
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA radical strategy to the synthesis of bicyclo[1.1.1]pentyl C-glycosides
Giulio
Goti
 *a,
Alessia
Marrese†
a,
Simone
Baldon†
a,
Patricia
Gómez Roibás
ab,
Giorgio
Pelosi
c,
Andrea
Sartorel
a and
Luca
Dell'Amico
*a
*a,
Alessia
Marrese†
a,
Simone
Baldon†
a,
Patricia
Gómez Roibás
ab,
Giorgio
Pelosi
c,
Andrea
Sartorel
a and
Luca
Dell'Amico
*a
aDepartment of Chemical Sciences, University of Padova, Via Francesco Marzolo 1, 35131 Padova, Italy. E-mail: giulio.goti@unipd.it; luca.dellamico@unipd.it
bCentro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS), Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain
cDepartment of Chemistry, Life Sciences and Environmental Sustainability, University of Parma, Parco Area delle Scienze 17, 43124 Parma, Italy
First published on 17th November 2025
Abstract
Aryl C-glycosides, in which carbohydrates are directly linked to aryl fragments through a stable C–C bond, are an important class of biologically active molecules widely found in nature. These compounds exhibit resistance to (enzymatic) hydrolysis, a property that has been successfully leveraged in the development of metabolically stable drugs. On the other hand, despite their potential, three-dimensional analogues of aryl C-glycosides remain largely overlooked. Here, we present a three-component radical strategy that grants access to this underexplored chemical space. Specifically, glycosyl bromides serve as a source of glycosyl radicals, which can react with [1.1.1]propellane and a suitable SOMOphile to afford bicyclopentyl C-glycosides. These C(sp3)-rich analogues replace a planar aryl ring with a three-dimensional bicyclopentyl moiety, which is expected to enhance physicochemical properties. The protocol is practical, mild, and amenable to scalable synthesis in continuous flow. Experimental and computational studies support a radical chain mechanism under kinetic control.
Introduction
Aryl C-glycosides are important compounds encompassing structurally diverse natural products and drugs.1 The distinctive feature of these molecules – i.e. the presence of a stable C–C bond connecting a saccharide unit to an aryl moiety – confers resistance towards hydrolytic enzymes, which makes them privileged glycomimetic candidates in drug development programs (Scheme 1a).2 Given their relevance, numerous synthetic methods for the efficient preparation of aryl C-glycosides have been developed in recent years, most of them achieving the formation of the aryl C-glycoside linkage through cationic, anionic, and radical glycosyl intermediates, or with the use of transition metal catalysts.1b,3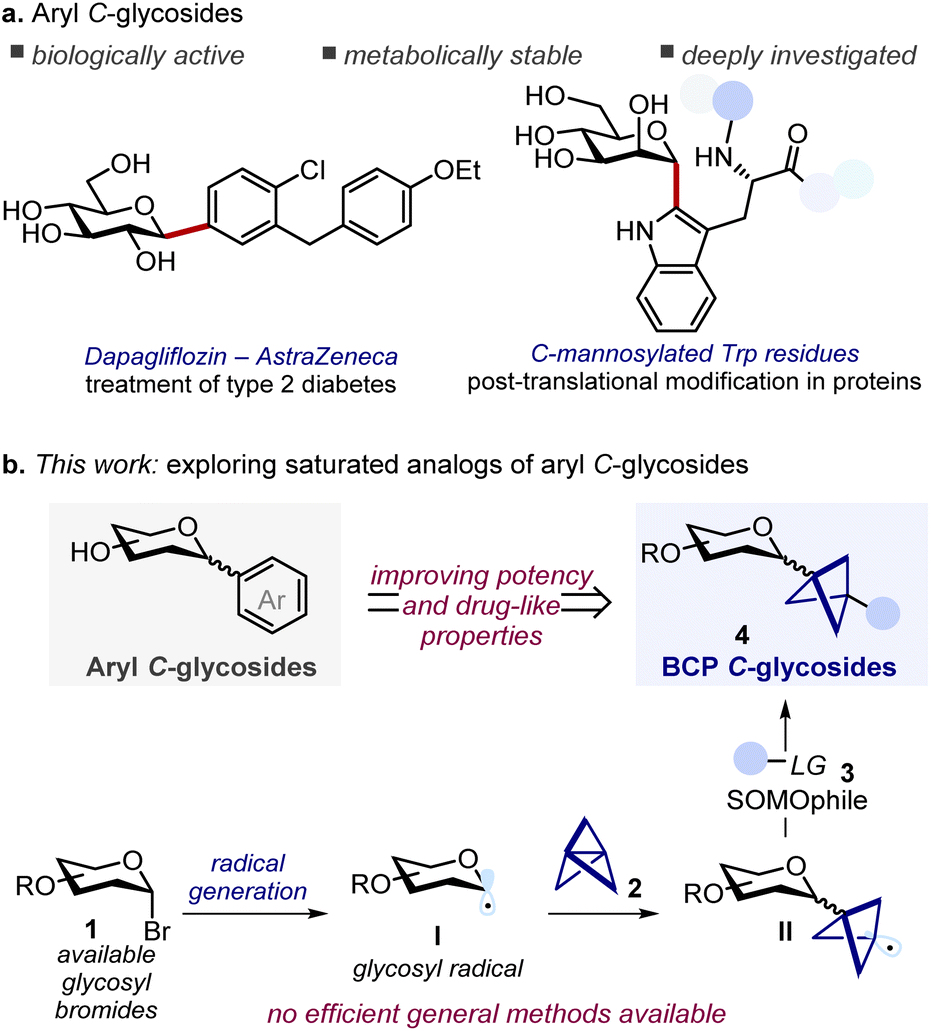 | ||
| Scheme 1 (a) Representative examples of aryl C-glycosides as natural products and drugs. (b) Three-component radical approach towards bicyclo[1.1.1]pentyl (BCP) C-glycosides as saturated analogs. Trp: tryptophan; LG: leaving group; SOMO: singly occupied molecular orbital; BCP: bicyclo[1.1.1]pentyl. | ||
On the other hand, the replacement of planar conjugated moieties with three-dimensional fragments has been established as a potent strategy in medicinal chemistry to improve the physicochemical properties of lead compounds and increase their success rate in clinical trials.4 Driven by this principle, in the past few years synthetic chemists have focused on the development of C(sp3) rich bioisosteres able to replace planar aromatic cores, which are highly represented motifs in drugs.5 Among these, the saturated bicyclo[1.1.1]pentane (BCP) is arguably the most studied bioisostere of phenyl rings.6 When 1,3-disubstituted at the bridgehead positions, this core acts as a rigid spacer projecting the substituents with exit vectors at approximately 180° that well mimic the spatial arrangement of para-disubstituted arenes. As such, bioisosteric replacement strategies with BCPs have been successfully applied in drug discovery programs, allowing the preparation of saturated analogs with unaltered targeting ability that show improved potency or enhanced physicochemical properties (e.g. solubility, and metabolic stability), and that can enable patent circumvention.7
Despite their potential, the development of saturated analogues of aryl C-glycosides has remained largely underdeveloped, which precludes the study of their properties and implementation in drug development campaigns. A possible explanation for this synthetic gap is the paucity of methods for the introduction of highly three-dimensional fragments at the anomeric position of saccharides. Indeed, while C-glycosylation strategies for the functionalization with alkyl, alkenyl, and alkynyl groups are well developed (either via polar or radical chemistry),3b,d,8 these approaches are mainly limited to the instalment of linear flexible carbon chains that do not have the requisites to properly mimicking the defined exit vectors provided by rigid aromatic scaffolds.
Restricting our focus to saturated C-glycosides featuring a BCP unit directly linked to the anomeric position, we envisioned that this moiety could be conveniently introduced by functionalization of [1.1.1]propellane (Scheme 1b).9 This strained precursor exhibits omniphilic reactivity, being able to react with electrophiles, strong nucleophiles, and radicals. Notably, radical addition to [1.1.1]propellane proceeds under remarkably mild conditions,10 making it particularly suited for functionalizing complex substrates such as glycosides.3d,11 In this regard, rare preparations of BCP C-glycosides have been independently reported by the Anderson and Molander groups by ring opening of [1.1.1]propellane with glycosyl radicals,10n,o although both protocols involve the use of either unstable radical precursors (e.g. glycosyl iodides), transition metal-based photocatalysts, or expensive SOMOphiles (e.g. Suginome reagent). Notably, although these methods targeted a broad range of alkyl iodide and bromide substrates, their application to C-glycosyl derivatives was limited. Both reported just a single glucoside derivative and, in one case, with an unusual β-stereoselectivity that we believe misassigned. Furthermore, the scalability and further derivatization of the C-glycoside products were not investigated. With this work we aim to address these limitations providing a general versatile strategy to the efficient synthesis of BCP C-glycosides to enable their study and characterization.12
Taking inspiration from the work of the Molander group, we recognized that readily available glucosyl bromide 1 could serve as a radical precursor to glycosyl radical I. This reactive intermediate would then engage in a radical addition to [1.1.1]propellane 2 giving the BCP radical II.10n,o Finally, reaction with a suitable SOMOphile 3 would deliver the desired BCP C-glycoside 4 (Scheme 1b). We reasoned that careful selection of the SOMOphile 3 would be key to the success of this three-component radical approach. Here, two key challenges must be considered: (i) glycosyl radical I has to discriminate between propellane 2 and SOMOphile 3, leading to the selective formation of the key intermediate II; (ii) II must then react selectively with SOMOphile 3 over propellane 2, thus preventing staffane side-products. Herein we detail our efforts in implementing this strategy to provide general and efficient access to saturated aryl C-glycoside analogs. Two distinct protocols for the hydroglycosylation and carboboration of [1.1.1]propellane were developed, providing synthetically useful sugar-based building blocks that enabled smooth diversification into a small library of BCP C-glycoside derivatives. A thorough mechanistic investigation is also presented.
Results and discussion
Strategy validation – hydroglycosylation of propellanes
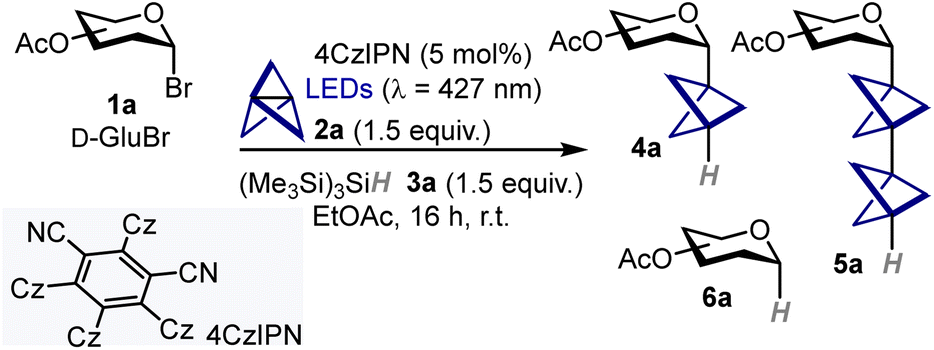
To test the feasibility of our approach, we began investigating the model reaction between acetobromo-α-D-glucose 1a (0.1 mmol), [1.1.1]propellane 2a, and tris(trimethylsilyl)silane 3a (Table 1).13 After optimization (for details, see SI, Section C.1) we found that using a slight excess of both propellane 2a and silane 3a (1.5 equiv.), along with 4CzIPN as photocatalyst (PC)14 under blue LED (λmax = 427 nm) irradiation provided BCP C-glucoside 4a in good yield and stereoselectivity (71% yield; α:β, 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as judged by 1H NMR analysis of the crude mixture (Table 1, entry 1), while minimizing the formation of staffane 5a and 1-deoxyglucoside 6a (9% and 20% yield, respectively). Notably, we found that the use of trialkylamines as hydrogen atom donors suppressed the formation of the reduced side-product 6a, although product 4a was obtained in diminished yield and with substantial formation of staffane 5a (Table 1, entries 2,3). Since this latter side-product is particularly difficult to purify by flash column chromatography from the desired C-glycoside 4a, we selected silane 3a as the best hydrogen atom transfer (HAT) donor species. Stoichiometry was also found to be a key parameter, as increasing the excess of either propellane 2a and silane 3a led to decreased performance (Table 1, entries 4–6). Pleasingly, only a slight decrease in yield was observed when performing the reaction on a 0.2 mmol scale (63% yield, Table 1, entry 7). Finally, control experiments revealed that the reaction can also proceed in the absence of the PC and light, albeit with lower efficiency and increased side-products formation (Table 1, entries 8–10). These results indicate that a radical chain reaction can be thermally initiated, although the process is relatively inefficient and the use of 4CzIPN as photoinitiator is beneficial.
:1) as judged by 1H NMR analysis of the crude mixture (Table 1, entry 1), while minimizing the formation of staffane 5a and 1-deoxyglucoside 6a (9% and 20% yield, respectively). Notably, we found that the use of trialkylamines as hydrogen atom donors suppressed the formation of the reduced side-product 6a, although product 4a was obtained in diminished yield and with substantial formation of staffane 5a (Table 1, entries 2,3). Since this latter side-product is particularly difficult to purify by flash column chromatography from the desired C-glycoside 4a, we selected silane 3a as the best hydrogen atom transfer (HAT) donor species. Stoichiometry was also found to be a key parameter, as increasing the excess of either propellane 2a and silane 3a led to decreased performance (Table 1, entries 4–6). Pleasingly, only a slight decrease in yield was observed when performing the reaction on a 0.2 mmol scale (63% yield, Table 1, entry 7). Finally, control experiments revealed that the reaction can also proceed in the absence of the PC and light, albeit with lower efficiency and increased side-products formation (Table 1, entries 8–10). These results indicate that a radical chain reaction can be thermally initiated, although the process is relatively inefficient and the use of 4CzIPN as photoinitiator is beneficial.
|
|
||||
|---|---|---|---|---|
| Entry | Deviation from standard conditions | 4a (%) | 5a (%) | 6a (%) |
| a Reactions were performed using 0.1 mmol of 1a, [1.1.1]propellane 2a as solution in Et2O, and 0.4 mL of EtOAc. b Determined by 1H NMR analysis of the crude mixture using trichloroethylene as the internal standard. 4CzIPN: 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene; Cz: carbazoyl; LED: light-emitting diode; r.t.: room temperature. | ||||
| 1 | — | 71 | 9 | 20 |
| 2 | iPr2EtN instead of (Me3Si)3SiH | 33 | 28 | — |
| 3 | Et3N instead of (Me3Si)3SiH | 37 | 17 | — |
| 4 | 3 equiv. of 2a | 48 | 24 | 23 |
| 5 | 3 equiv. of 3a | 53 | 11 | 36 |
| 6 | 3 equiv. of 2a and 3a | 59 | 11 | 28 |
| 7 | 0.2 mmol | 63 | 7 | 30 |
| 8 | No 4CzIPN | 44 | 8 | 45 |
| 9 | No light | 34 | 13 | 48 |
| 10 | No PC, no light, 40 °C | 40 | 18 | 21 |
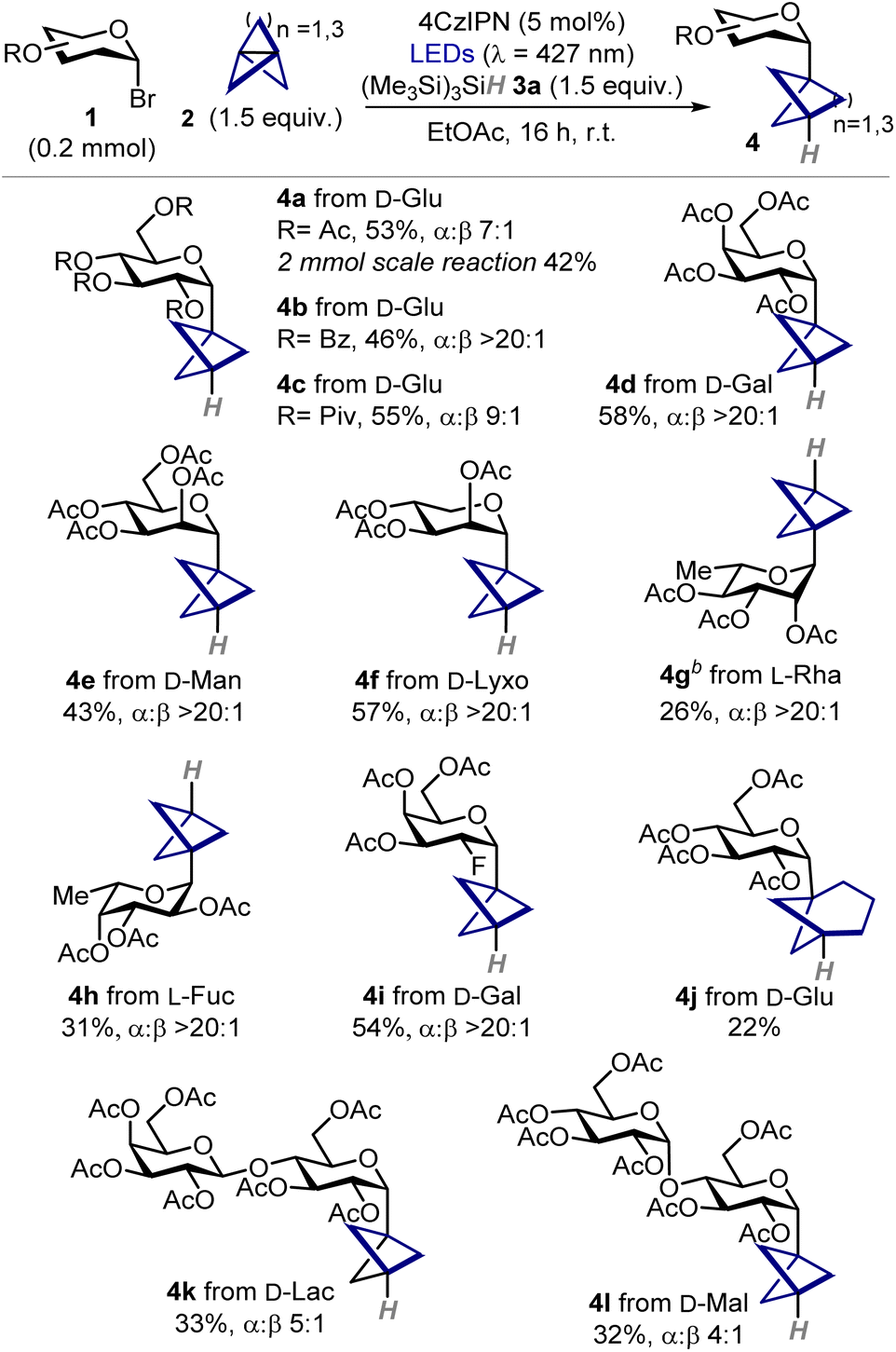
With the best conditions in hand, we sought to evaluate the applicability and generality of this three-component hydroglycosylation protocol (Table 2). First, we demonstrated that the protocol is scalable to a 2 mmol reaction under batch conditions, giving 4a in 42% yield after isolation (336 mg). Then, we varied the protecting groups of D-glucosyl bromide showing that benzoyl and pivaloyl esters are also well tolerated (4b–c). To our delight, the reaction was amenable to the functionalization of several hexoses, pentoses, and deoxysugars in their pyranosidic form, enabling the preparation of BCP C-glycosides of D-galactose, D-mannose, D-lyxose, L-rhamnose, and L-fucose in moderate to good yields and excellent a stereoselectivity (4d–h). A fluorinated D-galactose derivative was found to be a competent substrate (4i), showcasing the suitability of the method for the preparation of innovative chemical probes.15 When [3.1.1]propellane 2b was used we successfully obtained C-glucoside 4j whose bicycloheptyl moiety mimics a meta-disubstituted arene, albeit the reaction occurred in somewhat decreased yield.16 Finally, disaccharides bearing either a 1,4-α and 1,4-β glycosidic linkage gave the corresponding products 4k,l in synthetically useful yields. Remarkably, all products were generally obtained as the α anomers with very high levels of stereoselectivity. In all cases, BCP C-glycosides 4 were formed as the major products, along with traces of staffanes 5 and minor formation of 1-deoxyglycosides 6, as judged by 1H NMR analysis of crude reaction mixtures.
| a Reactions performed on a 0.2 mmol scale using 4CzIPN (5 mol%), propellane 2 as solution in Et2O (1.5 equiv.), and (Me3Si)3SiH 3a (1.5 equiv.). Yields refer to isolated products 4. α:β ratios were determined by 1H NMR analysis of the crude mixture using trichloroethylene as the internal standard. b Reaction performed at 0.3 mmol scale. 4CzIPN: 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene; LED: light-emitting diode; r.t.: room temperature. |
|---|
|
Carboboration of [1.1.1]propellane towards BCP C-glycoside boronates
Next, we wondered whether this radical strategy could be expanded to other classes of SOMOphiles to access BCP C-glycosides of increased synthetic value. To our delight, after intense screening and optimization of key parameters we identified diboron(4) reagents as suitable reaction partners (for details see SI, Section C.2).10o,17 Specifically, we found that glycosyl bromides 1 react in presence of an excess of [1.1.1]propellane 2a and bis(1,3-propanediolate)diboron (B2pro2) 3b to give BCP C-glycoside boronates 7 with good yield and good to excellent stereoselectivity (Table 3). Notably, this reaction occurs under thermal conditions – i.e., in the absence of light or PC – with perfect selectivity for boronate 7. Since isolation by flash chromatography proved unsuccessful due to degradation on silica gel, the Bpro moiety was instrumental, as it could be smoothly converted into the corresponding MIDA ester 8, enabling purification and full characterization.18 This protocol could be applied to both peracetylated and perbenzoylated glucosides (8a,b) as well as a variety of hexo- and pentopyranoses (8c–8f) (Table 3). While these boronates were generally obtained with good to excellent stereoselectivity for the α anomer, a lower α:β ratio of 2:1 was observed for D-xylose derivative 8e, likely due to its higher conformational freedom.19 Deoxy sugars from the L-series also reacted well (8g,h). This enabled us to grow crystals suitable for X-ray analysis of the L-fucose derivative 8h, confirming the α configuration of the anomeric center and determining key structural parameters within the BCP moiety (see SI, Section E). Finally, we were able to successfully apply the protocol to a fluoro D-galacto derivative (8i) and to D-maltose and D-lactose disaccharides (8j,k).
|
a Reactions performed on a 0.2 mmol scale using [1.1.1]propellane 2 as solution in Et2O (3.0 equiv.), and B2pro23b (3.0 equiv.). Yields outside brackets refer to 7 as determined by 1H NMR. Yields in brackets refer to isolated products 8. α:β ratios were determined by 1H NMR analysis of the crude mixture using trichloroethylene as the internal standard.
b Carboboration run for 2 h.
c Carboboration run for 5 h.
d Reaction run using 4 equiv. of both 2a and B2pro2. B2pro2: bis(1,3-propanediolate)diboron; MIDA: N-methyliminodiacetic acid.
|
|---|

|
Continuous flow scale-up and derivatization studies
To explore the synthetic potential of BCP C-glycosyl boronates, we scaled up the synthesis of glucoside 8a and took advantage of the boronate group as a handle for further diversification.20 Initial attempts to increase the reaction scale under batch conditions resulted in poor conversions due to slower kinetics. However, adapting the process to continuous flow using a homemade reactor enabled us to perform the reaction on a 2.0 mmol scale without loss of efficiency (see SI, Section F.2).21 Notably, the use of B2pro2 was crucial, as it ensured complete solubility of the reaction mixture, thereby facilitating a smooth transition to flow conditions. Finally, transesterification with MIDA afforded glucosyl boronate 8a in 54% overall yield (Scheme 2).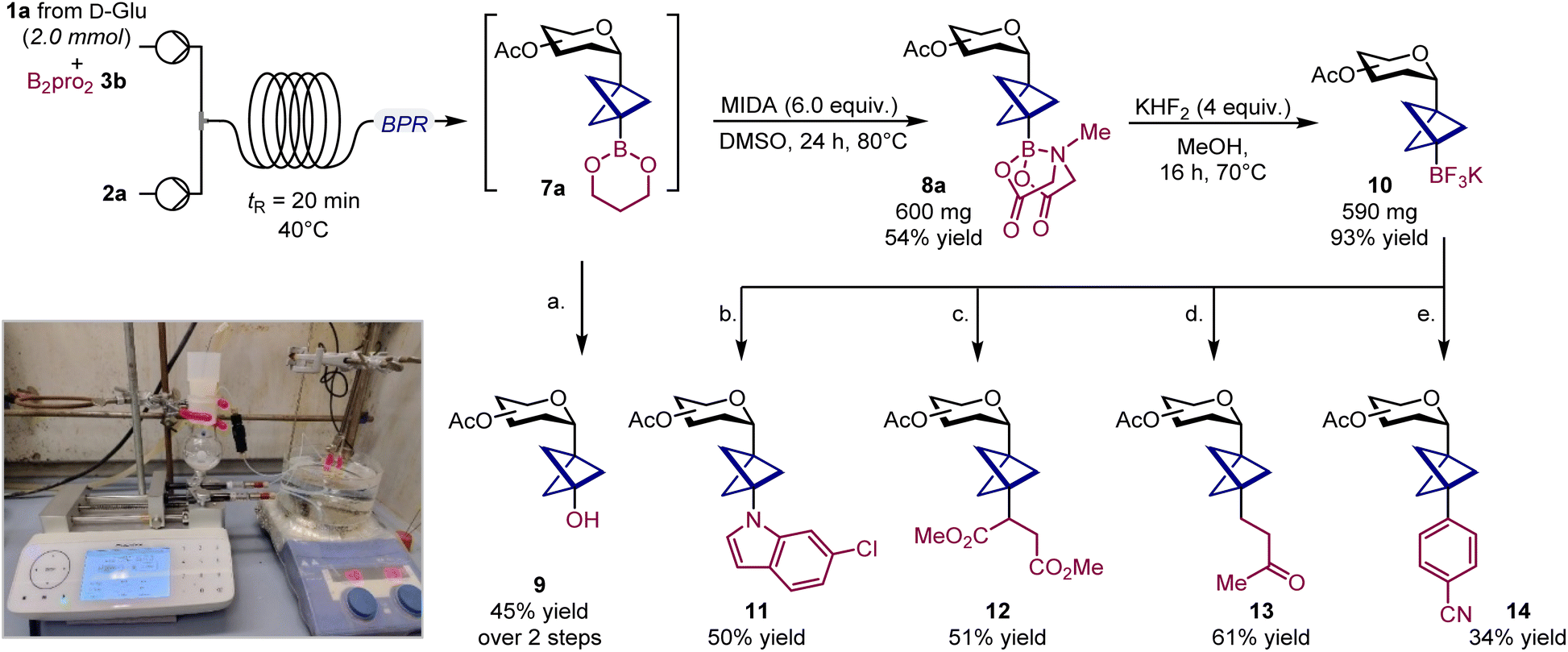 | ||
| Scheme 2 Reaction scale-up and product derivatization. Scale up performed on 2.0 mmol scale using [1.1.1]propellane 2a as solution in Et2O (3.0 equiv.), and B2pro23b (3.0 equiv.). Yields refer to isolated products. (a) Oxone (10 equiv.), K3PO4 (3 equiv.), H2O (5 equiv.), THF, 50 °C. (b) 10 (2 equiv.), 6-chloroindole (1 equiv.), Ir[dF(CF3)ppy]2(bpy)PF6 (2 mol%), Cu(acac)2 (50 mol%), Cs2CO3 (3 equiv.), LEDs λ = 456 nm, 1,4-dioxane. (c) 10 (1 equiv.), Ir[dF(CF3)ppy]2(bpy)PF6 (2 mol%), Na2HPO4·2H2O (3 equiv.), dimethyl fumarate (1.5 equiv.), LEDs λ = 456 nm, THF. 12 was obtained as a mixture with 1:1 d.r. (d) 10 (1 equiv.), Ir[dF(CF3)ppy]2(bpy)PF6 (2 mol%), Na2HPO4·2H2O (3 equiv.), Cu(CH3CN)4BF4 (12.5 mol%), methyl vinyl ketone (1.5 equiv.), LEDs λ = 456 nm, THF. (e) Ir[dF(CF3)ppy]2(dtbpy)PF6 (5 mol%), 4-bromobenzonitrile (1 equiv.), 10 (1.5 equiv.), Na2CO3 (2 equiv.), Ni(dtbpy)2Br2 (10 mol%), LEDs λ = 440 nm, 1,4-dioxane:DMA, 4:1. B2pro2: bis(1,3-propanediolate)diboron; BPR: back pressure regulator; MIDA: N-methyliminodiacetic acid. | ||
While treating crude boronate 7 with oxone smoothly provides access to tertiary alcohol 9 (45% yield over two steps), MIDA ester 8a could be efficiently converted into the corresponding tetrafluoroborate salt 10 (93% yield) (Scheme 2). Pleasingly, this key radical source participated in a Cu-catalyzed metallaphotoredox Chan–Lam reaction with 6-chloroindole, giving the corresponding C–N cross-coupled product 11.10o Alternatively, 10 could also be employed in a Giese-type addition to dimethyl fumarate (12), and to methyl vinyl ketone when using a Cu(I) salt as a co-catalyst (13).22 Finally, a Ni-catalyzed metallaphotoredox protocol enabled the forging of a C(sp3)–C(sp2) bond, affording the cross-coupled product 14.17d
Mechanistic investigation
After having assessed the generality of the developed methods, we aimed to elucidate the involved reaction mechanisms, starting from the hydroglycosylation protocol (Scheme 3a).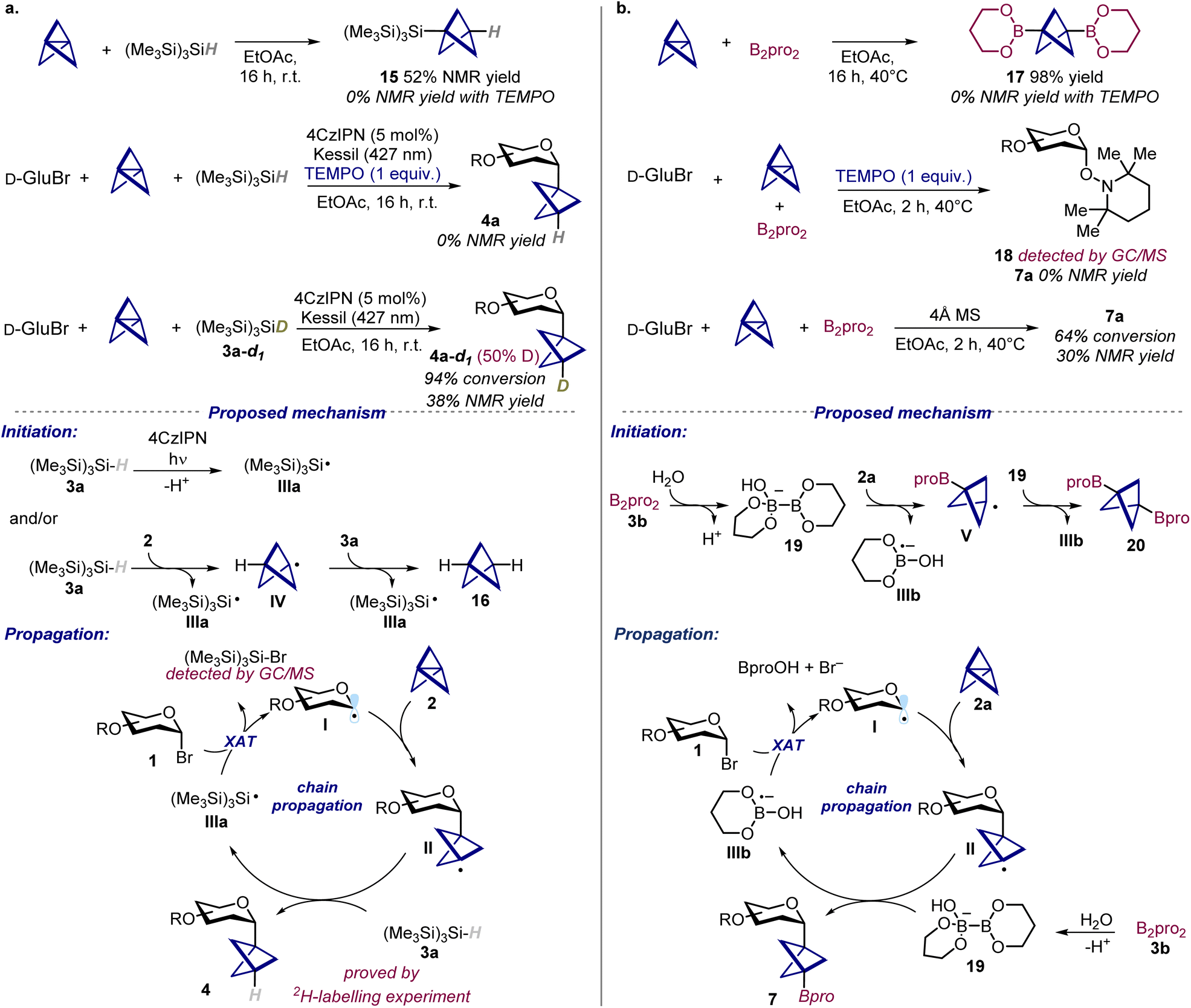 | ||
| Scheme 3 Mechanistic probes and proposed mechanism for the (a) hydroglycosylation and (b) carboboration of propellanes. 4CzIPN: 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene; TEMPO: 2,2,6,6-tetramethyl-1-piperidinyloxy. | ||
To gather insights on a possible thermal initiation pathway, we first studied the behavior of [1.1.1]propellane 2a upon mixing it with silane (Me3Si)3SiH 3a. Interestingly, we found that these two species can react together to give the addition product 15, a compound that could also be identified as side-product in the model reaction crude mixture. This addition reaction is completely inhibited in the presence of TEMPO (2,2,6,6-tetrameth-yl-1-piperidinyloxy) and a TEMPO adduct strongly support the involvement of silyl radicals (for details see SI, Section G).
Along this line, when the model reaction was performed in the presence of TEMPO, the formation of the BCP C-glycoside 4a was also completely suppressed, further supporting the radical nature of the transformation. A 2H-labelling experiment performed using the deuterated silane 3a-d1 resulted in the formation of glycoside 4a with 50% deuterium incorporation (as judged by 1H NMR), which unequivocally demonstrated the role of (Me3Si)3SiH 3a as HAT donor. Based on these findings, we propose the following mechanism for the hydroglycosylation reaction (Scheme 3a bottom). A main initiation pathway involves the oxidation of silane 3a (Eox ((Me3Si)3SiH˙+/(Me3Si)3SiH) = +0.73 V vs. SCE in CH3CN)23 from the photoexcited 4CzIPN* (Ered1/2 (PC*/PC˙−) = +1.43 V vs. SCE in CH3CN),14b which upon deprotonation leads to the formation of silyl radical IIIa.23 Oxidative quenching of the photoexcited 4CzIPN* (Eox1/2 (PC˙+/PC*) = −1.18 V vs. SCE in CH3CN)14b from glycosyl bromide 1 (Ered (1a/1a˙−) = −2.45 V vs. SCE in CH3CN)24 can be excluded on the basis of unfavorable thermodynamics. This mechanistic pathway is supported by Stern–Volmer quenching studies, which revealed (Me3Si)3SiH 3a as the only reaction component able to quench the photoexcited 4CzIPN* (for details see SI, Section G).
Besides, since control experiments revealed that the reaction can also proceed in the absence of light, a further thermal initiation pathway is likely operative. In this regard, we propose that (Me3Si)3SiH 3a reacts with propellane 2 to give silyl radical IIIa and BCP radical IV. The latter can further react with another molecule of 3a to give 16 and a second silyl radical IIIa. Then, silyl radical IIIa can act as a chain carrier undergoing halogen atom transfer (XAT) with glycosyl bromide 1,25 giving the corresponding glycosyl radical I and (Me3Si)3SiBr (whose formation was confirmed by GC/MS analysis). Finally, addition of I to propellane 2 leads to BCP radical II, which subsequently reacts with 3a to afford the BCP C-glycoside 4 and regenerate silyl radical IIIa, thereby sustaining a radical-chain. In this scenario, the side-product 15 is formed through a competitive addition reaction of silyl radical IIIa to propellane 2. Although the reaction can also proceed thermally, the use of 4CzIPN provides an additional pathway to reinitiate the chain reactions once they are terminated, which explains the improved outcome when the reaction is carried out under light and in the presence of the photocatalyst.
We next investigated the mechanism for the carboboration of propellane (Scheme 3b). Following the same approach, we first demonstrated that the SOMOphile B2pro2 can react with propellane 2a to give the bisboronate 17, which likely represent the main initiation pathway for the reaction (vide infra). Such addition process, as well as the model carboboration reaction were completely inhibited when performed using TEMPO. In the latter case we were also able to detect by GC/MS the TEMPO adduct 18, which proves the formation of glycosyl radicals as intermediates. Then, we interrogated the ability of B2pro2 to behave as a SOMOphile in the optimized reaction conditions. This diboron(4) compound is able to intercept radical species upon coordination from a Lewis base.17b Here, we showed that the reaction is strongly inhibited when performed using 4 Å molecular sieves, which suggest that water might activate B2pro2. Based on these observations, we propose a radical mechanism initiated by reaction between B2pro23b and propellane 2a. Specifically, we propose that coordination of B2pro2 by water might lead to the formation of the active ate-complex 19 that reacts with propellane 2 giving boryl radical anion IIIb and BCP radical V.17b,26 Addition of V to 19 then gives the bisboronate side-product 17.27 On the other hand, we speculate that the resulting boryl radical anion IIIb can then undergo XAT with glycosyl bromide 1, forming glycosyl radical I. Finally, subsequent addition of radical I to propellane 2 and activated 19 leads to the desired BCP boronate 7 and restores intermediate IIIb, thus propagating the chain reaction.
Next, seeking to determine the origin of the α stereoselectivity, we performed DFT calculations investigating the reaction between peracetylated D-glucosyl radical Ia and [1.1.1]propellane 2a. This key reactive intermediate populates three main conformations of the six-membered ring, namely the chair 4C1, the inverted chair 1C4, and the boat B2,5.3i These conformers exhibit relative free energy values spanning a narrow range of 1.74 kcal mol−1, with the B2,5-Ia conformer being the most stable. They also show similar calculated global electrophilicity (1.03 < ω < 1.08 eV), consistent with nucleophilic α-oxo carbon centered radicals.28 The addition of Ia to 2a to give BCP radical IIa is exergonic, with a calculated ΔG0 of −12.4 and −14.8 kcal mol−1 for the α and β anomer, respectively.29 Experimental data show the α anomer to be the favored one, suggesting that the stereoselectivity is likely determined by kinetic factors. Indeed, among the possible transition states, the lowest energy one was found to be TSα, which shows a 4C1 conformation (incipient C⋯C bond distance of 2.297 Å, Fig. 1) while favoring α stereoselectivity (ΔG‡ = +9.3 kcal mol−1).30 For comparison, the related TSβ is associated to ΔG‡ = +12.2 kcal mol−1 for the pathway to β-IIa anomer; other TS adopting different conformations (namely 1C4 or B2,5) were found at higher energies for both α and β pathways (SI, section H). Spin density maps of both TS clearly show that the anomeric radical is partially delocalized over the endocyclic oxygen of the sugar ring, an effect that is more pronounced for the TSα. This is indicative of a better orbital overlap between the lone pair of the oxygen atom and the σ*‡ orbital of the incipient C–C bond.19 Such stabilizing effect – i.e. the kinetic anomeric effect in radical glycosylation reactions – accounts for the α selectivity observed in the addition of glucosyl radical to [1.1.1]propellane.
 | ||
| Fig. 1 Energy diagram for the addition of peracetylated glucosyl radical Ia to [1.1.1]propellane 2. DFT calculation at M062X/6-311+G**//M062X/6-311+G** level of theory, including a continuum solvation model for Et2O using the integral equation formalism variant (IEFPCM). | ||
An analogous DFT calculation was performed for the reaction between the peracetylated xylosyl radical Ie and [1.1.1]propellane 2a, which revealed a similar trend of diastereoselectivity with the α-IIe anomer being the most favoured by a calculated ΔΔG‡ of 1.5 kcal mol−1. This value is lower than the calculated ΔΔG‡ of 2.9 kcal mol−1 for α-IIa, thus being consistent with the lower stereoselectivity experimentally observed for the addition of this xylosyl derivative to [1.1.1]propellane 2a (see SI, section H for details). These results are significant, as the stereoselectivity of glycosyl radical addition reactions has rarely been investigated by DFT calculations, and our findings align well with the available precedents.31
Conclusions
In summary, we have developed a mild radical approach for the preparation of saturated analogs of aryl C-glycosides. The generality of our strategy was proved in a hydroglycosylation protocol enabling the synthesis of glycosylated BCP derivatives, where the BCP moiety mimics an unsubstituted phenyl ring. By extending this reactivity to the use of diboron(4) SOMOphiles we provided access to difunctionalized glycosylated BCP boronates. This method stands out for its practicality, scalability, and synthetic versatility of the products, which enables straightforward access to a variety of BCP C-glycoside derivatives. Experimental and computational mechanistic studies revealed that both protocols proceed through a radical chain mechanism under a Curtin–Hammett scenario, with the stereodetermining addition of glycosyl radicals to [1.1.1]propellane being controlled by the kinetic anomeric effect. Altogether, this study introduces the replacement of planar arenes with highly three-dimensional fragments for the preparation of aryl C-glycoside analogues and paves the way for new developments in C-glycosylation chemistry.Author contributions
GG designed the project. GG, AM, SB, PGR performed the experiments. GP performed X-ray analyses. AS performed DFT calculations. LD supervised the work and secured fundings. GG and LD wrote the manuscript with contributions from all the authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data that support the findings of this study are available free of charge in the supporting information (SI). Supplementary information: includes details of experimental procedures, full characterization data, copies of NMR spectra, X-ray analysis, and DFT calculations. See DOI: https://doi.org/10.1039/d5sc07328f.CCDC 2422982 contains the supplementary crystallographic data for this paper.32
Acknowledgements
This work was supported by the European Union H2020 research and innovation program with the ERC StG grant for SYNPHOCAT, No. 101040025 (LD); and by MUR (Ministero dell’Università) PRIN 2020927WY3_00 (LD). GG thanks MUR for a Young Researchers, Seal of Excellence fellowship funded by the European Union – NextGenerationEU (PhotoFix-Bio – C93C22007640006). SB acknowledges UniPD for a doctoral fellowship. PGR thanks CiQUS for a fellowship within the International Mobility Program 2024 framework financed by the Xunta de Galicia and the European Union.References
- (a) S. I. Elshahawi, K. A. Shaaban, M. K. Kharel and J. S. Thorson, Chem. Soc. Rev., 2015, 44, 7591–7697 RSC; (b) K. Kitamura, Y. Ando, T. Matsumoto and K. Suzuki, Chem. Rev., 2018, 118, 1495–1598 CrossRef CAS PubMed.
- (a) J. Štambaský, M. Hocek and P. Kočovský, Chem. Rev., 2009, 109, 6729–6764 CrossRef; (b) É. Bokor, S. Kun, D. Goyard, M. Tóth, J.-P. Praly, S. Vidal and L. Somsák, Two aryl C-glycoside derivatives, dapagliflozin and empagliflozin, are among the top 50 best-selling drugs of 2023, Chem. Rev., 2017, 117, 1687–1764 CrossRef , https://www.drugdiscoverytrends.com/best-selling-pharmaceuticals-2023/, For details, see: ; (c) Boehringer Ingelheim – 2023 Highlights, https://annualreport.boehringer-ingelheim.com/2023/download/BOE_AR23_Highlights_2023_EN_safe.pdf; (d) AstraZeneca Annual Report and Form 20-F Information 2024, https://www.astrazeneca.com/content/dam/az/Investor_Relations/annual-report-2024/pdf/AstraZeneca_AR_2024.pdf.
- Selected reviews on the synthesis of aryl C-glycosides: (a) Y. Du, R. J. Linhardt and I. R. Vlahov, Tetrahedron, 1998, 54, 9913–9959 CrossRef CAS; (b) Y. Yang and B. Yu, Chem. Rev., 2017, 117, 12281–12356 CrossRef CAS PubMed; (c) L.-Y. Xu, N.-L. Fan and X.-G. Hu, Org. Biomol. Chem., 2020, 18, 5095–5109 RSC; (d) Y. Jiang, Y. Zhang, B. C. Lee and M. J. Koh, Angew. Chem., Int. Ed., 2023, 62, e202305138 CrossRef CAS PubMed; (e) X.-Y. Gou, X.-Y. Zhu, B.-S. Zhang and Y.-M. Liang, Chem.–Eur. J., 2023, 29, e202203351 CrossRef CAS , Selected modern synthetic methods to aryl C-glycosides: ; (f) H. Gong and M. R. Gagné, J. Am. Chem. Soc., 2008, 130, 12177–12183 CrossRef CAS; (g) S. H. Lemaire, N. Ioannis, T. Xiao, J. Li, E. Digard, C. Gozlan, R. Liu, A. Gavryushin, C. Diène, Y. Wang, V. Farina and P. Knochel, Org. Lett., 2012, 14, 1480–1483 CrossRef CAS PubMed; (h) L. Nicolas, P. Angibaud, I. Stansfield, P. Bonnet, L. Meerpoel, S. Reymond and J. Cossy, Angew. Chem., Int. Ed., 2012, 51, 11101–11104 CrossRef CAS; (i) L. Adak, S. Kawamura, G. Toma, T. Takenaka, K. Isozaki, H. Takaya, A. Orita, H. C. Li, T. K. M. Shing and M. Nakamura, J. Am. Chem. Soc., 2017, 139, 10693–10701 CrossRef CAS; (j) A. Dumoulin, J. K. Matsui, A. Gutierrez-Bonet and G. A. Molander, Angew. Chem., Int. Ed., 2018, 57, 6614–6618 CrossRef CAS; (k) Y. Wei, B. Ben-Zvi and T. Diao, Angew. Chem., Int. Ed., 2021, 60, 9433–9438 CrossRef CAS PubMed; (l) Z. D. Mou, J. X. Wang, X. Zhang and D. Niu, Adv. Synth. Catal., 2021, 363, 3025–3029 CrossRef CAS; (m) C. Zhang, S.-Y. Xu, H. Zuo, X. Zhang, Q.-D. Dang and D. Niu, Nat. Synth., 2023, 2, 251–260 CrossRef CAS.
- (a) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed; (b) T. J. Ritchie and S. J. Macdonald, Drug Discovery Today, 2009, 14, 1011–1020 CrossRef CAS; (c) F. Lovering, MedChemComm, 2013, 4, 515–519 RSC.
- (a) N. A. Meanwell, J. Med. Chem., 2011, 54, 2529–2591 CrossRef CAS PubMed; (b) N. Brown, Bioisosteres in Medicinal Chemistry, Wiley-VCH Verlag & Co. KGaA, Weinheim, Germany, 2012 CrossRef; (c) N. A. Meanwell, J. Agric. Food Chem., 2023, 71, 18087–18122 CrossRef CAS PubMed.
- (a) P. K. Mykhailiuk, Org. Biomol. Chem., 2019, 17, 2839–2849 RSC; (b) M. A. M. Subbaiah and N. A. Meanwell, J. Med. Chem., 2021, 64, 14046–14128 CrossRef CAS PubMed.
- (a) R. Pellicciari, M. Raimondo, M. Marinozzi, B. Natalini, G. Costantino and C. Thomsen, J. Med. Chem., 1996, 39, 2874–2876 CrossRef CAS; (b) A. F. Stepan, C. Subramanyam, I. V. Efremov, J. K. Dutra, T. J. O'Sullivan, K. J. DiRico, W. S. McDonald, A. Won, P. H. Dorff, C. E. Nolan, S. L. Becker, L. R. Pustilnik, D. R. Riddell, G. W. Kauffman, B. L. Kormos, L. Zhang, Y. Lu, S. H. Capetta, M. E. Green, K. Karki, E. Sibley, K. P. Atchison, A. J. Hallgren, C. E. Oborski, A. E. Robshaw, B. Sneed and C. J. O'Donnell, J. Med. Chem., 2012, 55, 3414–3424 CrossRef CAS; (c) N. D. Measom, K. D. Down, D. J. Hirst, C. Jamieson, E. S. Manas, V. K. Patel and D. O. Somers, ACS Med. Chem. Lett., 2017, 8, 43–48 CrossRef CAS; (d) E. G. Tse, S. D. Houston, C. M. Williams, G. P. Savage, L. M. Rendina, I. Hallyburton, M. Anderson, R. Sharma, G. S. Walker, R. S. Obach and M. H. Todd, J. Med. Chem., 2020, 63, 11585–11601 CrossRef CAS.
- (a) R. S. Andrews, J. J. Becker and M. R. Gagné, Angew. Chem., Int. Ed., 2010, 49, 7274–7276 CrossRef CAS; (b) Q. Wang, B. C. Lee, T. J. Tan, Y. Jiang, W. H. Ser and M. J. Koh, Nat. Synth., 2022, 1, 967–974 CrossRef CAS; (c) Q. Wang, Q. Sun, Y. Jiang, H. Zhang, L. Yu, C. Tian, G. Chen and M. J. Koh, Nat. Synth., 2022, 1, 235–244 CrossRef CAS; (d) S. Xu, Y. Ping, M. Xu, G. Wu, Y. Ke, R. Miao, X. Qi and W. Kong, Nat. Chem., 2024, 16, 2054–2065 CrossRef CAS; (e) Y. Jiang, Y. Wei, Q.-Y. Zhou, G.-Q. Sun, X.-P. Fu, N. Levin, Y. Zhang, W.-Q. Liu, N. Song, S. Mohammed, B. G. Davis and M. J. Koh, Nature, 2024, 631, 319–327 CrossRef CAS PubMed.
- (a) K. B. Wiberg and F. H. Walker, J. Am. Chem. Soc., 1982, 104, 5239–5240 CrossRef; (b) W. Wu, J. Gu, J. Song, S. Shaik and P. C. Hiberty, Angew. Chem., Int. Ed., 2009, 48, 1407–1410 CrossRef CAS PubMed; (c) A. J. Sterling, A. B. Durr, R. C. Smith, E. A. Anderson and F. Duarte, Chem. Sci., 2020, 11, 4895–4903 RSC , For a review on propellanes, see: ; (d) A. M. Dilmac, E. Spuling, A. de Meijere and S. Brase, Angew. Chem., Int. Ed., 2017, 56, 5684–5718 CrossRef CAS.
- For reviews on the use of [1.1.1]propellane in synthetic methods, see: (a) K. B. Wiberg and S. T. Waddell, J. Am. Chem. Soc., 1990, 112, 2194–2216 CrossRef CAS; (b) M. Uchiyama and J. Kanazawa, Synlett, 2018, 30, 1–11 Search PubMed; (c) J. Turkowska, J. Durka and D. Gryko, Chem. Commun., 2020, 56, 5718–5734 RSC; (d) B. R. Shire and E. A. Anderson, JACS Au, 2023, 3, 1539–1553 CrossRef CAS PubMed; (e) P. Bellotti and F. Glorius, J. Am. Chem. Soc., 2023, 145, 20716–20732 CrossRef CAS PubMed; (f) S. Cuadros, J. Paut, E. Anselmi, G. Dagousset, E. Magnier and L. Dell'Amico, Angew. Chem., Int. Ed., 2024, 63, e202317333 CrossRef CAS , Selected modern approaches to the 1,3-difunctionalization of [1.1.1]propellane: ; (g) R. Gianatassio, J. M. Lopchuk, J. Wang, C.-M. Pan, L. R. Malins, L. Prieto, T. A. Brandt, M. R. Collins, G. M. Gallego, N. W. Sach, J. E. Spangler, H. Zhu, J. Zhu and P. S. Baran, Science, 2016, 351, 241–246 CrossRef CAS PubMed; (h) R. A. Shelp and P. J. Walsh, Angew. Chem., Int. Ed., 2018, 57, 15857–15861 CrossRef CAS; (i) K. Schwarzer, H. Zipse, K. Karaghiosoff and P. Knochel, Angew. Chem., Int. Ed., 2020, 59, 20235–20241 CrossRef; (j) J. Kanazawa, K. Maeda and M. Uchiyama, J. Am. Chem. Soc., 2017, 139, 17791–17794 CrossRef CAS PubMed; (k) J. H. Kim, A. Ruffoni, Y. S. S. Al-Faiyz, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2020, 59, 8225–8231 CrossRef CAS; (l) S. Cuadros, G. Goti, G. Barison, A. Raulli, T. Bortolato, G. Pelosi, P. Costa and L. Dell'Amico, Angew. Chem., Int. Ed., 2023, 62, e202303585 CrossRef; (m) V. Ripenko, V. Sham, V. Levchenko, S. Holovchuk, D. Vysochyn, I. Klymov, D. Kyslyi, S. Veselovych, S. Zhersh, Y. Dmytriv, A. Tolmachev, I. Sadkova, I. Pishel, K. Horbatok, V. Kosach, Y. Nikandrova and P. K. Mykhailiuk, Nat. Synth., 2024, 3, 1538–1549 CrossRef CAS PubMed , For rare examples of glycosyl radical addition to [1.1.1]propellane, see: ; (n) J. Nugent, C. Arroniz, B. R. Shire, A. J. Sterling, H. D. Pickford, M. L. J. Wong, S. J. Mansfield, D. F. J. Caputo, B. Owen, J. J. Mousseau, F. Duarte and E. A. Anderson, ACS Catal., 2019, 9, 9568–9574 CrossRef CAS; (o) W. Dong, E. Yen-Pon, L. Li, A. Bhattacharjee, A. Jolit and G. A. Molander, Nat. Chem., 2022, 14, 1068–1077 CrossRef CAS PubMed.
- Selected book chapters and reviews on radical chemistry for carbohydrate functionalization: (a) A. J. Pearce, J. M. Mallet and P. Sinaÿ, in Radicals in Organic Synthesis, ed. P. Renaud and M. P. Sibi, WILEY-VCH Verlag GmbH, 2001, pp. 538–577, DOI:10.1002/9783527618293.ch52; (b) I. Pérez-Martín and E. Suárez, in Encyclopedia of Radicals in Chemistry, Biology and Materials, ed. C. Chatgilialoglu and A. Studer, John Wiley & Sons, Ltd, 2012, pp. 1131–1174 Search PubMed; (c) C. E. Suh, H. M. Carder and A. E. Wendlandt, ACS Chem. Biol., 2021, 16, 1814–1828 CrossRef CAS; (d) A. Shatskiy, E. V. Stepanova and M. D. Kärkäs, Nat. Rev. Chem., 2022, 6, 782–805 CrossRef CAS PubMed; (e) G. Goti, ChemCatChem, 2022, 14, e202200290 CrossRef CAS; (f) W. Shang and D. Niu, Acc. Chem. Res., 2023, 56, 2473–2488 CrossRef CAS , Selected references on modern synthetic methods: ; (g) V. Dimakos, H. Y. Su, G. E. Garrett and M. S. Taylor, J. Am. Chem. Soc., 2019, 141, 5149–5153 CrossRef CAS PubMed; (h) Y. Wang, H. M. Carder and A. E. Wendlandt, Nature, 2020, 578, 403–408 CrossRef CAS PubMed; (i) W. Yao, G. Zhao, Y. Wu, L. Zhou, U. Mukherjee, P. Liu and M.-Y. Ngai, J. Am. Chem. Soc., 2022, 144, 3353–3359 CrossRef CAS PubMed.
- During the submission and revision process of this manuscript, Ackermann and co-workers reported an electrochemical approach to the synthesis of related bicyclo[1.1.1]pentyl C-glycosides: J. Liu, R. Purushothaman, F. Hinrichs, M. Surke, S. Warratz and L. Ackermann, J. Am. Chem. Soc., 2025, 147, 34813–34822, DOI:10.1021/jacs.5c10732 , Please note that the preprint version of our manuscripts was deposited in ChemRxiv on the 21st of March 2025 (see ChemRxiv 2025; DOI:10.26434/chemrxiv-2025-0czlj)..
- C. Chatgilialoglu, C. Ferreri, Y. Landais and V. I. Timokhin, Chem. Rev., 2018, 118, 6516–6572 CrossRef CAS.
- (a) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS; (b) E. Speckmeier, T. G. Fischer and K. Zeitler, J. Am. Chem. Soc., 2018, 140, 15353–15365 CrossRef CAS; (c) T. Bortolato, S. Cuadros, G. Simionato and L. Dell'Amico, Chem. Commun., 2022, 58, 1263–1283 RSC.
- B. Linclau, A. Arda, N. C. Reichardt, M. Sollogoub, L. Unione, S. P. Vincent and J. Jimenez-Barbero, Chem. Soc. Rev., 2020, 49, 3863–3888 RSC.
- (a) N. Frank, J. Nugent, B. R. Shire, H. D. Pickford, P. Rabe, A. J. Sterling, T. Zarganes-Tzitzikas, T. Grimes, A. L. Thompson, R. C. Smith, C. J. Schofield, P. E. Brennan, F. Duarte and E. A. Anderson, Nature, 2022, 611, 721–726 CrossRef CAS PubMed; (b) T. Iida, J. Kanazawa, T. Matsunaga, K. Miyamoto, K. Hirano and M. Uchiyama, J. Am. Chem. Soc., 2022, 144, 21848–21852 CrossRef CAS PubMed.
- (a) E. C. Neeve, S. J. Geier, I. A. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS PubMed; (b) F. W. Friese and A. Studer, Chem. Sci., 2019, 10, 8503–8518 RSC; (c) M. Kondo, J. Kanazawa, T. Ichikawa, T. Shimokawa, Y. Nagashima, K. Miyamoto and M. Uchiyama, Angew. Chem., Int. Ed., 2020, 59, 1970–1974 CrossRef CAS PubMed; (d) M. D. VanHeyst, J. Qi, A. J. Roecker, J. M. E. Hughes, L. Cheng, Z. Zhao and J. Yin, Org. Lett., 2020, 22, 1648–1654 CrossRef CAS PubMed; (e) Y. Yang, J. Tsien, J. M. E. Hughes, B. K. Peters, R. R. Merchant and T. Qin, Nat. Chem., 2021, 13, 950–955 CrossRef CAS PubMed; (f) I. F. Yu, J. L. Manske, A. Dieguez-Vazquez, A. Misale, A. E. Pashenko, P. K. Mykhailiuk, S. V. Ryabukhin, D. M. Volochnyuk and J. F. Hartwig, Nat. Chem., 2023, 15, 685–693 CrossRef CAS PubMed; (g) L. Yi, D. Kong, A. Prabhakar Kale, R. Alshehri, H. Yue, A. Gizatullin, B. Maity, R. Kancherla, L. Cavallo and M. Rueping, Angew. Chem., Int. Ed., 2024, 63, e202411961 CrossRef CAS PubMed.
- H. Yoshida, M. Seki, I. Kageyuki, I. Osaka, S. Hatano and M. Abe, ACS Omega, 2017, 2, 5911–5916 CrossRef CAS PubMed.
- (a) H. Abe, S. Shuto and A. Matsuda, J. Am. Chem. Soc., 2001, 123, 11870–11882 CrossRef CAS; (b) H. Abe, M. Terauchi, A. Matsuda and S. Shuto, J. Org. Chem., 2003, 68, 7439–7447 CrossRef CAS PubMed.
- (a) C. Sandford and V. K. Aggarwal, Chem. Commun., 2017, 53, 5481–5494 RSC; (b) K. Duan, X. Yan, Y. Liu and Z. Li, Adv. Synth. Catal., 2018, 360, 2781–2795 CrossRef CAS.
- (a) J. Wegner, S. Ceylan and A. Kirschning, Adv. Synth. Catal., 2012, 354, 17–57 CrossRef CAS; (b) M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed; (c) L. Capaldo, Z. Wen and T. Noel, Chem. Sci., 2023, 14, 4230–4247 RSC.
- E. Wheatley, H. Melnychenko and M. Silvi, J. Am. Chem. Soc., 2024, 146, 34285–34291 CrossRef CAS PubMed.
- J. Zhu, W. C. Cui, S. Wang and Z. J. Yao, Org. Lett., 2018, 20, 3174–3178 CrossRef CAS PubMed.
- S. B. Rondinini, P. R. Mussini, F. Crippa and G. Sello, Electrochem. Commun., 2000, 2, 491–496 CrossRef CAS.
- F. Juliá, T. Constantin and D. Leonori, Chem. Rev., 2022, 122, 2292–2352 CrossRef PubMed.
- (a) L. Capaldo, T. Noël and D. Ravelli, Chem Catal., 2022, 2, 957–966 CAS; (b) S. B. Thorpe, J. A. Calderone and W. L. Santos, Org. Lett., 2012, 14, 1918–1921 CrossRef CAS PubMed.
- J. R. Pinchman, C. D. Hopkins, K. D. Bunker and P. Q. Huang, WO/2018/039232, 2018.
- (a) F. Parsaee, M. C. Senarathna, P. B. Kannangara, S. N. Alexander, P. D. E. Arche and E. R. Welin, Nat. Rev. Chem., 2021, 5, 486–499 CrossRef CAS PubMed; (b) J. J. A. Garwood, A. D. Chen and D. A. Nagib, J. Am. Chem. Soc., 2024, 146, 28034–28059 CAS.
- A conformational analysis was conducted for I and for both the α and β anomers of II, and the energies are given considering a weighted average of the conformers, according to Boltzmann distribution. While for α-II an almost equal contribution of 4C1 and 1C4 conformations was found, the β-II anomer is described almost exclusively by the 4C1 conformation (see SI, Section H).
- A calculated ΔG‡ of +17 kcal mol−1 was reported for the addition of a nucleophilic α-amido radical to 2a, see ref. 17g. The global electrophilicity ω of α-amido radicals is expected to range between 0.5 and 0.8 eV (see ref. 28b).
- (a) L. Xia, W. Fan, X.-A. Yuan and S. Yu, ACS Catal., 2021, 11, 9397–9406 CrossRef CAS; (b) Y. Jiang, K. Yang, Y. Wei, Q. Wang, S.-J. Li, Y. Lan and M. J. Koh, Angew. Chem., Int. Ed., 2022, 61, e202211043 CrossRef CAS PubMed.
- CCDC 2422982: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2mb9q0.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |