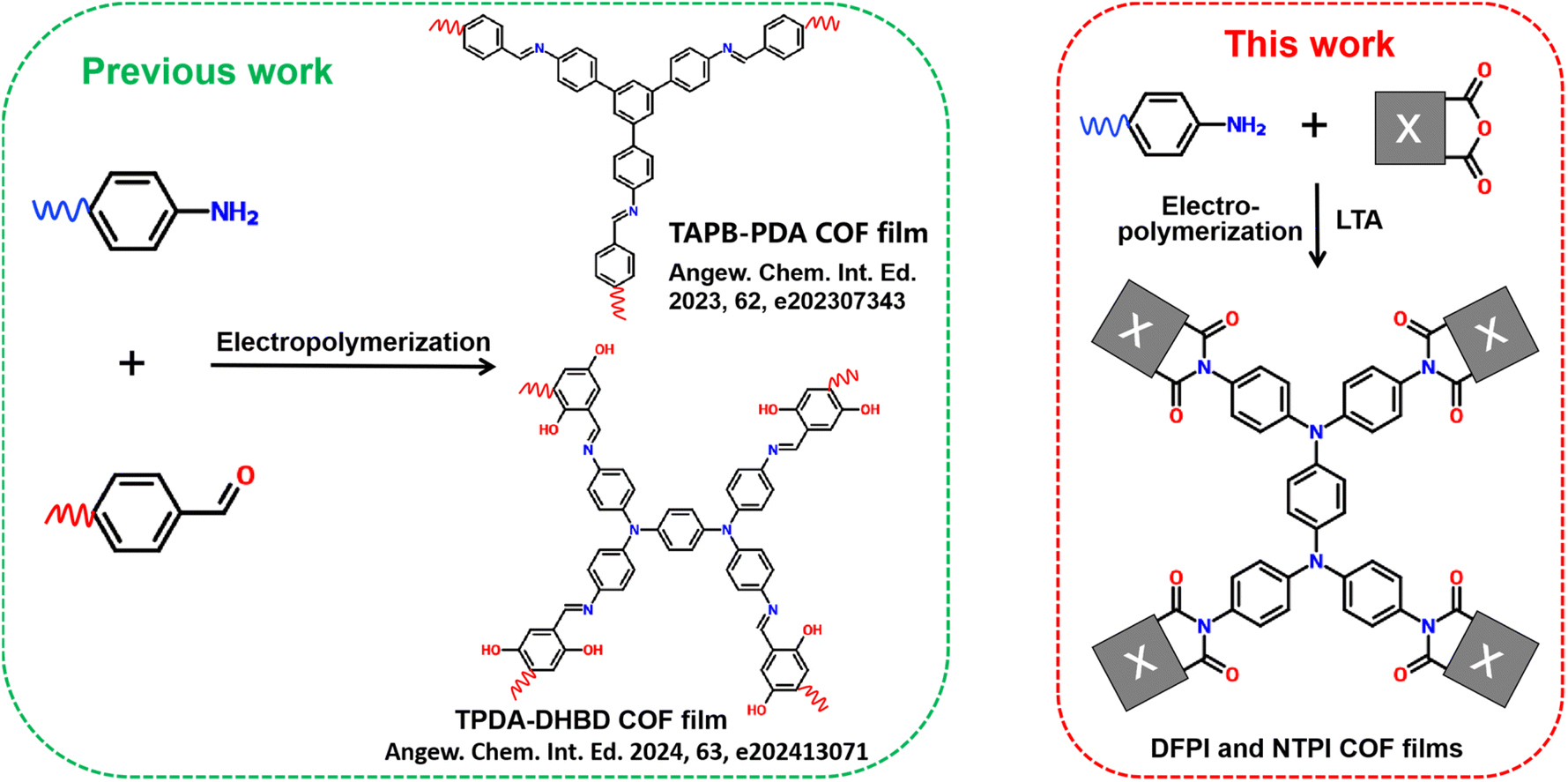
Electrochromic energy-storage materials (EESMs),1 which exhibit reversible optical changes signatures (e.g., in transmittance and reflectivity) during electrochemical charge–discharge processes, have attracted considerable attention in the field of smart windows,2,3 electrochemical visual energy-storage batteries,4–6 displays,7,8 and camouflage technologies.9,10 In recent years, inorganic compounds,11–14 organic molecules,15–19 and organometallic complexes20–22 have been explored for constructing EESMs. To achieve high performance, EESMs must possess suitable chemical structures, fast reaction kinetics, and high chemical stability. Covalent organic frameworks (COFs), a class of crystalline porous materials composed of covalently linked organic building blocks,23 have emerged as a promising platform for separation membranes,24 photo-electrocatalysis,25,26 electrochemical energy storage,27,28 and EESMs.29,30 Their appeal lies in the tunability of molecular structures, inherently high electron/ion transport and excellent chemical stability of the porous covalent crystalline frameworks. Nevertheless, the exploration of the EESMs based on COFs remains in its infancy. In 2019, Wang et al.31 first fabricated a COF3PATT electrochromic film via a solvothermal method, which exhibited superior switching speed and stability compared to amorphous polymer films of the same composition. Thereafter, only a limited number of studies have reported conventional imine-based COF electrochromic films.32–36 Additionally, “donor–acceptor” type and semi-isomerism imine-based COFs have been designed and synthesized to enhance electrochromic performance.37,38 Despite these efforts, existing COF-based electrochromic films still face two major challenges. Firstly, the performance of COF electrochromic materials requires further enhancement through molecular structure design. Although imine-based COFs have been employed as electrochromic materials, it remains difficult to simultaneously achieve a highly transparent state, high optical contrast, and bipolar electrochromic behavior. Secondly, most imine-based COF films are still synthesized via conventional solvothermal methods, which involve harsh reaction conditions, tedious procedures, long processing times, and limited scalability factors that severely hinder practical application. Therefore, it is both urgent and highly important to develop high-performance COF-based EESMs through rational molecular design alongside simpler and more efficient synthetic routes.The synthesis of covalent organic frameworks (COFs) can be facilitated by various external energy sources, including microwave irradiation,39,40 ultrasound,41 mechanical force,42 light,43 plasma,44 electric fields, and electron beams.45 Among these methods, interfacial electrochemical polymerization has garnered significant interest for the construction of COF films on electrode surfaces due to its simplicity, rapid processing, controllability, and the integration of polymerization and film formation into a single step. In 2019, De Feyter et al. reported the electric-field-mediated synthesis of boroxine-based COF films on a highly oriented pyrolytic graphite (HOPG) surface. This method enables localized initiation of on-surface polymerization at room temperature.45 More recently, Nejati, Inagi, Jiang, Qiu, and their colleagues have successfully fabricated a series of imine-based COF films via electrochemical interfacial polymerization (Scheme 1).36,46–48 Although several boroxine- and imine-based COF films have been prepared using electrochemical interfacial polymerization, their practical application in high-performance electrochromic energy storage materials (EESMs) has been limited by insufficient chemical stability and difficulties in achieving a colorless transparent state. By contrast, triphenylamine-based polyimides have demonstrated robust electrochemical stability, colorless transparency, high optical contrast, and bipolar electrochromic energy storage behavior. Our research group has also reported triphenylamine polyimide COF (TPI COF) films that exhibit high-performance bipolar electrochromic energy storage.29,30 Nevertheless, to the best of our knowledge, the preparation of imide-based COF films via a simple and rapid electrochemical interfacial polymerization route remains unreported and represents a considerable challenge. Therefore, it is of great scientific and practical importance to develop TPI COF films through electrochemical interfacial polymerization to achieve advanced electrochromic energy storage performance.
 | ||
| Scheme 1 Diagrams of the structure and synthesis of (a) imine-based COF and (b) imide-based COF films. | ||
In this work, two triphenylamine polyimide covalent organic framework (TPI COF) films, denoted as DFPI and NTPI were first synthesized on electrode surfaces via electrochemical interfacial polymerization using triphenylamine derivatives and dicarboxylic anhydride monomers, followed by a low-temperature annealing (LTA) step (Scheme 1). This approach offers a straightforward and efficient route, significantly reducing the preparation time from several days to just a few hours while successfully constructing TPI COF films. Both DFPI and NTPI COF films exhibited a colorless neutral state and demonstrated bipolar electrochemical redox behavior. Furthermore, they displayed high-performance electrochromic energy storage properties, including bipolar four color state (light red, transparent, yellow–green, and blue) switching, high optical contrast, and substantial specific capacitance. Notably, the electropolymerization and electrochromic mechanisms of the TPI COF films were systematically elucidated. The process begins with the oxidative nucleophilic substitution of TPPDA at the anode, generating an electrogenerated acid (EGA). This is followed by an EGA-catalyzed polycondensation between the triphenylamine derivatives and dicarboxylic anhydride monomers, leading to the formation of the TPI COF framework. The anodic electrochromism is attributed to the formation of nitrogen-centered cationic radicals through electrochemical p-doping of the triphenylamine units, whereas the cathodic electrochromism arises from oxygen-centered anion radicals generated via n-doping of the carbonyl groups. This work establishes a simple and rapid method for fabricating TPI COF films, paving the way for their application in high-performance optoelectronics, electrochemical sensing, separation membranes, and advanced electrochromic energy storage systems.
Results and discussion
Composition and structures of DFPI and NTPI COF films
The DFPI and NTPI COF films were directly fabricated on ITO glass substrates through interfacial electropolymerization followed by low-temperature annealing (LTA). As depicted in Fig. S1 and S2, no discernible 1H NMR signals corresponding to amino hydrogen were observed in the chemical shift range of 3.0–5.0 ppm.49 Furthermore, the hydrogen environments in both DFPI and NTPI were clearly identified, confirming the successful formation of the TPI COF via interfacial electropolymerization. To enhance the crystallinity of the DFPI and NTPI COF films, a low-temperature annealing treatment was applied. As illustrated in Fig. 1c and d, the absorption peaks observed at approximately 1710 cm−1 for the DFPI COF and 1730 cm−1 for the NTPI COF after annealing were assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of the five- and six-membered imide-based rings, respectively.29 Additionally, a fresh peak attributed to the C–N–C vibration emerged around 1310 cm−1, while the characteristic N–H stretching vibration of TPPDA at 3380 cm−1 was absent, indicating complete imidization.50 These results collectively demonstrate the successful synthesis of both DFPI and NTPI COF films through the integrated approach of interfacial electropolymerization and low-temperature annealing.
O stretching vibrations of the five- and six-membered imide-based rings, respectively.29 Additionally, a fresh peak attributed to the C–N–C vibration emerged around 1310 cm−1, while the characteristic N–H stretching vibration of TPPDA at 3380 cm−1 was absent, indicating complete imidization.50 These results collectively demonstrate the successful synthesis of both DFPI and NTPI COF films through the integrated approach of interfacial electropolymerization and low-temperature annealing.
 | ||
| Fig. 1 (a) Synthetic scheme and (b) route diagram of DFPI and NTPI COF films by interfacial electropolymerization integrated with low-temperature annealing; (c) the FT-IR spectra of TPPDA, DFD and DFPI COF; (d) the FT-IR spectra of TPPDA, NTCDA and NTPI COF; XRD patterns of DFPI and NTPI COF (e) powders and (f) films; (g) HRTEM image of the NTPI COF film (inset shows the electronic diffraction pattern). | ||
To evaluate the crystallinity of DFPI and NTPI COFs, their X-ray diffraction (XRD) plots were acquired. As shown in Fig. 1e, both COF powders exhibit diffraction signals corresponding to crystalline ordering within the low-angle range of 1–10°. DFPI displays a broadened peak centered around 8.89°, reflecting its limited long-range structural periodicity, while NTPI presents two discernible peaks at 6.93° and 8.92°, demonstrating relatively higher crystallinity compared to DFPI. This is attributed to the high planarity of the NTCDA skeleton.51 It should be emphasized that the characteristic peaks of the COFs do not appear in the PXRD patterns of the original monomers (Fig. S3), which confirms that the diffraction signals originate from the COF phase rather than residual monomers. Fig. 1f shows that a broad peak occurred in the range of 8–10° for the DFPI COF film, suggesting that low crystallinity was acquired. By contrast, a sharp peak at 8.73° appeared for the NTPI COF film, indicating that relatively higher crystallinity was obtained, which was consistent with that of the DFPI and NTPI COF powders.
High-resolution transmission electron microscopy (HRTEM) was further used to obtain the crystal structures of the DFPI and NTPI COF films. As illustrated in Fig. 1g and S4, both films exhibited clear lattice fringes and electron diffraction patterns, confirming their crystallinity. The NTPI COF film demonstrated higher crystallinity than the DFPI COF film, which aligned with the results of their XRD patterns. The measured lattice spacings were 0.331 nm for the DFPI COF and 0.312 nm for the NTPI COF. The larger interplanar distance in the DFPI COF may result from the three-dimensional warped structure of the DFD unit.52 Collectively, these results demonstrated the successful growth of crystalline triphenylamine-based polyimide (DFPI and NTPI) COF films on ITO glass via interfacial electropolymerization combined with low-temperature annealing. In addition, the BET surface areas and pore size distributions of DFPI and NTPI COF films were also determined from N2 adsorption–desorption isotherms measured at 77 K (Fig. S5). Their BET surface areas were 18.79 and 22.67 m2 g−1, respectively. Pore size analysis revealed that the DFPI COF primarily contained mesopores in the range of 2–20 nm, while the NTPI COF exhibited a hierarchical porous structure, containing micropores of 1–2 nm and mesopores of 2–15 nm. This nanoporous architecture was anticipated to facilitate ion transport and provide ample space to accommodate volumetric variations during ion insertion and extraction, thereby enhancing the optical contrast, response speed, and charge–discharge kinetics of EESMs.30,32–36
The surface morphologies of DFPI and NTPI COF films were meticulously characterized using scanning electron microscopy (SEM). As illustrated in Fig. 2a and d, both the DFPI and NTPI COF films displayed uniform morphology at low magnification. Specifically, the DFPI COF films exhibited a sandwall-like structure composed of small nanoparticles (Fig. 2a). The NTPI COF films showed a rougher micromorphology, featuring a nano-textured structure constructed from both spherical nanoparticles and rod-like structures with a diameter of approximately 200 nm (Fig. 2d). Among these components, the rod-like structures can be regarded as secondary morphologies formed by the ordered assembly and stacking of spherical nanoparticles as the basic building units. Energy-dispersive X-ray spectroscopy (EDS) confirmed the homogeneous distribution of N, O, and C elements across both the DFPI and NTPI COF films (Fig. 2b and e), with no evidence of localized enrichment or depletion. Compared to the films prepared without low-temperature annealing (Fig. S6 and S7), the annealed DFPI and NTPI COF films exhibited larger nanoparticle sizes, attributing to molecular reorganization during annealing, which enhanced crystallinity and structural stability. Atomic force microscopy (AFM) was further used to obtain surface topography (Fig. 2f and S8). The measured surface roughness values were approximately ±60 nm for DFPI and ±200 nm for NTPI COF films, which were consistent with the SEM observations. Additionally, the growth mechanism of the DFPI COF film displayed both surface and bottom layers consisting of small nanoparticles. In contrast, the NTPI COF film exhibited a mixed growth mechanism: the bottom layer showed island-layered growth with small nanoparticles, while the surface layer underwent island growth with larger nanoparticles (Fig. 2h). Cross-sectional SEM images revealed film thicknesses of approximately 230 nm for DFPI and 360 nm for NTPI COF films (Fig. 2c and S9).
 | ||
| Fig. 2 SEM images and EDS of (a) and (b) DFPI COF and (d) and (e) NTPI COF films; (c) cross-sectional SEM and (f) AFM images of NTPI COF films; ex situ SEM images of (g) DFPI COF and (h) NTPI COF films at electropolymerization for 10 s, 50 s, 100 s, 150 s, and 200. | ||
Electropolymerization mechanism of DFPI and NTPI COF films
High-quality DFPI and NTPI COF films were prepared through electropolymerization followed by low-temperature annealing, and their underlying electropolymerization mechanism was systematically investigated. Cyclic voltammetry (CV) curves of all raw monomer (DFD, NTCDA, TPPDA, DFD/TPPDA, and NTCDA/TPPDA) for the preparation of DFPI and NTPI COF films were investigated. As shown in Fig. 3a, the dicarboxylic anhydride monomers (DFD and NTCDA) exhibited no redox activity within the potential window of −0.2 V to +1.2 V. By contrast, TPPDA displayed a distinct onset oxidation potential at approximately +0.1 V. Linear sweep voltammetry (LSV) results further supported these observations, as demonstrated in Fig. S10. These results indicated that the electropolymerization of DFD/TPPDA and NTCDA/TPPDA was initiated by the electrochemical oxidation of TPPDA, which was consistent with the previous report.52 | ||
| Fig. 3 (a) First-cycle CV curves of DFD, NTCDA, TPPDA, DFD/TPPDA, and NTCDA/TPPDA electrolyte solution; (b) the in situ UV-vis spectra of NTCDA/TPPDA electrolyte solution; the in situ Raman spectra of (d) TPPDA electrolyte solution and (e) and (f) NTCDA/TPPDA electrolyte solution at different voltages; (c) the in situ UV-vis spectra of NTCDA/TPPDA electrolyte solution; the in situ Raman spectra of (g) DFD/TPPDA electrolyte solution (h) and (i) NTCDA/TPPDA electrolyte solution during electropolymerization at +1.0 V voltage for 200 s; (j) the proposed electropolmerization mechanism of DFPI and NTPI COF films. | ||
To determine the lowest critical electrodeposition voltage for DFPI and NTPI COF films, in situ UV-vis and Raman spectroscopy studies were performed at various electropolymerization voltages in DFD/TPPDA and NTCDA/TPPDA solutions, respectively. Since copper substrates did not exhibit significant Raman signals (Fig. 3e), detailed mechanistic studies were conducted using copper as the working electrode for in situ electropolymerization of DFPI and NTPI COF films. UV-vis spectra of the NTCDA/TPPDA and DFD/TPPDA electrolyte solutions showed no notable changes within the voltage range of 0.0 V to +0.6 V (Fig. 3b and S11b). However, upon increasing the voltage from +0.8 V to +1.6 V, a broad absorption band emerged around 1100 nm, attributed to intravalent charge transfer (ICT) within TPPDA+˙ radicals.29,53 An additional band appeared at approximately 723 nm when the voltage was raised from +1.0 V to +1.6 V, corresponding to the formation of TPPDA2+˙ species.29 Concurrently, DFPI and NTPI COF films were progressively deposited on the ITO substrate at voltages above +0.8 V, as visually confirmed by the inset photographs. These results indicated that a minimum voltage of +0.8 V is required for the successful electropolymerization of DFPI and NTPI COF films. Although the in situ UV-vis spectra of TPPDA alone (Fig. S11a) were similar to those of the monomer mixtures, electrodeposition of poly-TPPDA on ITO required a higher voltage (>+1.2 V), as shown in the inset of Fig. S11a. This difference may be attributed to the moderate solubility of TPPDA oligomers in dichloromethane (DCM). To avoid concurrent deposition of poly-TPPDA during the DFPI or NTPI COF film growth, the electropolymerization voltage was maintained at +1.0 V. In situ Raman spectroscopy was further employed to probe structural changes in DFD, NTCDA, TPPDA, and their mixtures under applied voltages (Fig. 3d–f and S12–S14). No significant Raman shifts or intensity changes were observed for DFD or NTCDA between 0.0 V and +1.2 V, confirming their electrochemical stability and the absence of deposition. In contrast, for TPPDA electrolyte solution, characteristic peaks emerged and intensified at applied voltages from +1.2 V to +1.8 V, including ring deformation modes at 810 cm−1 (benzene-type, B) and 905 cm−1 (quinone-type, Q), C–H bending at 1156 and 1176 cm−1, C–N stretching at 1275 and 1346 cm−1 (cationic and neutral forms, respectively), and CC stretching at 1586 and 1610 cm−1 (B and Q),54,55 indicating electrodeposition of poly-TPPDA. For DFD/TPPDA and NTCDA/TPPDA mixtures; similar Raman features appeared and intensified at potentials as low as +0.8 V, further confirming the electrodeposition of DFPI and NTPI COF films at this threshold. These results demonstrate that TPPDA+˙ radicals were essential intermediates in the electropolymerization process. Based on previous studies on aromatic diamine electropolymerization,21,56 it could be inferred that TPPDA+˙ radicals were unstable and underwent electrophilic substitution on the benzene ring, accompanied by generation of electrogenerated acid (EGA). Meanwhile, C–N bonds in TPPDA oligomers convert to CN linkages, accompanied by EGA formation on the ITO surface. At higher potentials, TPPDA+˙ could further polymerize into poly-TPPDA, also releasing EGA. The presence of EGA catalyzed the polycondensation between TPPDA and dicarboxylic anhydride monomers (DFD or NTCDA), reducing the reaction energy barrier and enabling interfacial electropolymerization at room temperature. A schematic illustration of the proposed electropolymerization mechanism is depicted in Fig. 3j.
To further elucidate the electropolymerization process of DFPI and NTPI COF films, in situ UV-vis and Raman spectra were acquired during potentiostatic deposition at +1.0 V for 200 s, as shown in Fig. 3c and S15. The UV-vis results revealed a gradual increase in absorption peaks around 1100 nm and 800 nm, corresponding to TPPDA+˙ and TPPDA2+˙ species, respectively, from 0 to 200 s. This spectral evolution was assigned to the gradual p-doping of TPPDA, as well as the growth of the COF frameworks (DFPI or NTPI). The inset in Fig. 3b clearly shows that the NTPI COF film began to deposit on the ITO substrate after approximately 50 s. Furthermore, the in situ Raman spectra (Fig. 3g–i) exhibited the emergence and gradual intensification of characteristic vibrational modes over time. For the DFPI COF, peaks at approximately 1169 cm−1 (C–H bending, quinone-type), 1270 cm−1 (C–N stretching, cationic), 1582 cm−1, and 1611 cm−1 (CC stretching, benzene- and quinone-type) became detectable after 20 s. Similarly, for NTPI COF, signals at around 1172 cm−1 (C–H, Q), 1280 cm−1 (C–N, cationic), 1578 cm−1, and 1611 cm−1 (CC, B and Q) appeared after 40 s. These results further confirmed that TPPDA+˙ radicals were essential intermediates in the electropolymerization process of both COF films. The electrogenerated acid (EGA), produced via nucleophilic substitution between TPPDA+˙ and neutral TPPDA, initiated and catalyzed the electropolymerization, enabling the formation of highly ordered DFPI and NTPI COF structures under mild electrochemical conditions.
Electrochemical properties
CV measurements of DFPI (Fig. S16a) and NTPI (Fig. S17a) COF films were conducted in a standard three-electrode system within both positive and negative potential windows. They exhibited two distinct pairs of redox peaks in the anodic region. The first and second oxidation peaks correspond to the formation of the monocationic radical (TPPA+˙) and dicationic radical (TPPA2+˙) of N,N,N′,N′-tetraphenyl-1,4-phenylenediamine (TPPA), respectively.29,53 In the cathodic region, two pairs of redox peaks were observed, attributed to the stepwise reduction leading to the monoanionic radical (DFD−˙) and dianionic radical (DFD2+˙) of the dicarboxylic anhydride unit.52 These results confirmed the bipolar electrochemical activity of both DFPI and NTPI COF films. To further identify the electrochemically active sites in the NTPI COF, the electrostatic potential (ESP) of a representative molecular model was computed using density functional theory (DFT). Fig. 4a shows that the highest positive ESP values were localized at the two nitrogen atoms of the TPPA moiety (excluding the amine group), indicating their tendency to lose electrons during electrochemical p-doping. Conversely, the most negative ESP values are located at the oxygen atoms of the NTCDA unit, suggesting their affinity for gaining electrons during n-doping. Based on the CV results and ESP analysis, the electrochemical redox mechanisms for DFPI and NTPI COFs are schematically illustrated in Fig.S16b and S17b, respectively. Furthermore, CV measurements were performed at various scan rates to evaluate the ion transport kinetics within the DFPI and NTPI COF films. The plots of the peak current density (jp) vs. square root of the scan rate (v1/2) are shown in Fig. S18 and 4b, c. The diffusion coefficient of ClO4− (DClO4−) was calculated using the Randles–Servcik equation:3,30| D1/2 = jp/(Cn3/2AC0v1/2) |
 | ||
| Fig. 4 (a) ESP of the molecular model of the NTPI COF; (b) positive CV curves of the NTPI COF film at different scan rates; (c) spectroelectrochemistry and electrochromic digital photographs of the NTPI COF film; (d) plots of jpvs. v1/2 of the NTPI COF film; (e) and (f) in situ Raman spectra of NTPI COF film electrochromism; (g) C 1s, (h) N 1s, and (i) Cl 2p XPS spectra of the NTPI COF film in the bleached and anodic colored states; (j) response time of DFPI and NTPI COF films; (k and l) electrochromic cycling life of the NTPI COF film. | ||
The charge transfer resistance (Rct) of DFPI and NTPI COF film electrodes was further evaluated through electrochemical impedance spectroscopy. As shown in Fig. S19, the Nyquist plots contain a semicircle in the high-frequency region and a linear segment in the low-frequency region. The equivalent circuit model used for fitting is presented, where Rct, Cdl, and Wo denote the charge transfer resistance, double-layer capacitance between the substrate and the COF film, and the Warburg diffusion element, respectively. The Rct values were determined to be 108.7 Ω for DFPI and 82.7 Ω for NTPI COF films (Table S1), indicating reasonably low charge transfer resistance in both the DFPI and NTPI COF films. The high diffusion coefficient of ClO4− ions (DClO4−) and low Rct values suggested efficient ion transport and charge transfer kinetics, which were conducive to rapid electrochromic switching performance.29,57
Electrochromic performance
Spectroelectrochemical analyses were used to elucidate the electrochromic features of DFPI and NTPI COF films. They displayed poor absorption across the visible to near-infrared regions in the neutral state, resulting in a highly transparent appearance, as shown in Fig. S20 and 4c. As the applied voltage increased from 0.0 V to approximately +0.8 V, the absorption band at 350 nm gradually decreased, while new bands emerged at 1020 nm for the DFPI COF and 1041 nm for the NTPI COF, accompanied by a color transition from transparent to yellow–green. These new absorption features were attributed to ICT within TPPA+˙ radical species. Upon further increasing the voltage from +0.8 V to +1.6 V, the films transitioned from yellow–green to blue, with the intensities of absorption peaks progressively increasing at 753 nm for DFPI and 736 nm for NTPI, corresponding to electronic transitions of TPPA2+˙ species.29,53 Meanwhile, the absorption peaks at 1020 and 1041 nm initially increased and then decreased within this voltage window. Under negative bias (0.0 V to −2.0 V), both the DFPI and NTPI COF films exhibited increased absorption between 400 nm and 600 nm, accompanied by a color transition from transparent to light red. These results demonstrate that DFPI and NTPI COF films displayed four color states (light red, transparent, yellow–green, and blue) and bipolar electrochromic behavior.To elucidate the electrochromic mechanisms of DFPI and NTPI COF films, in situ Raman spectroscopy was performed during electrochromic cycling. As shown in Fig. 4e and f, the peaks at 1269 cm−1 (C–N, cationic) and 1610 cm−1 (CC, quinone-type) intensified gradually as the voltage increased from +0.7 V to +1.6 V, while the peak at 1166 cm−1 (C–H, quinone-type) increased notably between +1.1 V and +1.6 V. These results indicated that the yellow–green state (+1.0 V) and blue state (+1.6 V) of the NTPI COF film were associated with the formation of TPPA+˙ and TPPA2+˙ species, respectively.54,55 Furthermore, the in situ Raman spectra exhibited excellent reversibility when switching between +1.6 V and −1.0 V, suggesting high structural stability during the electrochromic process. Similar Raman behavior was observed for the DFPI COF film (Fig. S21).
To further corroborate the electrochromic mechanism, ex situ XPS analysis was conducted on both bleaching (0.0 V) and coloring states. In the bleached state, the C 1s spectra of DFPI and NTPI COF films were deconvoluted into three components, 284.83, 285.69, and 288.60 eV for the DFPI COF, and 284.81, 285.79, and 288.39 eV for the NTPI COF, corresponding to CC, C–C, and CO bonds, respectively.58 Under anodic coloring (+1.6 V), the C–C peak exhibited a slight blue shift due to the formation of quinoid-type structures (TPPA+˙ and TPPA2+˙), as illustrated in Fig. 4g and S22a. The N 1s spectra in the bleached state displayed peaks at 399.96 and 402.31 eV for the DFPI COF and 399.79 and 402.21 eV for the NTPI COF, assigned to C–N–C (neutral) and C–N+˙–C (cationic) species, respectively (Fig. 4h and S22b).29,30 The persistence of the C–N+˙–C peak in the bleached state was attributed to residual ClO4− anions complexed with the cationic nitrogen sites, as confirmed by the presence of Cl 2p signals (Fig. 4i and S22c). Upon anodic polarization, the intensity of the C–N–C peak decreased while that of C–N+˙–C increased, consistent with the oxidation of triphenylamine units. Concurrently, enhanced intensities of Cl 2p and O 1s peaks (Fig. S22d and S23) indicated the insertion of ClO4− anions during coloring, aligning with the in situ Raman results. For the cathodic electrochromic process, XPS spectra were acquired at −2.0 V. The C 1s peak corresponding to CO (288.34 eV for DFPI and 288.39 eV for NTPI) decreased in intensity, while a new peak emerged at 286.82 eV (C–O−˙ for the DFPI COF) and 286.83 eV (C–O−˙ for the NTPI COF), suggesting reduction of carbonyl groups.58,59
Deconvolution of the O 1s region revealed components at 532.91 eV and 531.64 eV for the DFPI COF and 533.03 eV and 531.51 eV for the NTPI COF, assigned to CO and C–O−˙ species, respectively,59 further confirming the formation of radical anions during cathodic polarization. These results were consistent with previously reported bipolar electrochemical energy storage mechanisms in triphenylamine-based polyimide films.27,29
The electrochromic performance of DFPI and NTPI COF films was further evaluated by measuring the switching times (coloration, tc; bleaching, tb) and optical contrast (ΔT). Fig. 4j presents the time-dependent transmittance curves recorded during the switching process. The results suggested that the ΔT values reached approximately 70.5% at 753 nm for DFPI and 70.0% at 736 nm for NTPI COF films. Notably, both films maintained high ΔT across visible and near-infrared regions. Moreover, the films exhibited reasonably fast switching speeds, with tc/tb values of 4.1/5.9 s for the DFPI COF and 3.8/5.6 s for the NTPI COF, demonstrating efficient electrochromic switching kinetics.
Cycling stability is a critical parameter for evaluating the practical applicability of EESMs. To assess the durability of DFPI and NTPI COF films, long-term coloring/bleaching tests were conducted using a UV-vis spectrometer coupled with an electrochromic cycling tester. The transmittance of the films was monitored under repeated square-wave voltage cycling between −1.6 and +1.6 V. Fig. S26a and 4k show that the transmittance of DFPI and NTPI COF films remained highly unchanged even after 100 electrochromic cycles. Furthermore, chronoamperometry was employed to further evaluate electrochromic cycling stability. Fig. S26b and 4l show negligible attenuation in peak current after 1000 cycles, demonstrating exceptional electrochromic cycling durability. The structural stability during cycling was also further investigated using in situ Raman. As illustrated in Fig. S27, the characteristic Raman peaks of both the DFPI and NTPI COF films remained unchanged after 500 cycles, with no emergence of new peaks or peak shifts, indicating no detectable chemical degradation over extended operation. Additionally, we also performed FT-IR spectroscopy analysis on the DFPI and NTPI COF films after 1000 cycles (Fig. S28). Both films retained clear C–N–C stretching vibration peaks around 1310 cm−1 after cycling, with no significant peak shift observed. This indicates that the imine bonds remained stable during the cycling process and no cleavage or degradation occurred. Meanwhile, the CO stretching vibration peaks of the imide ring, which were at 1710 cm−1 for the DFPI COF and 1730 cm−1 for the NTPI COF, showed no obvious changes. This phenomenon is attributed to the reversible interactions between electrolyte ions and carbonyl groups during the cycling process. The FT-IR results further confirmed that both the DFPI and NTPI COFs remained stable after 1000 cycles. The remarkable cycling stability of DFPI and NTPI COF films could be attributed to their robust chemical structures, which remained intact throughout repeated electrochemical redox processes. The coloration efficiency (η) of DFPI and NTPI COF films was also acquired according to the previous reported method:1
| η = (log(Tb/Tc))/Q |
Energy-storage performance
DFPI and NTPI COF films represent favorable candidates for high-performance EESMs, due to their outstanding electrochemical redox activity and high structural stability. A detailed investigation into the electrochemical energy storage performance and mechanism of DFPI and NTPI COF films was conducted. The kinetic characteristics of charge storage were evaluated by analyzing the relationship between current density (i) and v using the equation:3,29| i = avb |
As shown in Fig. 5a, the calculated b values of peaks 2/2′ were 0.885/0.867 for the DFPI, and 0.699/0.796 for the NTPI COF films. These results indicated a mixed charge storage mechanism involving both diffusion-controlled and capacitive processes during the electrochromic energy-storage operation of the films.30 The contributions of diffusion- and capacitive-controlled processes were quantitatively analyzed using the equation:29,30
| i(v) = k1v + k2v1/2 |
| Csp = IΔt/m |
 | ||
Fig. 5 (a) Plots of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ipvs. logv for the electrochemical redox peaks of the NTPI and DFPI COF films; normalized contribution ratios of capacitive and diffusion-controlled currents versus scan rates of (b) DFPI and (c) NTPI COF films; (d) GCD curves of DFPI and NTPI COF films at 0.1, 0.2, 0.4, and 0.5 mA cm2; (e) the plots of Cspvs. current density of DFPI and NTPI COF films; (f) comparison of COF-based electrochromic energy-storage performance; (g) optical transmittance spectra of the flexible ECDs based on the NTPI COF in the bleached and colored states (insets show electrochromic digital photographs); (h) electrochromic response and (i) long-term cycling stability of the flexible ECDs based on DFPI and NTPI COF films. ipvs. logv for the electrochemical redox peaks of the NTPI and DFPI COF films; normalized contribution ratios of capacitive and diffusion-controlled currents versus scan rates of (b) DFPI and (c) NTPI COF films; (d) GCD curves of DFPI and NTPI COF films at 0.1, 0.2, 0.4, and 0.5 mA cm2; (e) the plots of Cspvs. current density of DFPI and NTPI COF films; (f) comparison of COF-based electrochromic energy-storage performance; (g) optical transmittance spectra of the flexible ECDs based on the NTPI COF in the bleached and colored states (insets show electrochromic digital photographs); (h) electrochromic response and (i) long-term cycling stability of the flexible ECDs based on DFPI and NTPI COF films. | ||
Flexible electrochromic devices
Considering that the imide-based COF films could be built on flexible transparent electrodes through electropolymerization combined with low-temperature annealing technology. To assess the latent applicability of DFPI or NTPI COF films, their flexible electrochromic devices (ECDs) were assembled, namely ECD 1 (structure: PET/ITO/DFPI COF/PC + TBAP/ITO/PET) and ECD 2 (structure: PET/ITO/NTPI COF/PC + TBAP/ITO/PET). The electrochromic performance of ECD 1 and 2 was investigated. Fig. S30 and 5g show that both ECD 1 and 2 displayed four color states (light red, transparent, yellow–green, and blue) and bipolar electrochromism. Their ΔT values were 53.2% at 753 nm and 56.2% at 736 nm, and the tb/tc values of ECD 1 and ECD 2 were 12.4/7.2 s and 13.9/5.0 s, respectively, as shown in Fig. 5h and S30. In addition, Fig. 5i illustrates that the ΔT of ECD 1 and ECD 2 remained unchanged after 2200 s cycles, indicating that suitable cycling stability was acquired. The triphenylamine-based imide COF film prepared by electro-polymerization combined with low-temperature annealing shows great potential in the field of high-performance flexible optoelectronics, electrochemical energy storage, and photoelectrocatalysis.Conclusions
In a word, we developed a facile electropolymerization combined with a low-temperature thermal annealing process to successfully prepare TPI COF (including DFPI and NTPI COFs) films on electrode surfaces. The results demonstrated that compared to DFD with a twisted spatial configuration, the planar-structured NTCDA and TPPDA more readily form NTPI COF films with higher crystallinity through this process. Both the DFPI and NTPI COF films exhibited multiple outstanding properties: a transparent bleached state, bipolar electrochromic behavior, four color states, a high optical contrast of 70%, robust cycling stability (maintaining unchanged performance after 1000 cycles), and relatively high specific capacitance values (80.9 mAh g−1 and 72.2 mAh g−1, respectively). Mechanistic studies revealed that the electropolymerization process initially involves anodic oxidative nucleophilic substitution of TPPDA to generate EGA, followed by EGA-catalyzed polycondensation between TPPDA and dianhydride to form the polyimide COF structure. Further electrochromic mechanism analysis indicated that the anodic coloration originates from TPPDA oxidation forming mono-/di-cation radicals, while the cathodic coloration results from carbonyl reduction generating oxygen anion radicals. The triphenylamine-based imide COF films prepared by electropolymerization combined with low-temperature annealing show great potential in the field of high-performance flexible optoelectronics, electrochemical energy storage, and photoelectrocatalysis.Author contributions
Jinming Zeng: methodology, conceptualization, writing – original draft preparation, supervision. Huiling Hou: methodology, data curation, formal analysis. Lei Huang: writing – original draft preparation, data curation. Zheng Xie: methodology. Qingqing Qiu: resources. Huan Li: validation. Dongfa Liu: writing – reviewing, editing. Putrakumar Balla: writing – reviewing. Tongxiang Liang: software. Ping Liu: investigation, resources.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc07413d.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22366016); the Jiangxi Provincial Natural Science Foundation, PR China (grant no. 20212BAB214022 and 20224BAB204034).References
- R. J. Mortimer, D. R. Rosseinsky and P. M. Monk, Electrochromic Mater. Devices, John Wiley & Sons, 2015, pp. 1–40 Search PubMed.
- J. Wang, X. Guo, C. Bian, Y. Zhong, J. Tu, P. S. Lee and G. Cai, Prog. Mater. Sci., 2025, 153, 101461 CrossRef CAS.
- Z. Jia, Y. Sui, L. Qian, X. Ren, Y. Zhao, R. Yao, L. Wang, D. Chao and C. Yang, Nat. Commun., 2024, 15, 6110 CrossRef CAS.
- L. Manjakkal, L. Pereira, E. K. Barimah, P. Grey, F. F. Franco, Z. Lin, G. Jose and R. A. Hogg, Prog. Mater. Sci., 2024, 142, 101244 CrossRef CAS.
- X. Guo, L. Chen, R. Liu, B. Fan, S. Cong, Z. Wang and Z. Zhao, Adv. Mater., 2025, 37, 2504575 CrossRef CAS PubMed.
- H. Miao, L. Chen, F. Xing, H. Li, T. Baumgartner and X. He, Chem. Sci., 2024, 15, 7576–7585 RSC.
- L. Lu, K. Wang, H. Wu, A. Qin and B. Z. Tang, Chem. Sci., 2021, 12, 7058–7065 RSC.
- J. Mei and I. Song, Nat. Electron., 2024, 7, 1082–1083 CrossRef.
- T. Chen, Q. Yang, C. Fang, S. Deng and B. Xu, Adv. Mater., 2025, 37, 2413665 CrossRef CAS.
- J. Wang, L. Zhang, Y. Ma, W. Xie, J. Dong, Y. Dong, W. Li and C. Zhang, Chem. Eng. J., 2024, 498, 155278 CrossRef CAS.
- G. Cai, M. Cui, V. Kumar, P. Darmawan, J. Wang, X. Wang, A. L.-S. Eh, K. Qian and P. S. Lee, Chem. Sci., 2016, 7, 1373–1382 RSC.
- D. Ma and J. Wang, Sci. China: Chem., 2017, 60, 54–62 CrossRef CAS.
- F. Zhao, B. Wang, B. Huang, W. Zhang, J. Chen, L. Liu, H. Wang, A. Y. Elezzabi, P. S. Lee, D. J. Milliron, W. W. Yu and H. Li, Nat. Rev. Clean Technol., 2025, 1, 396–412 CrossRef.
- Y. Ding, Z. Mei, X. Wu, W. Zhang, Y. Zhang, A. Xi, D. Gao, F. Lan, J. Xu, X. Diao and R. Zhang, Adv. Funct. Mater., 2025, 35, 2500122 CrossRef CAS.
- J. Cho, T. Y. Yun, H. Y. Noh, S. H. Baek, M. Nam, B. Kim, H. C. Moon and D.-H. Ko, Adv. Funct. Mater., 2020, 30, 1909601 CrossRef CAS.
- J. Feng, T.-F. Liu and R. Cao, Angew. Chem., Int. Ed., 2020, 59, 22392–22396 Search PubMed.
- L. Huang, S. Cao, Y. Liu, J. Chen, H. Li, Y. Liang, T. Yang and B. Zou, Adv. Funct. Mater., 2025, 35, 2500064 CrossRef CAS.
- Y. Li, N. Li, G. Li, Y. Qiao, M. Zhang, L. Zhang, Q.-H. Guo and G. He, J. Am. Chem. Soc., 2023, 145, 9118–9128 CrossRef CAS PubMed.
- L. R. Savagian, A. M. Österholm, J. F. Ponder Jr, K. J. Barth, J. Rivnay and J. R. Reynolds, Adv. Mater., 2018, 30, 1804647 CrossRef PubMed.
- Q. Zhao, J. Yang, Q. Wang, Y.-W. Zhang and J. Wang, Adv. Mater., 2025, 37, 2413452 CrossRef CAS.
- G. Cai, P. Cui, W. Shi, S. Morris, S. N. Lou, J. Chen, J.-H. Ciou, V. K. Paidi, K.-S. Lee, S. Li and P. S. Lee, Adv. Sci., 2020, 7, 1903109 CrossRef CAS.
- C. Li, H. Zhang, F. Lang, Y. Liu, L. Xu, X.-J. Xi, Y. Li, J. Pang, H.-C. Zhou and X.-H. Bu, Nat. Commun., 2025, 16, 1–11 Search PubMed.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- S. Yuan, X. Li, J. Zhu, G. Zhang, P. Van Puyvelde and B. Van der Bruggen, Chem. Soc. Rev., 2019, 48, 2665–2681 RSC.
- S. Yang, S. Bi, L. Zhai and Q. Xu, Chem. Sci., 2025, 34, 15611–15619 RSC.
- S. S. A. Shah, M. S. Javed, T. Najam, M. A. Nazir, A. ur Rehman, A. Rauf, M. Sohail, F. Verpoort and S.-J. Bao, Mater. Today, 2023, 67, 229–255 CrossRef CAS.
- H. Li, M. Cao, Z. Fu, Q. Ma, L. Zhang, R. Wang, F. Liang, T. Zhou and C. Zhang, Chem. Sci., 2024, 15, 4341–4348 RSC.
- S. Xu, J. Wu, X. Wang and Q. Zhang, Chem. Sci., 2023, 14, 13601–13628 RSC.
- L. Huang, R. Guo, Q. Qiu, H. Li, P. Balla, J. Zeng, T. Liang, X. Qi and P. Liu, Chem. Eng. J., 2024, 497, 155018 CrossRef CAS.
- H. Xie, Q. Qiu, H. Li, P. Liu, P. Balla, X. Qi, T. Liang and J. Zeng, ACS Appl. Mater. Interfaces, 2024, 17, 2180–2189 CrossRef.
- Q. Hao, Z.-J. Li, C. Lu, B. Sun, Y.-W. Zhong, L.-J. Wan and D. Wang, J. Am. Chem. Soc., 2019, 141, 19831–19838 CrossRef CAS PubMed.
- J. Hou, Y. Li, H. Tan, M. Chen, J. Wu, Y. Zhang, X. Lu, Y. Wang, X. Wang, C. Hua, J. Chu, M. Gong and S. Xiong, J. Colloid Interface Sci., 2025, 699, 138107 CrossRef CAS PubMed.
- Q. Hao, Z.-J. Li, B. Bai, X. Zhang, Y.-W. Zhong, L.-J. Wan and D. Wang, Angew. Chem., 2021, 133, 12606–12611 CrossRef.
- D. Bessinger, K. Muggli, M. Beetz, F. Auras and T. Bein, J. Am. Chem. Soc., 2021, 143, 7351–7357 CrossRef CAS PubMed.
- G. Kumar Silori, S.-C. Chien, L.-C. Lin and K.-C. Ho, Angew. Chem., 2025, 137, e202416046 CrossRef.
- Y.-A. Wang, Q. Wu, X. Wang, M. Jiang, R. Zhang, X.-J. Chen, R.-P. Liang and J.-D. Qiu, Angew. Chem., Int. Ed., 2024, 63, e202413071 CrossRef CAS.
- F. Yu, W. Liu, S.-W. Ke, M. Kurmoo, J.-L. Zuo and Q. Zhang, Nat. Commun., 2020, 11, 5534 CrossRef CAS.
- B. Bao, R. Li, Y. Hao, R. Xiao, C. Hou, Y. Li, Q. Zhang, K. Li and H. Wang, Chem. Mater., 2024, 36, 2880–2887 Search PubMed.
- J. Hu, Z. Huang and Y. Liu, Angew. Chem., Int. Ed., 2023, 62, e202306999 CrossRef CAS.
- A. Dey, S. Chakraborty, A. Singh, F. A. Rahimi, S. Biswas, T. Mandal and T. K. Maji, Angew. Chem., Int. Ed., 2024, 63, e202403093 CrossRef CAS PubMed.
- W. Zhao, P. Yan, H. Yang, M. Bahri, A. M. James, H. Chen, L. Liu, B. Li, Z. Pang, R. Clowes, N. D. Browning, J. W. Ward, Y. Wu and A. I. Cooper, Nat. Synth., 2022, 1, 87–95 CrossRef CAS.
- S. T. Emmerling, L. S. Germann, P. A. Julien, I. Moudrakovski, M. Etter, T. Friščić, R. E. Dinnebier and B. V. Lotsch, Chem, 2021, 7, 1639–1652 CAS.
- S. Kim, C. Park, M. Lee, I. Song, J. Kim, M. Lee, J. Jung, Y. Kim, H. Lim and H. C. Choi, Adv. Funct. Mater., 2017, 27, 1700925 CrossRef.
- J. He, X. Jiang, F. Xu, C. Li, Z. Long, H. Chen and X. Hou, Angew. Chem., 2021, 133, 10072–10077 CrossRef.
- Z.-F. Cai, G. Zhan, L. Daukiya, S. Eyley, W. Thielemans, K. Severin and S. De Feyter, J. Am. Chem. Soc., 2019, 141, 11404–11408 CrossRef CAS.
- E. Tavakoli, A. Kakekhani, S. Kaviani, P. Tan, M. M. Ghaleni, M. A. Zaeem, A. M. Rappe and S. Nejati, J. Am. Chem. Soc., 2019, 141, 19560–19564 Search PubMed.
- T. Shirokura, T. Hirohata, K. Sato, E. Villani, K. Sekiya, Y.-A. Chien, T. Kurioka, R. Hifumi, Y. Hattori, M. Sone, I. Tomita and S. Hattori, Angew. Chem., 2023, 135, e202307343 Search PubMed.
- M. Wang, Y. Wang, J. Zhao, J. Zou, X. Liang, Z. Zhu, J. Zhu, H. Wang, Y. Wang, F. Pan and Z. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202219084 CrossRef CAS.
- G.-S. Liou and C.-W. Chang, Macromolecules, 2008, 41, 1667–1674 CrossRef CAS.
- M. Chafiq, A. Chaouiki and Y. G. Ko, Energy Storage Mater., 2023, 63, 103014 CrossRef.
- L. Guo, J. Zhang, Q. Huang, W. Zhou and S. Jin, Chin. Chem. Lett., 2022, 33, 2856–2866 Search PubMed.
- T. G. Yun, J. Lee, H. S. Kim, J. Y. Cheong, S. H. Kim, Y. Kim, S. Lee and I.-D. Kim, Adv. Mater., 2023, 35, 2301141 Search PubMed.
- H.-J. Yen and G.-S. Liou, Prog. Polym. Sci., 2019, 89, 250–287 CrossRef CAS.
- M. Lapkowski, K. Berrada, S. Quillard, G. Louarn, S. Lefrant and A. Pron, Macromolecules, 1995, 28, 1233–1238 CrossRef CAS.
- S. Pluczyk, P. Zassowski, C. Quinton, P. Audebert, V. Alain-Rizzo and M. Lapkowski, J. Phys. Chem. C, 2016, 120, 4382–4391 Search PubMed.
- X.-G. Li, M.-R. Huang, W. Duan and Y.-L. Yang, Chem. Rev., 2002, 102, 2925–3030 CrossRef CAS PubMed.
- S. Zhang, Y. Peng, J. Zhao, Z. Fan, B. Ding, J. Y. Lee, X. Zhang and Y. Xuan, Adv. Opt. Mater., 2023, 11, 2202115 CrossRef CAS.
- J. Zheng, X. Liu, W. Li, W. Li, X. Feng and W. Chen, Nano Res., 2023, 16, 9538–9545 CrossRef CAS.
- X. Lian, Z. Ma, Z. Zhang, J. Yang, Y. Liu, C. Gu, R. Guo, Y. Wang, X. Ye, S. Sun, Y. Zheng, H. Ding, J. Hu, X. Cao, H. Mao, J. Zhu and S. Li, Appl. Surf. Sci., 2020, 524, 146396 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |