
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimple s-block metal hydrides for selective hydrogenation of quinoline compounds
Shengyuan
Zhang†
ab,
Hong
Wen†
a,
Qianru
Wang
 *ab,
Yongli
Cai
ab,
Zhao
Li
ab,
Liang
Liu
c,
Xuanwei
Chang
ab,
Jianping
Guo
ab and
Ping
Chen
*ab
*ab,
Yongli
Cai
ab,
Zhao
Li
ab,
Liang
Liu
c,
Xuanwei
Chang
ab,
Jianping
Guo
ab and
Ping
Chen
*ab
aDalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China. E-mail: wqr92@dicp.ac.cn; pchen@dicp.ac.cn
bCenter of Materials and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing, China
cCollege of Chemistry, Central China Normal University, Wuhan, China
First published on 11th November 2025
Abstract
For many decades, selective hydrogenation of N-heterocycles using H2 has been considered the exclusive domain of d-block metals. Herein, a series of solid-state s-block metal hydrides is demonstrated to function as transition-metal-free catalysts for the hydrogenation of quinoline, affording high yields of 1,2,3,4-tetrahydroquinoline. Under relatively mild conditions, the catalytic performance of the representative barium hydride (BaH2) is good enough to compete with some recently developed Fe, Co, Ni catalysts. Moreover, this transformation is remarkably simple and does not need any sophisticated ligands, co-catalysts, and additives compared with the existing methodology. Mechanistic studies reveal that the quinoline substrate can activate BaH2in situ, generating a catalytically active organobarium hydride species that effects the hydrogenation process. With the aid of theoretical modeling, the synergistic scenario between the Lewis acidic barium cation and the nucleophilic hydride anion in regioselective hydrogenation of quinoline is delineated, which shows a working principle distinct from the conventional transition metal catalysis. These findings provide insights into the discovery of new catalysts and/or reactivity that is complementary to that of transition metals.
1. Introduction
1,2,3,4-Tetrahydroquinoline is a common structural motif that exists in numerous biologically active natural products and pharmacologically relevant therapeutic agents. Because of the significance of this scaffold in drug discovery and medicinal chemistry, some synthetic methods such as reductive cyclization, Beckmann rearrangement, and hydrogenation of quinoline, have been established during the past few decades.1 Among these processes, selective hydrogenation of quinoline is straightforward and promising because of its high atom efficiency together with the easy accessibility of the raw material. Relevant homogeneous or heterogeneous catalytic systems traditionally fall under the purview of noble metals (Ir, Rh, Ru, Pd, etc.) owing to their superior activity and selectivity.1a,2 Considering the growing awareness of the toxicity and limited availability of such metals, current research efforts focus on using earth-abundant transition metal (Mn, Fe, Co, etc.) based catalysts.3 Particularly noteworthy is that these transition metal-catalyzed processes often involve H–H bond cleavage to generate a metal hydride intermediate that can play a role in facilitating hydrogen transfer to the unsaturated nitrogen heterocycle of quinoline.3b,i,4By virtue of their unique bonding characteristics and reaction chemistry, s-block metal hydrides, either in inorganic or organic form, have been demonstrated to show great promise in bond activation and catalytic processes that involve N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, C–H, H–H, C
N, C–H, H–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, etc.5 Their lack of partially filled valence d-orbitals makes them operate through working principles different from the transition-metal catalysis that strongly relies on backbonding interactions toward substrate activation. In terms of the hydrogenation of unsaturated CC or CN bonds, several early reports have indicated that solid-state main group hydrides such as KH, CaH2, and LiAlH4 show limited activity for the hydrogenation of ethylene or conjugated dienes.6 However, the reaction processes and mechanisms that are responsible for these transformations remains obscure. Recently, breakthroughs in ligand design have enabled a series of molecular calcium complexes to be used in catalyzing the hydrogenation of alkenes or imines.7 The key to such catalysis is suggested to be the formation of active calcium hydride species, which can be generated upon hydrogenolysis of the organocalcium precursors.
C, etc.5 Their lack of partially filled valence d-orbitals makes them operate through working principles different from the transition-metal catalysis that strongly relies on backbonding interactions toward substrate activation. In terms of the hydrogenation of unsaturated CC or CN bonds, several early reports have indicated that solid-state main group hydrides such as KH, CaH2, and LiAlH4 show limited activity for the hydrogenation of ethylene or conjugated dienes.6 However, the reaction processes and mechanisms that are responsible for these transformations remains obscure. Recently, breakthroughs in ligand design have enabled a series of molecular calcium complexes to be used in catalyzing the hydrogenation of alkenes or imines.7 The key to such catalysis is suggested to be the formation of active calcium hydride species, which can be generated upon hydrogenolysis of the organocalcium precursors.
Hydrogenation of quinoline to 1,2,3,4-tetrahydroquinoline is a transformation involving both CN and CC bond activation, but it is also confronted with challenges arising from the inherent aromatic stability of heteroarenes as well as the chemo- and regio-selectivity issues. Herein, we demonstrate a series of simple, easily accessible, solid-state s-block metal hydrides as catalysts for selective hydrogenation of quinoline under mild and ligand-free conditions. Among them, barium hydride BaH2 performs extraordinarily well with both conversion and selectivity higher than 99%, achieving gram scale preparation of 1,2,3,4-tetraquinoline effectively. With a combination of experimental and theoretical studies, we find that the key to achieving efficient hydrogenation the formation of a soluble barium hydride active species in situ under operating conditions, which behaves differently from the conventional transition-metal-based systems in substrate coordination and bond activation. This study offers an alternative class of s-block hydrogenation catalysts, which are unlike the existing molecular s-block catalysts that generally rely on specially designed ligands to stabilize and solubilize structural motifs, and the mechanistic insights arising from this case study would facilitate our understanding of catalytic hydrogenation mediated by s-block metal elements, which is far from being on par with d-block metal catalyzed processes.
2. Results and discussion
2 1. Catalytic performance for the hydrogenation of quinoline and analogues
Hydrogenation reactions need the inputs of electrons and hydrogens for activating unsaturated bonds and forming new H–C/N/O bonds, which could be supplied by hydridic hydrogens.8 Based on the recent success of using s-block metal hydrides in catalyzing the ammonia synthesis reaction,9 we initiated our studies by exploring the reactivity of simple alkali or alkaline earth metal hydrides (AMHs) in the benchmark hydrogenation of quinoline. In a preliminary set of experiments with 20 mol% AMH materials (with respect to the quinoline substrate) under 50 bar H2 at 150 °C for 24 h in THF, the desired product 1,2,3,4-tetrahydroquinoline could be obtained but with different yields depending on AMH (Fig. 1a). It was seen that the use of alkali metal hydrides led to almost full conversion of quinoline, and applying alkaline earth metal hydrides showed impressive selectivity towards 1,2,3,4-tetrahydroquinoline. The differences likely arise from the balance between reactivity and coordination ability of these hydrides, with alkali hydrides being more reactive but less selective, and alkaline earth hydrides providing better selectivity through controlled substrate coordination. Among them, barium-containing hydrides (BaH2 and LiBaH3) perform extraordinarily well, affording almost full conversion and excellent yield. Taking the simple and efficient BaH2 as the objective, we carried out further optimization experiments mainly on the critical reaction parameters such as solvent, temperature, pressure, and catalyst concentration (see Tables S1 and S2). The key findings are listed as follows: (1) even at temperatures below 100 °C and pressures below 30 bar, 99% yield can be attained in the presence of 20 mol% BaH2; (2) applying a reduced catalyst amount (2.5 mol% BaH2, 150 °C, and 50 bar H2) shows the feasibility of a gram scale synthesis of 1,2,3,4-tetrahydroquinoline within 24 hours; (3) good yields (>99%) were obtained only with THF or mixed solvents containing THF, in which the BaH2 powder dissolved after the reaction. In comparison, testing other strong-polar or non-polar solvents resulted in poor reactivity along with the formation of colorless precipitates. Such a comparison indicates the unique role of THF during the transformation; (4) little conversion (<1%) was observed when carrying out the reaction in the presence of argon instead of a hydrogen atmosphere, which excludes the contributions from transfer hydrogenation with THF or BaH2. With the above optimized conditions in hand, the catalyst stability was then investigated. As shown in Fig. 1b, excellent yields of 1,2,3,4-tetrahydroquinoline were maintained at >95% after repeating the reaction 12 times. By comparing the reactivity of BaH2 with that of the commonly used d-block metal-based catalysts, it is seen that such a transition-metal free compound shows comparable performances with some of the recently developed Fe or Co catalysts under similar reaction conditions (Table S3). | ||
| Fig. 1 Catalytic performance. (a) 1,2,3,4-Tetrahydroquinoline yield of a series of s-block metal hydrides for the hydrogenation of quinoline. Reaction conditions: quinoline (4 mmol), AMH (20 mol%), THF (8 mL), H2 (50 bar), 150 °C, 48 h for LiH, NaH, and KH, 24 h for BaH2, SrH2, and LiBaH3; yield of 1,2,3,4 tetrahydroquinoline was calculated by GC using dodecane as the internal standard. (b) Long-life catalytic performance of the BaH2-catalyzed hydrogenation of quinoline. (c) Substrate scope of selective hydrogenation over the BaH2 sample. Reaction conditions: substrates (4 mmol), BaH2 (2.5 mol%), THF (8 mL), H2 (50 bar), 150 °C, 24 h; a48 h; b72 h; cBaH2 (20 mol%). Yield of the product is given. | ||
Moreover, substituted quinolines as well as other N-heteroarenes were explored under the developed protocol for selective hydrogenation, and the synthetic scope and limitation are summarized in Fig. 1c. Quinolines bearing a methyl group on either the benzene or the heteroarene ring reacted smoothly, affording the corresponding products in high yields. The steric effect is obvious. For the substituted quinolines with a more sterically hindered group such as isopropyl and phenyl substituents, longer reaction times and/or higher BaH2 concentrations were required to achieve high conversion. Quinoline derivatives featuring acidic protons such as hydroxyquinolines and aminoquinolines are not amenable to hydrogenation under catalytic conditions; however hydrogenation can be carried out when the protons are replaced. For example, N,N-dimethylquinolin-8-amine can be selectively converted to the hydrogenation product in good yield. The tolerance of this catalytic protocol was further studied by exploring several quinolines bearing redox-sensitive substituents such as nitrile, ester, ketone, but no desired product was observed. This could be due to the tendency of acidic protons or reducible groups to react with hydridic hydrogen on the catalyst, forming stable organobarium salts that do not work for hydrogenation. Finally, other N-heteroarenes such as isoquinoline, benzo(h)quinoline, and acridine were hydrogenated selectively with the present catalytic system, and the corresponding products could be obtained in good yields under optimized conditions.
2 2. Identification and characterization of the catalytically active species
The efficacy of BaH2 in selective hydrogenation of quinoline motivated us to identify and determine the actual catalytic active species that could evolve in situ under working conditions, especially considering that the reactivity of solid saline hydrides can be increased by taking them out of their crystal lattice and bringing them into a soluble form.10 Indeed, such a hypothesis can be partially supported by our experimental observation: the initially insoluble BaH2 powder “disappeared” and a yellowish-brown solution appeared at the end of the reaction, indicating the formation of soluble Ba species during hydrogenation. To better understand the reaction process, we recorded the time course of quinoline conversion and 1,2,3,4-tetrahydroquinoline yield over BaH2 sample. As depicted in Fig. 2a, the formation of the desired product follows the significant consumption of quinoline (around 40% conversion), and the maximum difference occurs at the initial stage of the reaction (0–2 h), which indicates that BaH2 experiences substrate-induced activation before catalyzing the synthesis of 1,2,3,4-tetrahydroquinoline. Unfortunately, attempts to isolate and characterize the in situ generated active species were not successful, as with most of previous studies on s-block metal mediated catalytic hydrogenations.11 This is mainly due to their sensitivity towards ligand exchange and hydrolysis leading to a mixture of species with undefined nature. However, we found that, with proper pretreatment, BaH2 can be activated for the selective hydrogenation of quinoline. Fig. 2b shows that a treatment of BaH2 with excess quinoline in THF at 150 °C for 2 h can lead to the immediate production of 1,2,3,4-tetrahydroquinoline at the beginning of hydrogenation, which eventually reaches similar levels before approaching full conversion, while no 1,2-dihydroquinoline is detected throughout the reaction. These results prompt us to propose quinoline-activated BaH2 as the potential active species for the selective hydrogenation of quinoline to 1,2,3,4-tetrahydroquinoline. | ||
| Fig. 2 Characterization of the catalytically active species. (a) Time course of quinoline conversions and 1,2,3,4-tetrahydroquinoline yields over the BaH2 sample. Reaction conditions: quinoline (4 mmol), BaH2 (20 mol%), THF (8 mL), H2 (50 bar), 100 °C; (b) time course of 1,2,3,4-tetraquinoline yields over BaH2 activated with quinoline in THF solvent under 9 bar Ar and 150 °C for 2 h. The inset picture and figure show the color change and the corresponding UV-vis spectra of BaH2 before and after quinoline activation. The reaction conditions remain the same as in (a); (c) 1H NMR spectra for the activation of BaH2 with quinoline at 150 °C in THF solvent (blue line), the reaction of quinoline-activated BaH2 with HBpin at room temperature (red line), and the reaction of quinoline-activated BaH2 with H2 at 150 °C (black line); (d) FT-IR spectra of quinoline-activated BaH2, quinoline-activated BaD2, and quinoline-activated BaH2 after being exposed to air (denoted as activated BaH2 (air)), and pristine BaH2 samples. The peaks observed in the 1800–1200 cm−1 region correspond to the stretching vibrations of CC, C–C, and C–N of the 1,2-dihyroquinolyl group. | ||
The composition and structure of such reactive species were analyzed by spectroscopic characterization and theoretical simulation. As shown in the inset of Fig. 2b, a visible color change was observed upon activating BaH2 with excess quinoline in THF, and the corresponding UV-vis spectrum displays a new absorption peak centered at ca. 412 nm, which might be relevant to the formation of 1,2-dihydroquinoline species according to the previous literature reports.12 Meanwhile, the 1H NMR spectrum of the activated BaH2 also displays a set of new peaks similar to that of 1,2-dihydroquinoline but without H resonance of the –NH group (Fig. S1 and the blue line in Fig. 2c). These results indicate that during the activation treatment, a certain 1,2-dihydroquinoline barium salt was formed via a 1,2-addition pathway involving Ba addition to 1-position nitrogen and hydride addition to 2-position carbon of the quinoline, as depicted in eqn (1).
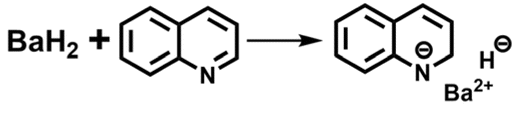 | (1) |
This is consistent with the previous observation on the reactivity of s-block metal hydrides with N-heterocycle compounds.13 Moreover, the formed 1,2-dihydroquinoline barium salt can be trapped by reacting with pinacolborane (HBpin), where the exclusive production of N-borylated-1,2-dihydroquinoline again evidences the 1,2-addition of BaH2 to quinoline (red line in Fig. 2c and eqn (2)).
 | (2) |
Upon hydrogenation with H2, the 1,2-dihydroquinoline species bound to barium can be converted to 1,2,3,4-tetrahydroquinoline (black line in Fig. 2c and eqn (3)).
 | (3) |
Our FT-IR observation confirms that hydride species is still bound to barium within the quinoline-activated BaH2. As shown in Fig. 2d, it exhibits a Ba–H stretching vibration centered at 1072 cm−1, which is confirmed by the Ba–D stretching band (peak frequency: 794 cm−1) in the deuterated sample. Note that hydride species are commonly air- or moisture-sensitive, and hence the corresponding Ba–H peak would disappear after being exposed to air. When compared to pristine BaH2, a similar Ba–H stretching range can be observed but somehow with a blue shift, which might be attributable to the activation treatment that leads to a shortened Ba–H bond after the formation of the Ba–N bond. As a supplementary proof, the hydride species also appears in the 1H NMR spectrum of quinoline-activated BaH2 in THF-d8, with a singlet resonance at 9.14 ppm (Fig. S2). Such a chemical shift is consistent with typical barium hydride complexes which feature hydride resonances in the range of 7.9–10.4 ppm.14 In addition to the dihydroquinolyl and hydrido species, such activated Ba species may further coordinate with THF by sharing the electron pair on oxygen, especially considering its unique solubility in THF-containing solvent. Nevertheless, no obvious change in the 1H NMR spectrum was observed between the coordinated THF and excess free THF, probably because their fast exchange would average out the chemical shifts. Given the results described above, the active species involved in the catalytic cycle were proposed to be a soluble barium cluster coordinated with hydrido, 1,2-dihydroquinolyl, and THF groups. Those experimental findings provide the basis for building a reasonable catalyst model, by which a relatively accurate mechanistic study can be carried out by means of theoretical simulations.
2 3. Mechanistic investigations and kinetic analyses
Although the use of s-block metals in catalysis has attracted increasing interest in the past two decades, the corresponding mechanistic studies are scarce. This is partly due to the dynamic behavior of these catalytic systems, which makes them difficult to isolate and characterize. In our preliminary theoretical study, a series of monomeric barium complexes were screened to simulate the catalytically active center, considering that less crowded monomeric species are easily accessible by reactants and intrinsically more reactive than dimers or larger aggregates.15 Taking into account the above experimental findings, a relatively stable monomeric Ba(H)(1,2-DHQ)(THF)4 cluster consisting of one hydride, one 1,2-DHQ, and four coordinating THF molecules was identified as the potential active center for catalytic quinoline hydrogenation (Fig. 3a). Its optimized structure shows a Ba–H bond length (2.60 Å) slightly shorter than that of BaH2 (2.61–3.02 Å), which correlates well with our FT-IR observation of a weak blue-shift effect (Fig. 2d). Based on this model catalyst, the full free energy diagram for the hydrogenation of quinoline was mapped out using DFT calculations (Fig. 3b). Generally, it is seen that the Lewis acidic barium cation and the reactive hydride group work collaboratively to mediate the hydrogenation cycle and hence serve as the active components of the catalyst, whereas the coordinated 1,2-DHQ anion and THF act as innocent spectators. | ||
| Fig. 3 Mechanistic study. The simulated active cluster (a), the calculated free energy profile (b), and the geometries of transition states TS1–2 and TS2–3 (c) for the quinoline hydrogenation with the BaH2 precatalyst. ΔG values at 150 °C in kcal mol−1 are corrected for THF solvent effects. The 1,2-DHQ anion and THF substituents of 0 are simplified as “L” in the reaction pathway. In the geometries shown, only hydridic H atoms and coordinated dihydrogen are shown and the distances are in Å. | ||
The catalytic cycle starts with the activation of quinoline through the facile dissociation of a coordinated THF (0 → 1), where the electronegative nitrogen atom of the heterocyclic ring coordinates to the barium center, largely determining the regioselective outcome of the catalytic event. What follows is an inner-sphere transfer of the hydride to 2-position carbon of the coordinated quinoline substrate, forming the thermodynamically more favorable complex 2via the transition state TS1–2. After the migratory insertion of CN to the Ba–H bond, a vacant coordination site becomes available for dihydrogen. The intermediate 2, bearing a strongly polarized Baδ+−Nδ− bond, behaves like an intramolecular frustrated Lewis pair and can cause the heterolytic cleavage of the H–H bond through a concerted σ-bond metathesis process, in which a polarized four-center transition state (TS2–3) is proposed to explain the regeneration of the catalytically active barium hydride species. This hydrogenolysis step (2 → TS2–3 → 3), with a free energy barrier of up to 19.2 kcal mol−1, was identified as the rate-determining step (RDS) of the whole catalytic cycle. Such a mechanistic feature not only correlates with our observation that applying high H2 pressure promotes the selective hydrogenation of quinoline, but also can be supported through a combined experimental and theoretical analysis of the inverse H/D isotope effect in the hydrogenation process.16 Experimentally, we identified a characteristic inverse isotope effect (kH2/kD2 ≈ 0.68) when H2 was replaced by D2 in the reactant stream (Fig. 4a). This is consistent with the theoretical predictions, in which an inverse H/D isotope effect related to H2 splitting (2 → TS2–3 → 3) as the RDS is confirmed (the corresponding detailed information is shown in Fig. S3 and Table S4). By contrast, a normal isotope effect (kBa–H/kBa–D ≈ 1.03) was observed when BaD2 instead of BaH2 was used as the catalyst precursor (Fig. 4b), suggesting that the hydride transfer step may not be the kinetically slowest step. Indeed, kinetic calculations on the hydride transfer step (1 → TS1–2 → 2) of the proposed reaction pathway also predict a normal isotope effect.
 | ||
| Fig. 4 H–D kinetic isotope effect observed in 2-methylquinoline hydrogenation at 150 °C and 50 bar H2. (a) Plot of relative conversion vs. reaction time with H2 or D2 as the reactant; (b) plot of relative conversion vs. reaction time with BaH2 or BaD2 as the catalyst precursor. Note that the changes in 2-methylquinoline conversion (i.e., relative conversion) with the reaction time are relative to the initial conversion under the given conditions. | ||
Intriguingly, the hydrogenated intermediate 3 tends to undergo a structural rotation forming intermediate 4 through coordination of the CC bond to the metal center. This leads to polarization of the π-electron density of the CC bond, thus giving an incentive for hydride nucleophilic attack. The second hydride transfer step (4 → TS4–5 → 5) occurs in a manner similar to the first hydride transfer step (1 → TS1–2 → 2), but with the formation of a Ba–C bond after insertion of CC moieties into the Ba–H bond. Likewise, this affords a vacant coordination site for activating another dihydrogen molecule. The polarized nature of the Baδ+–Cδ− bond in intermediate 5 allows the subsequent heterolytic cleavage of H2 to proceed through the assembly of a polarized four-center transition state TS5–6. Such a σ-bond metathesis step finally gives the hydrogenated product 1,2,3,4-tetrahydroquinoline, while regenerating the starting barium hydride complex.
According to the proposed reaction mechanism, it can be learned that the working principle of the Baδ+–Xδ− (X = H, C, N) unit towards CN, CC, and H–H bond activation is mainly dictated by the Lewis acidity of the electropositive metal cation as well as the basicity and nucleophilic nature of the bound electronegative substituent, X. As a consequence, the catalytic activity of this organobarium hydride complex is not based on redox chemistry but instead relies on basic dipolar transformations. Within the catalytic cycle, the polarized Baδ+–Xδ− bonds tend to engage in a succession of two key elementary steps (Fig. 5a), that is (1) Baδ+–Hδ− induces the polarization of π-electrons of the unsaturated CC/CN bond to facilitate hydride nucleophilic attack; (2) Baδ+–Nδ−/Baδ+–Cδ− leads to heterolytic cleavage of the σ-bonded H–H via σ-bond metathesis, wherein regeneration of reactive hydride species does not involve oxidation or reduction of the metal site. These mechanistic features markedly set this organobarium hydride catalyst apart from the conventional transition metal (TM)-based hydrogenation catalysts, in which a series of redox steps such as oxidative addition and reductive elimination, as well as homolytic cleavage of H2 are usually involved in the reaction pathway (Fig. 5b). This is understandable since the lack of partially filled valence d-orbitals make s-block metals generally favor only one oxidation state (+II for barium), which limits their reactivity to redox-neutral pathways. To be specific, transition metal catalysts are able to backdonate d-electrons in antibonding orbitals, thus activating the substrate. In contrast, the reported organobarium hydride catalyst principally reacts through Lewis acidic substrate polarization followed by nucleophilic attack.
 | ||
| Fig. 5 Proposed reaction models for the hydrogenation of quinoline to 1,2,3,4-tetrahydroquinoline (py-THQ) over the alkali or alkaline earth metal hydride complex (H–AM–L) catalyst (a), and transition metal (TM) catalyst (b). | ||
Heavier alkaline earth metals such as Ca, Sr, and Ba are characterized by their high electropositivity and polarizability, and hence normally rely on the Lewis acidity of the metal center to bind the reaction substrate via electrostatic interactions.17 Such an electrostatic activation mode can be reflected by the interaction between quinoline and barium illustrated in Fig. 3b, which features electrostatic Ba2+⋯Nδ+Cδ− or Ba2+⋯Cδ+Cδ− contacts. Nevertheless, there are reports highlighting the relevance of the inner (n − 1)d orbitals of heavier alkaline earth in covalent bonding during substrate activation, despite the fact that these empty d-orbitals are relatively high in energy. This is exemplified by several recent observations such as N2 activation at low-valent calcium and benzene activation in Ba(benzene)3.18 These intriguing findings encouraged us to delve deeper into the bonding situation of the reported organobarium hydride for quinoline activation. Herein, the energy decomposition with natural orbitals for chemical valence (EDA-NOCV) analysis was performed to unravel the key interactions in 1 and 4. Table 1 reports the computed interaction energy (ΔEint) and its decomposition into Pauli (ΔEPauli), dispersion (ΔEdisp), electrostatic (ΔEelstat), and orbital (ΔEorb) interaction energy components. The results reflect a high degree of electrostatic character in the interaction between barium and nitrogen of quinoline (1), as well as the interaction between barium and carbon of 1,2-dihydroquinoline (4). Further NOCV orbital projections indicate a small but discernible covalent contribution associated with Ba 5d orbital participation (Table S5).
| Fragments | ΔEint | ΔEPauli | ΔEdisp | ΔEelstat | ΔEorb |
|---|---|---|---|---|---|

|
−21.7 | 46.5 | −10.4 | −23.9 (65.3%) | −12.7 (34.7%) |

|
−31.1 | 67.6 | −17.8 | −32.7 (63.7%) | −18.6 (36.3%) |
3. Conclusion
In summary, selective hydrogenation of quinoline to 1,2,3,4-tetrahydroquinoline with catalytic instead of stoichiometric quantities of alkali or alkaline earth metal hydrides is demonstrated, among which BaH2 performs relatively active and stable under mild conditions. Its simplicity and facile synthetic accessibility make BaH2 stand out from the recently developed metal-based catalysts, which generally require the utilization of sophisticated ligands or construction of special structure during catalyst preparation. Experimental studies suggested that solid-state BaH2 is merely a catalyst precursor, which can be in situ transformed into a more active soluble barium hydride species upon reaction with the quinoline substrate. The mechanistic function of each component of this catalytic active species is delineated by simulating the whole reaction pathway, in which a different substrate activation mode from the transition metal catalysis is highlighted. Additionally, the simulated kinetic performances agree well with the experimental results, strengthening the rationality of the proposed reaction mechanism. The scientific findings derived from this study would be conducive to the design of s-block metal hydride compounds aimed at fine-tuning their reactivity and related catalytic applications.4. Materials and methods
4 1. General information
All material handling was performed in a glovebox filled with purified argon to maintain a low water vapor concentration (<1 ppm) and a low oxygen concentration (<1 ppm). H2 (99.999%), D2 (99.999%), and Ar (99.999%) were purchased from Dalian Special Gas Co., Ltd. All reagents were obtained commercially from various chemical companies and were used without further purification, unless otherwise mentioned. Dry and oxygen-free solvents (THF, toluene, benzene, cyclohexane, 1,4-dioxane and 2-methyltetrahydrofuran, methylcyclohexane and n-hexane) were dried over 4 Å molecular sieves before use. Deuterated organic solvents (THF-d8) were purchased from J&K and dried over sodium/potassium alloy.4 2. Materials preparation
BaH2 or SrH2 was synthesized via hydrogenation of barium metal (Macklin, ≥99% trace metals basis, shot diameter: ∼2 cm) or strontium metal (Aladdin, ≥99.5% trace metals basis) at 20 bar H2 pressure and room temperature for 2 days followed by heat treatment at 400 °C for 5 h. The corresponding deuteride samples were obtained following a similar approach, except that hydrogenation was carried out in pure D2 instead of H2. LiBaH3 was synthesized by ball milling a mixture of LiH and BaH2 solid powders in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at room temperature under an an argon atmosphere for 12 h. All of these alkali and alkaline earth metal hydrides, including the synthesized BaH2 and LiBaH3 as well as the purchased LiH (Alfa, 99.4%) and NaH (Alfa, 57–63% oil dispersion), were ball-milled in an Ar-filled vessel on a Retsch planetary ball mill (PM 400) at 200 revolutions per minute (rpm) for 3 h before the hydrogenation test.
:1 at room temperature under an an argon atmosphere for 12 h. All of these alkali and alkaline earth metal hydrides, including the synthesized BaH2 and LiBaH3 as well as the purchased LiH (Alfa, 99.4%) and NaH (Alfa, 57–63% oil dispersion), were ball-milled in an Ar-filled vessel on a Retsch planetary ball mill (PM 400) at 200 revolutions per minute (rpm) for 3 h before the hydrogenation test.
4 3. Characterization
Fourier transform infrared (FTIR) measurements were conducted on a Brucker Tensor II unit with a scan resolution of 4 cm−1 and an accumulation of 32 scans per measurement. UV-visible spectra measurements were conducted on a Jasco V-750 spectrometer in diffuse reflection mode. An airtight sample cell was used to avoid air exposure. Solution NMR spectroscopy was performed at room temperature on a JEOL spectrometer operating at 400 MHz. Solution 1H NMR spectra were recorded with THF-d8 as the deuterated reagent. GC-MS measurements were performed on a Shimadzu GCMS-QP2020 NX with an Electron Ionization (EI) source. Gas chromatography (GC) analyses were conducted on a Fu Li 9790-II equipped with a flame ionization detector (FID) and a capillary column (OV-1701). To determine the GC yield, calibration curves were generated using dodecane as the internal standard.4 4. Catalysis
4 5. Calculations
DFT calculations were performed with the Gaussian 09 package. The molecular geometries of all complexes were optimized at the B3LYP-D3 (BI) level with the SMD solvent model for THF. The SDD basis set was used for the Ba atom, and the def2-SVP basis set was used for other atoms. Frequency calculations at the same level of theory were also performed to identify all the stationary points as either minima (zero imaginary frequencies) or transition states (one imaginary frequency) and to obtain free energies at 150 °C. The intrinsic reaction coordinate (IRC) analysis was carried out to confirm that all stationary points are smoothly connected to each other. EDA-NOCV analyses were additionally performed using the ADF (AMS 2023) program at the BP86/TZ2P level with scalar relativistic ZORA corrections. Neutral moieties, [H–Ba–L] and the corresponding ligand (quinoline or 1,2-dihydroquinoline), were defined as the interacting fragments, and the frozen-core approximation was applied to the [1s–4d] shells of Ba.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Data availability
The data supporting the findings of this study are available within the article and its supplementary information (SI) or from the corresponding authors upon reasonable request. Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc07195j.Acknowledgements
The authors are grateful for the financial support from the National Natural Science Foundation of China (Grant No. 22379139) and the Dalian Science and Technology Innovation Fund (2024RY033). The authors also thank Dr Gaoxiang Wang at Sun Yat-Sen University for sample preparation and testing.References
- (a) V. Sridharan, P. A. Suryavanshi and J. C. Menéndez, Advances in the Chemistry of Tetrahydroquinolines, Chem. Rev., 2011, 111, 7157–7259 CrossRef CAS; (b) Z. Wei, F. Shao and J. Wang, Recent advances in heterogeneous catalytic hydrogenation and dehydrogenation of N-heterocycles, Chin. J. Catal., 2019, 40, 980–1002 CrossRef CAS.
- (a) R. H. Fish, A. D. Thormodsen and G. A. Cremer, Homogeneous catalytic hydrogenation. 1. Regiospecific reductions of polynuclear aromatic and polynuclear heteroaromatic nitrogen compounds catalyzed by transition metal carbonyl hydrides, J. Am. Chem. Soc., 1982, 104, 5234–5237 CrossRef CAS; (b) W.-B. Wang, S.-M. Lu, P.-Y. Yang, X.-W. Han and Y.-G. Zhou, Highly Enantioselective Iridium-Catalyzed Hydrogenation of Heteroaromatic Compounds, Quinolines, J. Am. Chem. Soc., 2003, 125, 10536–10537 CrossRef CAS PubMed; (c) H. Zhou, Z. Li, Z. Wang, T. Wang, L. Xu, Y. He, Q.-H. Fan, J. Pan, L. Gu and A. S. C. Chan, Hydrogenation of Quinolines Using a Recyclable Phosphine-Free Chiral Cationic Ruthenium Catalyst: Enhancement of Catalyst Stability and Selectivity in an Ionic Liquid, Angew. Chem., Int. Ed., 2008, 47, 8464–8467 CrossRef CAS; (d) R. Yamaguchi, C. Ikeda, Y. Takahashi and K.-i. Fujita, Homogeneous Catalytic System for Reversible Dehydrogenation−Hydrogenation Reactions of Nitrogen Heterocycles with Reversible Interconversion of Catalytic Species, J. Am. Chem. Soc., 2009, 131, 8410–8412 CrossRef CAS PubMed; (e) C. Wang, C. Li, X. Wu, A. Pettman and J. Xiao, pH-Regulated Asymmetric Transfer Hydrogenation of Quinolines in Water, Angew. Chem., Int. Ed., 2009, 48, 6524–6528 CrossRef CAS PubMed; (f) H. Mao, C. Chen, X. Liao and B. Shi, Catalytic hydrogenation of quinoline over recyclable palladium nanoparticles supported on tannin grafted collagen fibers, J. Mol. Catal. A: Chem., 2011, 341, 51–56 CrossRef CAS; (g) N. A. Beckers, S. Huynh, X. Zhang, E. J. Luber and J. M. Buriak, Screening of Heterogeneous Multimetallic Nanoparticle Catalysts Supported on Metal Oxides for Mono-, Poly-, and Heteroaromatic Hydrogenation Activity, ACS Catal., 2012, 2, 1524–1534 CrossRef CAS; (h) D. Ren, L. He, L. Yu, R.-S. Ding, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, An Unusual Chemoselective Hydrogenation of Quinoline Compounds Using Supported Gold Catalysts, J. Am. Chem. Soc., 2012, 134, 17592–17598 CrossRef CAS.
- (a) G. Zhu, K. Pang and G. Parkin, New Modes for Coordination of Aromatic Heterocyclic Nitrogen Compounds to Molybdenum: Catalytic Hydrogenation of Quinoline, Isoquinoline, and Quinoxaline by Mo(PMe3)4H4, J. Am. Chem. Soc., 2008, 130, 1564–1565 CrossRef CAS PubMed; (b) S. Chakraborty, W. W. Brennessel and W. D. Jones, A Molecular Iron Catalyst for the Acceptorless Dehydrogenation and Hydrogenation of N-Heterocycles, J. Am. Chem. Soc., 2014, 136, 8564–8567 CrossRef CAS PubMed; (c) F. Chen, A.-E. Surkus, L. He, M.-M. Pohl, J. Radnik, C. Topf, K. Junge and M. Beller, Selective Catalytic Hydrogenation of Heteroarenes with N-Graphene-Modified Cobalt Nanoparticles (Co3O4–Co/NGr@α-Al2O3), J. Am. Chem. Soc., 2015, 137, 11718–11724 CrossRef CAS; (d) R. Adam, J. R. Cabrero-Antonino, A. Spannenberg, K. Junge, R. Jackstell and M. Beller, A General and Highly Selective Cobalt-Catalyzed Hydrogenation of N-Heteroarenes under Mild Reaction Conditions, Angew. Chem., Int. Ed., 2017, 56, 3216–3220 CrossRef CAS; (e) I. Sorribes, L. Liu, A. Doménech-Carbó and A. Corma, Nanolayered Cobalt–Molybdenum Sulfides as Highly Chemo- and Regioselective Catalysts for the Hydrogenation of Quinoline Derivatives, ACS Catal., 2018, 8, 4545–4557 CrossRef CAS; (f) B. Sahoo, C. Kreyenschulte, G. Agostini, H. Lund, S. Bachmann, M. Scalone, K. Junge and M. Beller, A robust iron catalyst for the selective hydrogenation of substituted (iso)quinolones, Chem. Sci., 2018, 9, 8134–8141 RSC; (g) P. Ryabchuk, G. Agostini, M.-M. Pohl, H. Lund, A. Agapova, H. Junge, K. Junge and M. Beller, Intermetallic nickel silicide nanocatalyst—A non-noble metal–based general hydrogenation catalyst, Sci. Adv., 2018, 4, eaat0761 CrossRef PubMed; (h) W. Gong, Q. Yuan, C. Chen, Y. Lv, Y. Lin, C. Liang, G. Wang, H. Zhang and H. Zhao, Liberating N-CNTs Confined Highly Dispersed Co–Nx Sites for Selective Hydrogenation of Quinolines, Adv. Mater., 2019, 31, 1906051 CrossRef CAS; (i) V. Papa, Y. Cao, A. Spannenberg, K. Junge and M. Beller, Development of a practical non-noble metal catalyst for hydrogenation of N-heteroarenes, Nat. Catal., 2020, 3, 135–142 CrossRef CAS; (j) M. Puche, L. Liu, P. Concepción, I. Sorribes and A. Corma, Tuning the Catalytic Performance of Cobalt Nanoparticles by Tungsten Doping for Efficient and Selective Hydrogenation of Quinolines under Mild Conditions, ACS Catal., 2021, 11, 8197–8210 CrossRef CAS PubMed; (k) Y. Shu, X. Zhou, J. Yang, F. Lan, W. Li, Z. Zhang, W. Li, Q. Guan and S. Ma, Fragmented Ultrathin Carbon Buffed Copper Clusters for Selective Hydrogenation of N-Heteroarenes under Ambient Pressure, J. Am. Chem. Soc., 2025, 147, 15578–15590 CrossRef CAS.
- (a) D. Ren, L. He, L. Yu, R. S. Ding, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, An Unusual Chemoselective Hydrogenation of Quinoline Compounds Using Supported Gold Catalysts, J. Am. Chem. Soc., 2012, 134, 17592–17598 CrossRef CAS PubMed; (b) Z. Luo, Y. Min, D. Nechiyil, W. Bacsa, Y. Tison, H. Martinez, P. Lecante, I. C. Gerber, P. Serp and M. R. Axet, Chemoselective reduction of quinoline over Rh–C60 nanocatalysts, Catal. Sci. Technol., 2019, 9, 6884–6898 RSC.
- (a) Y. Guan, H. Wen, K. Cui, Q. Wang, W. Gao, Y. Cai, Z. Cheng, Q. Pei, Z. Li, H. Cao, T. He, J. Guo and P. Chen, Light-driven ammonia synthesis under mild conditions using lithium hydride, Nat. Chem., 2024, 16, 373–379 CrossRef CAS; (b) Y. Guan, C. Liu, Q. Wang, W. Gao, H. A. Hansen, J. Guo, T. Vegge and P. Chen, Transition-Metal-Free Barium Hydride Mediates Dinitrogen Fixation and Ammonia Synthesis, Angew. Chem., Int. Ed., 2022, 61, e202205805 CrossRef CAS; (c) F. Chang, I. Tezsevin, J. W. de Rijk, J. D. Meeldijk, J. P. Hofmann, S. Er, P. Ngene and P. E. de Jongh, Potassium hydride-intercalated graphite as an efficient heterogeneous catalyst for ammonia synthesis, Nat. Catal., 2022, 5, 222–230 CrossRef CAS; (d) Y. Cai, L. Rao, Y. Wang, F. Chang, T. He, Y. Zhao, J. Yu, H. Wen, J. Hao, A. Wu, B.-T. Guan, J. Guo and P. Chen, Fabrication of atomically dispersed barium hydride catalysts for the synthesis of deuterated alkylarenes, Nat. Commun., 2025, 16, 1868 CrossRef CAS PubMed; (e) D. Mukherjee, D. Schuhknecht and J. Okuda, Hydrido Complexes of Calcium: A New Family of Molecular Alkaline-Earth-Metal Compounds, Angew. Chem., Int. Ed., 2018, 57, 9590–9602 CrossRef CAS; (f) D. D. Zhai, H. Z. Du, X. Y. Zhang, Y. F. Liu and B. T. Guan, Potassium Yttrium Ate Complexes: Synergistic Effect Enabled Reversible H2 Activation and Catalytic Hydrogenation, ACS Catal., 2019, 9, 8766–8771 CrossRef CAS; (g) M. M. D. Roy, A. A. Omaña, A. S. S. Wilson, M. S. Hill, S. Aldridge and E. Rivard, Molecular Main Group Metal Hydrides, Chem. Rev., 2021, 121, 12784–12965 CrossRef CAS PubMed.
- (a) L. Wright and S. Weller, The Catalytic Activity of Barium and Calcium Hydrides. I. An Exploratory Study, J. Am. Chem. Soc., 1954, 76, 5305–5308 CrossRef CAS; (b) L. H. Slaugh, Metal hydrides. Hydrogenation and isomerization catalysts, J. Org. Chem., 1967, 32, 108–113 CrossRef CAS; (c) M. Yamaguchi, Y. Hiraki, M. Wada and K. Tarama, The Catalytic Hydrogenation of Ethylene over Calcium Hydride and the Role of the Hydrogen of the Hydride, Bull. Chem. Soc. Jpn., 1975, 48, 1345–1348 CrossRef CAS.
- (a) J. Spielmann, F. Buch and S. Harder, Early Main-Group Metal Catalysts for the Hydrogenation of Alkenes with H2, Angew. Chem., Int. Ed., 2008, 47, 9434–9438 CrossRef CAS; (b) D. Schuhknecht, C. Lhotzky, T. P. Spaniol, L. Maron and J. Okuda, Calcium Hydride Cation [CaH]+ Stabilized by an NNNN-type Macrocyclic Ligand: A Selective Catalyst for Olefin Hydrogenation, Angew. Chem., Int. Ed., 2017, 56, 12367–12371 CrossRef CAS; (c) H. Bauer, M. Alonso, C. Färber, H. Elsen, J. Pahl, A. Causero, G. Ballmann, F. De Proft and S. Harder, Imine hydrogenation with simple alkaline earth metal catalysts, Nat. Catal., 2018, 1, 40–47 CrossRef CAS; (d) H. Bauer, M. Alonso, C. Fischer, B. Rösch, H. Elsen and S. Harder, Simple Alkaline-Earth Metal Catalysts for Effective Alkene Hydrogenation, Angew. Chem., Int. Ed., 2018, 57, 15177–15182 CrossRef CAS PubMed.
- (a) Q. Wang, J. Guo and P. Chen, The Power of Hydrides, Joule, 2020, 4, 705–709 CrossRef; (b) Q. Wang, Y. Guan, J. Guo and P. Chen, Hydrides mediate nitrogen fixation, Cell Rep. Phys. Sci., 2022, 3, 100779 CrossRef CAS.
- J. Guo, Y. Cai, W. Gao and P. Chen, Hydrides for Dinitrogen Conversion, ACS Catal., 2025, 14805–14812 CrossRef CAS.
- S. Harder, Molecular early main group metal hydrides: synthetic challenge, structures and applications, Chem. Commun., 2012, 48, 11165–11177 RSC.
- S. Harder, Early Main Group Metal Catalysis, 2020 Search PubMed.
- (a) W. P. Neumann, Über Neue Möglichkeiten zur Hydrierung von Stickstoff-Heterocyclen, Adv. Cycloaddit., 1958, 618, 90–105 CAS; (b) W. S. Johnson and B. G. Buell, 1,2-Dihydroquinoline, J. Am. Chem. Soc., 1952, 74, 4517–4520 CrossRef CAS.
- (a) S. K. Mitsutaka Natsume, Y. Kanda and K. Kiuchi, Reduction of quinoline and isoquinoline with sodium hydride, Tetrahedron Lett., 1973, 14, 2335–2338 CrossRef; (b) E. C. Ashby and A. B. Goel, Reaction of magnesium hydride with pyridine: Formation of H3Mg2NC5H6, HMgNC5H6 and Mg(NC5H6)2, J. Organomet. Chem., 1981, 204, 139–145 CrossRef CAS; (c) T. Liu, J. He and Y. Zhang, Regioselective 1,2-hydroboration of N-heteroarenes using a potassium-based catalyst, Org. Chem. Front., 2019, 6, 2749–2755 RSC.
- (a) M. Wiesinger, C. Knüpfer, H. Elsen, J. Mai, J. Langer and S. Harder, Heterometallic Mg−Ba Hydride Clusters in Hydrogenation Catalysis, ChemCatChem, 2021, 13, 4567–4577 CrossRef CAS; (b) Y. Guan, W. Zhang, Q. Wang, C. Weidenthaler, A. Wu, W. Gao, Q. Pei, H. Yan, J. Cui, H. Wu, S. Feng, R. Wang, H. Cao, X. Ju, L. Liu, T. He, J. Guo and P. Chen, Barium chromium nitride-hydride for ammonia synthesis, Chem Catal., 2021, 1, 1042–1054 CAS.
- (a) J. Martin, J. Eyselein, S. Grams and S. Harder, Hydrogen Isotope Exchange with Superbulky Alkaline Earth Metal Amide Catalysts, ACS Catal., 2020, 10, 7792–7799 CrossRef CAS; (b) B. De Tobel, T. A. Hamlin, C. Fonseca Guerra, S. Harder, F. De Proft and M. Alonso, Role of alkaline-earth metal in catalysed imine hydrogenations, Polyhedron, 2024, 248, 116751 CrossRef CAS.
- (a) T. Hascall, D. Rabinovich, V. J. Murphy, M. D. Beachy, R. A. Friesner and G. Parkin, Mechanistic and Theoretical Analysis of the Oxidative Addition of H2 to Six-Coordinate Molybdenum and Tungsten Complexes M(PMe3)4X2 (M = Mo, W; X = F, Cl, Br, I): An Inverse Equilibrium Isotope Effect and an Unprecedented Halide Dependence, J. Am. Chem. Soc., 1999, 121, 11402–11417 CrossRef CAS; (b) R. Qin, L. Zhou, P. Liu, Y. Gong, K. Liu, C. Xu, Y. Zhao, L. Gu, G. Fu and N. Zheng, Alkali ions secure hydrides for catalytic hydrogenation, Nat. Catal., 2020, 3, 703–709 CrossRef CAS.
- (a) M. S. Hill, D. J. Liptrot and C. Weetman, Alkaline earths as main group reagents in molecular catalysis, Chem. Soc. Rev., 2016, 45, 972–988 RSC; (b) J. Penafiel, L. Maron and S. Harder, Early Main Group Metal Catalysis: How Important is the Metal?, Angew. Chem., 2014, 127, 203–208 CrossRef.
- (a) B. Rösch, T. X. Gentner, J. Langer, C. Färber, J. Eyselein, L. Zhao, C. Ding, G. Frenking and S. Harder, Dinitrogen complexation and reduction at low-valent calcium, Science, 2021, 371, 1125–1128 CrossRef; (b) Q. Wang, S. Pan, Y.-B. Wu, G. Deng, J.-H. Bian, G. Wang, L. Zhao, M. Zhou and G. Frenking, Transition-Metal Chemistry of Alkaline-Earth Elements: The Trisbenzene Complexes M(Bz)3 (M=Sr, Ba), Angew. Chem., Int. Ed., 2019, 58, 17365–17374 CrossRef CAS PubMed; (c) X. Wu, L. Zhao, J. Jin, S. Pan, W. Li, X. Jin, G. Wang, M. Zhou and G. Frenking, Observation of alkaline earth complexes M(CO)8 (M = Ca, Sr, or Ba) that mimic transition metals, Science, 2018, 361, 912–916 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |