
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLow-oxidation state dirhodium complex produced by four-electron reduction of dirhodium(II) complex supported by a flexible macrocyclic ligand
Liping
Yan
a,
Takumi
Moriyama
b,
Yutaro
Akita
b,
Satoshi
Muratsugu
 bc,
Mizuki
Tada
bcd,
Makoto
Yamashita
*a and
Yuma
Morimoto
*a
bc,
Mizuki
Tada
bcd,
Makoto
Yamashita
*a and
Yuma
Morimoto
*a
aDepartment of Chemistry, School of Science, Institute of Science Tokyo, Ookayama, Meguro-ku, Tokyo 152-8551, Japan. E-mail: yamashita.m.6dbb@m.isct.ac.jp; yuma.morimoto@chem.sci.isct.ac.jp
bDepartment of Chemistry, Graduate School of Science, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Aichi 464-8602, Japan
cIntegrated Research Consortium on Chemical Sciences (IRCCS), Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Aichi 464-8602, Japan
dResearch Center for Materials Science (RCMS), Graduate School of Science, Nagoya Unicersity, Furo-cho, Chikusa-ku, Nagoya, Aichi 464-8602, Japan
First published on 9th December 2025
Abstract
Dinuclear rhodium complexes represent a distinct class of transition-metal compounds, exhibiting unique reactivity enabled by close metal–metal proximity. However, despite extensive investigations, access to highly reduced Rh2 complexes has remained a challenge due to their intrinsic instability. Herein, we report the synthesis and characterization of a series of dirhodium complexes in the formal oxidation states of RhII2, RhI2, and Rh02, supported by a flexible macrocyclic bis(pyridinediimine) (PDI2) ligand. Single-crystal X-ray diffraction analysis revealed the crystal structure of the Rh2 complexes. The PDI2 ligand system flexibly adapts both structure and electronics across redox states, facilitating reversible multielectron interconversion. Structural analysis, UV-vis-NIR spectroscopy, Rh L3-edge absorption spectroscopy, and DFT calculations collectively demonstrate that the RhI2 and Rh02 complexes are stabilized by delocalization of electron density onto the redox-active PDI2 ligand. Moreover, molecular orbital analysis indicates that the Rh02 complex features a Rh–Rh bond mediated by 5p orbital interactions. This work offers fundamental insights into the electronic structure and bonding of dirhodium complexes in low oxidation states and expands the design principles for redox-active bimetallic systems.
Introduction
Dinuclear metal complexes have attracted considerable attention due to their distinct reactivity compared to mononuclear complexes. To realize such systems, dinucleating ligands have been developed that hold metal centres in fixed and well-defined proximity.1,2 These ligands enable the construction of bimetallic structures that serve as minimal models for metal–metal cooperation, providing valuable platforms for investigating mechanisms involving multiple metal centres. Generally, metal–metal bonding in these complexes leads to a unique reactivity that is inaccessible to mononuclear systems, through concerted multi-electron redox processes and multiple orbital interactions. A representative example is the dinickel-catalysed [4 + 1] cycloaddition reaction of vinylidenes and dienes, where catalytic activity arises specifically from cooperative substrate activation by the two nickel centres.3Among dinuclear metal complexes, RhII2 complexes have been extensively studied due to their characteristic electronic structure and catalytic reactivities. Paddlewheel-type dirhodium(II) complexes supported by bridging carboxylate ligands are prototypical examples, featuring low-lying σ* orbitals of the Rh–Rh bond, which facilitate efficient group–transfer reactions.4–6 Although the electrochemical properties of RhII2 complexes have been extensively studied, the isolation of their oxidation or reduction products remains quite rare.7–9 Doyle and co-workers reported the oxidation of RhII2 complex supported by four caprolactamate ligands, affording a (RhII–Ph)2 complex that retains a paddlewheel-type framework but lacks a formal Rh–Rh bond (Fig. 1A).7 This system represents the first crystallographically characterized RhII2 complex, and illustrates how ligand exchange can alter metal–metal interaction and its redox behaviour. Nitrogen-ligated dirhodium(II) complexes represent rare examples that undergo redox cycling between RhII2 and RhI2 states under electrochemical conditions, although the RhI2 state was not characterized spectroscopically and structurally (Fig. 1B).10 A common feature of precedent dinuclear Rh complexes in Fig. 1A and B is their rigid coordination environment, which controls both the oxidation state and the reactivity of the complexes. In other words, such rigidity often limits the accessible range of oxidation states for the Rh2 core. The oxidation state of a Rh complex is strongly influenced by its coordination geometry. Thus, flexible ligand frameworks have the potential to expand the accessible range of oxidation states, thereby enabling more diverse reactivity patterns and catalytic transformations.
 | ||
| Fig. 1 Dirhodium systems assembled with dinucleating ligands. | ||
The isolation and characterization of the further reduced state, Rh02 remain a challenge. In 1980, Kubiak and Eisenberg reported a dinuclear Rh0 complex [Rh2(CO)2(dpm)2] (dpm = bis(diphenylphosphino)methane). Although its low solubility and high reactivity hindered detailed structural and spectroscopic characterization, the authors proposed that it contains a Rh0–Rh0 bonded dinuclear core (Fig. 1C).11 This Rh02 complex exhibits high catalytic activity in the water–gas shift reaction. This study highlights both the challenges associated with characterizing highly reactive Rh02 species and the importance of constructing dinuclear cores to achieve desirable catalytic functions. The Nakajima and Tanase group utilized a tetraphosphine ligand to achieve a redox cycle involving a RhI2 complex and a mixed-valent RhI–RhIII complex with a change in the Rh–Rh distance from 2.67 to 3.82 depending on their oxidation state and coordination geometry.12 These studies indicate that the flexibility of the methylene linker between phosphorus donor atoms supports geometrical adaptation upon changes in the oxidation state of the Rh2 core.
The pyridine-diimine (PDI) ligand is well known for its strong coordination ability and high electron-accepting character, which enable it to stabilize metal centres in low oxidation states.13–15 This ligand motif has also been employed to support dinuclear metal complexes by tethering two metal centres through linker units.16,17 The Tomson group has demonstrated that the PDI2 ligand,† a macrocyclic ligand composed of two PDI units connected by propylene linkers, is effective in stabilizing dinuclear metal complexes. They have synthesized dinuclear complexes of iron, cobalt, and nickel, and reported on the distinctive reactivity associated with dinuclear metal systems.18–21 Notably, they successfully isolated a formal Co02 complex by leveraging the highly electron-accepting nature of PDI2 ligand.22 In these complexes, the linker connecting the PDI units appropriately controls the metal–metal distance, and the high electron-accepting ability of the PDI moieties is also considered to contribute significantly to the stabilization of the dinuclear structures. However, to our knowledge, PDI2-type ligands have not been applied to 4d transition metals in catalytically active or low-valent complexes, although related Ag(I) complexes have been reported.23 Using the PDI2 ligand system, we herein report the synthesis of the first crystallographically characterized formal Rh02 complex and its RhI2 and RhII2 precursors, along with their NMR and UV-vis-NIR spectroscopic information, which are supported by DFT calculations (Fig. 1D). Furthermore, we have employed Rh L3-edge XANES to directly prove the electronic structure of the Rh complexes, enabling us to discuss electron-sharing interactions between the Rh centres and the electronically flexible ligands, PDI2, hydride and ethylene.
Results and discussion
Synthesis of dinuclear Rh complexes
A treatment of Sr2+-coordinated PDI2 (ref. 24) with two equivalents of [Rh(C2H4)2Cl]2 in THF promptly generated dark-red precipitates of the cationic dinuclear Rh trichloride complex [RhII2(µ-Cl)Cl2(PDI2)](OTf) (1) (72% yield, Scheme 1).‡ The formation of 1 was confirmed by single-crystal X-ray crystallography (Fig. 2a). The crystal structure of 1 showed a dirhodium core with a bridging chloride, two terminal chlorides, and the macrocyclic ligand in a folded geometry, where two pyridine units stacked (Fig. S26 and Table S2 in SI). Each Rh centre in 1 is six-coordinate, despite deviations from the ideal right angles due to the formation of rigid Rh2Cl three-membered rings. The interatomic Rh–Rh distance of 2.5804(5) Å falls within the typical range observed for RhII–RhII complexes (2.3–2.6 Å) with a covalent Rh–Rh bond, furnishing stable 18 e− electronic structures on each Rh atom.9 The 1H NMR spectrum of 1 in acetonitrile exhibited diamagnetic signals in C2v symmetry as found in its crystal structure (Fig. S1). By treating 1 with AgOTf, all the chlorides were replaced with CH3CN to generate dinuclear complex [RhII2(NCCH3)4(PDI2)](OTf)4 (2) (Scheme 1). Single crystal X-ray diffraction analysis showed that the exchange of the ligand elongated the Rh–Rh bond length [2.7019(4) Å] of 2 compared to that of 1 (Fig. 2b) and provided a nearly ideal octahedral coordination environment with the bond angles around the Rh centre being close to 90° (Fig. 2b and Table S2). The coordination environment around each Rh centre in 2 is similar to that of a paddle-wheel Rh2 complex,25 which is considered to be in the RhII oxidation state, being consistent with its diamagnetic behavior observed in the 1H NMR spectrum (Fig. S7).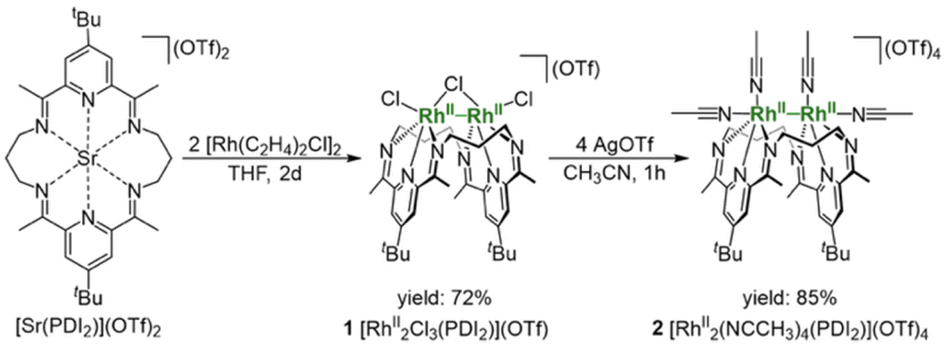 | ||
| Scheme 1 Synthesis of [RhII2Cl3(PDI2)](OTf) (1) and [RhII2(NCCH3)4(PDI2)](OTf)4 (2). | ||
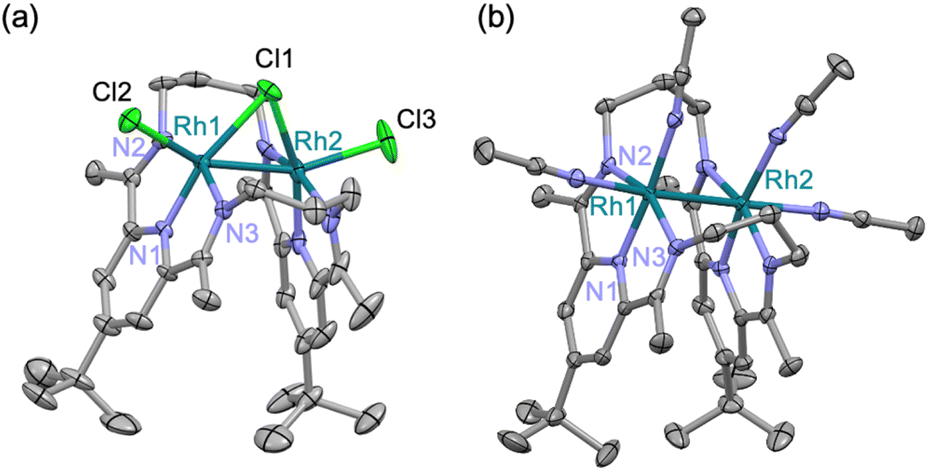 | ||
| Fig. 2 ORTEP diagrams of crystal structure of 1 and 2. Thermal ellipsoids are set at the 50% probability. Hydrogen atoms and OTf are omitted for clarity. Selected bond length of 1: Rh1–Rh2: 2.5804(5) Å; 2: Rh1–Rh2: 2.7019(4) Å. | ||
Treatment of 1 with 4 equivalents of LiHBEt3 generated a black di(µ-hydrido)dirhodium complex 3 (Scheme 2). In the 1H NMR spectrum of 3, a triplet signal corresponding to two equivalent hydrides was observed at −9.80 ppm (1JRhH = 28 Hz) (Fig. S11). Black needle-shaped crystals grown at −30 °C were analysed by a single-crystal X-ray diffraction analysis (Fig. 3a). The ligand adopted a planar geometry with two pyridine units being nearly coplanar and oriented toward each other, accompanied by a drastic elongation of the Rh–Rh distance to 2.8264(8) Å in comparison with those of 1 and 2. The structural change is attributed to the reduction of RhII to RhII, wherein the Rh–Rh anti-bonding orbital (σ*) is filled with two electrons, resulting in the cleavage of the Rh–Rh bond. The hydride ligands in 3 were placed in the differential Fourier map. The generated RhI centre is stabilized by adopting a trigonal bipyramidal (TBP) structure with two hydride ligands on the equatorial positions. The di(µ-hydrido) bridged structure is consistent to the triplet signal observed in 1H NMR as mentioned above, although the position of two µ-hydride cannot be discussed in detail due to the quality of diffraction data. This trigonal bipyramidal structure is in contrast to that of [Co2(µ-H)2(PDI2)] has two-hydride ligands occupied apical and equatorial positions of the square-pyramidal cobalt centre (Fig. S31).22 Notably, 3 was oxidized by a proton donor to regenerate RhII2 (Scheme 2); treatment of 3 with HCl in THF regenerated 1 as red precipitates with liberation of H2, as confirmed by 1H NMR spectrum (Fig. S23).
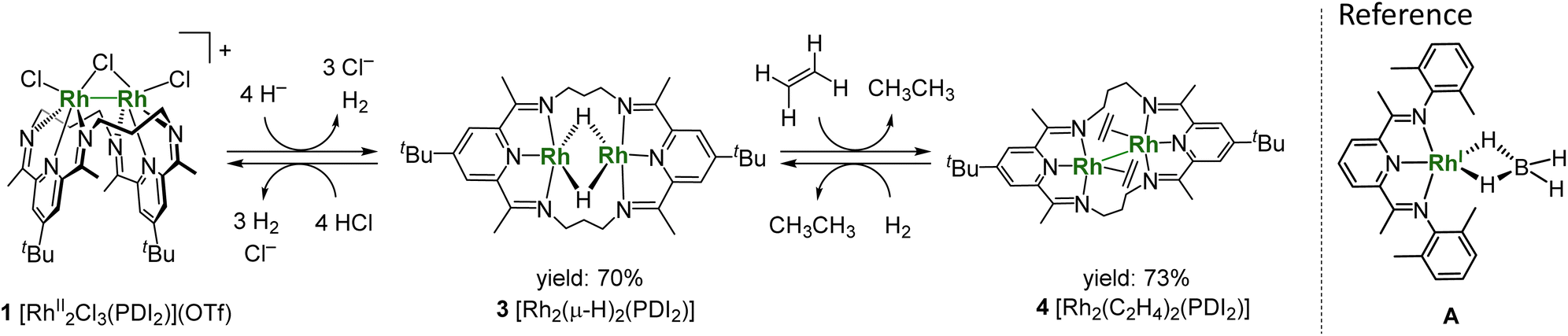 | ||
| Scheme 2 Interconversion between 1, 2 and 3, and structure for A. | ||
 | ||
| Fig. 3 ORTEP diagrams of crystal structure of (a) 3 and (b) 4. Thermal ellipsoids are set at the 50% probability. Hydrogen atoms are omitted for clarity. Selected bond length of 3: Rh1–Rh2: 2.8264(8) Å; (c) bond lengths (Å) of Rh–C3–C4 core structure for 4 (units omitted in the figure). | ||
Complex 3 hydrogenated ethylene under atmospheric pressure to give ethane and is converted to Rh02(H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)2(PDI2) (4), as confirmed by the 1H NMR spectrum of the resulting solution. It should be noted that a mononuclear Rh(XylPDI)BH4 complex A [XylPDI = 1,1′-(pyridine-2,6-diyl)bis(N-(2,6-dimethylphenyl)-ethane-1-imine)] (Scheme 2) shows no reactivity toward ethylene,26 in contrast to the bridging hydride reactivity observed for 3. Single crystals of 4 suitable for X-ray diffraction were successfully obtained from the resulting solution. The crystal structure revealed that the complex carries no counter ion, indicating that both rhodium centres are formally in the zero-oxidation state. Each Rh centre adopts a square planar geometry, and the molecule possesses an inversion centre located between the two Rh atoms. The Rh–Rh distance is slightly elongated to 2.8439(6) Å compared to that in 3. Notably, this Rh–Rh distance is substantially shorter than the Co–Co distance (2.957 Å) in the formal Co02 complex,22 despite the larger atomic radius of Rh, indicating the presence of Rh–Rh interaction. The C3–C4 distance of ethylene ligand [1.416(6) Å] is elongated from the free ethylene (1.331 Å) due to the strong backdonation from the Rh centres (Fig. 3c). Among the 137 Rh–ethylene complexes in the Cambridge Crystallographic Data Centre (CSD) dataset (mean = 1.382 Å, standard deviation = 0.045 Å), the C–C bond distance obtained in this study (1.416 Å) falls within the longer region of the distribution, but slightly shorter than the average value of metallacyclopropane C–C bonds (25 data points, mean = 1.441 Å, standard deviation = 0.057 Å) (Fig. S32).27–30 The Rh–C3–C4 triangle was slightly distorted with two different Rh–C distances of 2.105(4) and 2.162(4) Å, which are falling into common values (2.05–2.19 Å) for ethylene ligand on Rh,31–36 due to the steric repulsion between the C4 atom with the other Rh centre (C4–Rh2: 3.036 Å). The ligand backbone adopts an offset geometry, where the two pyridines are non-coplanar but in parallel, reflecting the structural flexibility of the PDI2 ligand. The 1H NMR spectrum of 4 shows two sets of signals, which were attributed to two conformers of 4, and their ratio was reversibly changed with varying the temperature (Fig. S21), indicating the presence of conformational equilibrium (Fig. S22). From a solution of 4, another conformer of 4, having a folded PDI2 ligand, was also isolated and structurally identified, although the data quality is not sufficiently good (Fig. S30). The reverse reaction for the formation of 4 was confirmed by treating 4 with H2, which cleanly regenerated 3 without any byproducts (Fig. S24). The occurrence of oxidative addition highlights the reactivity of 4 as a formal Rh02 species. The alkene hydrogenation activity was also demonstrated with a small excess of cyclohexane (20 equivs.). Introduction of hydrogen gas (1.0 atm) into the THF solution of 3 at room temperature produced cyclohexane (TON = 17) as a sole product after one week. The electronic structure of 4 is discussed in detail in the following sections.
CH2)2(PDI2) (4), as confirmed by the 1H NMR spectrum of the resulting solution. It should be noted that a mononuclear Rh(XylPDI)BH4 complex A [XylPDI = 1,1′-(pyridine-2,6-diyl)bis(N-(2,6-dimethylphenyl)-ethane-1-imine)] (Scheme 2) shows no reactivity toward ethylene,26 in contrast to the bridging hydride reactivity observed for 3. Single crystals of 4 suitable for X-ray diffraction were successfully obtained from the resulting solution. The crystal structure revealed that the complex carries no counter ion, indicating that both rhodium centres are formally in the zero-oxidation state. Each Rh centre adopts a square planar geometry, and the molecule possesses an inversion centre located between the two Rh atoms. The Rh–Rh distance is slightly elongated to 2.8439(6) Å compared to that in 3. Notably, this Rh–Rh distance is substantially shorter than the Co–Co distance (2.957 Å) in the formal Co02 complex,22 despite the larger atomic radius of Rh, indicating the presence of Rh–Rh interaction. The C3–C4 distance of ethylene ligand [1.416(6) Å] is elongated from the free ethylene (1.331 Å) due to the strong backdonation from the Rh centres (Fig. 3c). Among the 137 Rh–ethylene complexes in the Cambridge Crystallographic Data Centre (CSD) dataset (mean = 1.382 Å, standard deviation = 0.045 Å), the C–C bond distance obtained in this study (1.416 Å) falls within the longer region of the distribution, but slightly shorter than the average value of metallacyclopropane C–C bonds (25 data points, mean = 1.441 Å, standard deviation = 0.057 Å) (Fig. S32).27–30 The Rh–C3–C4 triangle was slightly distorted with two different Rh–C distances of 2.105(4) and 2.162(4) Å, which are falling into common values (2.05–2.19 Å) for ethylene ligand on Rh,31–36 due to the steric repulsion between the C4 atom with the other Rh centre (C4–Rh2: 3.036 Å). The ligand backbone adopts an offset geometry, where the two pyridines are non-coplanar but in parallel, reflecting the structural flexibility of the PDI2 ligand. The 1H NMR spectrum of 4 shows two sets of signals, which were attributed to two conformers of 4, and their ratio was reversibly changed with varying the temperature (Fig. S21), indicating the presence of conformational equilibrium (Fig. S22). From a solution of 4, another conformer of 4, having a folded PDI2 ligand, was also isolated and structurally identified, although the data quality is not sufficiently good (Fig. S30). The reverse reaction for the formation of 4 was confirmed by treating 4 with H2, which cleanly regenerated 3 without any byproducts (Fig. S24). The occurrence of oxidative addition highlights the reactivity of 4 as a formal Rh02 species. The alkene hydrogenation activity was also demonstrated with a small excess of cyclohexane (20 equivs.). Introduction of hydrogen gas (1.0 atm) into the THF solution of 3 at room temperature produced cyclohexane (TON = 17) as a sole product after one week. The electronic structure of 4 is discussed in detail in the following sections.
Electronic structure of Rh2 complexes
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1) (Fig. 4, black trace), assigned to an intra-Rh2 core σ → σ* transition based on time-dependent density functional theory (TD-DFT) calculations (Fig. S42 and Table S4). This type of transition is well known in RhII2 complexes, as exemplified by [RhII2(O2CCH3)4] which exhibits a sharp band at 221 nm.37 As well, absorption band around 460 nm (ε = 10400 M−1 cm−1) was assigned to a metal–metal-to-ligand charge transfer (MMLCT) transition from the Rh2 core to the PDI2 ligand. Complex 2 displays two distinct absorption bands: σ → σ*(Rh–Rh) at 269 nm (ε = 29200 M−1 cm−1) and π* → σ* (Rh–Rh) at 293 nm (ε = 24700 M−1 cm−1) (Fig. 4a, blue trace) (Fig. S43 and Table S5). The MMLCT band at 380 (ε = 6700 M−1 cm−1) and 450 nm (ε = 10500 M−1 cm−1) became weaker upon the ligand exchange.
000 M−1 cm−1) (Fig. 4, black trace), assigned to an intra-Rh2 core σ → σ* transition based on time-dependent density functional theory (TD-DFT) calculations (Fig. S42 and Table S4). This type of transition is well known in RhII2 complexes, as exemplified by [RhII2(O2CCH3)4] which exhibits a sharp band at 221 nm.37 As well, absorption band around 460 nm (ε = 10400 M−1 cm−1) was assigned to a metal–metal-to-ligand charge transfer (MMLCT) transition from the Rh2 core to the PDI2 ligand. Complex 2 displays two distinct absorption bands: σ → σ*(Rh–Rh) at 269 nm (ε = 29200 M−1 cm−1) and π* → σ* (Rh–Rh) at 293 nm (ε = 24700 M−1 cm−1) (Fig. 4a, blue trace) (Fig. S43 and Table S5). The MMLCT band at 380 (ε = 6700 M−1 cm−1) and 450 nm (ε = 10500 M−1 cm−1) became weaker upon the ligand exchange.
 | ||
| Fig. 4 UV-vis-NIR absorption spectrum of (a) 1 (black) and 2 (blue) (25.0 µM) measured in MeCN, and (b) 3 (red) and A (black) (100 µM) measured in THF; 4 (blue) (100 µM) measured in C6H6UV. | ||
Dirhodium dihydride 3 exhibits an intense absorption band with a maximum at 1180 nm (ε = 12000 M−1 cm−1). This characteristic absorption band is assigned to the ligand-to-ligand transition (LLT), which is caused by the migration of electron density from the RhI2 centre to the PDI2 ligand, forming a formally reduced ligand PDI22−. Reference compound A shows corresponding LLT transition band at 680 nm (Fig. 4b, black trace), and this marked red shift observed for 3 compared to A is induced by the connection of each PDI orbitals over Rh2H2 core, which effectively compress the HOMO–LUMO energy gap (vide infra). The absorption bands of 3 at 420 nm (10900 M−1 cm−1) and A at 420 nm (3650 M−1 cm−1) are assignable to MMLCT.
The C6H6 solution of complex 4 also exhibits absorption extending into the NIR region, indicating effective orbital conjugation across the two PDI units with help of Rh2 orbital, as observed for complex 3 (vide infra). Rather complicated spectrum was interpreted taking the conformer of 4 into account under the support of TD-DFT. The absorption band at 411 nm (ε = 7500 M−1 cm−1) and 867 (ε = 4600 M−1 cm−1) nm were ascribed to the MMLCT and LLT in offset conformer of 4. The bands at 388 nm (ε = 7400 M−1 cm−1), 719 nm (ε = 4300 M−1 cm−1), and 1205 nm (ε = 1900 M−1 cm−1) were assigned to MMLCT and two modes for LLT in folded conformer of 4 (Fig. 4b, blue trace). In the folded conformer, the interaction between stacked pyridine moieties generates two absorption modes of LLT. Lowering the measurement temperature decreased the absorption at 867 nm with concomitant increase of the absorption at 719 nm (Fig. S25), further confirming the presence of conformers as observed in NMR spectra. Absorption spectra of 3 and 4 suggested the characteristic electronic structure of their ligand system, therefore, we further conducted the structural analyses on the ligand systems.
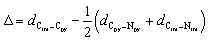 | (1) |
 | ||
| Fig. 5 (a) Averaged bond lengths of pyridine–diimine moiety. (b) Number line for the Δ value of Rh complexes. | ||
Complexes 2–4, with reference compounds A were subjected to L3-edge XANES measurements. All spectra were corrected for intensity saturation and then fitted using Lorentzian functions. Complex 2 showed a peak at 3009.39 eV, while those of 3, 4, and A observed at similar energies (3009.05, 3009.38, and 3008.69 eV, respectively, Fig. 6a). The rising edge energies were determined with the derivative of the spectra (Fig. S34 and Table S3), which are located between those of authentic samples RhIII(acac)3 (3008.69 eV) and Rh0 powder (3006.24 eV) (Fig. 6b). Complex 2 exhibited the edge energy (3007.99 eV) slightly smaller than that of RhIII(acac)3, consistent to its formal oxidation state. Edge energies of formal RhI complex 3 and A were indistinguishable (3007.28 eV) and higher than that of Rh0 powder. The absorption edge energy of 3 is consistent with the formal oxidation state, RhI2. However, structural analysis indicates that the ligand of 3 is reduced to PDI22− (vide infra). Therefore, structure and XAS spectrum of 3 indicate the bridging hydride ligands lose a considerable amount of electron, giving electronic structure of RhI2(H0)2(PDI22−). The edge energy of compound 4 lies between those of 2 and 3, indicating the actual oxidation state of 4 is somewhat higher than oxidation state, RhI2. Based on structural analysis, the ligand in 4 is assigned as PDI22−. The Rh2 core donates a significant amount of electron density to both the ethylene and PDI2 ligands, leading to an electronic structure that can be described as RhI2(C2H4)2(PDI22−) ↔ RhIII2(C2H42−)2(PDI22−), which is consistent with the elongated C–C bonds of ethylene ligands. The electronic structures of 3 and 4 were further investigated using DFT calculations.
 | ||
| Fig. 6 (a) Rh L3-edge XANES spectra of complexes 2 (green), 3 (blue), 4 (red), A (light blue) Rh0 powder (black dot) and RhIII(acac)3 (orange dot), Inset: expanded spectra for absorption edge. (b) Number line of absorption edge energies for Rh complexes obtained by deferential of spectra. | ||
 | ||
| Fig. 7 (a) The HOMO of 3 calculated with DFT at the level of UB3LYP-D3BJ/def2-SVP, (b) its schematic diagram with selected components; (c) partial Molecular graph of 3 with bond critical points (green dots), and bond paths (solid lines) calculated by AIM analysis based on electron density obtained at the UB3LYP/WTBS level of theory with D3BJ dispersion correction. | ||
Quantum theory of atoms in molecule (QTAIM) analysis indicates the absence of a direct Rh–Rh bond in complex 3, aligning with the expected d8 configuration of the Rh centres (Fig. 7c and S47). Taken together with the structural parameter of the ligand (Δ), L3-edge XANES data, and orbital analysis, these result support the resonance structure RhI2(H0)2(PDI22−) as the predominant contributor where the hydride (H−) ligands lose its charge, as shown in Scheme 3.41,42
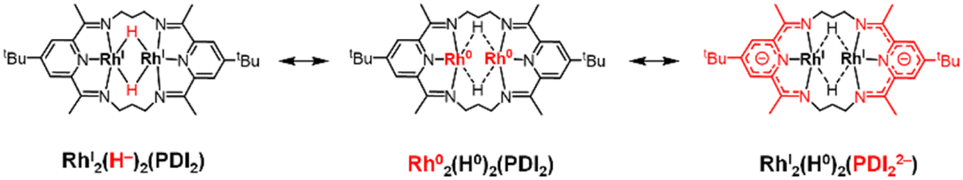 | ||
| Scheme 3 Resonance structures of 3. | ||
The HOMO of 4 is mainly composed of the ligand as in of 3. However, in contrast to 3, the HOMO of 4 does not conjugate with the Rh 4d-orbital. Other than the HOMO, eight occupied orbitals and two unoccupied orbitals based on the Rh d-orbital (Fig. S39), indicating a 4d8 configuration for each Rh centre as well as 3. This supports the presence of RhI2(PDI22−) resonance structures as a major contributor, which is consistent with its Δ parameter value and L3-edge XANES data (Scheme 4). Interestingly, although the HOMO of 4 remains primarily ligand-centred, it exhibits a notable 7.7% contribution from Rh 5p orbital, located between the two Rh centres (Fig. 8a). Participation of p orbital should be caused by high-lying dx2−y2 orbital of each Rh centre, resulted from their tetragonal geometry, and the p orbitals effectively conjugate molecular orbitals on each PDI moiety. QTAIM analyses of 4 identified a bond critical point (BCP) between the two Rh centres (Fig. 8b), in contrast to the case of 3 with no BCP between the two Rh centres. Despite lacking a direct Rh–Rh bond, complex 3 shows a slightly shorter Rh–Rh distance (0.018 Å) than complex 4, likely due to enhanced electron sharing between the Rh centres mediated by the two bridging hydride ligands. Since all Rh–Rh bonding interactions via d-orbitals are cancelled by occupation of their corresponding antibonding orbitals, this BCP is ascribed to Rh(5p)–Rh(5p) interaction (Fig. S48 and Table S12). This supports that the existence of Rh02(CH2CH2)2(PDI2) resonance structure with Rh 4d8 5p1 electronic configurations (Scheme 4).
 | ||
| Scheme 4 Resonance structures of 4. | ||
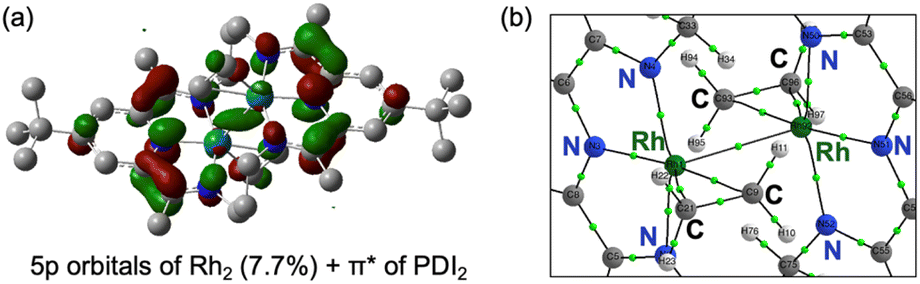 | ||
| Fig. 8 (a) The HOMO of [Rh2(C2H4)2(PDI2)] (4) calculated with DFT at the level of UB3LYP-D3BJ/def2-SVP. (b) Partial molecular graph of 4 with bond critical points (green spheres), and bond paths (solid lines) calculated by AIM analysis based on electron density obtained at the UB3LYP/WTBS level of theory with D3BJ dispersion correction. | ||
Conclusions
We successfully achieved the first isolation and characterization of a dinuclear formal Rh0 complex, stabilized by the electron-accepting ability of the PDI2 ligand. Starting from a RhII2 species, we synthesized a di(µ-hydrido) complex in the RhI2 state, and a di(ethylene) complex in the Rh02 state. These transformations represent a fully reversible four-electron redox process.These complexes were identified and characterized using UV-vis-NIR spectroscopy, 1H and 13C NMR spectroscopy, Rh L3-edge XANES, and single-crystal X-ray diffraction analysis. The PDI2 ligand functions as a structurally adaptive scaffold that responds to changes in the oxidation state of the rhodium centres by adjusting the Rh–Rh distance, Rh coordination geometries (octahedral, trigonal bipyramidal, and square planar), and orientation of PDI unit, thereby modulating energies of d-orbitals and their overlap. This adaptive coordination behaviour enables the stabilization of dinuclear Rh complexes across multiple redox states.
The non-innocent nature of the PDI unit facilitates access to the RhI2 and Rh02 oxidation states by accepting electron density. Despite substantial stabilization through electron delocalization, the formal Rh02 complex reacts with H2 to form the RhI2H2 complex with concurrent formation of CH3CH3, demonstrating its reactivity as a formal Rh02 species. Notably, theoretical calculations revealed that the formal Rh02 complex exhibits an electronic structure in which the Rh–Rh σ-bond arise from Rh 5p orbital overlap, rather than conventional 4d-based bonding.
Overall, this study demonstrates that ligand frameworks with both structural and redox flexibility can endow dinuclear complexes with electronic flexibility, offering a conceptual foundation for stabilizing highly reduced metal–metal bonded species. Ongoing studies are exploring the reactivity of this formal Rh02 complex, and the results will be reported in due course.
Author contributions
The manuscript was written through contributions of all authors. LY performed the experimental work. TM, YA, SM and MT directed the XANES work. LY, YM and MY conducted computational works. LY and YM conceptualized the research. YM and MY found the funds and administrated the project and supervised the work. LY and YM wrote the initial version of the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
CCDC 2464500 (1), 2464501 (2), 2464503 (3) and 2464504 (4) contain the supplementary crystallographic data for this paper.43a–dThe data that support the findings of this study are available in the supplementary information (SI) of this article. Supplementary information: data for this article, including chemical syntheses, NMR for 1–4 (Fig. S1–S20), X-ray crystallographic analysis data (Table S1), and selected bond distances and angles (Table S2 and Fig. S31), statistical analysis of Rh(ethylene) complex, S32 DFT calculation results (orbital analysis: Fig. S35–S41, TD-DFT: Fig. S42–S46 and Tables S4–S8, AIM analysis: Fig. S47, S48 and Tables S9–S12, NPA charges: Fig. S49). See DOI: https://doi.org/10.1039/d5sc07037f.
Acknowledgements
This research was supported by JSPS KAKENHI (grants 23K23363, 25K01785, 24H00455 and 23K26666). L.P. is grateful for financial support by the China Scholarship Council (CSC, grant no. 202308050049). Computations were carried out using resources of the Research Center for Computational Science, Okazaki, Japan, under the project number 25-IMS-C005. XAFS measurements were performed with the approval of PF-PAC (no. 2023G161 and 2023G162), and we thank Dr Yasuhiro Niwa for the support.Notes and references
- G. Li, D. Zhu, X. Wang, Z. Su and M. R. Bryce, Dinuclear Metal Complexes: Multifunctional Properties and Applications, Chem. Soc. Rev., 2020, 49, 765–838 RSC.
- Z. Freixa, P. W. N. M. van Leeuwen and P. Kalck, in Modes of Cooperative Effects in Dinuclear Complexes, Topics in Organometallic Chemistry, ed. P. Kalck, Springer, Cham, 2023, vol. 70, pp. 1–66 Search PubMed.
- Y.-Y. Zhou and C. Uyeda, Catalytic Reductive [4+1]-Cycloadditions of Vinylidenes and Dienes, Science, 2019, 363, 857–862 CrossRef CAS PubMed.
- K. Liao, S. Negretti, D. G. Musaev, J. Bacsa and H. M. Davies, Site-selective and Stereoselective Functionalization of Unactivated C–H Bonds, Nature, 2016, 533, 230–234 CrossRef CAS.
- K. P. Kornecki, J. F. Briones, V. Boyarskikh, F. Fullilove, J. Autschbach, K. E. Schrote, K. M. Lancaster, H. M. L. Davies and J. F. Berry, Direct Spectroscopic Characterization of a Transitory Dirhodium Donor–Acceptor Carbene Complex, Science, 2013, 342, 351–354 CrossRef CAS PubMed.
- A. F. Trindade, J. A. S. Coelho, C. A. M. Afonso, L. F. Veiros and P. M. P. Gois, Fine Tuning of Dirhodium(II) Complexes: Exploring the Axial Modification, ACS Catal., 2012, 2, 370–383 CrossRef CAS.
- J. M. Nichols, J. Wolf, P. Zavalij., B. Varughese and M. P. Doyle, Bis(phenyl)dirhodium(III) Caprolactamate: A Dinuclear Paddlewheel Complex with No Metal–Metal Bond, J. Am. Chem. Soc., 2007, 129, 3504–3505 CrossRef CAS PubMed.
- J. H. Xie, J. M. Nichols, C. Lubek and M. P. Doyle, Synthesis of Bis(σ-aryl)dirhodium(III) Caprolactamates by Oxidative Arylation with Arylboronic Acids, Chem. Commun., 2008, 2671–2673, 10.1039/b806283h.
- Y. Kataoka, N. Yano, M. Mikuriya and M. Handa, Paddlewheel-type Dirhodium Complexes with N,N’-Bridging Ligands, Coord. Chem. Rev., 2023, 479, 214997–215017 CrossRef CAS.
- T. J. Whittemore, A. Millet, H. J. Sayre, C. Xue, B. S. Dolinar, E. G. White, K. R. Dunbar and C. Turro, Tunable Rh2(II,II) Light Absorbers as Excited-State Electron Donors and Acceptors Accessible with Red/Near-Infrared Irradiation, J. Am. Chem. Soc., 2018, 140, 5161–5170 CrossRef CAS PubMed.
- C. P. Kubiak and R. Eisenberg, Molecular A Frames. Synthesis from Binuclear Rh(0) Precursors and Catalytic Activity in the Water Gas Shift Reaction and Alkyne Hydrogenation, J. Am. Chem. Soc., 1980, 102, 3637–3639 CrossRef CAS.
- T. Nakajima, M. Maeda, A. Matsui, M. Nishigaki, M. Kotani and T. Tanase, Unsymmetric Dinuclear RhI2 and RhIRhIII Complexes Supported by Tetraphosphine Ligands and Their Reactivity of Oxidative Protonation and Reductive Dechlorination, Inorg. Chem., 2022, 61, 1102–1117 CrossRef CAS.
- C. Römelt, T. Weyhermüller and K. Wieghardt, Structural Characteristics of Redox-active pyridine-1,6-diimine Complexes: Electronic Structures and Ligand Oxidation Levels, Coord. Chem. Rev., 2019, 380, 287–317 CrossRef.
- V. C. Gibson, C. Redshaw and G. A. Solan, Bis(imino)pyridines: Surprisingly Reactive Ligands and a Gateway to New Families of Catalysts, Chem. Rev., 2007, 107, 1745–1776 CrossRef CAS.
- Z. Flisak and W.-H. Sun, Progression of Diiminopyridines: From Single Application to Catalytic Versatility, ACS Catal., 2015, 5, 4713–4724 CrossRef CAS.
- M. G. B. Drew, J. Nelson, F. Esho, V. McKee and S. M. Nelson, Dicopper(II) Complexes of a Macrocyclic Ligand containing Single Hydroxo-, Methoxo-, or 1,1-Azido-bridges: Synthesis, Magnetic Properties, Electron Spin Resonance Spectra, and the Crystal and Molecular Structure of a µ-Hydroxo-derivative, J. Chem. Soc., Dalton Trans., 1982, 1837–1843 RSC.
- M. Stephan, W. Dammann and P. Burger, Synthesis and reactivity of dinuclear copper(I) pyridine diimine complexes, Dalton Trans., 2022, 51, 13396–13404 RSC.
- Q. Wang, S. Zhang, P. Cui, A. B. Weberg, L. M. Thierer, B. C. Manor, M. R. Gau, P. J. Carroll and N. C. Tomson, Interdependent Metal–Metal Bonding and Ligand Redox-Activity in a Series of Dinuclear Macrocyclic Complexes of Iron, Cobalt, and Nickel, Inorg. Chem., 2020, 59, 4200–4214 CrossRef CAS.
- P. Cui, Q. Wang, S. P. McCollom, B. C. Manor, P. J. Carroll and N. C. Tomson, Ring-Size-Modulated Reactivity of Putative Dicobalt-Bridging Nitrides: C–H Activation versus Phosphinimide Formation, Angew. Chem., Int. Ed., 2017, 56, 15979–15983 CrossRef CAS PubMed.
- S. Zhang, P. Cui, T. Liu, Q. Wang, T. J. Longo, L. M. Thierer, B. C. Manor, M. R. Gau, P. J. Carroll, G. C. Papaefthymiou and N. C. Tomson, N–H Bond Formation at a Diiron Bridging Nitride, Angew. Chem., Int. Ed., 2020, 59, 15215–15219 CrossRef CAS PubMed.
- T. Liu, R. P. Murphy, P. J. Carroll, M. R. Gau and N. C. Tomson, C–C σ-Bond Oxidative Addition and Hydrofunctionalization by a Macrocycle-supported Diiron Complex, J. Am. Chem. Soc., 2022, 144, 14037–14041 CrossRef CAS.
- A. Z. Spentzos, S. A. Ford, B. Muldowney, T. Liu, P. Cui, M. R. Gau, P. J. Carroll and N. C. Tomson, Reactivity Profile of a Formally Dicobalt(0) Complex Bound by a Redox-Active Macrocycle, Organometallics, 2024, 43, 1425–1437 CrossRef CAS.
- Q. Wang, M. R. Gau, B. C. Manor, P. J. Carroll and N. C. Tomson, Metallophilic and Inter-Ligand Interactions in Diargentous Compounds Bound by a Geometrically Flexible Macrocycle, Eur. J. Inorg. Chem., 2023, 26, e202300335 CrossRef CAS.
- L. M. Thierer, Q. Wang, S. H. Brooks, P. Cui, J. Qi, M. R. Gau, B. C. Manor, P. J. Carroll and N. C. Tomson, Pyridyldiimine Macrocyclic Ligands: Influences of Template Ion, Linker Length and Imine Substitution on Ligand Synthesis, Structure and Redox Properties, Polyhedron, 2021, 198, 115044–115059 CrossRef CAS.
- A. R. Barron, G. Wilkinson, M. Motevalli and M. B. Hursthouse, Synthesis of Rhodium(II) Pyrazolate Complexes: Crystal Structure of Tetra-µ-3,5-dimethylpyrazolato Dirhodium(II) Bis-acetonitrile, (Rh–Rh), Polyhedron, 1985, 4, 1131–1134 CrossRef CAS.
- S. Nückel and P. Burger, Synthetic Access to Square-Planar Terdentate Pyridine-Diimine Rhodium(I) and Iridium(I) Methyl Complexes: Successful Detour via Reactive Triflate and Methoxide Complexes, Organometallics, 2001, 20, 4345–4359 CrossRef.
- M. J. S. Dewar and G. P. Ford, Relationship between olefinic .pi. complexes and three-membered rings, J. Am. Chem. Soc., 1979, 101, 783–791 CrossRef CAS.
- P. T. Cheng and S. C. Nybur, The Crystal and Molecular Structure of Bis(triphenyphosphine)(ethylene)platinum, (PPh3)2PtC2H4, Can. J. Chem., 1971, 50, 912–916 CrossRef.
- P. T. Cheng, C. D. Cook, S. C. Nyburg and K. Y. Wan, The Molecular Structures and Proton Magnetic Resonance Spectra of Ethylene Complexes of Nickel and Platinum, Inorg. Chem., 1971, 10, 2200–2213 CrossRef.
- C. D. Cook, K. Y. Wan, U. Gelius, K. Hamrin, G. Johansson, E. Olsson, H. Siegbahn, C. Nordling and K. Siegbahn, Electron Spectroscopy of Platinum Complexes, J. Am. Chem. Soc., 1971, 93, 1904–1909 CrossRef CAS.
- M. Zhang, J. Chen, X. Wang, S. C. Zheng and X. Zhao, Tridentate Sulfoxide-N-olefin Hybrid Ligands in Rhodium-Catalyzed Asymmetric Allylic Substitution, Org. Lett., 2024, 26, 1970–1974 CrossRef CAS PubMed.
- S. Takegasa, M. M. Lee, K. Tokuhiro, R. Nakano and M. Yamashita, Rhodium-Catalyzed Acrylate Synthesis from Carbon Dioxide and Ethylene by using a Guanidine-Based Pincer Ligand: Perturbing Occupied d-Orbitals by pπ–dπ Repulsion Makes a Difference, Chem, 2022, 28, e202201870 CrossRef CAS.
- A. Johnson, C. G. Royle, C. N. Brodie, A. J. Martinez-Martinez, S. B. Duckett and A. S. Weller, η 2-Alkene Complexes of [Rh(PONOP-iPr)(L)]+ Cations (L = COD, NBD, Ethene). Intramolecular Alkene-Assisted Hydrogenation and Dihydrogen Complex [Rh(PONOP-iPr)(η-H2)]+, Inorg. Chem., 2021, 60, 13903–13912 CrossRef CAS PubMed.
- S. Gu, C. B. Musgrave, Z. M. Gehman, K. Zhang, D. A. Dickie, W. A. Goddard and T. B. Gunnoe, Rhodium and Iridium Complexes Bearing “Capping Arene” Ligands: Synthesis and Characterization, Organometallics, 2021, 40, 2808–2825 CrossRef CAS.
- E. L. Dias, M. Brookhart and P. S. White, Stable, Cationic Alkyl–Olefin Complexes of Ruthenium(II) and Rhodium(III): Effects of Ligand Geometry upon Olefin Insertion/Alkyl Migration, Organometallics, 2000, 19, 4995–5004 CrossRef CAS.
- J. A. Evsns and D. R. Russe, The Crystal Structures of Ethylene and Tetrafluoroethylene Complexes of Rhodium(I), Chem. Commun., 1971, 4, 197–198 Search PubMed.
- V. M. Miskowski, W. P. Schaefer, B. Sadeghi, B. D. Santarsiero and H. B. Gray, Polarized Electronic Spectra of Dirhodium(II) Tetraacetate, Inorg. Chem., 1984, 23, 1154–1162 CrossRef CAS.
- M. Kirchhof, K. Gugeler, F. R. Fischer, M. Nowakowski, A. Bauer, S. Alvarez-Barcia, K. Abitaev, M. Schnierle, Y. Qawasmi, W. Frey, A. Baro, D. P. Estes, T. Sottmann, M. R. Ringenberg, B. Plietker, M. Bauer, J. Kästner and S. Laschat, Experimental and Theoretical Study on the Role of Monomeric vs. Dimeric Rhodium Oxazolidinone Norbornadiene Complexes in Catalytic Asymmetric 1,2- and 1,4-Additions, Organometallics, 2020, 39, 3131–3145 CrossRef CAS.
- J. Liang, A. Levina, J. Jia, P. Kappen, C. Glover, B. Johannessen and P. A. Lay, Reactivity and Transformation of Antimetastatic and Cytotoxic Rhodium(III)-Dimethyl Sulfoxide Complexes in Biological Fluids: an XAS Speciation Study, Inorg. Chem., 2019, 58, 4880–4893 CrossRef CAS.
- Z. Majer, S. Bősze, I. Szabó, V. G. Mihucz, A. Gaál, G. Szilvágyi, G. Pepponi, F. Meirer, P. Wobrauschek, N. Szoboszlai, D. Ingerle and C. Streli, Study of Dinuclear Rh(II) Complexes of Phenylalanine Derivatives as Potential Anticancer Agents by Using X-ray Fluorescence and X-ray Absorption, Microchem. J., 2015, 120, 51–57 CrossRef CAS.
- S. Matsuishi, K. Hayashi, M. Hirano and H. Hosono, Hydride Ion as Photoelectron Donor in Microporous Crystal, J. Am. Chem. Soc., 2005, 127, 12454–12455 CrossRef CAS.
- W. Qianru, G. Yeqin, G. Jianping and C. Ping, Hydrides mediate nitrogen fixation, Cell Rep. Phys. Sci., 2022, 3, 100779 CrossRef.
- (a) CCDC 2464500: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2nqj0y; (b) CCDC 2464501: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2nqj1z; (c) CCDC 2464503: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2nqj31; (d) CCDC 2464504: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2nqj42.
Footnotes |
| † PDI2 = 14,94-di-tert-butyl-2,8,10,16-tetramethyl-3,7,11,15-tetraaza-1,9(2,6)-dipyridinacyclohexadecaphane-2,7,10,15-tetraene. |
| ‡ It is assumed that the disproportionation reaction of the Rh(I) source afforded a dark-red Rh(II)2 complex, together with an insoluble black material, which may be presumed to be Rh(0). |
| This journal is © The Royal Society of Chemistry 2026 |