
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrecise synthesis of telechelic rodlike polyisocyanides: versatile building blocks for fabricating polymer frameworks with controllable pore-apertures
Yang
Zong
a,
Run-Tan
Gao
a,
Na
Liu
*b,
Shixing
Lei
c,
Zhan-Ting
Li
 c and
Zong-Quan
Wu
*a
c and
Zong-Quan
Wu
*a
aState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, 2699 Qianjin Street, Changchun, Jilin 130012, China. E-mail: zqwu@jlu.edu.cn
bThe School of Pharmaceutical Sciences, Jilin University, 1266 Fujin Road, Changchun, Jilin 130021, China. E-mail: liuna606@jlu.edu.cn
cState Key Laboratory of Organometallic Chemistry, Shanghai-Hong Kong Joint Laboratory in Chemical Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, Shanghai 200032, China
First published on 10th November 2025
Abstract
The controlled synthesis of telechelic polymers with precisely functionalized chain-ends and predictable main-chain structures is highly desirable. Herein, a series of air-stable trans-bis(phenylethynyl)palladium catalysts were designed and efficiently synthesized, which initiate the polymerization of phenyl isocyanides via a living chain-growth process, resulting in polyisocyanides with precise control of molecular weights (Mns) and narrow molecular weight distributions (Đ). The substituents of the catalyst can regulate the polymerization rate while serving as entire chain-end functional groups of the resulting polymers. Given the precise control over the length of rigid polyisocyanides, these polyisocyanides are an ideal building block for constructing covalent polymer frameworks (CPFms) with tuneable pore apertures and functionalities. As a proof of concept, water-soluble CPFs with tuneable pore-apertures were prepared and the ordering of the resulting CPFs was systematically verified by dynamic light scattering (DLS), high-resolution transmission electron microscopy (HR-TEM), and small-angle X-ray scattering (synchrotron radiation facility). Moreover, the pore aperture can be directly controlled by tuning the length of the polyisocyanide link. Owing to the tuneable pore size and charge attraction effects, the CPFs with pore apertures matching the target single-stranded deoxyribonucleic acid (ssDNA) exhibit good performance on gene delivery. The percentage of delivered ssDNA into cells is up to ca. 98% (21 and 35 units).
Introduction
In nature, biological macromolecules such as nucleic acids,1 polypeptides,2 and proteins3 are strictly controlled in terms of their chain length, sequence, and chirality to ensure the accurate transmission of genetic information and the proper execution of biological functions.4–6 For polymer chemists, the controlled synthesis of polymers is a crucial method for preparing functional materials.7–9 Benefiting from the development of living polymerization methods, functional polymers with precise structures have found extensive applications in numerous fields, including catalysis,10,11 drug delivery,12,13 circularly polarized luminescence and so forth.14–16 Moreover, to further modulate the properties of polymers, chain-end functionalization plays a significant role in polymer chemistry.17,18 By introducing functional chain-end groups, it is not only possible to control the physical properties of polymers, such as the glass transition, but also to endow the polymers with novel characteristics such as multiple stimuli-responsiveness and self-assembly.19,20 However, due to the influence of the chain length and molecular weight distribution, post-polymerization chain-end functionalization of polymers often faces challenges such as incomplete functionalization, difficult purification, and the need for large excesses of reactive functional groups.17 Therefore, developing an efficient synthetic method to improve the efficiency of chain-end functionalization is highly necessary. Such polymers have great potential in constructing functional materials, such as amorphous polymer networks and crystalline frameworks, depending on the rigidity of the backbone.Over the past two decades, porous frameworks have developed rapidly as an important component of functional materials.21–23 As the special example of porous materials, Omar M. Yaghi and others proposed framework chemistry such as metal–organic frameworks (MOFs), covalent organic frameworks (COFs), supramolecular organic frameworks (SOFs), and hydrogen-bonded organic frameworks (HOFs).24–30 For porous frameworks, regulating pore size and introducing functional groups are of significant importance for practical applications because they may directly influence molecular accessibility, selectivity, host–guest interactions, and structural stability.31–33 The pore apertures dictate the size of target molecules that may enter the pores, and provide the surface and space to carry out the functions, such as the storage, separation, and conversion. To precisely control the pore aperture, Yaghi et al. synthesized a family of MOFs with pore apertures ranging from 14 to 98 angstroms using organic links containing many phenylene rings.34 Feng et al. constructed a series of COFs with record pore apertures ranging from 7.7 nm to 10.0 nm by using oligomeric ethynylbenzene links.35 These MOFs and COFs with large pore apertures realized the remarkable application in biomacromolecule encapsulation and separation. Inspired by these studies, controlled synthesis of mesoporous frameworks has a highly promising strategy in protein/gene delivery36–38 and peptide/protein discrimination.39,40 In this research area, nucleic acid-based (DNA and RNA) drugs have found applications in various fields, particularly in biomedicine.41,42 In order to exhibit their pharmacological effect, nucleotide-based drugs should be delivered intracellularly. Nevertheless, the negatively charged and hydrophobic properties of the phospholipid bilayer that forms the cell membrane make this process difficult.43 Water-soluble cationic polymers efficiently bind and load short nucleic acid chains through size-matching steric effects and electrostatic interactions, and deliver them into cells via endocytosis.44,45 Moreover, the diversity of nucleic acid lengths and species may demand a modular, water-soluble, framework-based delivery platform with facile tunability. However, these frameworks with large pore apertures were usually prepared using long and rigid aromatic linkers, which require multiple and complex organic syntheses and are time-consuming. Moreover, controlling the pore aperture of frameworks with atomic precision has rarely been achieved to date, to the best of our knowledge. Therefore, developing a convenient method for facilely constructing porous frameworks with controllable and large pore apertures in an atomically precise manner is highly desired.
The insertion polymerization of isocyanide gives polyisocyanide with precise control over the chain length and extremely low distribution. Owing to this C1 polymerization, the chain length increased by one carbon atom via one monomer insertion, which facilitates atomic control over the chain length. The reported alkyne–Pd(II) catalysts facilitate the controlled synthesis of helical polyisocyanides with controlled molecular weight (Mn) and narrow dispersity (Đ).46–48 The functional Pd(II)-catalysts can afford helical polyisocyanides with a functional group installed on the initiating end, while post-polymerization functionalization can modify the terminating end of polyisocyanide. However, this strategy involves cumbersome and time-consuming experimental efforts, posing a major practical limitation.49–51
Controlled synthesis of rodlike helical polyisocyanides bearing functionalities on both chain ends is in great demand, as they can provide useful links for constructing porous frameworks. Herein, we designed and synthesized a class of bis(phenylethynyl)palladium catalysts that can efficiently catalyse living polymerization of various isocyanides, leading to the rodlike stereoregular polyisocyanides with controlled Mns and narrow Đ. The functional phenylethynyl groups of the catalyst were installed at both the initiating and terminating chain ends. Thus, a variety of well-defined end-functionalized polyisocyanides carrying benzene, benzaldehyde, and anisole on each chain end were readily prepared. The benzaldehydes end-functionalized polyisocyanides bearing hydrophilic methyl triglycol on the pendants were used as length-tuneable links for constructing water-soluble covalent polymer frameworks (CPFs) with modulable pore apertures. The periodic structure was undoubtedly confirmed by high-resolution transmission electron microscopy (HR-TEM) and synchrotron small angle X-ray scattering (SAXS). Benefiting from the tuneable pore aperture and electrostatic interactions, these CPFs can accurately recognize ssDNAs of different lengths and achieve efficient delivery into cells, with the percentage of delivered cells reaching up to 98%. Collectively, this study demonstrates that bis(phenylethynyl)palladium catalysts enable the facile synthesis of well-defined helical polyisocyanides with functional groups at both chain ends.
Results and discussion
Synthesis of end-functionalized polyisocyanides
A family of bis(phenylethynyl)palladium catalysts 1a–c with various substituents at the para position of the benzene ring was synthesized through the reaction of the corresponding phenylethynyl with trans-bis(triethyl phosphine)palladium dichloride in dichloromethane using triethylamine as the base at room temperature (Scheme 1 and Fig. S1–S9 in the SI). The resulting Pd(II)-catalysts show excellent solubility in common organic solvents and are air-stable. To assess the catalytic performance of 1a–c, isocyanide 2a was employed as a model monomer and was polymerized by 1a in THF at room temperature ([2a]0 = 0.35 M, [2a]0/[Pd]0 = 75). However, the polymerization was quite slow, and almost no polymeric product was isolated after 20 h. Then, the polymerization solution was heated to 55 °C. Following overnight polymerization, the solution was directly precipitated into methanol, resulting in a deep yellow solid. The successfully obtained polymer 1a-poly-2a75(Pd) (the subscript is the theoretical degree of polymerization (DP)) was characterized by size-exclusion chromatography (SEC), which confirmed its average Mn of 22.6 kg mol−1 and a narrow Đ of 1.08 (Fig. 1a). To investigate whether the polymerization exhibited a living character, polymerizations of 2a catalysed by 1a with varying [2a]0/[Pd]0 ratios were conducted. As depicted in Fig. 1a, all the generated 1a-poly-2an(Pd) polymers exhibited a single-peak SEC distribution and shifted to higher Mn-regions as the [2a]0/[Pd]0 ratio increased. Moreover, Mn increased linearly with [2a]0/[Pd]0, and the Đ values for the generated 1a-poly-2an(Pd)s were less than 1.15 (Fig. 1b), confirming the living nature of the polymerization. | ||
| Scheme 1 (a) Synthesis of the catalysts and the living polymerization of isocyanides. (b) Living block polymerization of isocyanides. | ||
 | ||
| Fig. 1 (a) SEC curves of 1a-poly-2an(Pd) prepared with different [2a]0/[Pd]0 values. (b) Plots of Mns and Đ values of 1a-poly-2an(Pd) as a function of [2a]0/[Pd]0 values. (c) Plots of 2a conversion vs. polymerization time of 1-initiated polymerization of 2a in THF at 55 °C. (d) First-order kinetic plots for the polymerization of 2a initiated by 1a, 1b, and 1c in THF at 55 °C ([2a]0 = 0.35 M, [2a]0/[Pd]0 = 75). | ||
Further experiments revealed that catalysts 1b and 1c could also catalyze the living polymerization of 2a, giving the desired polyisocyanides with controlled Mns and low Đ in high yield. The polymerization process was monitored by analyzing the SEC of aliquots collected at appropriate time intervals to track the changes in the Mn of the resulting polymers and to determine the monomer conversions. The time-dependent SEC for the polymerization of 2a using 1a, 1b and 1c is summarized in Fig. S10 in the SI. Within four hours, the monomer conversions for the polymerizations catalyzed by 1a, 1b, and 1c are 67%, 51% and 75%, respectively (Fig. 1c). Meanwhile, as shown in Fig. 1d, the plots revealed that the isocyanide polymerizations catalyzed by 1a–c all follow first-order kinetics, as a clear linear relationship was observed between −ln([M]/[M]0) and polymerization time. The apparent polymerization rate constants of the isocyanide polymerization catalyzed by 1a, 1b, and 1c are 7.93 × 10−5, 4.87 × 10−5, 9.35 × 10−5 s−1, respectively. That is, the polymerization rate of 2a using 1c, carrying an electron-donating (methoxy) substituent, is approximately 1.9 times higher than that using 1b, which bears an electron-withdrawing (aldehyde) moiety, indicating that the substituents on the phenylethynyl group of the catalysts influence the polymerization activity. Taking advantage of these catalysts, a variety of telechelic polyisocyanides with different Mn and low Đ were prepared using isocyanide monomers 2a, 2b, 2L and 2D (Table S1, SI).
The structure and regularity of the isolated polymers were further investigated. The proton nuclear magnetic resonance (1H NMR) spectrum clearly demonstrated the occurrence of polymerization. For instance, the resonances assignable to the terminal benzaldehyde group and the methylene group in the isocyanide side chain were clearly observed in the 1H NMR spectrum of 1b-poly-2a50(Pd). The integral ratio of the terminal –CHO group to the methylene groups of the pendant is ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :48, which agrees well with the theoretical DP of 50, further supporting the high controllability of the polymerization (Fig. 2a). The stereoregularity of 1b-poly-2a50(Pd) was determined by calculating the half-bandwidth of the main chain imino carbon signal in 13C NMR spectra. A rather sharp singlet resonance at δ 162.60 ppm, corresponding to the imino carbons of the polymer backbone, was observed (Fig. 2b). The estimated half-bandwidth was just 31 Hz, suggesting that 1b-poly-2a50(Pd) had a high degree of isotactic backbone.52 The 31P NMR spectrum revealed that 1b-poly-2a50(Pd) still retained a metallic palladium moiety (Fig. S11, SI), because the resonance of PEt3 was clearly observed. To further confirm the living nature of the polymerization, a block copolymerization of 2b with 1a-poly-2a50(Pd) (Mn = 16.2 kg mol−1, Đ = 1.11) was conducted in THF at 55 °C ([2b]0/[Pd]0 = 15). SEC analysis of the resulting 1a-poly(2a50-b-2b15)(Pd) revealed a monomodal distribution that shifted to a higher Mn-region (Fig. S12, SI). Accordingly, the Mn increased to 21.4 kg mol−1 while retaining a low Đ of 1.12. The 1H NMR spectrum of the block copolymer displayed characteristic resonances from both the poly-2a50 and poly-2b15 segments (Fig. S13, SI). Integral analysis indicated that the ratio of the poly-2a50 to poly-2b15 blocks was 50/13, which is approximately consistent with the feed ratio of the monomers. Furthermore, FT-IR analysis corroborated the formation of the expected block copolymer structure (Fig. S14, SI).
:48, which agrees well with the theoretical DP of 50, further supporting the high controllability of the polymerization (Fig. 2a). The stereoregularity of 1b-poly-2a50(Pd) was determined by calculating the half-bandwidth of the main chain imino carbon signal in 13C NMR spectra. A rather sharp singlet resonance at δ 162.60 ppm, corresponding to the imino carbons of the polymer backbone, was observed (Fig. 2b). The estimated half-bandwidth was just 31 Hz, suggesting that 1b-poly-2a50(Pd) had a high degree of isotactic backbone.52 The 31P NMR spectrum revealed that 1b-poly-2a50(Pd) still retained a metallic palladium moiety (Fig. S11, SI), because the resonance of PEt3 was clearly observed. To further confirm the living nature of the polymerization, a block copolymerization of 2b with 1a-poly-2a50(Pd) (Mn = 16.2 kg mol−1, Đ = 1.11) was conducted in THF at 55 °C ([2b]0/[Pd]0 = 15). SEC analysis of the resulting 1a-poly(2a50-b-2b15)(Pd) revealed a monomodal distribution that shifted to a higher Mn-region (Fig. S12, SI). Accordingly, the Mn increased to 21.4 kg mol−1 while retaining a low Đ of 1.12. The 1H NMR spectrum of the block copolymer displayed characteristic resonances from both the poly-2a50 and poly-2b15 segments (Fig. S13, SI). Integral analysis indicated that the ratio of the poly-2a50 to poly-2b15 blocks was 50/13, which is approximately consistent with the feed ratio of the monomers. Furthermore, FT-IR analysis corroborated the formation of the expected block copolymer structure (Fig. S14, SI).
 | ||
| Fig. 2 (a) 1H NMR spectra (500 MHz) and (b) 13C NMR spectra (125 MHz) of 1b-poly-2a50(Pd) recorded in CDCl3. | ||
This class of catalysts was also applicable to the polymerization of chiral isocyanide monomers and led to optically active helical polyisocyanides with preferred one-handed helicity. For instance, the polymerization of chiral isocyanide 2L and 2D catalyzed by 1b resulted in helical polyisocyanides 1b-poly-2Ln(Pd) and 1b-poly-2Dn(Pd), respectively. The excess of one-handed helical sense was assessed by circular dichroism (CD) and UV-vis absorption analyses. The 1b-poly-2Ln(Pd) and 1b-poly-2Dn(Pd) respectively showed negative and positive CD in the absorption region of the poly(phenyl isocyanide) backbone, indicative of the preferred left- and right-handed helices (Fig. S15, SI).47 Based on the analyses described above, the polymerization probably occurred via the isocyanide coordination to the Pd(II) center bound to the phosphine ligand. Subsequently, the monomers underwent ordered insertion at both ends of the palladium center, resulting in the formation of a linear polyisocyanide (Fig. S16, SI). Moreover, the 1H NMR spectrum of the prepared polyisocyanide oligomer 1b-poly-2c2(Pd) with low DP showed simple and symmetric signals, which further confirmed the insertion of isocyanides into both sides of the Pd(II)-center. Due to the living nature of this Pd(II)-initiated polymerization, the initiation process is faster than chain extension, that is, the insertion of isocyanide into Pd–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C is easier than into Pd–(C
C is easier than into Pd–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–R) (Fig. S17–S20, SI). Thus, the polymerization occurs via the insertion of isocyanides into both sides of the Pd(II)-catalyst.
N–R) (Fig. S17–S20, SI). Thus, the polymerization occurs via the insertion of isocyanides into both sides of the Pd(II)-catalyst.
The Pd(II)-residues embedded in the polymer backbone may have toxic effects on applications related to biology and life sciences. To remove the Pd(II)-residue, the isolated polyisocyanide 1b-poly-2a15(Pd) was treated with a bidentate phosphine Wei-Phos (5.0 eq. to Pd) in the presence of triethylamine and cuprous iodide to facilitate the reductive elimination (Scheme 1).19 It was revealed that the Pd(II)-residue was completely removed, as confirmed by a set of analyses of the resulting polyisocyanides. Prior to the palladium removal reaction, the structural integrity of the synthesized polyisocyanides was investigated by mass spectroscopy (MS). The 1b-poly-2a15(Pd) was analyzed by matrix-assisted laser desorption/ionization spectroscopy (MALDI) in combination with time-of-flight (TOF) detection mass spectrometry (MS). It exhibited a maximum peak at 4643.83, corresponding to the molecular weight of 14-mer with integrity terminal benzaldehyde groups and a Pd(PEt3)2 moiety (Fig. 3a). Furthermore, the molecular weight difference between adjacent highest peaks is 287, which is equivalent to the molecular weight of a 2a monomer, indicating the successful insertion of isocyanides. Following the Pd(II)-residue removal reaction, the MALDI-TOF spectrum of 1b-poly-2a15 showed the highest peak with an m/z value of 4279.78, corresponding to 1b-poly-2a14 (Fig. 3b).
 | ||
| Fig. 3 MALDI-TOF-MS spectra of (a) 1b-poly-2a15(Pd) and (b) 1b-poly-2a15. | ||
After the reductive elimination of Pd(PEt3)2 from 1b-poly-2a15(Pd), the Mn of the resulting 1b-poly-2a15 was 4.5 kg mol−1, slightly lower than its precursor (4.7 kg mol−1), while maintaining the narrow distribution with Đ = 1.08, as determined by SEC (Fig. 4a). The preservation of the characteristic stretching vibration peaks in the FT-IR spectra of 1b-poly-2a15(Pd) and 1b-poly-2a15 indicates the structural similarity of the polymer before and after palladium removal (Fig. 4b). No 31P signal could be detected on the 31P NMR spectrum of 1b-poly-2a15, suggesting that the Pd(PEt3)2 moiety of 1b-poly-2a15(Pd) was clearly removed (Fig. S21, SI). Accordingly, the resonances at 2.01 and 1.26 ppm attributed to the ethyl groups of P(CH2CH3)3 in 1b-poly-2a15(Pd) could not be observed in the 1H NMR spectrum of 1b-poly-2a15 (Fig. S22, SI). Energy dispersive X-ray spectroscopy (EDS) mapping intuitively showed the disappearance of palladium elements after reductive elimination (Fig. 4c, d, S23 and S24 in SI). Collectively, these studies clearly confirmed the complete removal of the Pd-complex embedded in the polymer backbone. Using this method, a series of telechelic polyisocyanides, such as 1a-poly-2an, 1b-poly-2an, 1c-poly-2an and 1a-poly-2bn, with defined chain end functionalities, predictable Mn, and low Đ were facilely prepared (Table S2 and Fig. S25–S30, SI).
 | ||
| Fig. 4 (a) SEC curves of 1b-poly-2a15(Pd) and 1b-poly-2a15. (b) FT-IR spectra of 1b-poly-2a15(Pd) and 1b-poly-2a15 measured at 25 °C using KBr. TEM-EDS elemental mapping images of 1b-poly-2a15(Pd) (c) and 1b-poly-2a15 (d). | ||
Synthesis of CPFs
The acylhydrazone bond, fabricated from the reaction of hydrazine and aldehyde, has been reported as an efficient and convenient dynamic covalent bond in the construction of flexible organic frameworks.36 To construct water-soluble CPFs, a series of 1b-poly-2bns with DPs of 10, 20, 30 and 40, were prepared using the method described above (Fig. S31 in SI). The 1b-poly-2bns of different chain lengths were treated with tetra-armed crosslinker tetrakis-hydrazide T1 in water while maintaining the stoichiometric ratio of 1:2 between the hydrazide groups of T1 and aldehyde groups of 1b-poly-2bn.53 The formation of CPFs was monitored by measuring the characteristic signal of CH(OD)2via1H NMR spectroscopy of the mixtures at appropriate time intervals. It was revealed that the CH(OD)2 signals completely disappeared within two hours for all the 1b-poly-2bns, indicating the completion of the reaction of acylhydrazone moiety with benzaldehyde (Fig. 5a, S32 and S33 in the SI).
 | ||
| Fig. 5 (a) Synthesis of water-soluble CPFms. (b) DLS profiles of T1 (2.0 mM) and 1b-poly-2bn (0.2 mg mL−1) in water at 25 °C. (c) DLS profiles of T1 (2.0 mM) and CPFms (0.2 mg mL−1) in water at 25 °C. | ||
The dynamic light scattering (DLS) experiment also corroborated the construction of the desired CPFms. The hydrodynamic diameter (DH) of T1 (2.0 mM) is only 0.9 nm, while the DH values of 1b-poly-2b10, 1b-poly-2b20, 1b-poly-2b30 and 1b-poly-2b40, in water (0.2 mg mL−1) were 9.9, 24.0, 27.9, and 33.2 nm, indicative of water soluble macromolecules. However, the DH values of the resulting CPFs were 125.6, 146.1, 169.9, and 267.2 nm for CPF10, CPF20, CPF30, and CPF40, respectively, constructed from 1a-poly-2bms with DPs of 10, 20, 30, and 40, respectively (Fig. 5b and c). Furthermore, upon varying the concentration of CPF10 from 0.1 to 1.5 mg mL−1 (Fig. S34, SI), the DH remained constant, indicating that the DH of the framework tends to stabilize beyond a certain concentration.
Molecular modeling studies elucidated potential framework configurations of water-soluble CPFms, where tetrahedral molecule T1 was employed to form stereoscopic and crosslinking support points, while the rodlike polyisocyanides of varying DPs contributed to the formation of porous frameworks with different pore apertures. The theoretical pore sizes calculated for CPF10, CPF20, CPF30, and CPF40 are 8.0, 11.6, 13.3, and 15.1 nm, respectively (Fig. S35, SI). The porous characteristics of these CPFms were clearly confirmed by HR-TEM observations. As shown in Fig. 6a, the HR-TEM image of CPF10 showed distinct square porous structures with a pore aperture of ca. 8.4 nm. As the DPs of the links increased, the HR-TEM image of CPF20 also showed a periodic square pore structure, and the pore aperture was increased to ca. 10.5 nm (Fig. 6b). For CPF30 and CPF40, with further increased pore sizes, the square pore structure could not be observed on HR-TEM images; however, the clear lattice fringes were observed. The pore apertures of CPF30 and CPF40 were estimated to be 14.6 nm and 16.7 nm, respectively (Fig. S36, SI). As the DP of 1b-poly-2bms increases, the enlargement of pore apertures makes the maintenance of CPFs more challenging, while the rigid structure of polyisocyanides stabilizes the framework structure. Interestingly, the pore aperture determined by HR-TEM was linearly correlated to the DP of the polyisocyanide links (Fig. 6c). The ordering of the CPFms of different pore apertures was further verified via synchrotron SAXS analysis. The water solution-phase synchrotron SAXS curves of CPF10 and CPF20 exhibit peaks at q ≈ 0.76 and 0.54 nm, respectively, which correspond to the {100} peak and indicate pore apertures of 8.27 nm for CPF10 and 11.64 nm for CPF20, closely agreeing with the HR-TEM observations (Fig. 6d and e).
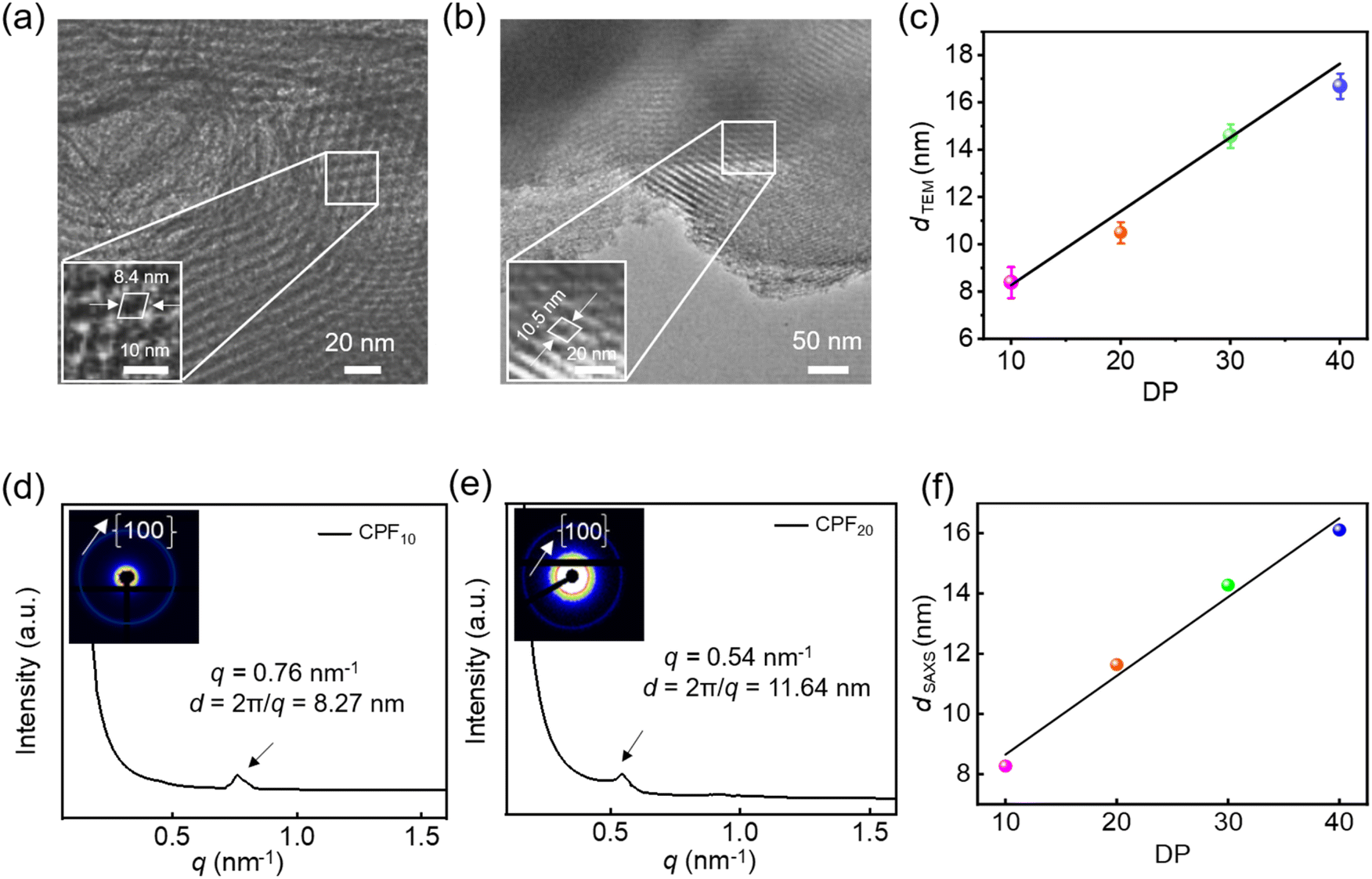 | ||
| Fig. 6 HR-TEM images of CPF10 (a) and CPF20 (b). (c) Plot of d-spacing (TEM) vs. the DP of the polyisocyanide link. Solution-phase synchrotron SAXS profiles of CPF10 (d) and CPF20 (e) recorded in H2O (10 mg mL−1). (f) Plot of d-spacing (SAXS) vs. the DP of the polyisocyanide link. | ||
Similarly, CPF30 and CPF40 exhibit peaks at q ≈ 0.44 and 0.39 nm on the SAXS profiles, corresponding to the pore apertures of 14.28 and 16.11 nm, respectively (Fig. S37, SI), generally constant with the HR-TEM observations. It is worth noting that the pore aperture of the constructed frameworks was linearly correlated to the length of the 1b-poly-2bm links (Fig. 6f). Since the length of 1b-poly-2bm was determined by the DP of the backbone, it can be facilely controlled by tuning the initial feed ratio of the isocyanide monomer to the catalyst. Thus, the pore aperture of the CPFms is precisely controllable.
DNA delivery
Based on the successful preparation of water-soluble CPFms with controlled pore apertures, we explored their application for DNA delivery. The isothermal titration calorimetry (ITC) experiments were first conducted in vitro to test the interaction between CPFms and ssDNA of 21 and 35 units (ssDNA21 and ssDNA35) (Fig. 7a). As depicted in Table 1, the encompassed thermodynamic information, such as the stoichiometry (N), apparent binding constants (Ka), enthalpy change (ΔH), and entropy change (ΔS), was summarized. Given the complexity of DNA-CPFms binding in real scenarios, a 1:1 binding model was adopted in ITC to simplify the analysis and ensure comparability with existing literature.43 Assuming each CPFms aperture exhibited uniform binding affinity to one entire ssDNA, the Ka values can be calculated.44 In the binding interaction between CPFms and ssDNAs in H2O, the Ka values are quite high and varying from 1.37 × 106 to 2.28 × 107 M−1 in H2O depending on the pore aperture of CPFms (Table 1 and Fig. 7b–d). In contrast, the binding between the individual T1 and the linear polymer 1b-poly-2b10 with Cy5-ssDNA was negligible as revealed by ITC analysis (Fig. S38–S40, SI).
 | ||
| Fig. 7 (a) Schematic representation of the in situ loading of ssDNAs into CPFms. (b–d) Isothermal titration thermograms of CPFms titrated by ssDNAs (b and c) and ssDNA35 (d) into the solution of CPF10 (b), CPF20 (c) and CPF30 (d) at 25 °C in H2O ([anion] = 0.1 mM; [cation] = 50 µmol). [Anion] represents the total phosphate concentration of the ssDNAs. [Cation] represents the total pyridinium concentration of CPFs. The solid lines represent the best nonlinear fit of the data to a 1:1 binding mode. | ||
| DNA | CPFm | K a (M−1) | ΔH (kcal mol−1) | −TΔS (kcal mol−1) | ΔG (kcal mol−1) |
|---|---|---|---|---|---|
| ssDNA21 | CPF10 | (2.28 ± 0.46) × 107 | −6.19 ± 0.055 | −8.94 | −15.13 ± 0.055 |
| CPF20 | (1.59 ± 0.66) × 107 | −2.84 ± 0.039 | −7.01 | −9.85 ± 0.039 | |
| CPF30 | (1.36 ± 0.22) × 107 | −4.14 ± 0.026 | −5.60 | −9.74 ± 0.026 | |
| CPF40 | (4.29 ± 0.90) × 106 | −2.83 ± 0.029 | −6.23 | −9.06 ± 0.029 | |
| ssDNA35 | CPF10 | (1.37 ± 0.34) × 106 | −3.91 ± 0.065 | −4.50 | −8.41 ± 0.065 |
| CPF20 | (2.47 ± 0.26) × 106 | −3.74 ± 0.043 | −4.71 | −8.45 ± 0.043 | |
| CPF30 | (3.63 ± 0.42) × 106 | −9.37 ± 0.202 | −0.82 | −10.19 ± 0.202 | |
| CPF40 | (1.54 ± 0.26) × 106 | −3.97 ± 0.047 | −4.50 | −8.47 ± 0.047 |
The experimental results also indicate that CPFms with different pore apertures have varying binding strengths with ssDNA, suggesting that the size-matching effect plays a crucial role in the binding. In all instances of CPFm binding to ssDNA, the entire binding process of ssDNA is a spontaneous behavior driven by both entropy and enthalpy, where the enthalpy contribution is likely primarily due to electrostatic interactions between ion pairs, and the entropy contribution is likely mainly due to the release of high-energy water to lower the freedom from the hydrophobic surfaces.
The above ITC experiments confirmed the binding affinity of the CPFms toward ssDNA21 and ssDNA35. Among them, CPF10 and CPF30 demonstrated remarkably high binding affinity and have great potential for ssDNA delivery. Subsequently, confocal laser scanning microscopy (CLSM) was employed to evaluate the capacity of CPF10 and CPF30 for delivering Cy5-ssDNA21 and Cy5-ssDNA35 into H9C2 cells (Fig. 8).
 | ||
| Fig. 8 CLSM images of H9C2 cells after incubation for 2 h with Cy5-ssDNA21, Cy5-ssDNA21@Lipo2000, Cy5-ssDNA21@CPF10, Cy5-ssDNA35@CPF10, and Cy5-ssDNA35@CPF30. The dosages of Cy5-ssDNA21 and Cy5-ssDNA35 were 2.5 µg mL−1, while those of Lipo2000, CPF10 and CPF30 were 15 µg mL−1. Nuclei and lysosomes were stained with Hoechst (blue) and Lyso-Tracker Green (green), respectively. | ||
The lysosomes and nuclei of the cells were labeled with Lyso-Tracker Green and Hoechst dye, respectively, and were post-incubated with Cy5-ssDNA21 (2.5 µg mL−1) and its in situ combinations with CPF10 and CPF30 for 2 h together; all imagery was captured. For comparative purposes, CPFms remained constant at 15 µg mL−1 across all variations. At the same time, the commercial reagent Lipo2000 was evaluated for its ssDNA delivery efficacy at an equivalent concentration, serving as a positive control. In the presence of the CPFms, fluorescence of the Cy5 probe was evident in all images, with Cy5-ssDNA21@CPF10 and Cy5-ssDNA35@CPF30 exhibiting more intense fluorescence. Conversely, in the absence of CPFms, no fluorescence signal from the Cy5 dye was detected, suggesting that due to the barrier imposed by the cell membrane, Cy5-ssDNA21 alone cannot be delivered into the cells. Under identical incubation times and dosages, weak fluorescence of Cy5 within the cells was observed with Lipo2000, indicating uneven delivery and low delivery efficiency. Compared to the commercial reagent Lipo2000, CPFms demonstrate superior delivery efficiency for Cy5-ssDNA. The CPFms with different pore sizes exhibit a size-matching effect with ssDNAs of varying chain lengths, which correlates with the binding affinity determined by ITC (Table 1). Additionally, the fluorescence signals of Cy5 dye and lysosome-stained Lyso-Tracker Green were significantly co-localized, verifying that Cy5-ssDNA21 and Cy5-ssDNA35 were internalized via endocytosis facilitated by CPF10 and CPF30 delivery. Conversely, no co-localization was detected between the Cy5 and Hoechst dyes, suggesting that CPFms did not transport DNA into cell nuclei within a 2 h incubation period.
Since the CLSM imaging experiments confirmed that CPF10 and CPF30 could effectively deliver Cy5-ssDNA21 and Cy5-ssDNA35 into cells, flow cytometry experiments were conducted to quantitatively assess their delivery capabilities. Cy5-ssDNA21 and CPF10 were used as preliminary examples to screen the incubation time in H9C2 cells. The percentage of cells that underwent DNA internalization was recorded to determine the optimal incubation time. The percentage of cells undergoing internalization increased with extended incubation time, reaching 35.9%, 81.3%, and 98.2% for 0.25, 1 and 2 h, respectively (Fig. 9a). A similar screening of the CPFms and DNA delivery ratios determined the optimal delivery mass ratio to be 8:1, indicating that the corresponding CPFm concentration for ssDNA21 (20 µg mL−1) delivery is 2.5 µg mL−1 (Fig. 9b). Note that lacking any delivery agent or only in the presence of 1b-poly-2bn, the precursor of CPFms, Cy5-ssDNA21 showed a poor internalization percentage (<2%), which again confirmed the necessity of a complete CPFms delivery vehicle. With CPF10 and CPF30 as the delivery agents, they demonstrated high delivery efficiency for both Cy5-ssDNA21 and Cy5-ssDNA35. Specifically, CPF10, with a smaller pore aperture, achieved an internalization efficiency of 96.7% for Cy5-ssDNA21 and 86.2% for Cy5-ssDNA35, respectively. In contrast, CPF30 with a larger pore aperture exhibited increased delivery trends, with an internalization percentage of 97.3% for Cy5-ssDNA35 and 79.7% for Cy5-ssDNA21. Compared to the commercial reagent Lipo2000, CPF10 and CPF30 both showed more than a threefold increase in delivery efficiency under the same experimental conditions (Fig. 9c and S41–S44 in the SI).
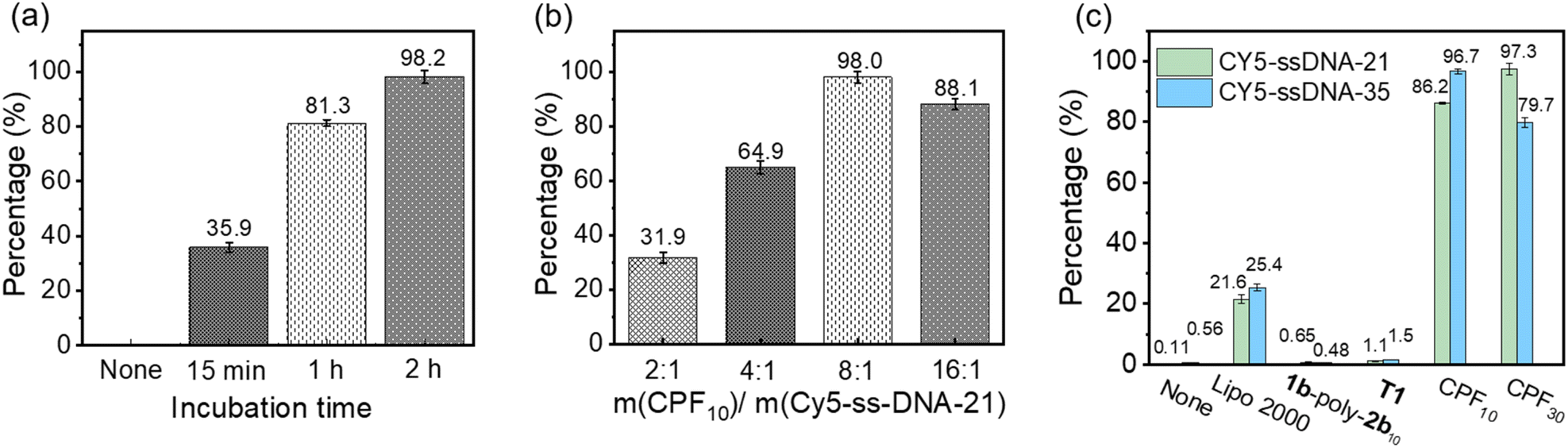 | ||
| Fig. 9 The efficiency of delivering Cy5-ssDNA21 (2.5 µg mL−1) to H9C2 cells with CPF10 as a carrier under different (a) incubation times and (b) experimental dosages. (c) Delivery of Cy5-ssDNA21 and Cy5-ssDNA35 (2.5 µg mL−1) into H9C2 cells by none, Lipo2000 (20 µg mL−1), 1b-poly-2b10 (20 µg mL−1), T1 (20 µg mL−1), CPF30 (20 µg mL−1) and CPF10 (20 µg mL−1) after incubation for 2 h. | ||
Conclusion
In summary, this work conveniently synthesized a class of air-stable bis(phenylethynyl)palladium catalysts, which can efficiently catalyze living insertion polymerization of various isocyanides. The resulting well-defined polyisocyanides possess controlled Mn and narrow Đ and bear intact chain-end functionalities. The activity of the Pd(II)-catalysts can be simply modulated by varying the substituents. Importantly, the Pd(II)-complex embedded in the polymer backbone can be easily removed via reductive elimination. Interestingly, owing to the rigid rodlike backbone, precisely controlled chain length, and relatively narrow distribution, these well-defined polyisocyanides were utilized to construct porous polymer frameworks with tunable pore structures. The polyisocyanides bearing benzaldehyde on each chain end reacted with a four-arm linker T1, affording a class of CPFms with ordered structures and tunable pore apertures, as confirmed by DLS, HR-TEM, and SAXS. The pore apertures increase progressively with the DP of the polyisocyanide backbone, indicative of pore-size controllability. The water-soluble CPFms were further employed for the delivery of ssDNA21 and ssDNA35 into H9C2 cells. The achieved delivery efficiency was up to 98%, which is more than three times higher than that of the commercial reagent Lipo2000. The present study developed a new kind of catalyst for the precise synthesis of well-defined telechelic polymers, which have great potential in the construction of ordered porous frameworks with tunable pore structures. In the future, by modulating the framework aperture and the functional groups of polyisocyanide pendants, it has the potential to become a versatile platform for the loading of biomacromolecules, enabling a variety of biological applications. Simultaneously, through the alteration of catalyst substituents, it holds promise as a universal link for constructing other porous materials and molecular cages.Author contributions
Z.-Q. W., N. L., and Z.-T. L. designed and directed the project, Y. Z., R.-T. G., and S. L. performed the experiments and analyzed the data. Z.-Q. W., N. L., and Y. Z. wrote the manuscript with input from all other authors.Conflicts of interest
There are no conflicts to declare.Data availability
The data that support the findings of this study are available in the supplementary information (SI) of this article. Supplementary information: experimental details and additional spectra. See DOI: https://doi.org/10.1039/d5sc07130e.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 22571117, 52573008, and 52273006), Major Research Plan of the National Natural Science Foundation of China (No. 92256201, and 92356302), and Science and Technology Development Plan of Jilin Province (20240101181JC). Z.-Q. Wu, S. Lei, and Z.-T. Li acknowledge the Shanghai Synchrotron Radiation Facility (SSRF) of BL16B1 (No. 31124.02.SSRF.BL16B1) and BL10U1 (No. 31124.02.SSRF.BL10U1) for the assistance on SAXS and the SSRF Key Research Project (No. 2023-SSRF-ZD-503466).Notes and references
- R. Kumar, C. F. S. Chalarca, M. R. Bockman, C. V. Bruggen, C. J. Grimme, R. J. Dalal, M. G. Hanson, J. K. Hexum and T. M. Reineke, Chem. Rev., 2021, 121, 11527–11652 CrossRef CAS PubMed.
- W. Zhao, Y. Lv, J. Li, Z. Feng, Y. Ni and N. Hadjichristidis, Angew. Chem., Int. Ed., 2021, 60, 889–895 CrossRef CAS.
- R. E. Thompson and T. W. Muir, Chem. Rev., 2020, 120, 3051–3126 CrossRef CAS.
- M. J. Lajoie, A. J. Rovner, D. B. Goodman, H.-R. Aerni, A. D. Haimovich, G. Kuznetsov, J. A. Mercer, H. H. Wang, P. A. Carr, J. A. Mosberg, N. Rohland, P. G. Schultz, J. M. Jacobson, J. Rinehart, G. M. Church and F. J. Isaacs, Science, 2013, 342, 357–360 CrossRef CAS PubMed.
- R. Hu, J. Wang, A. J. Qin and B. Z. Tang, Biomacromolecules, 2022, 23, 2185–2196 CrossRef CAS PubMed.
- H. Almeida, G. Traverso, B. Sarmento and J. das Neves, Nat. Rev. Bioeng., 2024, 2, 609–625 CrossRef CAS.
- D. A. Corbin and G. M. Miyake, Chem. Rev., 2022, 122, 1830–1874 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- Y. Song, Y. Kim, Y. Noh, V. K. Singh, S. K. Behera, A. Abudulimu, K. Chung, R. Wannemacher, J. Gierschner, L. Lüer and M. S. Kwon, Macromolecules, 2019, 52, 5538–5545 CrossRef CAS.
- Y. Yoshinaga, T. Yamamoto and M. Suginome, J. Am. Chem. Soc., 2020, 142, 18317–18323 CrossRef CAS.
- Y. Yoshinaga, T. Yamamoto and M. Suginome, ACS Macro Lett., 2017, 6, 705–710 CrossRef CAS.
- M. A. van Dongen, C. A. Dougherty and M. M. Banaszak Holl, Biomacromolecules, 2014, 15, 3215–3234 CrossRef CAS PubMed.
- M. A. Beach, U. Nayanathara, Y. Gao, C. Zhang, Y. Xiong, Y. Wang and G. K. Such, Chem. Rev., 2024, 124, 5505–5616 CrossRef CAS.
- B. H. Liu, Y. Zong, N. Liu and Z.-Q. Wu, Sci. China: Chem., 2024, 67, 3247–3257 CrossRef CAS.
- S. Ma, J. Jiang, Z. Liu, Y. Jiang, Z. Wu and M. Liu, Nanoscale, 2020, 12, 7895–7901 RSC.
- D. Yang, P. Duan, L. Zhang and M. Liu, Nat. Commun., 2017, 8, 15727 CrossRef CAS PubMed.
- J. Kim, H. Y. Jung and M. J. Park, Macromolecules, 2020, 53, 746–763 CrossRef CAS.
- S. Pispas and N. Hadjichristidis, Macromolecules, 1994, 27, 1891–1896 CrossRef CAS.
- X.-H. Xu, W.-B. Liu, X. Song, L. Zhou, N. Liu, Y.-Y. Zhu and Z.-Q. Wu, Polym. Chem., 2021, 12, 4838–4845 RSC.
- R.-T. Gao, S.-Y. Li, Y. Zong, Z. Chen, N. Liu and Z.-Q. Wu, Angew. Chem., Int. Ed., 2024, 63, e202410010 CrossRef CAS PubMed.
- X.-H. Xu, Y.-X. Li, L. Zhou, N. Liu and Z.-Q. Wu, Chem. Sci., 2022, 13, 1111–1118 RSC.
- W. Song, Y. Zhang, C. H. Tran, H. K. Choi, D.-G. Yu and I. Kim, Prog. Polym. Sci., 2023, 142, 101691 CrossRef CAS.
- S. Kitagawa, Acc. Chem. Res., 2017, 50, 514–516 CrossRef CAS PubMed.
- O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 15507–15509 CrossRef CAS PubMed.
- R. Freund, O. Zaremba, G. Arnauts, R. Ameloot, G. Skorupskii, M. Dincă, A. Bavykina, J. Gascon, A. Ejsmont, J. Goscianska, M. Kalmutzki, U. Lachelt, E. Ploetz, C. S. Diercks and S. Wuttke, Angew. Chem., Int. Ed., 2021, 60, 23975–24001 CrossRef CAS PubMed.
- S. D. Diwakara, W. S. Ong, Y. H. Wijesundara, R. L. Gearhart, F. C. Herbert, S. G. Fisher, G. T. McCandless, S. B. Alahakoon, J. J. Gassensmith, S. C. Dodani and R. A. Smaldone, J. Am. Chem. Soc., 2022, 144, 2468–2473 CrossRef CAS PubMed.
- B. Wang, R.-B. Lin, Z. Zhang, S. Xiang and B. Chen, J. Am. Chem. Soc., 2020, 142, 14399–14416 CrossRef CAS.
- K.-D. Zhang, J. Tian, D. Hanifi, Y. Zhang, A. C.-H. Sue, T.-Y. Zhou, L. Zhang, X. Zhao, Y. Liu and Z.-T. Li, J. Am. Chem. Soc., 2013, 135, 17913–17918 CrossRef CAS PubMed.
- B. Han, H. Wang, C. Wang, H. Wu, W. Zhou, B. Chen and J. Jiang, J. Am. Chem. Soc., 2019, 141, 8737–8740 CrossRef CAS PubMed.
- M. Zhao, Y. Huang, Y. Peng, Z. Huang, Q. Ma and H. Zhang, Chem. Soc. Rev., 2018, 47, 6267–6295 RSC.
- A. Thomas, Angew. Chem., Int. Ed., 2010, 49, 8328–8344 CrossRef CAS PubMed.
- Y. Yuan and G. Zhu, ACS Cent. Sci., 2019, 5, 409–418 CrossRef CAS.
- Y. Gu, J. Zhao and J. A. Johnson, Angew. Chem., Int. Ed., 2020, 59, 5022–5049 CrossRef CAS PubMed.
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gandara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018–1023 CrossRef CAS PubMed.
- Z. Mu, Y. Zhu, B. Li, A. Dong, B. Wang and X. Feng, J. Am. Chem. Soc., 2022, 144, 5145–5154 CrossRef CAS PubMed.
- J.-L. Lin, Z.-K. Wang, Z.-Y. Xu, L. Wei, Y.-C. Zhang, H. Wang, D.-W. Zhang, W. Zhou, Y.-B. Zhang, Y. Liu and Z.-T. Li, J. Am. Chem. Soc., 2020, 142, 3577–3582 CrossRef CAS PubMed.
- X. Chen, Q. Zheng, W. Cai, J. Sheng and M. Wang, ACS Appl. Mater. Interfaces, 2023, 15, 54346–54352 CrossRef CAS.
- Z.-T. Li, S.-B. Yu, Y. Liu, J. Tian and D.-W. Zhang, Acc. Chem. Res., 2022, 55, 2316–2325 CrossRef CAS.
- S. Yu, F. Lin, J. Tian, J. Yu, D. Zhang and Z.-T. Li, Chem. Soc. Rev., 2022, 51, 434–449 RSC.
- Y. Li, Q. Li, X. Miao, C. Qin, D. Chu and L. Cao, Angew. Chem., Int. Ed., 2021, 60, 6744–6751 CrossRef CAS.
- X. Tan, F. Jia, P. Wang and K. Zhang, J. Controlled Release, 2020, 323, 240–252 CrossRef CAS.
- X. Tan, B. B. Li, X. Lu, F. Jia, C. Santori, P. Menon, H. Li, B. Zhang, J. J. Zhao and K. Zhang, J. Am. Chem. Soc., 2015, 137, 6112–6115 CrossRef CAS PubMed.
- Z. Wang, J. Lin, Y. Zhang, C. Yang, Y. Zhao, Z. Leng, H. Wang, D. Zhang, J. Zhu and Z.-T. Li, Mater. Chem. Front., 2021, 5, 869–875 RSC.
- B. Yang, X. Zhang, J. Li, J. Tian, Y. Wu, F. Yu, R. Wang, H. Wang, D. Zhang, Y. Liu, L. Zhou and Z.-T. Li, CCS Chem., 2019, 1, 156–165 CrossRef CAS.
- B. Shi, M. Zheng, W. Tao, R. Chung, D. Jin, D. Ghaffari and O. C. Farokhzad, Biomacromolecules, 2017, 18, 2231–2246 CrossRef CAS PubMed.
- Y.-X. Xue, Y.-Y. Zhu, L.-M. Gao, X.-Y. He, N. Liu, W.-Y. Zhang, J. Yin, Y. Ding, H. Zhou and Z.-Q. Wu, J. Am. Chem. Soc., 2014, 136, 4706–4713 CrossRef CAS PubMed.
- N. Liu, L. Zhou and Z.-Q. Wu, Acc. Chem. Res., 2021, 54, 3953–3967 CrossRef CAS.
- N. Liu, R.-T. Gao and Z.-Q. Wu, Acc. Chem. Res., 2023, 56, 2954–2967 CrossRef CAS.
- K. Onitsuka, M. Yamamoto, T. Mori, F. Takei and S. Takahashi, Organometallics, 2006, 25, 1270–1278 CrossRef CAS.
- K. Onitsuka, K.-I. Yabe, N. Ohshiro, A. Shimizu, R. Okumura, F. Takei and S. Takahashi, Macromolecules, 2004, 37, 8204–8211 CrossRef CAS.
- A. Croom, R. Tarallo and M. Weck, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2766–2773 CrossRef CAS.
- M. M. Green, R. A. Gross, F. C. Schilling, K. Zero and C. Crosby, Macromolecules, 1988, 21, 1839–1846 CrossRef CAS.
- J.-D. Sun, Q. Li, W.-W. Haoyang, D.-W. Zhang, H. Wang, W. Zhou, D. Ma, J.-L. Hou and Z.-T. Li, Mol. Pharmaceutics, 2022, 19, 953–962 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |