
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the formation of medium-sized cyclic amines within self-assembled yoctoliter inner-spaces
Yahya A.
Ismaiel
 ,
Douglas
Rogers
and
Bruce C.
Gibb
*
,
Douglas
Rogers
and
Bruce C.
Gibb
*
Department of Chemistry, Tulane University, New Orleans, LA 70118, USA. E-mail: bgibb@tulane.edu
First published on 4th November 2025
Abstract
Medium-sized (7-11-membered) rings are difficult to cyclize for both enthalpic and entropic reasons. Moreover, SN2 cyclizations to such products in the greenest of solvent (H2O) are complicated by competing substrate hydrolysis. Here we explore the utility of the yoctoliter (10−24 L) inner-space of a dimeric container — assembled via the hydrophobic effect — to catalyze the formation of strained, 7–11-membered cyclic amines. Specifically, we examine the ability of the dimeric capsule of deep-cavity cavitand octa-acid 1 to promote cyclization processes, by leveraging relatively large pseudo-halide sulfonate leaving groups to minimize product inhibition and engender catalysis. We find that strained 7-membered azepane can be formed catalytically, with reaction rates dependent on the conformation or motif of the bound guest. We also find that the hydrolysis rate of a bound guest can be up to four orders of magnitude slower than the free state, and in such watertight complexes strained 11-membered aza-cycloundecane can also be formed. More generally, our results provide the first examples of SN2 cyclizations to medium-sized cyclic amines in water, and provide benchmarks in quantifying the degree of water tightness of water-based container molecules.
Introduction
Pivotal to replicating the catalytic efficiencies observed in Nature's enzymes is an understanding of the roles played by the shape and structural dynamics of binding sites, as well as the electronics of the functionality crucial for reaction. To explore these and related topics, supramolecular chemists have most recently turned to the rules of self-assembly to devise a wide range of yoctoliter (10−24 L) supramolecular containers for bringing about substrate encapsulation and conversion.1 Whether such reactors are stoichiometric or truly catalytic, metal–ligand coordination2 and hydrogen bonding3,4 have played preeminent roles here, providing a wealth of design guidelines. Thus the resulting yoctoliter containers have facilitated exploring: (1) the modulation of reaction molecularity; (2) strategies for substrate conformational control to engender high energy pre-reaction complexes; (3) design approaches to engendering a high effective molarity of the reacting species, and; (4) the non-covalent contributions for minimizing the energy barrier to reaction by stabilizing and/or redistributing charge in the lead-up to reaction and transition state (TS‡).Water provides a challenging environment to explore yoctoliter container design and catalysis, but whilst the small size and cohesivity of water leads to unique solvation properties that are still not fully understood,5 the hydrophobic effect can nevertheless be leveraged to considerable effect to bring about assembly, substrate encapsulation, and reaction. Water-soluble deep-cavity cavitands have been particularly effective here, both when they form open 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 cavitand–guest complexes,4,6–8 and when they self-assemble to form fully encapsulating, dimeric host–guest container complexes.8–11
:1 cavitand–guest complexes,4,6–8 and when they self-assemble to form fully encapsulating, dimeric host–guest container complexes.8–11
Previously, we have investigated the properties of dimer capsules of deep-cavitand such as 1 (Fig. 1). Thus, the dimer 12 can form around one to three guests depending on their size, encapsulating species that total between six and twenty-six non-hydrogen atoms.12–14 These container complexes can possess lifetimes of tens of seconds, but also exhibit a fast, dynamic ‘breathing’ whereby the equatorial interface partially opens to allow the entry and egress of small guests; including adventitious water.15 Guests modulate this breathing, and hence the degree of capsule watertightness,16 imbuing a heterogeneity to the inner-space whereby the pole regions possess a lower ‘dielectric’ than the equatorial region.10 Additionally, the sixteen water solubilizing groups on the outer surface of the capsule engender a significant electrostatic potential field that can affect the physicochemical properties of the inner-space, and hence any bound guest.10,17
 | ||
| Fig. 1 Structures of cavitand host 1, the amino sulfonate guests (G1–G12), and the cyclized products azepane, azocane, azonane and aza-cycloundecane. | ||
One advantage of working in water is the ease by which guests too long for a binding pocket fold into U-shaped conformations (motifs) to open up reaction pathways rarely observed in solution. In essence, because of the confining nature of the inner-space, the hydrophobic effect provides a thermodynamic driving force for the guest to adopt high-energy conformations rarely sampled in free solution. Such motifs approximate to the structure of a substrate on the cusp of undergoing an intramolecular cyclization reaction, and correspondingly have been leveraged to considerable effect for the formation of macrocycles composed of up to 19-membered rings.6,9,17,18
In wishing to explore SN2 cyclizations within the 12 capsule further, we were mindful of several interrelated points. First, although such assemblies have been investigated for their ability to form macrocycles, there was no evidence as to whether they could form difficult-to-cyclize medium sized (7-11-membered) rings. Second, product inhibition is common in fully encapsulating yoctoliter spaces, suggesting that once the entropy of cyclization has been paid, a relatively high product affinity is not unusual; which strategies can skirt this issue? Third, although capsule watertightness for ideal guests is relatively assured,19 there is little to no understanding of how guest size, shape, or functionality can affect the degree of water ingress into their inner-spaces.
Given these points, we selected the amino-sulfonate guests shown in Fig. 1 to: (1) explore the feasibility of catalytically forming azepane, azocane, azonane and aza-cycloundecane; (2) investigate how the size of the sulfonate leaving group and therefore the change in the volume of the guest upon reaction (ΔV) affected turnover, and; (3) probe the watertightness of the capsule by assessing the degree to which guest hydrolysis (rather than cyclization) occurred.
Results
Host and guest synthesis
Host 1 was synthesized as previously described.20 The synthesis (SI, Section 2) of guests G1–G12 is shown in Scheme 1. Briefly, for the requisite suite of amino alcohols, two were commercially available and two were synthesized by Gabriel synthesis using the corresponding bromides (Scheme 1a). These two precursors, in combination with commercially available 6-aminohexan-1-ol and 8-aminooctan-1-ol, were converted to the required amino-sulfonates G1–G12via a three step process of protection using Boc2O, reaction with the requisite sulfonyl chloride, and deprotection with HCl (Scheme 1b). For the final step, under the conditions selected no significant sulfonate–chloride exchange was noted. | ||
| Scheme 1 (a) Synthesis of 7-aminoheptan-1-ol and 10-aminodecan-1-ol. (b) The protection, sulfonation, and deprotection strategy used to synthesize amino sulfonates G1–G12 from the precursor amino alcohols. | ||
Host–guest complex characterization: identifying the guest motif
All guests formed the expected capsular 2:1 host–guest complexes with host 1. To characterize how the different guests packed in the yoctoliter inner-space of 12, 12.5 µL of a 20 mM stock solution of the guest ammonium salt (1 equiv.) was added to 0.5 mL of a D2O solution of 1.0 mM host 1 (2 equiv.) in 10 mM phosphate buffer (pH = 11.3, corrected) and the resulting mixture vortexed for 1 min. Complexation was uniformly rapid because of the high solubility of the salt, and no issues were noted with the deprotonation of the guest by the excess buffer before or during encapsulation.
Characterization of each complex involved three types of NMR experiments: (1) 1H NMR studies of the free and bound host and guest; (2) COSY NMR analysis of the free and bound guest; and; (3) NOESY NMR experiments of each host–guest complex. By this procedure three kinds of guest motifs were identified, epitomized by guests G3, G7 and G12 (Fig. 2). In the figure, the Δδ plots for G3, G7 and G12 are compared to n-alkanes of the same number of atoms in the mainchain. As discussed further below, because the pocket of each cavitand approximates to a truncated cone, in general protons that reside more towards the pole regions of the capsule are more upfield shifted. With this, these Δδ plots often provide sufficient information to identify the guest motif. However, in select cases it is necessary to also perform 1H–1H NOESY NMR analysis to confirm the motif. In such experiments the two inward-pointing hydrogen atoms of the cavitands Hb and Hc (Fig. 1) were used, and in examining the intensities of cross-peaks between these and the different guest atoms, the intensity of the intra-host He–Hf NOE of each capsule was used to normalize the data. Values ranging from one quarter to three times the reference were obtained. In Fig. 2 these are shown as white (NOEs with Hb) and blue (NOEs with Hc) disks, centered on the guest group in question and possessing diameters proportional to the normalized intensity. Full details of the Δδ plots and NOESY analysis for all guests G1–G12 with host 12 are provided in the SI (Section 4). As Fig. 2 summarizes, three general motifs were observed: a J-motif in which the sole terminal group anchoring to a capsule pole was the amino group (J(NH2) motif), an extended motif (E-motif) in which the two termini occupy the poles, and a (weak; see discussion) J-motif in which the methyl was the anchoring terminal group (J(CH3) motif).
 | ||
| Fig. 2 Motif identification for guests G3 (a), G7 (b), and G12 (c) bound to the octa-carboxylate dimer represented as the scarlet capsule. Shown for each guest is a 1H NMR spectroscopy Δδ plot revealing the shift in each guest signal between the free and the bound states (Δδ = δbound − δfree). Studies were carried out in D2O, and involved 1 mM complex solutions in 10 mM phosphate buffer, pH = 11.3 (corrected). In all plots the –NH2 terminal is labeled as atom # 1, and all mainchain atoms labeled consecutively. The red guest structure under each plot shows the terminal atom numbering, and the mainchain N, O, and S atoms are also indicated (red) on the x-axis of each plot. The amino terminus was non-reporting because of N–H exchange with the D2O solvent, and breaks in the data caused by the non-reporting O and S atoms are shown as dashed blue lines. For comparison, the data for each guest is plotted with that of the corresponding n-alkane possessing the same number of mainchain atoms. Also shown for each guest is a rendition of the inferred guest motif/conformation within each capsule. Each motif is further supported by the observed 1H–1H NOEs between the host Hb or Hc atoms (Fig. 1, and indicated in each scarlet capsule in blue and white respectively) and the guest. The NOE cross-peak intensities involving both Hb (white disks) and Hc (blue disks) with the different guest atoms were normalized by reference to the intra-cavitand He–Hf NOE of each capsule, giving relative values ranging from 0.25–3.0× the reference. These intensities are represented by the relative diameter of each disk on its respective group. | ||
Further information of the guest motif was obtained by repeating the 1H NMR experiments of the host–guest complexes in 95:5H2O/D2O to minimize the replacement of acidic hydrogen atoms with deuterium and so observe the amino group signal in the bound guest region. This allowed us to observe bound amino signal for nine guests, but not G9, G11 and G12. Table 1 summarizes the obtained data, revealing that the Δδ values for the amino groups can be very large; over −5 ppm in some cases.
| Guest | Mainchain atoms | Motif | Δδ value (NH2, ppm) | Δδ value (CH3, ppm) |
|---|---|---|---|---|
|
a Δδ = δbound − δfree, where the bound state corresponds to a 1 mM host–guest complex with 12 in 10 mM phosphate buffer solution (95:5H2O/D2O), and the free state the guest (1 mM) in CHCl3.
b The kinetics of proton exchange on the amino group prevented observation of its 1H NMR signal (see, Discussion).
|
||||
| G1 | 10 | E | −3.90 | −3.46 |
| G2 | 11 | J (NH2) | −4.76 | −2.09 |
| G3 | 12 | J (NH2) | −4.96 | −2.54 |
| G4 | 13 | E | −5.07 | −3.43 |
| G5 | 14 | E | −5.05 | −4.23 |
| G6 | 11 | E | −4.48 | −4.10 |
| G7 | 12 | E | −4.97 | −4.77 |
| G8 | 14 | J (NH2) | −4.93 | −1.80 |
| G9 | 16 | J (NH2) | –b | −1.87 |
| G10 | 14 | E | −5.15 | −4.70 |
| G11 | 16 | J (NH2) | –b | −2.33 |
| G12 | 18 | J (CH3) | –b | −3.68 |
Bound guest reactivity
We examined the reactivity of guests G1–G12 inside the capsule formed by host 1. In these experiments 1 mM solutions of the preformed complexes in 10 mM sodium phosphate/D2O buffer (pH = 11.3, corrected) were heated to 60 °C. Two possible reaction outcomes were assumed: cyclization and hydrolysis (Scheme 2).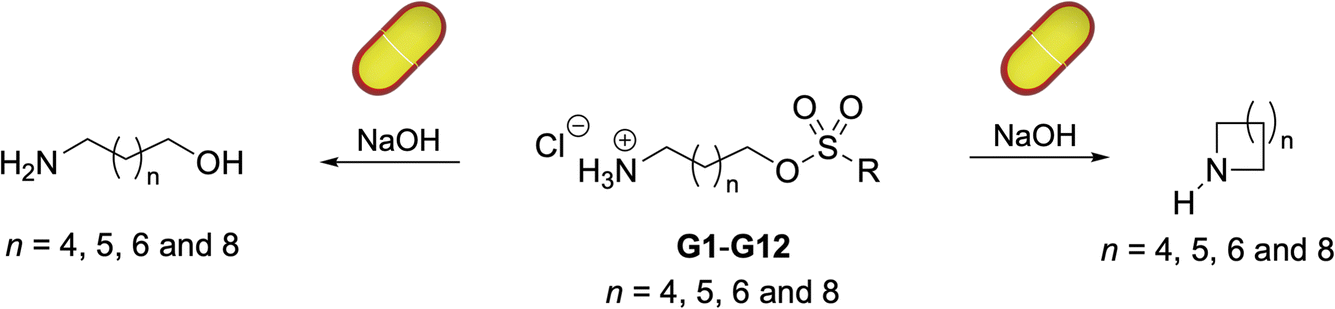 | ||
| Scheme 2 Cyclization and hydrolysis of amino sulfonates within the container capsule 12. | ||
Reaction rates were generally sufficiently slow to allow the extent of reaction to be monitored by 1H NMR simply by periodically removing samples from a heating block and carrying out NMR analysis at 60 °C (G1–G5 and G12) or 25 °C (when signals were too broad for analysis at higher temperature). Monitoring the progress of reaction utilized different methyl and/or methylene signals in the starting material and product depending on the reaction under study. For example, the methyl group of the alkylsulfonyl group often served as a metric for the amount of remaining starting material, but in the case of methanesulfonyl guests this was not possible because the α-CH3 to the sulfonyl group underwent relatively rapid hydrogen–deuterium exchange leading to signal disappearance. Thus, in the case of guest G1, the α-CH2 to the sulfonate oxygen was used to gauge the amount of remaining starting material, whilst the α-CH2 adjacent to the N-atom was used to monitor the appearance of cyclized product. This problem of exchanging acidic C–H groups, combined with occasional signal overlap, demanded a bespoke approach to analysis for many substrates (see SI Section 5). Regardless, in all cases data was gathered in triplicate. For guests G1–G5 first order cyclization was observed. The approximate reaction times and obtained rate constant (kcyc) are given in Table 2. In each case, confirmation of the product was made using 1H NMR by comparison of the authentic product (azepane, Fig. 1) complex with that obtained at the end of the reaction. The product was also confirmed by isolation and 1H NMR and MS (ESI) analysis. Both by examination of the product complex and MS analysis of the isolated product, each reaction proceeded quantitatively (no hydrolysis observed).
| Guest | Approximate reaction time (hrs) | Rate constant (kcyc, s−1 × 105) |
|---|---|---|
| a Rate constants and errors obtained from the average of triplicate experiments. | ||
| G1 | 20 | 7.92 ± 0.2 |
| G2 | 48 | 3.76 ± 0.1 |
| G3 | 48 | 2.62 ± 0.2 |
| G4 | 20 | 6.42 ± 0.3 |
| G5 | 20 | 6.23 ± 0.2 |
Considering the differences in cyclization rate noted for homologues G3 and G4, we carried out Eyring analysis to determine the reaction thermodynamic parameters at 333–346 K (G3) and 323–339 K (G4). In both cases rates were determined at five temperatures, with data collected in triplicate. Reaction progress was monitored by 1H NMR using the ‘Hd’ and ‘He’ peaks of the host. The data calculated at 298 K are given in Table 3.
| Kinetic parameters (298 K) | G3 | G4 |
|---|---|---|
| a Error propagation gave < 5% error in each thermodynamic parameters. | ||
| k (s−1) | 8.56 × 10−7 | 8.72 × 10−6 |
| Half-life (s) | 8.10 × 105 | 7.95 × 104 |
| ΔG‡ (kJ mol−1) | 107.6 ± 3.76 | 101.9 ± 2.90 |
| ΔH‡ (kJ mol−1) | 77.0 ± 8.41 | 45.9 ± 4.20 |
| −TΔS‡ (kJ mol−1) | 30.6 ± 1.25 | 56.0 ± 2.1 |
For guests G6–G12, confirmation of each product utilized: (1) 1H NMR by comparison of the authentic hydrolysis product (7-aminoheptanol, 8-aminooctanol, or 10-aminodecanol) complex with that obtained, and; (2) MS analysis of the isolated product. Pseudo first order hydrolysis rates were observed for all guests, but G10 also gave a secondary minor product complex identified as aza-cycloundecane (20% yield). Table 4 shows the obtained hydrolysis data.
| Guest | Approximate hydrolysis time (d) | Rate constant (k × 107, s−1) |
|---|---|---|
| a Rate constants and errors obtained from the average of triplicate experiments. | ||
| G6 | 60 | 7.07 ± 0.1 |
| G7 | 90 | 3.71 ± 0.2 |
| G8 | 60 | 4.74 ± 0.3 |
| G9 | 10 | 30.1 ± 0.3 |
| G10 | 100 | 1.28 ± 0.4 |
| G11 | 10 | 29.1 ± 0.2 |
| G12 | 3 | 512 ± 0.3 |
We also measured the rates of hydrolysis of free guests (Table 5). We utilized G1, G3, G5 and G7 to give representative data as these smaller guests were the best behaved in aqueous solution. Reactions were carried out using 1.0 mM solutions of each in either D2O, or 10 mM sodium phosphate in D2O (pH = 11.3, corrected) at 60 °C. In the reaction of G1, 1H NMR spectroscopy was used to monitor the disappearance of the 1H NMR signal of either the methylene adjacent to the sulfonate oxygen (for reaction in D2O) or the methylene adjacent to the amino group (in phosphate buffer). For guests G3, G5 and G7 the signals from the methylene adjacent to the sulfonate oxygen could be utilized in both sets of conditions.
| Guest | Rate constant in D2O (k × 104, s−1) | Rate constant in bufferb (k, × 103, s−1) |
|---|---|---|
| a Rate constants and errors obtained from the average of triplicate experiments. b Buffer was 10 mM phosphate (pH = 11.3, corrected). | ||
| G1 | 2.28 ± 0.2 | 1.45 ± 0.3 |
| G3 | 1.53 ± 0.3 | 1.53 ± 0.2 |
| G5 | 1.80 ± 0.3 | 1.35 ± 0.4 |
| G7 | 1.59 ± 0.2 | 1.46 ± 0.3 |
As the rates of hydrolysis were an order slower in D2O versus buffer, where it was comparable to the rate of cyclization of encapsulated G5, we opted to probe cyclization using sub-stoichiometric amounts of the octa-sodium salt of 12 in the absence of excess base. For these experiments we formed 0.6 mL samples of a 1.0 mM solution of the ammonium salt of G5 containing 1 mM Na2CO3 in D2O (pH 7.3). This deprotonated the guest, leaving one equivalent of NaHCO3 for reacting with the RSO3H generated from reaction. Cyclization itself was initiated with the addition of an aliquot of 1 from a 2 mM stock solution in 16 mM bicarbonate buffer (pH 7.6). Each reaction mixture was heated at 60 °C for 24 hours, after which time 1H NMR spectroscopy used to determine the ratio of azepane to hydrolysis product. By this approach, 2, 5 and 10 mol% capsule were found to lead to 3:7, 3:7 and 1:1 ratios of azepane and hydrolysis product 7-aminohexanol.
Discussion
Bound guest motif
A comparison of the guest 1H NMR signals in the free and bound states — a process that typically involves COSY NMR analysis to ensure signal assignment — provides Δδ values for each reporting atom. The resulting Δδ plots (listed reporting atom vs. each Δδ value) yield detail of the time-averaged preferred motif of bound guests. A summary of Δδ plot interpretation is given in the SI.Ideally, to study the motif of guests G1–G12 it is necessary to determine the Δδ for both the NH2 and the alkylsulfonyl CH3 termini. Duplication of the initial NMR studies of the complexes in 95:5H2O/D2O demonstrated that our initial supposition that the proton exchange rate on the amino group would be close to the NMR timescale — rendering these signals unobservable21 — was only partially correct. As Table 1 summarizes, guests with ≤ 14 mainchain atoms provided observable bound NH2 signals. We attribute an observable NH2 signal for guests G1-G8 and G10 to two structural features rendering it inaccessible to bulk water: (1) a relatively deep position in the least water-accessible volume of the container, and; (2) a relatively watertight capsule in which the rims of both cavitands are in intimate contact with each other. In such cases, NH2 exchange with bulk water is relatively slow and its signal observable. Prior work has demonstrated that partial opening of the capsule to allow the entry and egress of small guests (such as water) — so-called “breathing” — occurs typically 105 times more rapidly than guest exchange.15 This breathing leads to a wetter equatorial region of the inner-space, a corresponding gradient of the effective dielectric, and modulation of the pKa of any acidic group in the guest.10 Thus the absence of an amino signal from bound G9 and G11 can be attributed to their larger size (16 mainchain atoms) promoting capsule ‘breathing’ and acceleration of NH2 exchange. This is also true for guest G12 (18 mainchain atoms), but here another factor is its motif that locates the NH2 group in the equatorial zone of the yoctoliter space (Fig. 2c).
Overall for guests G1–G12 we identified three packing motifs: extended (E) motifs, and two J-motifs in which there is a turn in the mainchain of the host and the sole terminal group anchoring to a pole region of the capsule was either the amino or methyl group (J(NH2) and J(CH3)). All guests that adopt an E-motif, and three that adopted a J(NH2)-motif, allowed the Δδ values of the NH2 termini to be determined (Table 1). These were large chemical shifts. For reference, maximal chemical shifts for the methyl termini of bound n-alkanes are typically around −4.5 ppm,14,22 whereas the majority of this set of guests had Δδ values for the NH2 group close to or in excess of −5.0 ppm. This difference between the free and the bound states suggests that the two polarized N–Hs of the NH2 group are close to the aromatic walls and form a relatively strong anchor via N–H⋯π hydrogen bonding.
As it was not possible to observe the NH2 signal from all guests, we used its α-CH2 as a proxy to calculate the asymmetry between the Δδ values of the termini. Fig. 3 plots the relationship between the number of atoms in the mainchain of the guest against the difference in Δδ value of the terminal CH3 and the NH2 α-methylene (ΔΔδ = ΔδCH3 − ΔδCH2). Based on these differences, the time-averaged motif was assigned to a guest as follows: positive values (J(NH2)-motif), small negative values (E-motif), and large negative ΔΔδ value (J(CH3)-motif). Each of these assignments was corroborated with NOE data. Guests that adopted E-motifs included all of the methyl sulfonyl derivatives G1, G6, G7 (Fig. 2b), G10, as well as G4 and G5. In contrast, G2, G3 (Fig. 2a) G8, G9, and G11 were observed to bind in a J(NH2)-motif. G12 was the sole guest adopting a J(CH3)-motif (Fig. 2c).
 | ||
| Fig. 3 Plot of the number of guest mainchain atoms versus the difference in the Δδ values reported by the terminal CH3 and the NH2 α-methylene (ΔΔδ = δCH3 − δCH2). Guests on the same vertical are constitutional isomers. The sets of guests sharing common methanesulfonyl (G1, G6, G7, and G10), n-propyl sulfonyl (G3, G8, and G11) and n-pentyl sulfonyl (G5, G9, and G12) leaving groups are highlighted with red connecting lines. | ||
Our interpretation as to why the E-motif and J(NH2)-motif are observed with guests of identical constitution or similar size is that a key structural feature is the polarized nature of the C–H bonds alpha to the sulfonyl (which leads to H-D exchange in the guest). The polarization of the sulfonyl α-CH2 group, or α-CH3 group in the case of methanesulfonyls, leads to C–H⋯π host–guest interactions stronger than those of simple alkanes. Consider for example constitutional isomers G3 (J(NH2)-motif) and G7 (E-motif), and how their motifs compare to their corresponding alkanes. With alkanes bound to 12, the switch from E- to J-motif is controlled by guest length and occurs at ∼17 mainchain atoms.23 Shorter guests adopt E-motifs with some degree of helical structure/compression.13,14,22,24 However, at ≥17 atoms the guest adopts a J-motif.23 In contrast, guests G3 and G7 are only 12 atoms long, but the former nevertheless adopts a J(NH2)-motif. We believe that the E- to J(NH2)-motif switch occurs with such short guests because of the acidic sulfonyl α-CH2 in G3 and the α-CH3 in G7. Thus, in G3 it is energetically preferred for the n-propyl group to form a turn so that its α-CH2 group anchors to the base of the pocket,25 rather than adopting an E-motif in which its terminal CH3 anchors.26 In contrast, constitutional isomer G7 can adopt a low energy E-motif using its acidic α-CH3 as anchor. This noted, at a certain length of alkyl sulfonyl this observed preference for acidic α-CH2 anchoring breaks down. Consider for example constitutional isomers G5 (E), G8 (J(NH2)), and G10 (E). Whilst the sulfonyl α-CH3 in G10 is a firm anchor, G8 prefers to anchor with its acidic α-CH2 group. However, in the case of G5, the n-pentyl sulfonyl preferentially binds with its terminal CH3 rather than with its α-CH2. Evidently, the flexibility of the n-pentyl group to pack into the pole is energetically preferred.27 This is also apparent in the homologous series G1–G5. Thus, after the motif switch between G1 (E) and G2 (J(NH2)) there is a gradual trend back to a E-motif as the guest increases in length and the preference for sulfonyl α-CH2 anchoring decreases.
The homologs with the n-pentyl sulfonyl group (G5, G9, and G12) demonstrated the greatest range of motifs. Smallest G5 adopts an E-motif controlled by the strong anchoring of the NH2 and the efficient packing of the n-pentyl chain. Add two CH2 groups to the mainchain, and the 16 atom long G9 cannot adopt an E-motif and instead adopts a J(NH2)-motif by anchoring with its NH2 and its sulfonyl α-CH2. Add two more CH2 groups and the 18 atom mainchain G12 switches to a J(CH3)-motif in which the anchors are the CH3 of the n-pentyl sulfonyl group and the midsection of the carbon chain between the NH2 and SO3R groups. We believe that G12 forms this singular J(CH3)-motif because the power of its NH2 group to act as anchor is overridden by geometrical considerations and steric crowding. Thus, typically the start of a turn in a J-motif is located 9–10 bonds from the anchor. This provides little issue for the J(CH3)-motif, but in the case of the J(NH2)-motif forces the bulbous sulfonate group to be located at or near the narrow polar region of the capsule (Fig. 4). As a result, the CH3 is energetically preferred as the anchor and the NH2 group left relatively exposed to bulk water and invisible to 1H NMR.
 | ||
| Fig. 4 The two extreme J-motifs for guest G12 within the 12 capsule. The J(CH3)-motif (left) is observed, whereas the J(NH2)-motif (right) in not. | ||
To recap, despite guests G1–G12 possessing similar structures, NMR analysis reveals three motifs within capsule 12. All of the methyl sulfonates examined use their two termini as anchors. In each case the Δδ plots demonstrate the NH2s have the greatest anchoring ability. Guests with alkyl sulfonate groups other than CH3 possess three latent anchors: the NH2, the sulfonyl α-CH2, and the terminal CH3. The balance between the non-covalent interactions that these anchors form with the capsule, the length of the guest, and the relative position of the sulfonyl group dictates which two anchors dominate. This leads to a relatively complex motif space for the different guests.
Bound guest reactivity
As discussed above, the three interrelated aims of this work were: (1) explore the formation of difficult-to-cyclize medium-sized rings; (2) investigate the effect of leaving group volume on minimizing product inhibition and maximizing the likelihood of turn-over, and (3) investigate the extent to which the different capsular complexes are waterproof, i.e., cyclization can occur in the absence of hydrolysis.Although the cyclization of α,ω-amino (pseudo)halides have not been determined for a wide number of ring sizes,28 it is uniformly the case that the ΔG‡ of cyclization of 7–11 membered (medium) rings is higher than that of common-sized rings.29 Both enthalpy and entropy contribute. Thus, ΔH‡ of cyclization is increased because of significant bond angle deformations, eclipsing conformations, and transannular ring strain within medium rings. Additionally, for rings larger than 7–8 atoms there is an increasing ΔS‡ penalty associated with the two ends of the molecule attaining a suitable geometry for reaction. To compensate for the high ΔG‡ values for the synthesis of medium rings we surmised that in much the same way as capsule 12 can promote the cyclization of 13–19 macrocycles,17 so it may be capable of binding guests G1–G12 in relatively high energy J-motifs to facilitate cyclization.
Regarding maximizing the likelihood of turn-over, prior cyclization reactions within capsules using halide leaving groups were found to generally suffer from product inhibition; in most cases the affinity of the cyclic product binding was higher than that of the J-motif precursor. In seeking to avoid this, we reasoned that the use of larger sulfonate leaving groups could be leveraged to cause a large (and adjustable), negative change in guest volume (ΔV) upon reaction. Thus, with alkanes of 12–14 atoms typically being ideal for capsule 12,13,22,23,30 we deduced that 7–11 membered ring products would have relatively low affinities compared to guests G1–G12, be readily displaced, and facilitate turnover.
The 7-membered ring of azepane (Fig. 1) — the product of cyclizing G1–G5 — represents the upper limit of “common rings” and possesses approximately the same degree of strain as 5-membered rings. In contrast, the products of cyclization of G6, G7/8/9 or G10/11/12, i.e., 8-membered azocane, 9-membered azonane, and 11-membered aza-cycloundecane (Fig. 1) were expected to possess roughly twice the ring strain of azepane. Moreover, as the entropy change upon cyclization becomes more penalizing as the number of bonds between the reacting termini increases, this was also expected to disfavor cyclization. In short, G1–G5 were expected to cyclize more readily than G7–G12.
As their 2:1 capsular complexes, guests G1–G5 cyclized smoothly and quantitively to give azepane (Fig. 5). The rates of cyclization (Table 2) followed the order: G1 > G4 ≈ G5 > G2 > G3. In comparing pairs of guests, the most striking differences involve the slowest reacting G3. Thus, adding one CH2 to the substrate (G4) resulted in a rate acceleration of 150%, whilst removing two CH2s (G1) resulted in a 200% rate acceleration. There are evidently at least two factors at play behind the non-monotonic trend in cyclization rates of G1–G5. We rationalize that the nature of the sulfonate anchor was central. G1 only has its CH3 sulfonate anchor, and is only ten-atoms in length; a length sufficient to span between the two anchor-points of the capsule, but only in a fully extended (all-trans dihedrals) E-motif. This leads to relatively poor anchoring of both termini (Table 1) and an increased likelihood of them attaining a suitable reaction trajectory. Hence G1 reacts quickly. In the case of 11- and 12-atom long G2 and G3, the width of the turn in the guest (e.g., Fig. 2a) prevents deep anchoring, forcing the amino group to be more deeply buried (Table 1) and potentially less available to act as a nucleophile. Simultaneously, the turn in the sulfonyl alkyl group may block access to the proximal electrophilic site. This may be by simple steric encumbrance, or may also involve the guest adopting a combination of dihedrals that turn the electrophilic CH2 towards the NH2 group, preventing backside SN2 attack. It is not possible to discern specifics from the time-averaged data provided by NMR spectroscopy, but evidently the combination of a deep NH2 and a sterically encumbered electrophilic center slows cyclization. In contrast, guests G4 and G5 exist in the capsule in E-motifs. Like G1, the E-motif dictates more deeply anchored NH2 groups, but this is the only impediment to cyclization. Thus, in an E-motif the electrophilic center of both G4 and G5 is pushed away from the narrow pole region by the length of the sulfonyl alkyl, and hence towards the wider and more hydrated equatorial zone of the yoctoliter space. As a result, cyclization of G4 and G5 are faster than G2 and G3, but slower than G1. Interestingly, the similar cyclization rates of G1, G4 and G5 suggest that deep anchoring of a NH2 group is not a major impediment to reaction, suggesting accessibility to the electrophilic methylene is more important in controlling reaction rate.
 | ||
| Fig. 5 Stack of 1H NMR spectra (600 MHz) showing the guest binding region for: (1) substrate G3 bound to 12; (2) product of reaction with G3 (including free propyl sulfonate), and; (3) Authentic azepane complex. | ||
Eyring analysis of the cyclization of G3 and G4 revealed that the faster reaction of G4 was rooted in ΔH‡, with a 40% lower enthalpy cost to cyclization (Table 3). This decrease was partially countered by an increased entropic cost to reach the TS‡. We attribute these differences to the starting motif for several reasons. First, both reactions form azepane, and so however the energetics of the product factors into the TS‡, the contributions from each are the same. Second, unlike thiol/thiolate cyclizations,17 amine cyclizations are not sensitive to the electrostatic potential field generated by the sixteen charges of the capsule.9 In other words, we have no evidence of unanticipated charge development in either encapsulated TS‡, and correspondingly conclude little difference in the charge distribution within each. Moreover, because of the exergonic nature of the SN2 mechanism, the early TS‡ (Hammond postulate) dictates that differences in the substrate energetics weigh more heavily. As there are essentially no differences in the charge distribution within each substrate (n-propyl versus n-butyl sulfonyl groups), we surmise that it is the motif that primarily dictates differences in ΔH‡ and TΔS‡. Furthermore, as the NH2 group of both guests are anchored to similar extents (Table 1) this is unlikely to be important. Given this, then it is the differences in the environment around the electrophilic CH2 under SN2 attack that are key. In other words, the Eyring analysis points to the turn in the sulfonyl group in G3 impeding nucleophilic attack (higher ΔH‡) but rigidifying the guest somewhat making cyclization less entropically costly (lower TΔS‡).
How do the rates of cyclization within the capsule compare to that in free solution? To address this question we attempted the cyclization of G5 in benzene (as a surrogate for the walls of the container). However, we saw no evidence of cyclization at 60 °C in a timeframe twice that of cyclization. Presumably reaction is facilitated by polar aprotic solvents,31 but we did not examine such radically different conditions as any inferences about rate changes would be limited in nature.
Separately, we confirmed that the rates of hydrolysis of the sulfonates were slower in base-free conditions (Table 5) and only ∼2.9 times faster than the rate of cyclization of G5 (Table 2). Given this we probed the catalytic properties of the octa-sodium salt of 12 in the absence of excess base. We examined three sets of conditions — 10, 5 and 2 mol% capsule — reacting each mixture at 60 °C for 24 hours and recording the ratio of hydrolysis product and azepane determined by integration of the relevant 1H NMR spectroscopy signals. The resulting azepane to 6-aminohexan-1-ol ratios obtained were respectively: 1:1, 3:7, and 3:7. In other words, at a low 2 mol% host the turnover number (TON) was ∼35.32 Without removal of the product, product inhibition would ultimately inhibit the capsule. However, considering the degree of hydrolysis observed, we anticipate that the TON value of catalyst 12 is >35, and only limited under the conditions investigated because of substrate hydrolysis.
Guest motif differences aside, G6–G12 were expected to cyclize much more slowly than guests G1–G5. Indeed, we saw no evidence of the formation of azocane or azonane, and only in the case of guest G10 was aza-cycloundecane isolated. Rather, the guests were found to undergo hydrolysis to generate the corresponding amino alcohol, with only G10 leading to both hydrolysis and cyclization in a 4:1 ratio.
Regarding guest hydrolysis, recall that the presence or absence of a bound NH21H NMR signal was strongly suggestive of differences in its accessibility to bulk water; deeply bound NH2 groups in smaller guests gave sharp signals, whereas larger guests and/or cases where the NH2 group was located at the equatorial region led to no signal. Of the seven guests G6–G12, three showed no amino signal (G9, G11 and G12), with one of the remaining four exceptions (G10) also giving cyclized product. This suggested that these larger guests led to more open capsules, a loss of watertightness, and hydrolyses out-competing slow cyclization.
To gauge the extend of watertightness of the different capsules, we examined the position of the 1H NMR signal from host proton Hc (Fig. 1). Located at the cavitand rim, Hc is very sensitive to capsule formation. Thus for the guest n-decane it undergoes a −0.40 ppm shift from the free to the bound state (Δδ = δcomplex − δfree) because in the dimer capsule the Hc proton of one cavitand is shielded by the aromaticity of the other. Moreover, as the n-alkane guest is increased in size, so this shift is attenuated (to ∼ −0.17 ppm in the case of n-heptadecane). This highlights how the signal of proton Hc can be used as a surrogate for defining the degree of capsule watertightness.
A plot of the corresponding Δδ value for Hcversus logkhyd of guests G6–G12 shows a reasonable linear correlation (Fig. 6, R2 = 0.85). The two series G7–G9 and G10–G12 both trend as expected, with hydrolyses rates increasing and Δδ values decreasing as the guest size increases. Of these guests, only G10 gives cyclized product because it forms the tightest capsule by the metric of its hydrolysis rate inside the capsule (and third tightest by the metric of Hc Δδ value), and because the product of cyclization (aza-cycloundecane) contains the least amount of strain (after azepane).
 | ||
| Fig. 6 Plot of the 1H NMR Δδ value for host (1) proton Hc (Δδ = δcomplex − δfree) versus the log of the hydrolysis rate constant (logkHyd) of the bound guest with the 12 capsule. | ||
The one outlier in Fig. 6 is arguably G6. Despite being smaller than G7, its complex is apparently more open as measured by its hydrolysis rate (but just as tight in terms of Hc Δδ value). The methanesulfonyl group of G6 is not so deeply bound into the pole region as G7 (Table 1). Thus, we conclude that when the electrophilic CH2 of a guests resides away from the pole it not only favors SN2 attack by the NH2 group, but also attack by adventitious water that enters the capsule. Presumably hydrolysis is not a problem for slightly shorter G1 because the rate of formation of 7-membered azepane is much quicker than 8-membered azocane.
How do the observed rates of hydrolysis compare to the hydrolysis rates in the free state? To gauge this we averaged the rate constants of hydrolysis given in Table 5 ![[( with combining macron]](https://www.rsc.org/images/entities/char_0028_0304.gif) khyd = 1.45 × 10−3 s−1). Thus, the quotient of the bound hydrolysis rates and average rates of hydrolysis in the free state gives a measure of the watertightness of each complex. For 12. GX these quotients were calculated to one-two significant figures to be: G6 (
khyd = 1.45 × 10−3 s−1). Thus, the quotient of the bound hydrolysis rates and average rates of hydrolysis in the free state gives a measure of the watertightness of each complex. For 12. GX these quotients were calculated to one-two significant figures to be: G6 (![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 100), G7 (
100), G7 (![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) 900), G8 (100), G9 (
900), G8 (100), G9 (![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 80), G10 (
80), G10 (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1,000), G11 500), and G12 30). In other words, hydrolysis of guest G10 is attenuated over 104 times relative to the free state, whilst guest G12 is protected less than 30 fold. A comparison of the constitutional isomeric complexes undergoing hydrolysis reveal that in smaller constitutional pair G8 (J(NH2)-motif) and G10 (E-motif), the former is protected 3.7 times more than the latter. In contrast, larger G9 and G11 (both J(NH2)-motifs) are protected to essentially the same extent. This confirms that the motif of a bound guest influences the watertightness of the complex and hence its rate of hydrolysis.
1,000), G11 500), and G12 30). In other words, hydrolysis of guest G10 is attenuated over 104 times relative to the free state, whilst guest G12 is protected less than 30 fold. A comparison of the constitutional isomeric complexes undergoing hydrolysis reveal that in smaller constitutional pair G8 (J(NH2)-motif) and G10 (E-motif), the former is protected 3.7 times more than the latter. In contrast, larger G9 and G11 (both J(NH2)-motifs) are protected to essentially the same extent. This confirms that the motif of a bound guest influences the watertightness of the complex and hence its rate of hydrolysis.
Conclusions
To explore cyclization reactions within the yoctoliter inner-space of the 12 capsule, we have targeted the cyclization of amino-sulfonate guests. These guests allowed the exploration of the catalytic formation of difficult-to-cyclize medium-sized rings (7-11-membered), whilst at the same time providing information concerning the watertightness of the inner-space engendered by the dimeric assembly of octa-acid 1. Thus, 12 catalytically forms 7-membered azepane at only 2 mol%, with a turnover that is only limited by substrate hydrolysis in free solution. On the other hand, the formation of 8–11 membered cyclic amines is considerably slower and as a result substrate hydrolysis largely outcompetes cyclization. In these instances hydrolysis occurs in the inner-space of 12, but at a rate up to 104 times slower than the free state. Thus, using the capsule stoichiometrically, only the most watertight of complex forms the strained 11-membered aza-cycloundecane product. These results demonstrate for the first time that the use of a relatively large leaving group allows catalytic turnover for the synthesis of strained 7-membered cyclic amines. In the case of the most highly strained 8–11 membered rings, amine cyclization is slow relative to bound guest hydrolysis and stoichiometric amounts of container are required to form aza-cycloundecane. Simultaneously then, these cyclizations provide a valuable benchmark for the degree to which capsular complexes of 12 are watertight, and hence the rates of any desired reaction that are needed to ensure water ingress and alternative side reactions are to be avoided. Further studies of catalytic cyclizations of difficult-to-cyclize medium-sized rings are ongoing, and will be reported in due course.Author contributions
YAI performed the described syntheses, complexations, motif determinations, and assessment of complex reactivity. DR assisted with the synthesis of the guests G1–G12. BCG designed the overall study, advised YAI, and wrote the manuscript.Conflicts of interest
The authors declare no conflict of interest.Data availability
The data that support the findings of this study are available in the supplementary information (SI) of the article. Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc07390a.Acknowledgements
This work was supported in part by a grant from the National Science Foundation (CHE-2305018). A significant portion of the reported NMR data was collected on a 600 MHz NMR spectrometer funded by an NSF Major Research Instrumentation Program under the award number MRI 2216220.References
- (a) G. Olivo, G. Capocasa, D. Del Giudice, O. Lanzalunga and S. Di Stefano, New horizons for catalysis disclosed by supramolecular chemistry, Chem. Soc. Rev., 2021, 50(13), 7681–7724, 10.1039/d1cs00175b; (b) R. Ham, C. J. Nielsen, S. Pullen and J. N. H. Reek, Supramolecular Coordination Cages for Artificial Photosynthesis and Synthetic Photocatalysis, Chem. Rev., 2023, 123(9), 5225–5261, DOI:10.1021/acs.chemrev.2c00759; (c) C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Supramolecular catalysis in metal-ligand cluster hosts, Chem. Rev., 2015, 115(9), 3012–3035, DOI:10.1021/cr4001226; (d) E. A. Kataev and C. Müller, Recent advances in molecular recognition in water: artificial receptors and supramolecular catalysis, Tetrahedron, 2014, 70, 137–167, DOI:10.1016/j.tet.2013.11.010; (e) M. Yoshizawa, J. K. Klosterman and M. Fujita, Functional Molecular Flasks: New Properties and Reactions within Discrete, Self-Assembled Hosts, Angew. Chem., 2009, 48, 3418–3438 CrossRef CAS; (f) K. I. Assaf and W. M. Nau, Cucurbiturils: from synthesis to high-affinity binding and catalysis, Chem. Soc. Rev., 2015, 44(2), 394–418, 10.1039/c4cs00273c; (g) J. Meeuwissen and J. N. Reek, Supramolecular catalysis beyond enzyme mimics, Nat. Chem., 2010, 2(8), 615–621, DOI:10.1038/nchem.744.
- (a) R. L. Spicer, A. D. Stergiou, T. A. Young, F. Duarte, M. D. Symes and P. J. Lusby, Host–Guest-Induced Electron Transfer Triggers Radical-Cation Catalysis, J. Am. Chem. Soc., 2020, 142(5), 2134–2139, DOI:10.1021/jacs.9b11273; (b) J. Wang, T. A. Young, F. Duarte and P. J. Lusby, Synergistic Noncovalent Catalysis Facilitates Base-Free Michael Addition, J. Am. Chem. Soc., 2020, 142(41), 17743–17750, DOI:10.1021/jacs.0c08639; (c) T. A. Young, V. Martí-Centelles, J. Wang, P. J. Lusby and F. Duarte, Rationalizing the Activity of an “Artificial Diels-Alderase”: Establishing Efficient and Accurate Protocols for Calculating Supramolecular Catalysis, J. Am. Chem. Soc., 2020, 142(3), 1300–1310, DOI:10.1021/jacs.9b10302; (d) V. Marti-Centelles, R. L. Spicer and P. J. Lusby, Non-covalent allosteric regulation of capsule catalysis, Chem. Sci., 2020, 11(12), 3236–3240, 10.1039/d0sc00341g; (e) C. Ngai, C. M. Sanchez-Marsetti, W. H. Harman and R. J. Hooley, Supramolecular Catalysis of the oxa-Pictet-Spengler Reaction with an Endohedrally Functionalized Self-Assembled Cage Complex, Angew Chem. Int. Ed. Engl., 2020, 59(52), 23505–23509, DOI:10.1002/anie.202009553; (f) C. M. Hong, D. M. Kaphan, R. G. Bergman, K. N. Raymond and F. D. Toste, Conformational Selection as the Mechanism of Guest Binding in a Flexible Supramolecular Host, J. Am. Chem. Soc., 2017, 139(23), 8013–8021, DOI:10.1021/jacs.7b03812; (g) M. D. Levin, D. M. Kaphan, C. M. Hong, R. G. Bergman, K. N. Raymond and F. D. Toste, Scope and Mechanism of Cooperativity at the Intersection of Organometallic and Supramolecular Catalysis, J. Am. Chem. Soc., 2016, 138(30), 9682–9693, DOI:10.1021/jacs.6b05442; (h) D. M. Kaphan, M. D. Levin, R. G. Bergman, K. N. Raymond and F. D. Toste, A supramolecular microenvironment strategy for transition metal catalysis, Science, 2015, 350, 1235–1238 CrossRef CAS; (i) C. Zhao, F. D. Toste, K. N. Raymond and R. G. Bergman, Nucleophilic Substitution Catalyzed by a Supramolecular Cavity Proceeds with Retention of Absolute Stereochemistry, J. Am. Chem. Soc., 2016, 136(41), 14409–14412, DOI:10.1021/ja508799p; (j) Z. J. Wang, C. J. Brown, R. G. Bergman, K. N. Raymond and F. D. Toste, Hydroalkoxylation catalyzed by a gold(I) complex encapsulated in a supramolecular host, J. Am. Chem. Soc., 2011, 133(19), 7358–7360, DOI:10.1021/ja202055v; (k) C. J. Brown, G. M. Miller, M. W. Johnson, R. G. Bergman and K. N. Raymond, High-Turnover Supramolecular Catalysis by a Protected Ruthenium(II) Complex in Aqueous Solution, J. Am. Chem. Soc., 2011, 133(31), 11964–11966, DOI:10.1021/ja205257x; (l) M. D. Pluth, R. G. Bergman and K. N. Raymond, The Acid Hydrolysis Mechanism of Acetals Catalyzed by a Supramolecular Assembly in Basic Solution, J. Org. Chem., 2009, 74, 58–63 CrossRef CAS PubMed; (m) M. D. Pluth, R. G. Bergman and K. N. Raymond, Acid Catalysis in Basic Solution: A Supramolecular Host Promotes Orthoformate Hydrolysis, Science, 2007, 316, 85–88 CrossRef CAS PubMed; (n) M. Yoshizawa, M. Tamura and M. Fujita, Diels-Alder in Aqueous Molecular Hosts: Unusual Regioselectivity and Efficient Catalysis, Science, 2006, 312, 251–254 CrossRef CAS PubMed; (o) W. Cullen, M. C. Misuraca, C. A. Hunter, N. H. Williams and M. D. Ward, Highly efficient catalysis of the Kemp elimination in the cavity of a cubic coordination cage, Nat. Chem., 2016, 8(3), 231–236, DOI:10.1038/nchem.2452; (p) D. Fiedler, R. G. Bergman and K. N. Raymond, Supramolecular Catalysis of a Unimolecular Transformation: Aza-Cope Rearrangement within a Self-Assembled Host, Angew. Chem., Int. Ed., 2004, 43(48), 6748–6751, DOI:10.1002/anie.200461776; (q) S. M. Bierschenk, R. G. Bergman, K. N. Raymond and F. D. Toste, A Nanovessel-Catalyzed Three-Component Aza-Darzens Reaction, J. Am. Chem. Soc., 2020, 142(2), 733–737, DOI:10.1021/jacs.9b13177; (r) S. M. Bierschenk, J. Y. Pan, N. S. Settineri, U. Warzok, R. G. Bergman, K. N. Raymond and F. D. Toste, Impact of Host Flexibility on Selectivity in a Supramolecular Host-Catalyzed Enantioselective aza-Darzens Reaction, J. Am. Chem. Soc., 2022, 144(25), 11425–11433, DOI:10.1021/jacs.2c04182; (s) D. M. Kaphan, F. D. Toste, R. G. Bergman and K. N. Raymond, Enabling New Modes of Reactivity via Constrictive Binding in a Supramolecular-Assembly-Catalyzed Aza-Prins Cyclization, J. Am. Chem. Soc., 2015, 137(29), 9202–9205, DOI:10.1021/jacs.5b01261.
- (a) I. Nemethova, D. Schmid and K. Tiefenbacher, Supramolecular Capsule Catalysis Enables the Exploration of Terpenoid Chemical Space Untapped by Nature, Angew Chem. Int. Ed. Engl., 2023, 62(14), e202218625, DOI:10.1002/anie.202218625; (b) T. R. Li, G. Piccini and K. Tiefenbacher, Supramolecular Capsule-Catalyzed Highly beta-Selective Furanosylation Independent of the S(N)1/S(N)2 Reaction Pathway, J. Am. Chem. Soc., 2023, 145(7), 4294–4303, DOI:10.1021/jacs.2c13641; (c) T.-R. Li, F. Huck, G. Piccini and K. Tiefenbacher, Mimicry of the proton wire mechanism of enzymes inside a supramolecular capsule enables β-selective O-glycosylations, Nat. Chem., 2022, 14(9), 985–994, DOI:10.1038/s41557-022-00981-6; (d) L. D. Syntrivanis, I. Nemethova, D. Schmid, S. Levi, A. Prescimone, F. Bissegger, D. T. Major and K. Tiefenbacher, Four-Step Access to the Sesquiterpene Natural Product Presilphiperfolan-1β-ol and Unnatural Derivatives via Supramolecular Catalysis, J. Am. Chem. Soc., 2020, 142(12), 5894–5900, DOI:10.1021/jacs.0c01464; (e) M. De Rosa, S. Gambaro, A. Soriente, P. Della Sala, V. Iuliano, C. Talotta, C. Gaeta, A. Rescifina and P. Neri, Carbocation catalysis in confined space: activation of trityl chloride inside the hexameric resorcinarene capsule, Chem. Sci., 2022, 13(29), 8618–8625, 10.1039/d2sc02901d; (f) H. Chen, T.-R. Li, N. Sakai, C. Besnard, L. Guénée, M. Pupier, J. Viger-Gravel, K. Tiefenbacher and S. Matile, Decoded fingerprints of hyperresponsive, expanding product space: polyether cascade cyclizations as tools to elucidate supramolecular catalysis, Chem. Sci., 2022, 13(35), 10273–10280, 10.1039/D2SC03991E; (g) L. Catti and K. Tiefenbacher, Brønsted Acid-Catalyzed Carbonyl-Olefin Metathesis inside a Self-Assembled Supramolecular Host, Angew. Chem., Int. Ed., 2018, 57(44), 14589–14592, DOI:10.1002/anie.201712141; (h) Q. Zhang and K. Tiefenbacher, Sesquiterpene Cyclizations inside the Hexameric Resorcinarene Capsule: Total Synthesis of δ-Selinene and Mechanistic Studies, Angew. Chem., Int. Ed., 2019, 58(36), 12688–12695, DOI:10.1002/anie.201906753; (i) Q. Zhang, L. Catti, J. Pleiss and K. Tiefenbacher, Terpene Cyclizations inside a Supramolecular Catalyst: Leaving-Group-Controlled Product Selectivity and Mechanistic Studies, J. Am. Chem. Soc., 2017, 139(33), 11482–11492, DOI:10.1021/jacs.7b04480; (j) R. J. Hooley and J. Rebek Jr, A Deep Cavitand Catalyzes the Diels-Alder Reaction of Bound Maleimides, Org. Biomol. Chem., 2007, 5, 3631–3636 RSC; (k) F. R. P. Crisostomo, A. Lkedo, S. R. Shenoy, T. Iwasawa and J. Rebek Jr, Recognition and Organocatalysis with a Synthetic Cavitand Receptor, J. Am. Chem. Soc., 2009, 131, 7402–7410 CrossRef; (l) S. Kamioka, D. Ajami and J. Rebek Jr, Autocatalysis and organocatalysis with synthetic structures, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 541–542 CrossRef CAS; (m) Q. Zhang, L. Catti, V. R. I. Kailac and K. Tiefenbacher, To catalyze or not to catalyze: elucidation of the subtle differences between the hexameric capsules of pyrogallolarene and resorcinarene, Chem. Sci., 2017, 8, 1653–1657, 10.1039/c6sc04565k; (n) Q. Zhang and K. Tiefenbacher, Terpene cyclization catalysed inside a self-assembled cavity, Nat. Chem., 2015, 7(3), 197–202, DOI:10.1038/nchem.2181; (o) Q. Zhang and K. Tiefenbacher, Hexameric resorcinarene capsule is a Bronsted acid: investigation and application to synthesis and catalysis, J. Am. Chem. Soc., 2013, 135(43), 16213–16219, DOI:10.1021/ja4080375.
- Q. Shi, M. P. Mower, D. G. Blackmond and J. Rebek Jr, Water-soluble cavitands promote hydrolyses of long-chain diesters, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(33), 9199–9203, DOI:10.1073/pnas.1610006113.
- (a) N. E. Ernst and B. C. Gibb, Water Runs Deep, in Supramolecular Chemistry in Water, ed. Kubik, S., Wiley-VCH, 2019; pp. 1–33 Search PubMed; (b) M. B. Hillyer and B. C. Gibb, Molecular Shape and the Hydrophobic Effect, Annu. Rev. Phys. Chem., 2016, 67, 307–329, DOI:10.1146/annurev-physchem-040215-112316; (c) D. Ben-Amotz, Water-Mediated Hydrophobic Interactions, Annu. Rev. Phys. Chem., 2016, 67, 617–638, DOI:10.1146/annurev-physchem-040215-112412; (d) G. Hummer, Under water's influence, Nat. Chem., 2010, 2, 906–907 CrossRef CAS PubMed; (e) G. Hummer, S. Garde, A. E. García and L. R. Pratt, New perspectives on hydrophobic effects, Chem. Phys., 2000, 258(2), 349–370, DOI:10.1016/S0301-0104(00)00115-4.
- (a) S. Mosca, Y. Yu, J. V. Gavette, K. D. Zhang and J. Rebek Jr, A Deep Cavitand Templates Lactam Formation in Water, J. Am. Chem. Soc., 2015, 137, 14582–14585, DOI:10.1021/jacs.5b10028; (b) N. W. Wu and J. Rebek Jr, Cavitands as Chaperones for Monofunctional and Ring-Forming Reactions in Water, J. Am. Chem. Soc., 2016, 138(24), 7512–7515, DOI:10.1021/jacs.6b04278; (c) N.-W. Wu, I. D. Petsalakis, G. Theodorakopoulos, Y. Yu and J. Rebek Jr., Cavitands as Containers for α,ω-Dienes and Chaperones for Olefin Metathesis, Angew. Chem., Int. Ed., 2018, 57(46), 15091–15095, DOI:10.1002/anie.201808265.
- (a) D. Masseroni, S. Mosca, M. P. Mower, D. G. Blackmond and J. Rebek Jr., Cavitands as Reaction Vessels and Blocking Groups for Selective Reactions in Water, Angew Chem. Int. Ed. Engl., 2016, 55(29), 8290–8293, DOI:10.1002/anie.201602355; (b) V. Angamuthu, M. Petroselli, F.-U. Rahman, Y. Yu and J. Rebek, Binding orientation and reactivity of alkyl α,ω-dibromides in water-soluble cavitands, Org. Biomol. Chem., 2019, 17(21), 5279–5282, 10.1039/C9OB01018A; (c) V. Angamuthu, F.-U. Rahman, M. Petroselli, Y. Li, Y. Yu and J. Rebek, Mono epoxidation of α,ω-dienes using NBS in a water-soluble cavitand, Org. Chem. Front., 2019, 6(18), 3220–3223, 10.1039/C9QO00849G; (d) M. Petroselli, V. Angamuthu, F.-U. Rahman, X. Zhao, Y. Yu and J. Rebek Jr, Radical Reactions in Cavitands Unveil the Effects of Affinity on Dynamic Supramolecular Systems, J. Am. Chem. Soc., 2020, 142(5), 2396–2403, DOI:10.1021/jacs.9b11595; (e) J.-M. Yang, Y.-Q. Chen, Y. Yu, P. Ballester and J. Rebek Jr, Rigidified Cavitand Hosts in Water: Bent Guests, Shape Selectivity, and Encapsulation, J. Am. Chem. Soc., 2021, 143(46), 19517–19524, DOI:10.1021/jacs.1c09226.
- (a) Y. Gao, Y. Zhu, M. Zhao, J. Rebek Jr. and Y. Yu, Selective Aliphatic Aldimine Formation and Stabilization by a Hydrophobic Capsule in Water, J. Am. Chem. Soc., 2025, 147(15), 12989–12995, DOI:10.1021/jacs.5c02779; (b) K. Kanagaraj, R. Wang, M.-K. Zhao, P. Ballester, J. Rebek Jr. and Y. Yu, Selective Binding and Isomerization of Oximes in a Self-Assembled Capsule, J. Am. Chem. Soc., 2023, 145(10), 5816–5823, DOI:10.1021/jacs.2c12907; (c) F.-U. Rahman, R. Wang, H.-B. Zhang, O. Brea, F. Himo, J. Rebek Jr. and Y. Yu, Binding and Assembly of a Benzotriazole Cavitand in Water, Angew. Chem., Int. Ed., 2022, 61(29), e202205534, DOI:10.1002/anie.202205534.
- W. Yao, K. Wang, Y. A. Ismaiel, R. Wang, X. Cai, M. Teeler and B. C. Gibb, Electrostatic Potential Field Effects on Amine Macrocyclizations within Yoctoliter Spaces: Supramolecular Electron Withdrawing/Donating Groups, J. Phys. Chem. B, 2021, 125(32), 9333–9340, DOI:10.1021/acs.jpcb.1c05238.
- X. Cai, R. Kataria and B. C. Gibb, Intrinsic and Extrinsic Control of the pKa of Thiol Guests inside Yoctoliter Containers, J. Am. Chem. Soc., 2020, 142(18), 8291–8298, DOI:10.1021/jacs.0c00907.
- (a) Y. A. Ismaiel and B. C. Gibb, Water-Soluble Yoctoliter Reaction Flasks, in Supramolecular Catalysis, ed. van Leeuwen, P. W. N. M. and Raynal, M., Wiley, 2022; pp. 519–536 Search PubMed; (b) L. S. Kaanumalle, C. L. D. Gibb, B. C. Gibb and V. Ramamurthy, Controlling Photochemistry with Distinct Hydrophobic Nanoenvironments, J. Am. Chem. Soc., 2004, 126(44), 14366–14367, DOI:10.1021/ja0450197; (c) L. S. Kaanumalle, C. L. D. Gibb, B. C. Gibb and V. Ramamurthy, A Hydrophobic Nanocapsule Controls the Photophysics of Aromatic Molecules by Suppressing Their Favored Solution Pathways, J. Am. Chem. Soc., 2005, 127(11), 3674–3675, DOI:10.1021/ja0425381; (d) A. Natarajan, L. S. Kaanumalle, S. Jockusch, C. L. D. Gibb, B. C. Gibb, N. J. Turro and V. Ramamurthy, Controlling Photoreactions with Restricted Spaces and Weak Intermolecular Forces: Remarkable Product Selectivity during Oxidation of Olefins by Singlet Oxygen, J. Am. Chem. Soc., 2007, 129, 4132–4133 CrossRef CAS PubMed; (e) C. L. D. Gibb, A. K. Sundaresan, V. Ramamurthy and B. C. Gibb, Templation of the Excited-State Chemistry of α-(n-Alkyl) Dibenzyl Ketones: How Guest Packing within a Nanoscale Supramolecular Capsule Influences Photochemistry, J. Am. Chem. Soc., 2008, 130(12), 4069–4080, DOI:10.1021/ja7107917; (f) K. Ito, T. Nishioka, M. Akita, A. Kuzume, K. Yamamoto and M. Yoshizawa, An aromatic micelle with bent pentacene-based panels: encapsulation of perylene bisimide dyes and graphene nanosheets, Chem. Sci., 2020, 11(26), 6752–6757, 10.1039/D0SC01748E; (g) L. Catti, N. Kishida, T. Kai, M. Akita and M. Yoshizawa, Polyaromatic nanocapsules as photoresponsive hosts in water, Nat. Commun., 2019, 10(1), 1948, DOI:10.1038/s41467-019-09928-x; (h) Y. Satoh, L. Catti, M. Akita and M. Yoshizawa, A Redox-Active Heterocyclic Capsule: Radical Generation, Oxygenation, and Guest Uptake/Release, J. Am. Chem. Soc., 2019, 141(31), 12268–12273, DOI:10.1021/jacs.9b03419.
- (a) C. L. Gibb and B. C. Gibb, Well-defined, organic nanoenvironments in water: the hydrophobic effect drives a capsular assembly, J. Am. Chem. Soc., 2004, 126(37), 11408–11409, DOI:10.1021/ja0475611; (b) C. L. D. Gibb and B. C. Gibb, Templated Assembly of Water-Soluble Nano-Capsules: Inter-Phase Sequestration, Storage, and Separation of Hydrocarbon Gases, J. Am. Chem. Soc., 2006, 128(51), 16498–16499, DOI:10.1021/ja0670916.
- C. L. D. Gibb and B. C. Gibb, Straight-Chain Alkanes Template the Assembly of Water-Soluble Nano-Capsules, Chem. Commun., 2007, 1635–1637 RSC.
- S. Liu, D. H. Russell, N. F. Zinnel and B. C. Gibb, Guest packing motifs within a supramolecular nanocapsule and a covalent analogue, J. Am. Chem. Soc., 2013, 135(11), 4314–4324, DOI:10.1021/ja310741q.
- (a) H. Tang, C. S. de Oliveira, G. Sonntag, C. L. D. Gibb, B. C. Gibb and C. Bohne, Dynamics of a Supramolecular Capsule Assembly with Pyrene, J. Am. Chem. Soc., 2012, 134(12), 5544–5547, DOI:10.1021/ja301278p; (b) S. S. Thomas, H. Tang, A. Gaudes, S. B. Baggesen, C. L. D. Gibb, B. C. Gibb and C. Bohne, Tuning the Binding Dynamics of a Guest–Octaacid Capsule through Noncovalent Anchoring, J. Phys. Chem. Lett., 2017, 8(12), 2573–2578, DOI:10.1021/acs.jpclett.7b00917.
- Although 'watertightness' can be defined in a number of different ways, as we discuss below, here we use the ratio of the hydrolysis rates of free and bound guest to assess how accessible the inner space of a complex is watertight.
- K. Wang, X. Cai, W. Yao, D. Tang, R. Kataria, H. S. Ashbaugh, L. D. Byers and B. C. Gibb, Electrostatic Control of Macrocyclization Reactions within Nanospaces, J. Am. Chem. Soc., 2019, 141(16), 6740–6747, DOI:10.1021/jacs.9b02287.
- (a) K. Wang, W. Yao, X. Cai and B. C. Gibb, Salt effects on the rates of a thiol cyclisation reaction within a yocto-litre inner-space, Supramol. Chem., 2023, 34(2), 87–93, DOI:10.1080/10610278.2023.2231120; (b) J. M. Yang, Y. Yu and J. Rebek Jr, Selective Macrocycle Formation in Cavitands, J. Am. Chem. Soc., 2021, 143(5), 2190–2193, DOI:10.1021/jacs.0c12302.
- S. Liu, H. Gan, A. T. Hermann, S. W. Rick and B. C. Gibb, Kinetic Resolution of Constitutional Isomers Controlled by Selective Protection inside a Supramolecular Nanocapsule, Nat. Chem., 2010, 2(10), 847–852, DOI:10.1038/nchem.751.
- (a) C. L. D. Gibb and B. C. Gibb, Well Defined, Organic Nano-Environments in Water: The Hydrophobic Effect Drives a Capsular Assembly, J. Am. Chem. Soc., 2004, 126, 11408–11409 CrossRef CAS PubMed; (b) C. L. D. Gibb, A. J. Hebert, Y. A. Ismaiel, P. Prusty, T. Wyshel and B. C. Gibb, An updated synthesis of octa-acid, Supramol. Chem., 2023, 34(11–12), 480–483, DOI:10.1080/10610278.2024.2379347; (c) M. B. Hillyer, C. L. Gibb, P. Sokkalingam, J. H. Jordan, S. E. Ioup, J. T. Mague and B. C. Gibb, Synthesis of Water-Soluble Deep-Cavity Cavitands, Org. Lett., 2016, 18(16), 4048–4051, DOI:10.1021/acs.orglett.6b01903.
- M. H. Levitt Spin Dynamics: Basics of Nuclear Magnetic Resonance; Wiley, 2013 CrossRef CAS; E. Demetriou, A. Kujawa and X. Golay, Pulse sequences for measuring exchange rates between proton species: From unlocalised NMR spectroscopy to chemical exchange saturation transfer imaging, Prog. Nucl. Magn. Reson. Spectrosc., 2020, 120–121, 25–71, DOI:10.1016/j.pnmrs.2020.06.001.
- J. W. Barnett, B. C. Gibb and H. S. Ashbaugh, Succession of Alkane Conformational Motifs Bound within Hydrophobic Supramolecular Capsular Assemblies, J. Phys. Chem. B, 2016, 120(39), 10394–10402, DOI:10.1021/acs.jpcb.6b06496.
- S. Liu, D. H. Russell, N. F. Zinnel and B. C. Gibb, Guest Packing Motifs within a Supramolecular Nanocapsule and a Covalent Analogue, J. Am. Chem. Soc., 2013, 135(11), 4314–4324, DOI:10.1021/ja310741q.
- (a) L. Trembleau and J. Rebek Jr, Helical Conformation of Alkanes in a Hydrophobic Cavitand, Science, 2003, 301, 1219–1220 CrossRef CAS; (b) A. Scarso, L. Trembleau and J. Rebek Jr, Encapsulation Induces Helical Folding of Alkanes, Angew. Chem., Int. Ed., 2003, 42, 5499–5502 CrossRef CAS; (c) D. Ajami and J. Rebek Jr, Coiled Molecules in Spring Loaded Devices, J. Am. Chem. Soc., 2006, 128, 15038–15039 CrossRef CAS PubMed; (d) A. Scarso, L. Trembleau and J. Rebek Jr, Helical Folding of Alkanes In a Self-Assembled, Cylinderical Capsule, J. Am. Chem. Soc., 2004, 126, 13512–13518 CrossRef CAS.
- The Δδ-plot for G3 (Figure 2a) highlights a common feature for all of the sulfonate guests examined possessing an alkylsulfonyl group larger than methyl; namely the signal from the β-protons of the alkylsulfonyl group shows a characteristic smaller upfield shift relative to the analogous n-alkane. Based on ab initio calculations (SI Section 10) of the NMR chemical shifts of simple sulfonates as a function of torsional angle (O–S(O)2–CH2–CH2) we attribute this ‘uptick’ in the Δδ data to the positioning of the methylene relative to the sulfonyl group in the turn conformation.
- Another possible NCI between host and guest that may be influencing the guest motif are weak hydrogen bonds between the sulfonyl (sp2) oxygens and the inward pointing C–Hb atoms (Figure 1).
- Guest G4 — the one example with an n-butyl chain — also adopts an E-motif, suggesting that like the n-pentyl group, the terminal Me of the n-butyl chain is a stronger anchor that its α-CH2 group.
- (a) G. Salomon, Kinetics of ring-formation and polymerisation in solution, Trans. Faraday Soc., 1936, 32(0), 153–175, 10.1039/TF9363200153; (b) G. Salomon, Die Kinetik der Bildung vielgliedriger Ringe. 3. Mitteilung zur Kenntnis der Bildungsleichtigkeit cyclischer Imine, Helv. Chim. Acta, 1936, 19(1), 743–793, DOI:10.1002/hlca.193601901106.
- (a) C. Galli and L. Mandolini, The Role of Ring Strain on the Ease of Ring Closure of Bifunctional Chain Molecules, Eur. J. Org. Chem., 2000, 2000(18), 3117–3125 CrossRef . https://doi.org/10.1002/1099-0690(200009)2000:18<3117::AID-EJOC3117>3.0.CO; (b) E. V. Anslyn; D. A. DoughertyModern Physical Organic Chemistry; University Science Books, 2006 Search PubMed.
- P. Suating, N. E. Ernst, B. D. Alagbe, H. A. Skinner, J. T. Mague, H. S. Ashbaugh and B. C. Gibb, On the Nature of Guest Complexation in Water: Triggered Wetting-Water-Mediated Binding, J. Phys. Chem. B, 2022, 126(16), 3150–3160, DOI:10.1021/acs.jpcb.2c00628.
- (a) In the last fifty years, access to azepane has typically been as a by-product in commercial processes employed for the manufacture of hexamethylenediamine (Nylon 6.6 synthesis). Alternatively, direct synthesis has typically involved heterogeneous catalysis ; (b) T. K. Bui, C. Concilio and G. Porzi, Cyclization of α,ω-aliphatic diamines and conversion of primary amines to symmetrical tertiary amines by a homogeneous ruthenium catalyst, J. Org. Chem., 1981, 46, 1759–1760, DOI:10.1021/jo00321a056 . or reduction of readily synthesized ε-caprolactam.; (c) T. Ayusawa, T. Shimodaira and J. Kanetaka, Production of Hexamethylenimine, Nat. Prod. R&D, 1976, 15, 295–298, DOI:10.1021/i360060a015.
- Correspondingly, at 5 and 10 mol% catalyst the TON = 14 and 5 respectively.
| This journal is © The Royal Society of Chemistry 2026 |