
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic diselenide/guanidine catalyzed dehydrophosphorylation of 2-nitrobenzhydrols to access C-stereogenic phosphinates
Jin-Yu
Gong†
a,
Pan-Pan
Zhou†
 a,
Yu-Hao
Qiao
a,
Zhi-Chao
Qi
a,
Qian-Ming
Zuo
a,
Qing-Xia
Fang
a and
Shang-Dong
Yang
*ab
a,
Yu-Hao
Qiao
a,
Zhi-Chao
Qi
a,
Qian-Ming
Zuo
a,
Qing-Xia
Fang
a and
Shang-Dong
Yang
*ab
aState Key Laboratory of Natural Product Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, 222 South Tianshui Road, Lanzhou 730000, China. E-mail: yangshd@lzu.edu.cn
bState Key Laboratory of Low Carbon Catalysis and Carbon Dioxide Utilization, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000, China
First published on 18th March 2026
Abstract
The development of a catalytic and mild variant of the Atherton–Todd reaction for the synthesis of chiral phosphorus-containing compounds presents substantial challenges. Herein, we report a method for accessing C-stereogenic phosphinates enabled by a synergistic diselenide/chiral guanidine catalytic system. This method avoids stoichiometric halogenating reagents, generating water as the sole byproduct. Mechanistic studies reveal that the electron-deficient diselenide catalyst serves as a recyclable alternative for halogenating agents, while the chiral guanidine activates the alcohol via Brønsted base catalysis. This strategy effectively achieves the kinetic resolution of secondary alcohols with similar steric substituents, offering a sustainable strategy to access phosphinates with stereogenic benzhydryl groups.
Introduction
Organophosphinates, as a pivotal class of compounds containing phosphorus–oxygen bonds, have attracted substantial attention due to their versatile applications in pharmaceuticals,1 agrochemicals,2 and organic synthesis.3 Conventional approaches for the synthesis of these compounds rely on the reaction between sensitive phosphoryl halides and alcohols or phenols. The Atherton–Todd (A–T) reaction offers a practical alternative by generating these sensitive electrophiles in situ from P(O)–H compounds upon treatment with stoichiometric halogenating agents and a base.4 Building upon this foundation, early improvements employed iodide/oxidant systems to expand the scope and practicality of this approach.5 More recently, Tf2O-mediated process has been introduced for the generation of electrophilic phosphorus species.6 (Fig. 1A). | ||
| Fig. 1 Dehydrophosphorylation for chiral phosphorus-containing compounds. | ||
While various efficient strategies have been established for the Atherton–Todd-type (A–T-type) reaction, the CCl4-mediated approach has uniquely proven applicable to the synthesis of chiral phosphorus-containing compounds in recent years (Fig. 1B). In 2019, Colobert and colleagues developed a diastereoselective Atherton–Todd reaction using sulfoxides as chiral auxiliaries to achieve dynamic kinetic resolution of racemic P(O)–H compounds.7 Subsequently, Wang and coworkers pioneered a catalytic protocol for the enantiodivergent synthesis of atropisomeric biaryls via a kinetic resolution process, guided by dipeptide-phosphonium salt-catalyzed Atherton–Todd reactions.8 On this basis, they further expanded the scope of chiral quaternary phosphonium salts catalysis, accessing sulfur-stereogenic sulfoximines9 and axially chiral phosphate-containing olefins.10 However, these advanced protocols still depend on stoichiometric CCl4, which even acts directly as the solvent component in certain protocols (i.e., in large excess). Therefore, the development of a catalytic and mild variant of the Atherton–Todd reaction for accessing chiral phosphorus-containing compounds becomes imperative.
Against this backdrop, diselenide catalysts11,12 offer a promising solution. Building on our recent studies in chiral diselenide-catalyzed intramolecular enantioselective desymmetrizing cyclization13 and our ongoing interest in organophosphorus chemistry,14 we sought to explore the dehydrogenative phosphinoylation of racemic alcohols with P(O)–H compounds for C-stereogenic phosphinates with catalytic amounts of diselenide. Current progress in this dehydrogenative coupling reaction is limited to racemic transformations mediated by electron-deficient aryl diselenide catalysts.11g However, extending this protocol to the synthesis of chiral phosphorus-containing compounds remains highly challenging, primarily due to the limited availability of electron-deficient chiral diselenide catalysts.11h–p To the best of our knowledge, there are no general methods for the efficient preparation of these chiral diselenide species. Consequently, new catalytic strategies must fundamentally bypass the need for chiral diselenide catalysts. Given the crucial role of base activation in alcohol functionalization, we propose a dual activation strategy combining achiral electron-deficient diselenide catalysts with chiral organo-superbase catalysts.15 Chiral organo-superbase catalysts serve as bifunctional catalysts that simultaneously mediate base activation and enantioselective control, while the electron-deficient diselenide facilitates the formation of electrophilic phosphorus intermediates from P(O)–H compounds, creating a synergistic interplay to promote dehydrogenative coupling of P(O)–H compounds with 2-nitrobenzhydrols. This synergistic strategy not only circumvents the need for stoichiometric CCl4 and chiral diselenide catalysts but, more importantly, effectively achieves the challenging kinetic resolution of secondary alcohols bearing substituents of similar steric demand, providing a sustainable route to C-stereogenic phosphinates with water as the sole byproduct (Fig. 1C).
Results and discussion
Optimization of reaction conditions
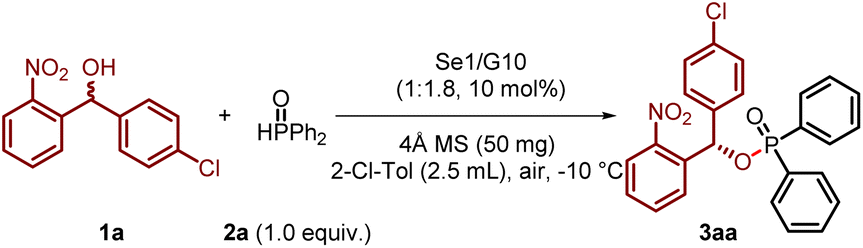
Compounds featuring a (2-nitrophenyl)methanol scaffold exhibit potent inhibitory activity against PqsD in Pseudomonas aeruginosa,16 and the nitro group of the substrate, as a strong electron-withdrawing group and hydrogen-bonding acceptor, can assist in chiral control while activating the substrate. In light of these considerations, we evaluated the proposed dual activation strategy using achiral diselenide catalysts and chiral guanidine catalysts.17,18 Using (4-chlorophenyl)(2-nitrophenyl)methanol (1a) and diphenylphosphine oxide (2a) as model substrates, we conducted the reactions in 2-chlorotoluene at −10 °C with 4 Å molecular sieves as the drying agent, as outlined in Table 1. Fortunately, the reaction proceeded efficiently when using diaryldiselenide with electron-withdrawing groups (Se1) and chiral guanidine catalyst (G10) with the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.8, delivering the desired phosphinate product 3aa in 86% yield and a 95:5 enantiomeric ratio (e.r.). During this process, we observed that the results were significantly influenced by the amino acid backbone, amide substituents, and amidine substituents of the guanidine catalysts. For instance, the use of bicyclic guanidine (G1), dialkyl-substituted catalysts (G2), and pipecolinic acid-based catalysts (G3) led to poor enantioselectivity and reactivity. Subsequently, we investigated the influence of amide substituents on N,N-diphenyl-substituted guanidines (G4–G7) based on a proline scaffold. A notable improvement was observed when using guanidine G7, which features an isopropyl group. Encouraged by these findings, we synthesized a series of guanidines containing cycloalkylamide substituents (G8–G10). Remarkably, these guanidines demonstrated significantly enhanced reactivity and enantiocontrol. Among them, the guanidine catalyst with a cyclohexyl amide substituent (G10) delivered the best results, as mentioned above.
:1.8, delivering the desired phosphinate product 3aa in 86% yield and a 95:5 enantiomeric ratio (e.r.). During this process, we observed that the results were significantly influenced by the amino acid backbone, amide substituents, and amidine substituents of the guanidine catalysts. For instance, the use of bicyclic guanidine (G1), dialkyl-substituted catalysts (G2), and pipecolinic acid-based catalysts (G3) led to poor enantioselectivity and reactivity. Subsequently, we investigated the influence of amide substituents on N,N-diphenyl-substituted guanidines (G4–G7) based on a proline scaffold. A notable improvement was observed when using guanidine G7, which features an isopropyl group. Encouraged by these findings, we synthesized a series of guanidines containing cycloalkylamide substituents (G8–G10). Remarkably, these guanidines demonstrated significantly enhanced reactivity and enantiocontrol. Among them, the guanidine catalyst with a cyclohexyl amide substituent (G10) delivered the best results, as mentioned above.
| a 1a (0.25 mmol), 2a (0.1 mmol), (ArSe)2 (10 mol%), G (18 mol%), 4 Å MS (50 mg), 2-Cl-Tol (2.5 mL), −10 °C, air. The yields given are isolated yields (based on 2a). Enantiomeric ratio (e.r.) was determined by HPLC analysis. |
|---|

|
Subsequently, we systematically investigated the reaction conditions using Se1 and G10 as catalysts, as summarized in Table 2. Notably, no desired product was observed when either the diaryldiselenide catalyst or the chiral guanidine catalyst was used alone (entries 2 and 3). The absence of a drying agent significantly decreased the yield of 3aa from 86% to 63% (entry 4), and replacing 4 Å molecular sieves with MgSO4 also led to a reduced yield (entry 5). These observations indicated that the presence of water significantly influenced the formation of 3aa, although it did not affect the enantiocontrol of the reaction. A survey of solvents revealed that aromatic chlorinated solvents provided good yield and enantioselectivity. Switching from 2-chlorotoluene to chlorobenzene or toluene slightly decreased the yield of 3aa (entries 6 and 7), while dichloromethane (DCM), 1,2-dichloroethane (DCE), and chloroform (CHCl3) proved ineffective for this transformation (entries 8 and 9), Furthermore, the enantioselectivity was found to be highly sensitive to temperature, with the enantiomeric ratio (e.r.) of 3aa decreasing as the temperature increased (entries 10 and 11). Although lower temperatures led to an improvement in enantioselectivity, the reaction only afforded a 60% yield (entry 12).
|
|
|||
|---|---|---|---|
| Entry | Variation from standard | Yield (%) | e.r. (%) |
| a 1a (0.25 mmol), 2a (0.1 mmol), Se1 (10 mol%), G10 (18 mol%), 4 Å MS (50 mg), 2-Cl-Tol (2.5 mL), −10 °C, air. The yields given are isolated yields (based on 2a). Enantiomeric ratio (e.r.) was determined by HPLC analysis. | |||
| 1 | None | 86 | 95:5 |
| 2 | Without (ArSe)2 catalyst | 0 | — |
| 3 | Without G10 catalyst | 0 | — |
| 4 | Without 4 Å MS | 63 | 95:5 |
| 5 | MgSO4 instead of 4 Å MS | 71 | 95:5 |
| 6 | PhCl instead of 2-Cl-Tol | 73 | 95.5:4.5 |
| 7 | PhCH3 instead of 2-Cl-Tol | 56 | 95:5 |
| 8 | DCM instead of 2-Cl-Tol | Trace | — |
| 9 | DCE or CHCl3 instead of 2-Cl-Tol | 0 | — |
| 10 | rt instead of −10 °C | 83 | 88:12 |
| 11 | 0 °C instead of −10 °C | 85 | 93:7 |
| 12 | −20 °C instead of −10 °C | 60 | 96:4 |
Substrates cope study
With the optimized reaction conditions established, we proceeded to evaluate the substrate scope of this novel reaction strategy. First, the scope of phosphine oxides was investigated. As shown in Scheme 1, a series of para-substituted phenyl rings, bearing either electron-donating or electron-withdrawing groups, afforded products (3aa–3ah) with excellent yields and enantiomeric excess (up to 97% yield and 97:3 e.r.). The absolute S-configuration of 3ga was determined by X-ray crystallographic analysis (CCDC 2426084), and the configurations of the other products were assigned by analogy.
 | ||
| Scheme 1 Scope of phosphine oxidesa. aReaction conditions: 1a (0.25 mmol), 2a-2o (0.1 mmol), Se1 (10 mol%), G10 (18 mol%), 4 Å MS (50 mg), 2-Cl-Tol (2.5 mL), −10 °C, air. The yields given are isolated yields (based on 2). Enantiomeric ratio (e.r.) was determined by HPLC analysis. bThe MgSO4 as drying agent instead of 4 Å MS. | ||
Notably, no product formation was observed with ortho-substituted phosphine oxides (3ai), which we attribute to significant steric hindrance that likely impedes the reaction between the phosphine oxide and diaryldiselenide. In contrast, meta-substituted phosphine oxides proved to be excellent substrates, delivering the desired phosphinate products (3aj and 3ak) in high yields with exceptional enantiocontrol. Additionally, β-naphthyl phosphine oxide demonstrated remarkable reactivity, affording the desired compound 3al in 89% yield with 96:4 enantiomeric ratio (e.r.). Furthermore, phosphine oxides incorporating heteroaromatic moieties, such as benzodioxole (3am), benzothiophene (3an) and benzofuran (3ao), were also found to be excellent substrates, yielding the corresponding products in moderate to high yields with consistently high enantioselectivity.
Subsequently, we further explored the scope of 2-nitrobenzhydrols. As shown in Scheme 2, a diverse range of substrates containing strong electron-withdrawing groups (F, Cl, CF3, OCF3, CN, CO2Me) at the para position of the phenyl ring were successfully transformed into the desired products (4bg–4fg), achieving high yields (78–97%) along with remarkable enantiomeric excess values (95:5–98:2 e.r.). This high performance highlights the beneficial role of electron-withdrawing groups in enhancing both reactivity and stereocontrol. In contrast, substrates featuring an unsubstituted benzene ring (4gg) and biphenyl (4hg) showed a moderate reduction in both yield and enantiomeric excess, further supporting the importance of electron-withdrawing groups. Consistent with this trend, substrates containing methyl (Me) substituents yielded only trace amounts of product (4ig). Additionally, substrates with meta-substituents on the aryl unit also afforded the desired products, 4jg and 4kg, in excellent yields with good to excellent enantiomeric excess values. However, the substrate bearing a sterically demanding ortho-Cl substituent produced the desired product (4lg) in only 30% yield with 74:26 enantiomeric ratio (e.r.). Furthermore, substitution patterns on the nitrobenzene ring were well tolerated under the standard reaction conditions (4mg–4og), suggesting flexibility in this moiety. Notably, the methodology was further evaluated using substrates containing alkyl groups of varying chain lengths as well as functionalized moieties, including terminal alkenyl, alkyne, unactivated ester, primary alkyl chloride/azide, and silyl/benzyl ether, which proved effective in the formation of chain tertiary carbon stereocenters in good yield and enantioselectivity (4pg–4zg).
 | ||
| Scheme 2 Scope of 2-nitrobenzhydrolsa. aReaction conditions: 1b–1z (0.25 mmol), 2g (0.1 mmol), Se1 (10 mol%), G10 (18 mol%), 4 Å MS (50 mg), 2-Cl-Tol (2.5 mL), −10 °C, air. The yields given are isolated yields (based on 2g). Enantiomeric ratio (e.r.) was determined by HPLC analysis. | ||
For the purpose of further validating the synthetic utility of this methodology, we extended the substrate scope to investigate its application in the late-stage functionalization of natural products and drug molecules (Scheme 3). Encouragingly, 2-nitrobenzhydrol derivatives derived from probenecid, gemfibrozil, and oxaprozin were successfully transformed into the corresponding phosphinates (6ag, 70%, 91:9 e.r.; 6bg, 83%, 96:4 e.r.; 6cg, 92%, 95:5 e.r.) with excellent yields and enantioselectivities. Although the substrate scope was relatively broad with respect to the other substituent of the secondary alcohol, the requirement that one substituent on the alcohol must be an ortho-nitrophenyl group imposed a limitation on the scope.
 | ||
| Scheme 3 Late-stage modification of complex molecule substrate scopea areaction conditions: 5a–5c (0.25 mmol), 2g (0.1 mmol), Se1 (10 mol%), G10 (18 mol%), 4 Å MS (50 mg), 2-Cl-Tol (2.5 mL), −20 °C, air. The yields given are isolated yields (based on 2g). Enantiomeric ratio (e.r.) was determined by HPLC analysis. | ||
Scale-up experiments and synthetic application
To further demonstrate the synthetic potential of this synergistic catalytic system, the reaction was successfully scaled up to 2 mmol, providing compounds 3ag and 3ga in excellent yields and enantioselectivities (3ag, 90%, 96.5:3.5 e.r.; 3ga, 54%, 95:5 e.r.). The enantiomeric ratio was further improved after recrystallization (3ag, 81%, >99.5:0.5 e.r.; 3ga, 43%, 98.5:1.5 e.r.; Scheme 4A). The enantioenriched phosphinate 3ga could be efficiently reduced to form chiral 2-nitrobenzhydrol 7 (84%, 97:3 e.r.), which exhibited potential for the development of innovative antibacterial therapies.16 Particularly practicable is the facile conversion of compound 7 into a diverse range of functionalized molecules (Scheme 4B). Initially, the direct acylation of 7 afforded hydroxyl-protected derivative 8 with 87% yield and 97:3 enantiomeric ratio (e.r.). Significantly, this structurally well-defined compound 8 serves as a versatile coupling reagent, demonstrating functionality as a key synthetic intermediate. Subsequently, the reduction of the nitro group was achieved using NaBH4/NiCl2·6H2O, which smoothly delivered the corresponding chiral amino-alcohol derivatives 9 with complete retention of configuration. Furthermore, the stereospecific Mitsunobu reaction of the secondary alcohol was successfully achieved under optimized conditions (DEAD, PPh3, THF, 0 °C), yielding configurationally inverted chiral azide 10 with 87% yield and 97:3 enantiomeric ratio (e.r.). The isophthalaldehyde-derived compound 11 undergoes efficient bisphosphorylation under standard conditions, affording the corresponding bisphosphorus compound 12 with 36% yield and 99.5:0.5 enantiomeric ratio (e.r.). This product serves as a promising precursor for the synthesis of a chiral PCP pincer type ligands, which have demonstrated significant potential in transition metal catalysis. However, it's a pity that the reaction exhibits limited diastereoselectivity (dr = 1:1.5).
 | ||
| Scheme 4 Transformations and applicationsa. aReaction conditions: (a) LiAlH4 (5.0 equiv.), THF, 0 °C, 30 min; (b) CH3COCl (1.2 equiv.), Et3N (1.5 equiv.), DCM, rt; (c) NaBH4 (10.0 equiv.), NiCl2·6H2O (1.0 equiv.), EtOH, 0 °C; (d) diphenylphosphoryl azide (1.2 equiv.), DEAD (2.0 equiv.), PPh3 (2.0 equiv.), THF, 0 °C. | ||
Mechanistic studies
To gain insight into the reaction mechanism, we prepared the P(O)-SeAr intermediate A in situ from 2a and Se1, which was identified by HRMS ([M + H]+m/z 494.9838). The 31P and 77Se NMR spectra of intermediate A each exhibited a doublet with coupling constants of identical magnitude (1JPSe = −346 Hz), consistent with the formation of a P–Se bond in Int-A.19 (Scheme 5A). After that, we used Int-A as the starting material to carry out the model reaction, which afforded the target product 3aa in 56% yield with 96:4 enantiomeric ratio (e.r.) (Scheme 5B(a)). In contrast, only a trace amount of 3aa was detected in the absence of G10 (Scheme 5B(b)). These results indicate that the chiral organo-superbase catalyst G10 is responsible for deprotonating secondary alcohols in this transformation. Further control experiments were performed to explore the impact of structural modifications. Under the standard reaction conditions, substrates without nitro groups (13) exhibited remarkably low reactivity, affording only trace amounts of the desired product 14 (Scheme 5B(c)). This pronounced substrate effect was further corroborated when the nitro group was placed at the 3-position (15), resulting in significantly diminished reaction efficiency and enantioselectivity (16, 24%, 74:26 e.r.) (Scheme 5B(d)). These results demonstrate that the nitro group, serving as a strong hydrogen-bond acceptor, significantly enhances the reacivity and enantioselective control. Comparative experiments were executed with the O derivative (G11) of the corresponding guanidine catalyst G10. Notably, the enantioselectivity stayed consistent under the same reaction conditions, suggesting the N–H moiety of the amide in the catalyst does not contribute to the regulation of enantioselectivity (Scheme 5B(e)). With the facts that the product enantiomeric ratio was influenced by the amount of alcohol and racemic 2-nitrobenzhydrols were employed in this dehydrophosphorylation reaction, we proposed that the reaction proceeds via a kinetic resolution process.20 To verify this hypothesis, some control experiments were conducted (see Scheme 5C, 5B). The reaction with 0.2 mmol of racemic alcohol 1a and 0.1 mmol of 2a was carried out under standard reaction conditions, 3aa was obtained in 71% yield and 94:6 e.r. (based on 2a), and the unreacted alcohol was recovered in a 57% yield with 69:31 e.r. (based on 1a). When equal amounts of 1a and 2a were used, 3aa was obtained in 48% yield with 90:10 e.r. (based on 2a), accompanied by the unreacted 1a being left with a 38% yield and 80:20 e.r. (based on 1a). These experimental results demonstrate that racemic alcohol 1a should go through a kinetic resolution in this synergistic catalysis protocol.
 | ||
| Scheme 5 Mechanistic studies. | ||
Based on the above experiments and literature reports,11g,17d,e,21 a plausible mechanism is proposed for this transformation, as illustrated in Scheme 6. The catalytic variant of the Atherton–Todd reaction enables the coupling of two formally nucleophilic reagents via a three-step process: (1) selenophosphorylation of the phosphorus substrate: the nucleophilic P(O)–H compound reacts rapidly with the diaryldiselenide to form P(O)-SeAr intermediate A, accompanied by the release of arylselenol. (2) Chiral guanidine catalyst G10 enables kinetic resolution of the racemic alcohol: density functional theory (DFT) calculations (Fig. S1–S3 in SI for details) indicate that the second step involves a concerted mechanism. When G10 interacts with Int-A and the substrate (S)-2-nitrobenzhydrol, molecular complex COM-S is formed; after which, the hydroxyl proton of the (S)-2-nitrobenzhydrol transfers to the catalyst, while the chiral alkoxide ion undergoes nucleophilic substitution at the P(O)-SeAr intermediate via the transition state TS-S, affording the P(O)-Nu product alongside the liberation of arylselenol and regeneration of the chiral guanidine catalyst. IGMH analysis of TS-S (Fig. S4 in SI) revealed that during the proton transfer process, the hydroxyl proton exhibits a distinct tendency to form an N–H bond with the catalyst, with a considerable degree of covalent character observed in this interaction. Simultaneously, there is a strong bonding tendency between the hydroxyl oxygen atom and the phosphorus atom. The system is further stabilized by various significant weak interactions, including π–π stacking and C–H⋯π interactions. (3) Regeneration of the diselenide catalyst: The liberated arylselenol intermediate undergoes aerobic oxidation under air conditions, regenerating the diaryldiselenide catalyst and completing the catalytic cycle.
 | ||
| Scheme 6 Proposed catalytic cycle. | ||
Conclusions
In conclusion, we have developed an efficient catalytic system for the dehydrophosphorylation of 2-nitrobenzhydrols with P(O)–H compounds through the synergistic action of diselenide and guanidine catalysts. The strategy leverages both the unique reactivity of the diaryldiselenide catalyst to activate P(O)–H bonds and the role of chiral guanidine to resolve secondary alcohols, offering a sustainable and efficient route to C-stereogenic phosphinates. The reaction proceeds under mild conditions, providing access to a diverse range of enantioenriched phosphinates with excellent functional group tolerance, and the resulting chiral phosphinates serve as versatile synthetic intermediates for further transformations. A limitation of the current system is that one substituent on the alcohol must be an ortho-nitrophenyl group. Further exploration of this catalytic strategy is in progress in our group, with the aim of synthesizing other important P-chirogenic compounds and broadening its utility in synthetic organic chemistry.Author contributions
Methodology, S.-D. Y.; investigation, J.-Y. G., Z.-C. Q. and Q.-X. F.; computational studies, P.-P. Z. and Y.-H. Q.; writing – original draft, S.- D. Y., J.-Y. G. and P.-P. Z.; writing – review & editing, S.-D. Y., J.-Y. G., Z.-C. Q., Q.-M. Z., Y.-H. Q. and P.-P. Z.; supervision, S.-D. Y.Conflicts of interest
There are no conflicts to declare.Data availability
CCDC 2426084 contains the supplementary crystallographic data for this paper.22All experimental procedures, characterisation data, mechanistic investigations, NMR spectra and HPLC spectra can be found in the supplementary information (SI). Supplementary information: general information, experimental procedures and characterization data of the synthesized compounds. See DOI: https://doi.org/10.1039/d5sc07228j.
Acknowledgements
We are grateful to the NSFC (No. 22171119 and 22371105) and The Science and Technology Major Program of Gansu Province of China (22ZD6FA006, 23ZDFA015, 24ZD13FA017) for financial support.Notes and references
-
(a) Ł. Joachimiak and K. M. Błażewska, J. Med. Chem., 2018, 61, 8536–8562 CrossRef PubMed
; (b) A. Babbs, A. Berg, M. Chatzopoulou, K. E. Davies, S. G. Davies, B. Edwards, D. J. Elsey, E. Emer, S. Guiraud, S. Harriman, C. Lecci, L. Moir, D. Peters, N. Robinson, J. A. Rowley, A. J. Russell, S. E. Squire, J. M. Tinsley, F. X. Wilson and G. M. Wynne, J.Med. Chem., 2020, 63, 7880–7891 CrossRef CAS PubMed
- M. Eto, Biosci., Biotechnol., Biochem., 1997, 61, 1–11 CrossRef CAS
-
(a) J.-L. Montchamp, Acc. Chem. Res., 2014, 47, 77–87 CrossRef CAS PubMed
- For the application of the Atherton–Todd reaction in phosphorylation, see:
(a) F. R. Atherton, H. T. Openshaw and A. R. Todd, J. Chem. Soc., 1945, 660–663 RSC
- For the iodide-mediated dehydrophosphorylation, see:
(a) J. Dhineshkumar and K. R. Prabhu, Org. Lett., 2013, 15, 6062–6065 CrossRef CAS PubMed
- For the Tf2O-mediated dehydrophosphorylation, see:
(a) Y. Unoh, K. Hirano and M. Miura, J. Am. Chem. Soc., 2017, 139, 6106–6109 CrossRef CAS PubMed
- A. Mohd, T. Anitha, K. R. Reddy, J. Wencel-Delord and F. Colobert, Eur. J. Org Chem., 2019, 2019, 7836–7841 CrossRef CAS
-
(a) S. Fang, J.-P. Tan, J. Pan, H. Zhang, Y. Chen, X. Ren and T. Wang, Angew. Chem., Int. Ed., 2021, 60, 14921–14930 CrossRef CAS PubMed
- S. Fang, Z. Liu, H. Zhang, J. Pan, Y. Chen, X. Ren and T. Wang, ACS Catal., 2021, 11, 13902–13912 CrossRef CAS
- S. Fang, J. He, Z. Liu, Z. Su, F. Guo and T. Wang, ACS Catal., 2023, 13, 13077–13088 CrossRef CAS
- Selenium-catalyzed organic reactions:
(a) T. Hori and K. B. Sharpless, J. Org. Chem., 1979, 44, 4204–4208 CrossRef CAS
-
(a) D. M. Freudendahl, S. Santoro, S. A. Shahzad, C. Santi and T. Wirth, Angew. Chem., Int. Ed., 2009, 48, 8409–8411 CrossRef CAS PubMed
- Z.-C. Qi, Y. Li, J. Wang, L. Ma, G.-W. Wang and S.-D. Yang, ACS Catal., 2023, 13, 13301–13309 CrossRef CAS
-
(a) S.-X. Li, Y.-N. Ma and S.-D. Yang, Org. Lett., 2017, 19, 1842–1845 CrossRef CAS PubMed
- Some Reviews of Chiral organo-superbases:
(a)
T. Ishikawa, Superbases for organic synthesis: guanidines, amidines and phosphazenes and related organocatalysts, Wiley Online Library, 2009 CrossRef
- M. P. Storz, G. Allegretta, B. Kirsch, M. Empting and R. W. Hartmann, Org. Biomol. Chem., 2014, 12, 6094–6104 RSC
- For selected examples of chiral guanidines and their derivatives in enantioselective reactions, see:
(a) D. Leow and C.-H. Tan, Asian J., 2009, 4, 488–507 CrossRef CAS PubMed
- For selected examples of the Resolution of Racemic alcohol Derivatives Catalyzed by Chiral Guanidine, see:
(a) K. Nakata and I. Shiina, Org. Biomol. Chem., 2011, 9, 7092–7096 RSC
- H. Duddeck, Prog. Nucl. Magn. Reson. Spectrosc., 1995, 27, 1–323 CrossRef CAS
-
(a) W.-Q. Cai, Q. Wei and Q.-W. Zhang, Org. Lett., 2022, 24, 1258–1262 CrossRef CAS PubMed
-
(a) T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672–12673 CrossRef CAS PubMed
-
CCDC 2426084: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2mfjsd
Footnote |
| † These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |