
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the electronic structure of molybdenum oxide nanoclusters with vanadium doping for electrochemical H2O2 production
Luyao
Zhang†
a,
Yingbo
Li†
a,
Shuhao
Zhang
b,
Jianghua
Wu
a,
Weiyang
Xue
 a,
Kun
Wang
a,
Haojie
Liu
a,
Boyuan
Yu
a,
Xin
Zhao
a,
Jiangfeng
Ni
*b and
Feng
Yang
*a
a,
Kun
Wang
a,
Haojie
Liu
a,
Boyuan
Yu
a,
Xin
Zhao
a,
Jiangfeng
Ni
*b and
Feng
Yang
*a
aDepartment of Chemistry, Southern University of Science and Technology, Shenzhen, 518055, China. E-mail: yangf3@sustech.edu.cn
bSchool of Physical Science and Technology, Center for Energy Conversion Materials & Physics (CECMP), Jiangsu Key Laboratory of Thin Films, Soochow University, Suzhou 215006, China. E-mail: jeffni@suda.edu.cn
First published on 17th February 2026
Abstract
Electrosynthesis of H2O2 has gained tremendous attention as a highly promising alternative strategy to the traditional anthraquinone technique. Nevertheless, there is still a lack of highly efficient, robust, and low-cost electrocatalysts to propel the industrialization of electrosynthesis of H2O2. Herein, we constructed carboxyl functionalized CNTs decorated with V-doped MoOx nanoclusters (MoVOx NCs/CNTs) with uniform subnanometer size (∼0.7 nm). The incorporation of V significantly modified the electronic structure of MoOx, enabling an impressive H2O2 electrosynthesis selectivity of up to 98% at 0.4 V, surpassing that of the undoped MoOx NCs/CNTs. The MoVOx NCs/CNTs catalyst retained 93% H2O2 electrosynthesis selectivity across a wide potential range (0.2–0.6 V). Moreover, the catalyst demonstrated excellent activity stability for 10 h with minimal decay. This work offers a pathway by rational design of a subnanometer catalyst for the electrocatalysis of O2 to H2O2.
Introduction
Since its first discovery by Thenard in 1818, hydrogen peroxide (H2O2) has been regarded as one of the 100 most important chemicals and found wide application in various areas, including paper bleaching, pharmaceutical synthesis, and wastewater degradation.1–8 Impressively, it was estimated that the global quantity demand of H2O2 would reach 6.0 million metric tons in 2024.9,10 Currently, over 95% of H2O2 stock solutions (∼30–70 wt%) strongly rely on the well-established anthraquinone oxidation process.11,12 It is an energy-intensive technique, involving sequential hydrogenation/autoxidation of 2-alkyl-9/10-anthraquinone under high pressure with the aid of expensive Pd/Ni catalysts, followed by complicated purification procedures.2,13–15 Consequently, the tremendous industrial waste generated and the high risk of storage/transportation remarkably deviate from our pursuit of green chemistry.16,17 Moreover, most applications typically require low concentrations of H2O2.Recently, with the rapid development of nanosized catalysts, electrosynthesis of H2O2 through a 2e− oxygen reduction reaction (ORR) pathway (O2 + 2H2O + 2e− → H2O2 + 2OH−) (U0 = 0.76 V vs. RHE, where RHE represents the reversible hydrogen electrode) has emerged as a highly appealing alternative strategy.5,14,18 Compared with the anthraquinone process, the electrochemical approach holds several advantages, including mild production conditions in ordinary atmospheric environments, suitability for on-site/on-demand production to avoid the risk of storage/transportation, environmental friendliness and organic waste emission, and the avoidance of explosive hazards by spatially separating H2 and O2 feeding.2,19–22 However, it is worth noting that the ORR also involves another thermodynamically favourable 4e− pathway that converts O2 to H2O (O2 + 2H2O + 4e− → 4OH−) (U0 = 0.40 V vs. RHE) which inevitably suppresses H2O2 production.23,24 Accordingly, one major challenge for electrosynthesis of H2O2 is to find an efficient and robust electrocatalyst with high selectivity towards the 2e− ORR.
Mechanistically, the ORR selectivity is highly dependent on the adsorption strength between the *OOH intermediate and the surface of catalysts.4,25 In detail, an intensive interaction could easily dissociate the O–O bond and induce the 4e− ORR, while too-weak cooperation may require a high overpotential to trigger the reaction.26 Therefore, the appropriate interaction strength is a critical knob for designing efficient 2e− ORR catalysts. Therefore, a great variety of catalysts have been developed, ranging from noble metals (Au–Pd),27 metal chalcogenides (CoSe2),7 conducting coordination compounds (cobalt–porphyrins),28 and carbon-based materials (N-doped oxo-functionalized graphene).29 In particular, metal-free carbonaceous materials have garnered tremendous attention due to their abundance, low cost, and high selectivity for the 2e− ORR.9,30,31 For instance, Lu et al. demonstrated that the as-prepared oxidized carbon nanotubes (CNTs) exhibited high activity for the 2e− ORR and claimed that the responsible active sites were the carbon atoms adjacent to carboxylic acid and other oxo-functional groups.32 Targeting further improving the performance of metal-free carbonaceous materials, introducing isolated metal single atoms into a carbon matrix to form single-atom catalysts (SACs) exhibits profound potential application in electrosynthesis of H2O2.33–38 Despite substantial progress, the catalyst design and the properties still need further revolutionization to meet the high criterion of practical application.
Nanocluster catalysts (Mx, where M and x refer to the metal and atom number, respectively) play a pivotal role in bridging the gap between SACs and nanoparticles. They not only share similar characteristics with SACs, such as maximized metal atom utilization and well-defined active sites, but also offer additional catalytic sites with adjacent metal atoms.33,39–41 As compared to Mx–Ny and Mx–Cy coordination structures, the highly electronegative O atom could induce accelerated electron transfer from Mx to Oy, potentially resulting in a favourable energy band structure and unexpected catalytic activity.42 Additionally, as a 4d transition metal, Mo possesses more intricate oxidation states and coordination numbers and has received less attention compared to the well-reported 3d transition metals (Fe, Co, Ni, etc.).43 On the other hand, polyoxometalates (POMs) are a type of well-defined anionic oxo–metal cluster with unique redox properties.44 Their charge density and coordination structures can be precisely tailored by adjusting their composition.45 As a typical POM, the above-mentioned characteristics make H3PMo12O40 an ideal precursor for constructing MoOx nanoclusters. Besides, doping a metal (e.g., V) with a lower d-orbital occupancy and electronegativity into MoOx nanoclusters may further optimize their electronic structure46–48 for reaction intermediate adsorption/desorption and stimulate the catalytic activity.
Herein, we prepared carboxyl functionalized CNTs decorated with vanadium-doped MoOx nanoclusters (MoVOx NCs/CNTs) through a mild thermal treatment using V-substituted H3PMo12O40 (H6PMo9V3O40) as a precursor. The MoVOx NCs were not only supported on the surface of CNTs, but also confined within the cavities of CNTs. Based on the advanced spectroscopic techniques, it was found that the doping of V could reduce the valence state of Mo in MoVOx NCs/CNTs, enabling Mo with a higher electronic cloud density, and thereby significantly modify the electronic structure of MoOx NCs. Consequently, MoVOx NCs/CNTs exhibit an impressive 2e− ORR selectivity of up to 98% at 0.4 V in 0.1 M KOH solution, which far exceeds that of their counterpart MoOx NCs/CNTs. Notably, MoVOx NCs/CNTs could retain more than 93% 2e− ORR selectivity in a wide potential range (0.2–0.6 V). Furthermore, MoVOx NCs/CNTs demonstrated excellent activity stability for 10 h with negligible decay.
Results and discussion
Preparation and characterization of MoVOx NCs/CNTs
V-substituted H3PMo12O40 (H6PMo9V3O40) was synthesized according to the literature. Within a POM unit, there are four distinct types of oxygen atoms: central oxygen (Oc), terminal oxygen (Ot), bridged oxygen between two octahedra sharing a corner (Ob), and bridged oxygen that shares an edge (Oe). Intense bands at 1058, 955, 881, and 768 cm−1 can be observed in the Fourier-transform infrared spectra (Fig. S1) of H3PMo12O40 and H6PMo9V3O40, corresponding to the vibrations of P–Oc, Mo–Ot, Mo–Ob–Mo, and Mo–Oe–Mo, respectively.49 These results confirm the formation of H6PMo9V3O40. Notably, the phenomenon of blue shift, characterized by a migration towards higher wavenumbers in H6PMo9V3O40, typically signifies alterations in the vibrational frequencies of chemical bonds or functional groups within a molecule. Such shifts may arise from the strengthening of chemical bonds, modifications in the intramolecular charge distribution, and intermolecular interactions after the doping of V.The synthesis procedures of MoOx NCs/CNTs and MoVOx NCs/CNTs are schematically displayed in Fig. S2. H3PMo12O40 or H6PMo9V3O40 and carboxyl functionalized CNTs were mixed and self-assembled by magnetic stirring for 48 h. The resultant powders were then subjected to a heat treatment at 500 °C for 30 min under a 10% H2/Ar atmosphere through a rapid heating-cooling strategy. During this process, H3PMo12O40 and H6PMo9V3O40 underwent thermal decomposition and reduction and were converted to MoOx and MoVOx nanoclusters, respectively. The morphology of the as-prepared MoOx NCs/CNTs and MoVOx NCs/CNTs was revealed by transmission electron microscopy (TEM). The high-angle annular dark-field scanning TEM (HAADF-STEM) images clearly demonstrated the presence of tiny nanoclusters both on outer walls and inner cavities of CNTs (Fig. 1a and S3a). The corresponding STEM-energy dispersive X-ray (EDX) elemental mapping further proved that the Mo, O, and V elements were uniformly distributed throughout CNTs, indicating the bright dots consisted of Mo, O, and V (Fig. 1b and S3b).
 | ||
| Fig. 1 Characterization of MoVOx NCs/CNTs. (a) and (b) HAADF-STEM image and the corresponding STEM-EDX elemental mapping of MoVOx NCs/CNTs. (c) and (d) High-resolution HAADF-STEM images, and (e) the corresponding cluster size distribution. (f) XRD patterns of CNTs, MoOx NCs/CNTs and MoVOx NCs/CNTs. The standard cards of MoO2 and MoO3 are also shown. | ||
To further elucidate the structure of these clusters, aberration-corrected STEM was conducted. The subnanometer clusters in MoVOx NCs/CNTs and MoOx NCs/CNTs were identified to be composed of Mo/V atoms with an average diameter of 0.67 nm and 0.75 nm, respectively (Fig. 1c–e and S3c, d). More aberration-corrected STEM images of MoVOx NCs/CNTs are shown in Fig. S4. The powder X-ray diffraction (XRD) patterns showed that instead of yielding sharp diffraction peaks, the phenomenon manifests as broad, diffuse background signals, implying the amorphous structure of tiny clusters in both MoVOx NCs/CNTs and MoOx NCs/CNTs (Fig. 1f). Moreover, no diffraction peaks can be ascribed to the characteristics of crystalized MoO2 or MoO3.
Electrocatalytic ORR activities towards H2O2
The electrocatalytic performance of the as-prepared hybrid catalysts for the 2e− ORR was evaluated using the typical rotating ring-disk electrode (RRDE) equipment, with an optimized catalyst loading of 0.1 mg cm−2. All potential reported in this work is calibrated to RHE (Fig. S5). Fig. 2a shows the linear sweep voltammetry (LSV) curves collected at 1600 rpm in O2-saturated 0.1 M KOH electrolyte, together with the H2O2 oxidation current collected using the Pt ring electrode at a constant potential of 1.2 V versus RHE. The MoVOx NCs/CNTs exhibit the highest ring current (up to 210 µA), which is substantially greater than that of MoOx NCs/CNTs and the CNTs benchmark. Additionally, there is only a minimal difference in disk current observed for all three samples. These findings indicate that MoVOx NCs/CNTs possess the highest 2e− ORR selectivity among the tested catalysts, making them capable of achieving the highest H2O2 yield. | ||
| Fig. 2 2e− ORR performance of MoVOx NCs/CNTs in 0.1 M KOH. (a) Disk and ring current of the three samples on the RRDE. (b) The corresponding electron transfer number n and H2O2 selection at various potentials. (c) The corresponding Tafel plots. (d) Stability measurement of MoVOx NCs/CNTs at a fixed disk potential of 0.4 V vs. RHE and the corresponding faradaic efficiency. (e) ORR LSV curves of MoVOx NCs/CNTs before and after 5000 ADT cycles. (f) Performance comparison of electrochemical H2O2 production in alkaline media on MoVOx NCs/CNTs and other recently reported catalysts based on RRDE performance. | ||
To accurately assess the selectivity for H2O2 production, the collection efficiency (N) of the RRDE was first calibrated using the redox reactions of potassium ferricyanide (Fig. S6). The experimentally determined collection efficiency is 34.2%, which was then used to calculate the selectivity for H2O2 production and the number of electrons transferred during the ORR. The calculated H2O2 selectivity and electron transfer number (n) were plotted as a function of potential (Fig. 2b). In the wide potential range of 0.2–0.6 V, the H2O2 selectivity of MoVOx NCs/CNTs consistently remained above 93%, indicating the highly selective 2e− ORR pathway. Notably, the selectivity further increased to 98% at 0.4 V vs. RHE. Moreover, the calculated electron transfer number was less than 2.2, further confirming the dominance of the 2e− ORR pathway during the ORR process. On the other hand, MoOx NCs/CNTs exhibited a lower H2O2 selectivity of ∼80% with a higher electron transfer number, indicating the critical role of V in promoting H2O2 generation. Additionally, the Tafel plots derived from LSV curves were obtained to examine the kinetic reaction rate. The Tafel slope of MoVOx NCs/CNTs (59.7 mV dec−1) is noticeably lower than that of MoOx NCs/CNTs (65.8 mV dec−1) and CNTs (78.6 mV dec−1), implying that MoVOx NCs/CNTs hold the best kinetic reaction rate (Fig. 2c). These results highlight the superior electrocatalysis performance of MoVOx NCs/CNTs in terms of electrosynthesis of H2O2.
Stability is another key indicator to assess the performance of catalysts for electrosynthesis of H2O2. We conducted a long-term stability test for MoVOx NCs/CNTs using a chronoamperometric test at a constant disk potential of 0.4 V vs. RHE. The H2O2 selectivity remained consistently around 98% with negligible fluctuation during the 10 h continuous operation (Fig. 2d), suggesting the robust electrocatalytic stability for the 2e− ORR. In addition, we also performed an accelerated degradation test (ADT) to further demonstrate the robust stability of MoVOx NCs/CNTs. The obtained LSV curves before and after the 5000 ADT cycles were essentially identical (Fig. 2e), confirming the impressive cycling stability of MoVOx NCs/CNTs. Specifically, MoVOx NCs/CNTs afforded an outstanding H2O2 selectivity of ∼98% at a current density of 1.0 mA cm−2, surpassing that of previously reported electrocatalysts (Fig. 2f and Table S1). RRDE measurements are highly reproducible across three independently prepared batches, with relative standard deviations below 5% (Fig. S7). Since catalyst durability is closely tied to structural and compositional integrity, we carried out comprehensive post-stability characterization studies to assess possible changes and to better understand the long-term performance. The electronic structure of MoVOx NCs/CNTs remains largely intact after electrochemical testing, with negligible alterations (Fig. S8a). We further tested the leaching amount of V during the stability test. We regularly monitored the V ion content in solution by inductively coupled plasma mass spectrometry (ICP-MS). The results show only 1.4% of V leaching after a 10 h long-term stability test, confirming that V leaching during electrochemical operation can be safely excluded (Fig. S8b). All the results indicated that MoVOx NCs/CNTs hold great promise as a highly efficient and robust 2e− ORR electrocatalyst in alkaline media.
It has been demonstrated that metal-free carbonaceous materials with abundant oxo-functional groups are promising, efficient 2e− ORR electrocatalysts in alkaline media.5,9,23,32 Given the reported activity of strongly oxidized CNTs for the 2e− ORR, we prepared further deep-oxidized CNTs as a control sample by refluxing blank carboxylated CNTs in concentrated HNO3 at 140 °C for 2.5 h following literature procedures.50 The normalized XPS survey spectra show a pronounced increase in the O 1s signal after oxidation (Fig. S9), with the oxygen content increasing from 7.7% for the blank carboxylated CNTs to 19.1% for the deep-oxidized CNTs, indicating a substantially more oxygen-rich CNT surface. Under identical electrode preparation and RRDE testing conditions, deep-oxidized CNTs deliver an H2O2 selectivity of ∼70% at 0.4 V in 0.1 M KOH, slightly higher than that of blank carboxylated CNTs support (∼60%) but still substantially lower than that of MoVOx NCs/CNTs (98%) (Fig. S10). These results suggest that increasing surface oxygen content can improve H2O2 selectivity to a limited extent. However, oxygen enrichment alone cannot account for the near-quantitative selectivity, supporting that the MoVOx nanoclusters play a key role in enabling highly selective 2e− ORR.
To evaluate the applicability of the MoVOx NCs/CNTs catalyst under more challenging acidic conditions, we further performed ORR measurements in an O2-saturated in 0.05 M H2SO4 at 1600 rpm. CNTs, MoOx NCs/CNTs, and MoVOx NCs/CNTs all remain ORR-active in acid and deliver measurable H2O2 production, evidenced by the resolved disk and ring current responses (Fig. S11). Compared with alkaline media, the reaction kinetics in acidic electrolyte are noticeably slower, as reflected by larger Tafel slopes and substantially reduced H2O2-related ring currents, which is consistent with the commonly observed limitations of non-precious-metal, carbon-based catalysts in acidic environments, where stronger proton adsorption, reduced site stability, and intensified competition from the 4e− ORR pathway often constrain performance.51,52 Notably, under identical testing conditions, the V-doped catalyst (MoVOx NCs/CNTs) still outperforms the undoped counterpart (MoOx NCs/CNTs) in acidic media. MoVOx NCs/CNTs retain an H2O2 selectivity of ∼60% over a wide potential window of 0.2–0.6 V (Fig. S11a and b) and exhibit a Tafel slope of 68.5 mV dec−1 (Fig. S11c). In addition, negligible performance decay is observed after 2000 accelerated ADT cycles, indicating good cycling stability in acid (Fig. S11d). These results suggest that the V-induced electronic modulation of MoOx nanoclusters remains operative in acidic media, leading to improved kinetics and H2O2 selectivity.
Electronic structures of catalysts
To elucidate the electrocatalysis mechanism of the as-prepared catalysts for the 2e− ORR, it is essential to investigate their electronic structures. Raman spectra showed that the relative intensity of D, G, and D′ bands for CNTs were almost the same (Fig. 3a and c), suggesting negligible changes in nanotube defects after depositing clusters as compared to CNTs. Consistently, the C 1s X-ray photoelectron spectroscopy (XPS) results of MoVOx NCs/CNTs and MoOx NCs/CNTs also exhibited similar CNT electronic structures (Fig. 3b, d and S12). These results imply that the synthetic process of nanoclusters did not significantly influence the content of surface oxo-functional groups in CNTs. | ||
| Fig. 3 Characterization of MoVOx NCs/CNTs. (a) Raman spectra. (b) High-resolution C 1s XPS spectra. The content of different carbon species in CNTs, MoOx NCs/CNTs and MoVOx NCs/CNTs determined using Raman (c) and C 1s XPS (d) spectra. The content of species is also shown. | ||
The Mo 3d XPS spectra of MoVOx NCs/CNTs and MoOx NCs/CNTs reveal a Mo5+ spin–orbit doublet at 232.67 and 235.78 eV, which are lower than the peaks of 233.95 and 236.16 eV for Mo6+ in the referenced MoO3 (Fig. 4a).53 Furthermore, as compared to undoped MoOx NCs/CNTs, the Mo 3d spectra of MoVOx NCs/CNTs slightly shift towards lower binding energy, suggesting a lower oxidation state of Mo in MoVOx NCs/CNT. This observation is further supported by theoretical calculations based on the Mo 3d spectra, where the average oxidation state in MoOx NCs/CNTs and MoVOx NCs/CNTs is 5.40 and 5.22 (Fig. S13a), respectively. This phenomenon may be attributed to the doping of V, which modifies the electronic structure of Mo species in MoVOx NCs/CNTs. Such changes in electron density can optimize the interaction strength between the surface of catalysts and the intermediate of *OOH, facilitating proton-coupled O2 reduction to *OOH and its following desorption.54 Meanwhile, based on the O 1s spectra, additional C–O–Mo species were observed in both MoOx NCs/CNTs and MoVOx NCs/CNTs (Fig. 4b), indicating the presence of strong interaction between the nanoclusters and CNTs.55 Furthermore, the introduction of V in MoVOx NCs/CNTs leads to the generation of more defective O species (Fig. S13b), which could act as additional active sites. This observation may also contribute to the enhanced electrocatalytic activity of MoVOx NCs/CNTs for the 2e− ORR.
 | ||
| Fig. 4 High-resolution (a) Mo 3d and (b) O 1s XPS spectra of MoOx NCs/CNTs and MoVOx NCs/CNTs. MoO3 is used as a reference. | ||
Insights into mechanisms
The surface area and pore structures play crucial roles in determining the performance of electrocatalysts. Normally, a larger surface area and the presence of mesopores could increase the number of exposed electroactive sites and facilitate mass transfer, thus improving the electrocatalytic performance.4,9,31 We carried out Brunauer–Emmett–Teller (BET) measurements to investigate the surface area and pore features of catalysts. The N2 adsorption/desorption isotherm curves displayed the typical features of mesoporous materials,5,56 with BET surface areas of 283.9 m2 g−1 for MoOx NCs/CNTs and 286.5 m2 g−1 for MoVOx NCs/CNTs, which were larger than that of the blank CNTs (246.5 m2 g−1), while the pore characteristics of MoOx NCs/CNTs and MoVOx NCs/CNTs are similar to those of blank CNTs (Fig. S14a). The content of clusters in CNTs was determined to be ∼9 wt% by thermal gravimetric analysis (TGA) (Fig. S14b). These results indicate that loading MoVOx nanoclusters does not impose a pronounced additional mass-transport limitation. These results imply that the enhanced electrocatalytic activity for the 2e− ORR is mainly attributed to the introduction of nanoclusters into the CNTs.To gain a deeper understanding of the remarkable performance of the MoVOx NCs/CNTs, electron energy loss spectroscopy (EELS) was employed as a powerful tool to unveil the local electronic structures.57,58 Notably, the region in Fig. 5a corresponds to the blank CNTs segment without nanoclusters, serving as a reference area. In Fig. 5b and c, two regions with typical morphology, where nanoclusters are not only anchored on the outer surface of CNTs but also confined within the cavities of CNTs, were selected. In EELS survey spectra, MoVOx NCs/CNTs show the existence of V L2,3-edge features (510–525 eV) compared to MoOx NCs/CNTs (Fig. 5d), proving the doping of V atoms in MoVOx NCs/CNTs, which is consistent with the corresponding EELS mapping (Fig. S15 and S16). The C K-edge EELS spectrum of MoVOx NCs/CNTs shows an upshift of the carbon energy loss peak (1.1 eV) compared with blank CNTs, while the undoped MoOx NCs/CNTs show negligible changes (Fig. 5e), indicating that the introduction of V could induce variations in the sp2 hybridization state of CNTs.59 It was found that the doping of V reduced the valence state of Mo in MoVOx NCs/CNTs (Fig. 5f), resulting in higher electronic cloud density at Mo sites,60 which is consistent with the XPS results (Fig. 4a).
 | ||
| Fig. 5 Insights into mechanisms. STEM-EELS images of CNTs (a), MoOx NCs/CNTs (b), and MoVOx NCs/CNTs (c). The EELS survey spectra (d) and C K-edge (e) and Mo M-edge (f) spectra as acquired in the region marked in (a)–(c), respectively. The structural model of MoOx (g). The calculated distribution of electron density difference of MoOx (h) and MoVOx (i). | ||
We performed the electron cloud density calculation of MoOx NCs and MoVOx NCs to unveil the effect of V doping on the electronic structure of MoOx NCs (Fig. S17). The calculated electron density distributions reveal higher electron density around Mo in the V-doped structure than in the undoped structure, indicating that V incorporation generates an electron-rich local environment around Mo (Fig. 5g–i). The Gibbs free energy of the key *OOH intermediate was then evaluated on the MoOx and MoVOx cluster surfaces. The V doping stabilizes *OOH, lowering ΔG(*OOH) from 0.85 eV on MoOx to 0.75 eV on MoVOx (Fig. S18), indicating that V doping promotes the kinetics of the entire redox process. This modulation is particularly relevant to the 2e−/4e− pathway competition after *OOH formation, thereby favouring H2O2 formation and enhancing selectivity, while the overall ORR rate (disk current) can remain largely unchanged because it is often governed by O2 transport and the initial O2 → *OOH activation step. The theoretical calculations corroborate the experimental results obtained from XPS and EELS. The combination of XPS, EELS analysis and the theoretical calculations proved that the doping of V could modify the electronic structure of MoOx nanoclusters. The modifications of electron density facilitate the enhancement of interaction strength between the surface of catalysts and the *OOH intermediate, thereby facilitating the 2e− ORR process.
Conclusions
Carboxyl functionalized CNTs decorated with V-doped MoOx nanoclusters (MoVOx NCs/CNTs) were prepared. Benefitting from the optimized electronic structure modified by V doping, the resulting MoVOx NCs/CNTs exhibited an impressive H2O2 electrosynthesis selectivity of up to 98% at 0.4 V in 0.1 M KOH solution, which far exceeds that of their counterpart MoOx NCs/CNTs. Importantly, MoVOx NCs/CNTs could retain more than 93% H2O2 electrosynthesis selectivity in a wide potential range from 0.2 V to 0.6 V. Furthermore, MoVOx NCs/CNTs demonstrated excellent activity stability for 10 h with negligible decay. These excellent results are attributed to the optimization of the electronic structure of MoVOx NCs/CNTs by V doping, which in turn affects the reaction pathways of the 2-electron and 4-electron reactions. Moreover, the catalyst demonstrates promising activity and stability even in acidic media, confirming the broad applicability of the electronic-structure tuning strategy. The work presents a new perspective on the development of high-performance nanocluster catalysts for the electrocatalysis of O2 to H2O2.Experimental section
Synthesis of H6PMo9V3O40
The synthesis of H6PMo9V3O40 follows a methodology similar to that in the previous report but with some modifications.61 Typically, 2.68 g Na2HPO4·7H2O was dissolved in 10 mL of water and mixed with 7.32 g NaVO3 that had been dissolved by heating in 40 mL of water. Subsequently, 1 mL of concentrated H2SO4 was introduced into the cooled mixture, yielding a vivid cherry red solution. This solution is then blended with 9.28 g of Na2MoO4 dissolved in 30 mL of water. While being vigorously stirred, 17 mL of concentrated H2SO4 was gradually incorporated into the mixture. The heated solution was allowed to naturally cool to room temperature. The resultant solution was subjected to 80 mL ethyl ether extraction, with the heteropoly etherate forming the intermediate layer. Upon passing an airstream through the solution, the heteropoly etherate was separated from the ether. Subsequently, rotary evaporation was conducted at 53 °C, resulting in the formation of a distinctive red solid. Following an overnight drying period at 40 °C, the compound was subjected to vacuum recrystallization within an anhydrous calcium chloride environment to further refine its structure.Synthesis of MoVOx NCs/CNTs and MoOx NCs/CNTs
Initially, 50 mg of CNTs were dispersed in 50 mL of DMF through sonication for 30 min. Subsequently, a solution containing 10 mg of H6PMo9V3O40 dissolved in 10 mL of DMF was gradually added drop by drop to the CNTs suspension. The resulting mixture underwent an additional 30 min of sonication to ensure thorough mixing. Then, the mixture was stirred at room temperature for a duration of 48 h, and the mixture was subjected to centrifugation, and the resulting precipitate was sequentially washed with DMF, ethanol, and deionized water. The final precipitate was freeze-dried at −60 °C to obtain the precursor powders. For the heat treatment of the precursor powders, a tube furnace was gradually heated to 500 °C under a 10% H2/Ar atmosphere (100 sccm). Once the temperature reached 500 °C, a quartz boat containing a specific amount of precursor powders was pushed into the center of the tube furnace and maintained for 30 min before being swiftly removed and allowed to cool to room temperature under the protective atmosphere of 10% H2/Ar. The resultant black powders obtained after this process were identified as MoVOx NCs/CNTs. The synthesis procedure of MoOx NCs/CNTs was almost the same as that of MoVOx NCs/CNTs except for using H3PMo12O40.Synthesis of deep-oxidized CNTs as a control catalyst
200 mg of CNTs were refluxed in 20 mL of concentrated HNO3 (68 wt%) at 140 °C for 2.5 h. After the treatment, the solid was collected by centrifugation and dried at 55 °C to obtain the deep-oxidized CNTs product.Characterization
The HAADF-STEM images were collected by aberration-corrected STEM (FEI Titan Cubed Themis G2) with an X-FEG electron gun and a DCOR aberration corrector operating at 300 kV. STEM-EDX mapping was executed on an FEI Talos F200X electron microscope with a HAADF detector operating at 200 kV. EDX spectra were obtained using a Bruker Super-X detection system. EELS spectra were collected through an aberration-corrected Titan G2 80–300 environmental TEM. It was equipped with a Gatan image filter (Quantum 936) with an energy dispersion of 0.25 eV operated at an acceleration voltage of 300 kV. The zero-loss EELS was acquired immediately after the obtainment of the core-loss EELS on individual nanoparticles. The as-obtained EELS data were further analyzed using a digital micrograph. The position of the core-loss EELS was corrected with the corresponding zero-loss peak followed by subtracting the extrapolated background from the edge of interest. A Fourier-ratio deconvolution was further performed to remove the effect of plural scattering. To facilitate the comparison of diverse spectra, we have normalized the spectra to the most prominent peak of the K-edge of carbon.Powder X-ray diffraction (XRD) was conducted on a Rigaku SmartLab X-ray diffractometer with Cu-Kα radiation (45 kV, 200 mA, λ = 1.54178 Å) at a scanning rate of 5° min−1 in the 2θ range of 10–80° at room temperature. XPS was performed on a PHI 5000 Versaprobe III photoelectron spectrometer with an Al Kα X-ray source. All peaks were calibrated with the Si 2p peak binding energy at 103.2 eV. The Raman spectra were collected through a Jovin Yvon-Horiba ARAMIS system with a 532 nm laser. The thermogravimetric analysis was conducted through a ZRT-A TGA instrument at a ramp rate of 10 °C min−1 from room temperature to 900 °C in air with a dry air flow rate of 30 sccm. Brunauer–Emmett–Teller (BET, 3Flex surface characterization analyzer) analysis was conducted to study the surface area and the porosity distribution of the samples. Fourier transform infrared spectra were recorded on a Thermo Scientific Nicolet iS 50. Inductively coupled plasma mass spectrometry (ICP-MS) was performed on an Agilent 7700.
Electrochemical measurements
All the electrochemical measurements were conducted on an electrochemical workstation (CHI 760E) coupled with a PINE rotating instrument in a standard three-electrode system. A rotating ring disk electrode (RRDE, 5.6 mm in diameter of the disk electrode), a graphite rod, and Ag/AgCl in saturated KCl were employed as the working electrode, counter electrode, and reference electrode, respectively. 0.1 M KOH aqueous solution was used as the electrolyte. All the electrode potential was referenced to the Ag/AgCl reference electrode, which was converted to the reversible hydrogen electrode (RHE) using the equation E(RHE) = E(Ag/AgCl) + 0.965 (Fig. S3). The catalyst ink was prepared by dispersing 1 mg active materials in 400 µL solution containing 195 µL isopropanol (IPA), 195 µL DIW, and 10 µL Nafion (5 wt%) solution, followed by ultrasonic treatment for about 60 min.To detect the ORR activity and H2O2 selectivity, linear sweep voltammetry (LSV) measurements were carried out at a scan rate of 5 mV s−1 with a catalyst loading of 0.1 mg cm−2 under 1600 rpm rotating speed. The scan range is from 0.1 to −0.8 V (vs. Ag/AgCl), and the Pt ring potential is set at 1.2 V (vs. RHE). Moreover, the working electrode should undergo more than 4 cycles of cyclic voltammetry (CV) at a rate of 100 mV s−1 and 4 cycles of CV (scanning rate 20 mV s−1) to stabilize the electrode before the LSV data were recorded. The following equations were used to determine the percent of H2O2 and the electron transfer number (n).

where Ir means the ring current, Id means the disk current, and N is the current collection efficiency of the platinum ring.
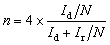
To understand the ORR stability, LSV measurements were conducted before and after 5000 CV cycles from 0.1 V to −0.8 V (vs. Ag/AgCl) at a scanning rate of 100 mV s−1. Chronopotentiometry measurement was also conducted at a constant potential of 0.4 V (vs. RHE) with a fixed Pt ring potential of 1.2 V (vs. RHE) in 0.1 M KOH and 0.05 M H2SO4 under 1600 rpm.
Computational calculations
We employed Castep within Materials Studio 2022 to calculate the electron density difference for MoOx and V-doped MoVOx. MoOx and MoVOx (25% V) nanocluster models were constructed based on the MoO3 crystal structure (Fig. S17). Within the density functional theory (DFT) framework, the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional was utilized to approximate the exchange-correlation potential. The calculated energy cutoff was set at 750 eV, with a 5 × 5 × 5 K-point mesh for both MoOx and MoVOx. During structural optimization, the maximum force per atom was required to decrease below 0.05 eV Å−1. The energy convergence criterion was set at 2 × 10−5 eV per atom, and the maximum displacement convergence criterion was set at 2 × 10−3 Å.The Gibbs free energy change can be expressed as
ΔG = ΔE + ΔEZPE − TΔS + kBT × ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 × pH 10 × pH |
Convergence testing of the MoO3 crystal structure
To ensure the numerical reliability of the computational results, we conducted systematic convergence tests on the truncated energy of plane waves and the k-point grid. The tests demonstrated that when the truncated energy reached 750 eV and the k-point grid reached 5 × 5 × 5, the total energy variation consistently fell below the convergence criterion of 1 meV per atom (Fig. S19).Author contributions
F. Y. and J. N. co-supervised the project and led the collaboration efforts. L. Z., Y. L., and F. Y. conceived and designed the experiments. L. Z. and Y. L. performed material synthesis and the characterizations. S. Z. and J. N. performed theoretical calculations. Y. L. wrote the manuscript and L. Z., Y. L., F. Y., S. Z., J. N. revised the manuscript. All authors analysed the data, discussed the results and approved the manuscript.Conflicts of interest
The authors declare no competing financial interest.Data availability
All data have been included in the main text and supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc07071f.Acknowledgements
The authors gratefully acknowledge the Core Research Facilities of Southern University of Science and Technology for characterization. This work was financially supported by the National Natural Science Foundation of China (22475093, 92461307, and 22222504), National Key Research and Development Program of China (2021YFA0717400), Shenzhen Basic Research Project (JCYJ20250604144531041), and State Key Laboratory of Advanced Fiber Materials (Donghua University) (KF2504). Y. B. Li acknowledges the financial support from the Shenzhen Basic Research Project (JCYJ20220530115215035) and China Postdoctoral Science Foundation (2022M711484).References
- Y. X. C. Xia, P. Zhu, L. Fan and H. Wang, Science, 2019, 366, 226–231 CrossRef PubMed.
- Y. Xia, X. Zhao, C. Xia, Z. Y. Wu, P. Zhu, J. Y. Kim, X. Bai, G. Gao, Y. Hu, J. Zhong, Y. Liu and H. Wang, Nat. Commun., 2021, 12, 4255 CrossRef PubMed.
- H. Sheng, E. D. Hermes, X. Yang, D. Ying, A. N. Janes, W. Li, J. R. Schmidt and S. Jin, ACS Catal., 2019, 9, 8433–8442 Search PubMed.
- P. Cao, X. Quan, X. Nie, K. Zhao, Y. Liu, S. Chen, H. Yu and J. G. Chen, Nat. Commun., 2023, 14, 172 Search PubMed.
- B. Q. Li, C. X. Zhao, J. N. Liu and Q. Zhang, Adv. Mater., 2019, 31, 1808173 Search PubMed.
- J. Gao, H. B. Yang, X. Huang, S. F. Hung, W. Cai, C. Jia, S. Miao, H. M. Chen, X. Yang, Y. Huang, T. Zhang and B. Liu, Chem, 2020, 6, 658–674 CAS.
- Y. R. Zheng, S. Hu, X. L. Zhang, H. Ju, Z. Wang, P. J. Tan, R. Wu, F. Y. Gao, T. Zhuang, X. Zheng, J. Zhu, M. R. Gao and S. H. Yu, Adv. Mater., 2022, 34, 2205414 Search PubMed.
- H. W. Kim, M. B. Ross, N. Kornienko, L. Zhang, J. Guo, P. Yang and B. D. McCloskey, Nat. Catal., 2018, 1, 282–290 Search PubMed.
- G.-F. Han, F. Li, W. Zou, M. Karamad, J.-P. Jeon, S.-W. Kim, S.-J. Kim, Y. Bu, Z. Fu, Y. Lu, S. Siahrostami and J.-B. Baek, Nat. Commun., 2020, 11, 2209 Search PubMed.
- M. Wang, N. Zhang, Y. Feng, Z. Hu, Q. Shao and X. Huang, Angew. Chem., Int. Ed., 2020, 59, 14373–14377 Search PubMed.
- C. Xia, J. Y. Kim and H. Wang, Nat. Catal., 2020, 3, 605–607 CrossRef CAS.
- G. Xia, Y. Tian, X. Yin, W. Yuan, X. Wu, Z. Yang, G. Yu, Y. Wang and M. Wu, Appl. Catal., B, 2021, 299, 120655 Search PubMed.
- N. Wang, S. Ma, P. Zuo, J. Duan and B. Hou, Adv. Sci., 2021, 8, 2100076 CrossRef CAS PubMed.
- J. Tang, T. Zhao, D. Solanki, X. Miao, W. Zhou and S. Hu, Joule, 2021, 5, 1432–1461 Search PubMed.
- Y. Zheng, X. Xu, J. Chen and Q. Wang, Appl. Catal., B, 2021, 285, 119788 CrossRef CAS.
- Y. J. Sa, J. H. Kim and S. H. Joo, Angew. Chem., Int. Ed., 2019, 58, 1100–1105 CrossRef CAS PubMed.
- R. Gao, L. Pan, Z. Li, C. Shi, Y. Yao, X. Zhang and J. J. Zou, Adv. Funct. Mater., 2020, 30, 1910539 CrossRef CAS.
- K. H. Wu, D. Wang, X. Lu, X. Zhang, Z. Xie, Y. Liu, B. J. Su, J. M. Chen, D. S. Su, W. Qi and S. Guo, Chem, 2020, 6, 1443–1458 Search PubMed.
- Y. Wang, G. I. N. Waterhouse, L. Shang and T. Zhang, Adv. Energy Mater., 2021, 11, 2003323 Search PubMed.
- S. Siahrostami, S. J. Villegas, A. H. Bagherzadeh Mostaghimi, S. Back, A. B. Farimani, H. Wang, K. A. Persson and J. Montoya, ACS Catal., 2020, 10, 7495–7511 Search PubMed.
- R. Shen, W. Chen, Q. Peng, S. Lu, L. Zheng, X. Cao, Y. Wang, W. Zhu, J. Zhang, Z. Zhuang, C. Chen, D. Wang and Y. Li, Chem, 2019, 5, 2099–2110 CAS.
- X. Zhang, X. Zhao, P. Zhu, Z. Adler, Z. Y. Wu, Y. Liu and H. Wang, Nat. Commun., 2022, 13, 2880 Search PubMed.
- Y. Wang, Y. Zhou, Y. Feng and X. Y. Yu, Adv. Funct. Mater., 2022, 32, 2110734 CrossRef CAS.
- F. Ma, S. Wang, X. Liang, C. Wang, F. Tong, Z. Wang, P. Wang, Y. Liu, Y. Dai, Z. Zheng and B. Huang, Appl. Catal., B, 2020, 279, 119371 CrossRef CAS.
- K. Jiang, S. Back, A. J. Akey, C. Xia, Y. Hu, W. Liang, D. Schaak, E. Stavitski, J. K. Nørskov, S. Siahrostami and H. Wang, Nat. Commun., 2019, 10, 3997 CrossRef PubMed.
- X. Guo, S. Lin, J. Gu, S. Zhang, Z. Chen and S. Huang, ACS Catal., 2019, 9, 11042–11054 CrossRef CAS.
- E. Pizzutilo, S. J. Freakley, S. Cherevko, S. Venkatesan, G. J. Hutchings, C. H. Liebscher, G. Dehm and K. J. J. Mayrhofer, ACS Catal., 2017, 7, 5699–5705 CrossRef CAS.
- X. Zhao, Q. Yin, X. Mao, C. Cheng, L. Zhang, L. Wang, T. F. Liu, Y. Li and Y. Li, Nat. Commun., 2022, 13, 2721 CrossRef CAS PubMed.
- L. Han, Y. Sun, S. Li, C. Cheng, C. E. Halbig, P. Feicht, J. L. Hübner, P. Strasser and S. Eigler, ACS Catal., 2019, 9, 1283–1288 CrossRef CAS.
- S. Xu, R. Lu, K. Sun, J. Tang, Y. Cen, L. Luo, Z. Wang, S. Tian and X. Sun, Adv. Sci., 2022, 9, e2201421 Search PubMed.
- D. Iglesias, A. Giuliani, M. Melchionna, S. Marchesan, A. Criado, L. Nasi, M. Bevilacqua, C. Tavagnacco, F. Vizza, M. Prato and P. Fornasiero, Chem, 2018, 4, 106–123 CAS.
- Z. Lu, G. Chen, S. Siahrostami, Z. Chen, K. Liu, J. Xie, L. Liao, T. Wu, D. Lin, Y. Liu, T. F. Jaramillo, J. K. Nørskov and Y. Cui, Nat. Catal., 2018, 1, 156–162 CrossRef CAS.
- Y. Sun, L. Silvioli, N. R. Sahraie, W. Ju, J. Li, A. Zitolo, S. Li, A. Bagger, L. Arnarson, X. Wang, T. Moeller, D. Bernsmeier, J. Rossmeisl, F. Jaouen and P. Strasser, J. Am. Chem. Soc., 2019, 141, 12372–12381 CrossRef CAS PubMed.
- Y. Wang, R. Shi, L. Shang, G. I. N. Waterhouse, J. Zhao, Q. Zhang, L. Gu and T. Zhang, Angew. Chem., Int. Ed., 2020, 59, 13057–13062 CrossRef CAS PubMed.
- C. Xiao, L. Cheng, Y. Zhu, G. Wang, L. Chen, Y. Wang, R. Chen, Y. Li and C. Li, Angew. Chem., Int. Ed., 2022, 61, e202206544 Search PubMed.
- Y. Tian, M. Li, Z. Wu, Q. Sun, D. Yuan, B. Johannessen, L. Xu, Y. Wang, Y. Dou, H. Zhao and S. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202212296 Search PubMed.
- X. Li, S. Tang, S. Dou, H. J. Fan, T. S. Choksi and X. Wang, Adv. Mater., 2022, 34, e2104891 CrossRef PubMed.
- Y. Jia, Z. Xue, J. Yang, Q. Liu, J. Xian, Y. Zhong, Y. Sun, X. Zhang, Q. Liu, D. Yao and G. Li, Angew. Chem., Int. Ed., 2022, 61, e202110838 CrossRef CAS PubMed.
- Q. Hu, S. Chen, T. Wagberg, H. Zhou, S. Li, Y. Li, Y. Tan, W. Hu, Y. Ding and X. B. Han, Angew. Chem., Int. Ed., 2023, 62, e202303290 Search PubMed.
- X. Zhao, F. Wang, X. Kong, R. Fang and Y. Li, Nat. Commun., 2022, 13, 2591 CrossRef CAS PubMed.
- L. Wang, J. Diao, M. Peng, Y. Chen, X. Cai, Y. Deng, F. Huang, X. Qin, D. Xiao, Z. Jiang, N. Wang, T. Sun, X. Wen, H. Liu and D. Ma, ACS Catal., 2021, 11, 11469–11477 CrossRef CAS.
- J. Zhang, M. Wang, T. Wan, H. Shi, A. Lv, W. Xiao and S. Jiao, Adv. Mater., 2022, 34, e2206960 CrossRef PubMed.
- Y. Wu, Y. Zhao, P. Zhai, C. Wang, J. Gao, L. Sun and J. Hou, Adv. Mater., 2022, 34, e2202523 Search PubMed.
- S. S. Wang and G. Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- Y. Liu, X. Wu, Z. Li, J. Zhang, S.-X. Liu, S. Liu, L. Gu, L. R. Zheng, J. Li, D. Wang and Y. Li, Nat. Commun., 2021, 12, 4205 CrossRef CAS PubMed.
- P. Li, X. Duan, Y. Kuang, Y. Li, G. Zhang, W. Liu and X. Sun, Adv. Energy Mater., 2018, 8, 1703341 CrossRef.
- T. Zhao, X. Shen, Y. Wang, R. K. Hocking, Y. Li, C. Rong, K. Dastafkan, Z. Su and C. Zhao, Adv. Funct. Mater., 2021, 31, 2100614 Search PubMed.
- L. Wu, S. Li, L. Li, H. Zhang, L. Tao, X. Geng, H. Yang, W. Zhou, C. Sun, D. Ju and B. An, Appl. Catal., B, 2023, 324, 122250 CrossRef CAS.
- D. Zhou and B.-H. Han, Adv. Funct. Mater., 2010, 20, 2717–2722 CrossRef CAS.
- Q. Chang, P. Zhang, A. H. B. Mostaghimi, X. Zhao, S. R. Denny, J. H. Lee, H. Gao, Y. Zhang, H. L. Xin, S. Siahrostami, J. G. Chen and Z. Chen, Nat. Commun., 2020, 11, 2178 CrossRef CAS PubMed.
- M. Yang, W. Song, C. Chen, X. Yang, Z. Zhuang, H. Zhang, F. Wang and L. Yu, Adv. Mater., 2025, 37, 2416401 CrossRef CAS PubMed.
- H. Huang, M. Sun, K. Chen, Y. Che, X. Tang, Z. Li, K. Nie, S. Qian, J. Fang, H. Wang, Y. Wu, Q. Hu, Y. Wang, X. Sun, J. He, Y.-X. Zhang, Z. Zhuang, L. Zhang and Z. Niu, Angew. Chem., Int. Ed., 2025, 64, e202511844 CrossRef CAS PubMed.
- C. Tang, Y. Jiao, B. Shi, J.-N. Liu, Z. Xie, X. Chen, Q. Zhang and S.-Z. Qiao, Angew. Chem., Int. Ed., 2020, 59, 9171–9176 CrossRef CAS PubMed.
- F. Xia, B. Li, Y. Liu, Y. Liu, S. Gao, K. Lu, J. Kaelin, R. Wang, T. J. Marks and Y. Cheng, Adv. Funct. Mater., 2021, 31, 2104716 CrossRef CAS.
- Y. Yao, Y. Zhu, C. Pan, C. Wang, S. Hu, W. Xiao, X. Chi, Y. Fang, J. Yang, H. Deng, S. Xiao, J. Li, Z. Luo and Y. Guo, J. Am. Chem. Soc., 2021, 143, 8720–8730 Search PubMed.
- R. Liu, K. Cao, A. H. Clark, P. Lu, M. Anjass, J. Biskupek, U. Kaiser, G. Zhang and C. Streb, Chem. Sci., 2019, 11, 1043–1051 Search PubMed.
- H. Liu, Y. Zhang, L. Zhang, X. Mu, L. Zhang, S. Zhu, K. Wang, B. Yu, Y. Jiang, J. Zhou and F. Yang, J. Am. Chem. Soc., 2024, 146, 20193–20204 Search PubMed.
- F. Yang, H. Zhao, W. Wang, L. Wang, L. Zhang, T. Liu, J. Sheng, S. Zhu, D. He, L. Lin, J. He, R. Wang and Y. Li, Chem. Sci., 2021, 12, 12651–12660 Search PubMed.
- X.-R. Wang, J.-Y. Liu, Z.-W. Liu, W.-C. Wang, J. Luo, X.-P. Han, X.-W. Du, S.-Z. Qiao and J. Yang, Adv. Mater., 2018, 30, 1800005 Search PubMed.
- C. Cai, M. Wang, S. Han, Q. Wang, Q. Zhang, Y. Zhu, X. Yang, D. Wu, X. Zu, G. E. Sterbinsky, Z. Feng and M. Gu, ACS Catal., 2020, 11, 123–130 Search PubMed.
- G. A. Tsigdinos and C. J. Hallada, Inorg. Chem., 1968, 7, 437–441 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |