
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiological semiconductors: self-assembled shell proteins as photoactive materials
Silky
Bedi
,
S. M.
Rose
 and
Sharmistha
Sinha
*
and
Sharmistha
Sinha
*
Chemical Biology Unit, Institute of Nanoscience and Technology, Sector-81, Mohali, 140306, India. E-mail: sinhas@inst.ac.in
First published on 13th November 2025
Abstract
Light-harvesting proteins are promising biocompatible materials for bioelectronics, yet their instability and inefficient charge transport hinder direct integration. External scaffolds offer support but introduce energy losses and delay response. Here, we identified a class of hyper-thermostable, self-assembling bacterial shell proteins that form disc-like structures with spatially organized tyrosine residues, facilitating efficient light absorption and charge transport. I–V profiling and ultraviolet photoelectron spectroscopy reveal their semiconducting behavior and low work function (<3 eV). These protein discs generate photocurrents under UV illumination without external bias and achieve external quantum efficiencies (∼0.5%) and response times (0.3 s) surpassing those of traditional photosynthetic proteins by an order of magnitude. Mutational analysis implicates a tyrosine-mediated electron transfer mechanism. These findings establish bacterial shell proteins as intrinsically stable, scaffold-free photoactive materials for next-generation bioelectronic applications.
Introduction
As the demand for self-powered, sustainable technologies grows, the field of bioelectronics is increasingly turning to biologically derived materials for device fabrication.1,2 Proteins and peptides, owing to their biocompatibility, structural diversity, and functional tunability, are emerging as attractive candidates for use in wearable electronics, organ-on-chip systems, and diagnostic platforms.3–5Nature already offers a blueprint for efficient energy conversion, with proteins playing central roles in respiration and photosynthesis. In these systems, proteins form the scaffolds for finely tuned electron transfer pathways, utilizing evolutionarily conserved mechanisms to move charges with high specificity and efficiency.6–8 Aromatic residues such as tyrosine and tryptophan often serve as transient redox centers, capable of mediating long-range electron transfer via hopping or tunnelling mechanisms.9–12 In light-sensitive proteins, such as those found in photosynthetic complexes or blue-light using flavin (BLUF) domains, these residues participate in photoinduced charge separation, allowing proteins to function as intrinsic photoactive materials.13–17 Such properties position proteins as promising components for next-generation light-responsive bioelectronic devices.
Despite these advantages, several fundamental challenges limit the practical integration of proteins into functional bioelectronic platforms. Proteins are often structurally fragile, susceptible to denaturation outside physiological conditions, and exhibit limited conductivity and short-range charge transport. Furthermore, most protein-based devices rely on artificial scaffolds or supports, which can introduce additional energy losses and delay response times.
In this context, bacterial microcompartment (BMC) shell proteins offer a compelling alternative. BMCs are protein-based organelles that encapsulate metabolic pathways in many prokaryotes.18 Their shells are composed of hexameric and pentameric protein subunits that self-assemble into highly ordered 2D sheets or disks (Fig. 1ai–iii). Remarkably, these protein assembled 2D sheets or disks can span up to several microns and maintain structural integrity at temperatures as high as 60 °C.19 These features make BMC shell proteins excellent candidates for integration into stable, scaffold-free bioelectronic materials.
 | ||
| Fig. 1 Asymmetric electron-density on shell proteins facilitating electron conduction. (ai) Monomeric unit of PduA (PDB ID: 3NGK) that assembles to form hexamers (aii). Hexamers undergo edge to edge association to form flat sheet assemblies (aiii); (bi) electron density map of PduA (PDB ID: 3NGK); (bii) PduB (PDB ID: 4I61); (biii) β-carboxysome shell protein CcmK1, structurally analogous to PduA shell protein of PduBMC (PDB ID: 4LIW); (biv) PduJ shell protein from Salmonella species (PDB ID: 5D6V). These images illustrate distinct electron density patterns on the protein surface facilitating electron conduction over the protein surface; (c) sheet assembly model of the PduA shell protein overlaid with its electron density pattern, highlighting potential electron conduction regions and its fabrication on the indium tin oxide (ITO) substrate for conductivity experiments. | ||
Beyond structural stability, BMC shell proteins display intriguing electronic characteristics. Across various species, their surfaces exhibit distinct electron density patterns,20 suggesting regions of electron-rich and electron-deficient character that may facilitate localized charge transfer. Supporting this, we found that BMC shell proteins such as PduA and PduBB′ can reduce gold ions into gold nanoparticles, indicating their intrinsic redox activity (Fig. S1). Such anisotropic charge distribution has been linked to their natural function as selectively permeable barriers, but it also hints at potential for directional electron flow across the protein surface.
Typically, BMC shell proteins across several species exhibit discrete electron density patterns across the protein surface (Fig. 1bi–iv). In this study, we investigate the photo-induced charge transport properties of two major shell proteins from the Salmonella propanediol utilization BMC (PduBMC): PduA and PduBB′ by drop casting them on an ITO-glass substrate (Fig. 1c). The rationale is the abundance and strategic positions of tyrosine residues within these proteins, which predictably arrange in a conjugated manner on the self-assembled extended sheet (Table S1). We hypothesize that this spatial arrangement enhances delocalization of photoexcited electrons and promotes long-range conductivity.
By combining structural insights, mutational analysis, and photoelectronic measurements, we demonstrate that these BMC shell proteins represent a unique class of robust, intrinsically photoactive biomaterials capable of generating significant photocurrent without the need for external scaffolding. This work paves the way for their application in stable, protein-based optoelectronic and bioelectronic devices.
Results
Intrinsic semiconducting and UV-responsive behaviour of self-assembled protein sheets
To investigate the potential of shell proteins as optoelectronic materials, we characterized the electrical and photoresponsive behaviour of a self-assembling bacterial protein that forms stable, two-dimensional nanosheets. Transmission Electron Microscopy (TEM), Field Emission Scanning Electron Microscopy (FESEM) and Confocal Laser Scanning Microscopy (CLSM) revealed extended sheet-like structures with lateral dimensions spanning several micrometres (Fig. 2ai–v). | ||
| Fig. 2 Electron conductivity and photocurrent generation in PduA shell protein: (a) TEM (i and ii), FESEM (iii and iv), and CLSM (v) images of PduA shell protein illustrate the sheet morphology of the protein; scale bars: 0.5 µm and 100 nm for TEM images, 100 nm for FESEM images, and 10 µm for CLSM images. (b) I–V characteristics of PduA, showing non-linear behavior, indicative of its semiconducting nature; (c) the measured work function of PduA, which falls within the typical range for semiconducting materials, indicating the ease of electron ejection from the surface of the PduA sheet; (d) photocurrent generation in PduA-fabricated samples under UV light illumination (254 nm) without the application of external bias, demonstrating the intrinsic light-harvesting capability of the PduA shell protein; (e) on–off cycles of photocurrent response recorded during alternating dark and UV light exposure, showing consistent and reversible photocurrent generation; (f and g) area used to calculate the response times for photocurrent rise τrise and τdecay, highlighting the fast and stable response of PduA to UV light, confirming its potential for photocurrent applications. | ||
We assessed the electronic properties of these PduA sheets by recording their I–V characteristics. The protein is drop-cast across a spacer defined channel on an indium tin oxide (ITO) substrate (Fig. 1c), creating a conductive bridge between electrodes. The resulting I–V profile exhibited a distinct non-linear response under an applied bias of ±7 V (Fig. 2b), indicative of semiconducting behaviour.
To further probe the electronic properties of PduA, we measured its work function using Ultraviolet Photoelectron Spectroscopy (UPS). Protein films were drop-cast on cleaned ITO substrates, with uniform coverage confirmed by Coomassie Brilliant Blue G250 staining (Fig. S2a). UPS spectra were recorded using a He I source (hν = 21.22 eV), a pass energy of 2 eV, and a step size of 0.05 eV. The secondary electron cutoff (Ecutoff) was identified from the low kinetic energy onset of the spectrum, and the Fermi edge (EF) was determined from the high kinetic energy onset. The work function (Φ) was then calculated using the standard equations as follows:
| Φ = hν − (EF − Ecutoff) |
From this analysis, the PduA-coated ITO surface exhibited a work function of 2.96 eV (Fig. 2c), significantly lower than that of bare ITO (4.23 eV, Fig. S2b) and within the typical range for semiconducting materials. Uniform film coverage and the exclusive contribution of the protein layer to the measured current were further confirmed by Coomassie staining (Fig. S2a) and control I–V measurements (Fig. S2c–e).
We next examined the protein's photoresponsivity by illuminating the sample with UV light (254 nm, 0.11 mW) in the absence of external bias. Under these conditions, a reproducible photocurrent of ∼20 nA was generated (Fig. 2d), with excellent stability over multiple light on/off cycles for more than 120 seconds (Fig. 2e). This bias-free photocurrent generation upon light illumination confirms the protein's intrinsic photoresponsivity. To benchmark this performance, we compared the shell proteins to globular protein, bovine serum albumin (BSA). Under identical conditions, BSA produced negligible current (0.5 mA) (Fig. S3a) and its higher work function (3.2 eV, Fig. S3) correlated with poor photoresponse (Fig. S3b and c). In summary, our results suggest that the ordered, planar architecture of the shell protein nanosheets facilitates charge transport more effectively than globular proteins. Finally, we quantified the protein's temporal response to light. To quantify the temporal response of the shell protein layer under illumination, the rise (τrise) and decay (τdecay) times of the photocurrent were determined from the transient current–time profiles (Fig. 2f and g). The shaded regions in the plots indicate the portions of the transient curves used for analysis: for τrise, the shaded region covers the increase of photocurrent from its baseline to the maximum value upon illumination, while for τdecay, the shaded region tracks the decrease of photocurrent from maximum back to baseline after the light is switched off. Together, these findings demonstrate that the self-assembled protein sheets possess intrinsic semiconducting and photoactive properties, enabling stable photocurrent generation without the need for synthetic scaffolds, cofactors, or external bias.
Mechanisms of photocurrent generation in shell proteins
To investigate the underlying mechanisms contributing to the current generation by the protein nano-sheets, we first examined temperature-dependent I–V characteristics of wild-type PduA. Current measurements conducted at 5 °C and 25 °C revealed negligible variation in conductivity (Fig. 3a), indicating a transport mechanism dominated by electron tunnelling rather than thermally activated hopping.21 Given the protein's photoresponsivity we sought mechanistic parallels with the natural electron transport systems. In biology, electron transport is frequently mediated by redox-active aromatic amino acids such as tyrosine and tryptophan. | ||
| Fig. 3 Mechanism of tyrosine-induced electron transfer in PduA shell protein: (a) temperature-dependent photocurrent generation in PduA representing electron tunneling as the conduction mechanism; (b) spatial arrangement of tyrosine residues within the PduA hexamer, illustrating the proximity between these residues, which facilitates efficient electron transfer across the protein surface; (c) photocurrent generation comparison, indicating a decrease in photocurrent when tyrosine is mutated to alanine (PduA Y35A), while the replacement of tyrosine with tryptophan (PduA Y35W) does not significantly impact photocurrent values, suggesting the importance of aromatic residue presence for maintaining photocurrent efficiency; (d) I–V curves for wild-type PduAWT and its mutants PduA Y35A (tyrosine to alanine) and PduA Y35W (tyrosine to tryptophan), showing no significant alteration in the semiconducting properties of the shell protein across mutations; (e) schematic of the PCET process in PduA shell protein, showing the formation of a tyrosyl radical from the tyrosine residue. The glutamate residues involved in hydrogen bonding with tyrosine participate in electron abstraction, initiating the PCET cycle. This mechanism highlights the role of tyrosine in enabling electron transfer within the PduA shell protein. | ||
For example, in photosystem II, a redox-active tyrosine residue (Yz) donates an electron to a chlorophyll radical (P680+) while concurrently transferring a proton to a nearby histidine (HisZ) through hydrogen bonding,22 exemplifying a proton-coupled electron transfer (PCET) mechanism. Similarly, in ribonucleotide reductase (RNR), tyrosine residues act as electron transporters to mediate PCET over long distances.23 In contrast to these, tryptophan plays a key role in light-induced electron transport in photoreceptors such as amphibian cryptochromes, where a chain of conserved tryptophan triad residues transmits photoinduced electrons from the surface to the buried flavin cofactor.24
Considering the central role of tryptophan and tyrosine in protein mediated electron transport, we examined the structural organization of the shell protein, PduA. This revealed that PduA has a conserved tyrosine residue at the 35th position (Y35) arranged in a symmetrical hexameric configuration (Fig. 3b). This spatial arrangement raises the possibility that Y35 may serve as a redox-active site, facilitating photoinduced electron transfer in a manner similar to natural protein-based PCET pathways. To assess the role of Y35 in photocurrent generation, we created a mutant, PduA [Y35A], replacing tyrosine at the 35th position with alanine (Table S2). This mutation led to a significant drop in photocurrent compared to wild-type PduA (Fig. 3c). This suggests that Y35 plays a key role in charge transfer. We also created a second mutant by replacing Y35 with tryptophan (Y35W, Table S3). Unlike the alanine mutant, the Y35W variant retained both semiconducting properties (Fig. 3d) and photocurrent output (Fig. 3c). These results indicate that both tyrosine and tryptophan can support photoinduced electron transport. This is likely due to their ability to initiate PCET.
Further analysis with the Ligand–Protein Contacts & Contacts of Structural Units (LPCCSU) server25 revealed that the hydroxyl group of the tyrosine residue forms hydrogen bonds with adjacent glutamate (E36) and isoleucine (I87) residues, suggesting a pre-organized environment for tyrosine mediated electron transfer. Glutamate-36 in PduA forms a hydrogen bond with Y35 through its side chain, while isoleucine makes the bond via its backbone carboxyl oxygen, as depicted in Fig. 3b. Interestingly, glutamate has been shown to play a critical role in PCET pathways in ribonucleotide reductase.27 In conclusion, we propose that Y35 in PduA generates electrons under UV light (254 nm) through a simultaneous proton transfer to the adjacent E36 residue. Together, these data support a model in which UV excitation induces electron transfer from Y35, coupled to proton donation to E36, thereby generating a photocurrent without the need for external chromophores. The formation of a tyrosyl radical under UV illumination, a hallmark of PCET initiation, is illustrated in Fig. 3e.26,27
To further validate the involvement of tyrosine-mediated electron transport, we examined the pH dependence of photocurrent generation, reasoning that proton availability would directly influence the efficiency of the transport process. We observed a marked increase in photocurrent at pH 11, relative to physiological pH 7.4 (Fig. S4). This trend correlates well with the pKa of tyrosyl radicals (∼10.1), above which the phenolic group exists predominantly in its deprotonated form. The enhanced current at pH 11 suggests that deprotonation of Y35 facilitates efficient electron transfer, consistent with PCET mechanisms where charge transfer is tightly coupled to proton release. While advanced techniques such as femtosecond transient absorption spectroscopy or electron paramagnetic resonance (EPR) could provide deeper mechanistic insight, our primary goal in this study was to uncover a previously unrecognized optoelectronic property of bacterial shell proteins. Nevertheless, the mechanistic evidence raises an intriguing possibility: could photocurrent be enhanced by increasing the number of redox-active tyrosines in the protein structure? This question motivates future protein engineering efforts aimed at tuning and optimizing charge transport in these bioelectronic materials. While tyrosine is essential, we next questioned if the specific sheet-like architecture of the shell proteins is equally vital for their electronic function. Control experiments with heat-denatured PduA and the intrinsically disordered protein (IDP) α-synuclein (which has four tyrosines) confirmed this structural dependence. Although, heat denaturation does not alter the polypeptide sequence, denatured PduA exhibited non-conducting behaviour under the applied voltage bias (±7 V) and remained non-responsive to light irradiation when compared to the PduA shell protein (Fig. S3a and b). Similarly, the IDP α-synuclein exhibited non-conducting behaviour under the applied bias and no photoresponse upon light irradiation (Fig. S3a and b). Thus, the intrinsic optoelectronic properties of the shell proteins arise not from tyrosine residues alone, but from their precise spatial organization within a highly ordered, two-dimensional protein sheet.
Enhanced photocurrent generation in tyrosine-rich PduBB′ shell protein
From our previous experiments, it is evident that both the presence of tyrosine residues and the sheet-like morphology are crucial for the electronic properties of shell proteins. We therefore asked whether a sheet-forming protein with a higher number of tyrosine residues would show enhanced electronic behaviour. To test this hypothesis, we investigated PduBB′, a shell protein that incorporates three tyrosine residues per BMC domain, compared to a single tyrosine in PduA.28 Similar to PduA, PduBB′ spontaneously assembles into flat, two-dimensional protein sheets, as confirmed by TEM, FESEM, and CLSM imaging (Fig. 4ai–v). Notably, the electron density map of PduBB′ reveals pronounced high-density surface regions (Fig. 1bii), suggestive of enhanced electronic delocalization. | ||
| Fig. 4 Electron conductivity and photocurrent generation of PduBB′ shell protein: (a)TEM (i and ii), FESEM (iii and iv), and CLSM (v) imaging of PduBB′ showing sheet forming tendency of the shell protein; scale bars: 500 nm for TEM, 1 µm for FESEM and 10 µM for CLSM images respectively; (b) I–V characteristics of PduBB′, showing a higher current flow compared to PduA, suggesting enhanced electron conductivity in the PduBB′ shell protein; (c) work function of PduBB′, (d) photocurrent generation observed in PduBB′ under UV light illumination (254 nm), demonstrating its capacity for light-driven electron transport; (e) on–off cycles of photocurrent response under alternating dark and UV light conditions, showing stable and reversible photocurrent generation; (f and g) τrise and τdecay time profiles for the PduBB′ photoresponse, indicating high light sensitivity and fast response times, suggesting PduBB′ as a promising candidate for light-harvesting application. | ||
I–V measurements under a ±7 V external bias exhibited significantly higher current flow in PduBB′ compared to PduA (Fig. 2b and 4b). Ultraviolet photoelectron spectroscopy (UPS) determined the work function of 2.68 eV for PduBB′, lower than that of PduA (2.96 eV) which correlates with its enhanced photocurrent output (Fig. 4c). Upon UV illumination (254 nm, ∼0.11 mW), PduBB′ generated photocurrents nearly three-times higher than PduA (Fig. 2d and 4d), with no external chromophores or redox agents required.
This performance enhancement supports our hypothesis that additional tyrosine residues enhance photocurrent generation, reinforcing the proposed mechanism of tyrosine mediated electron transport for photocurrent generation in shell proteins. Moreover, PduBB′ demonstrated consistent, high photocurrent generation across multiple on-and-off cycles of UV illumination (Fig. 4e), with high photoresponsivity indicated by shorter rise (τrise = 0.35 s) and decay times (τdecay = 0.74 s) compared to PduA (Fig. 4f and g).
Together, these results highlight a structure–function relationship in shell proteins, where redox-active residue content directly modulates photocurrent output. The superior performance of PduBB′ confirms that tyrosine-mediated electron transport is the primary mechanism driving photocurrent generation, and suggests a clear route toward engineering protein-based materials for high-efficiency, sustainable optoelectronic devices.
To further evaluate the nature of photo-induced charge transfer in shell proteins, we introduced catalase, a redox-active protein known to accept electrons in photoelectrochemical systems as an external electron scavenger.29 When added to PduBB′ at micromolar concentrations (0.1–0.5 µM), catalase caused a progressive decrease in photocurrent amplitude (Fig. S5a and b). This attenuation is attributed to catalase intercepting photo-generated electrons before they reach the electrode, thereby reducing the measurable current. The sensitivity of PduBB′ to catalase supports the presence of mobile, photo-generated electrons within the protein matrix that can be selectively quenched by external electron acceptors.
Energy band structure of shell protein sheets
To explore the energy levels in the shell proteins, we prepared an energy band diagram by analyzing the UPS data, complemented by the Tauc plot derived from the absorbance spectra of the proteins (Fig. 5 and S6). | ||
| Fig. 5 Energy band diagram of shell proteins PduA and PduBB′: a plausible energy band diagram prepared using work function values calculated from UPS data complemented by the Tauc plot derived from the absorbance spectra of shell proteins. | ||
UPS analysis yielded work functions of 2.96 eV for PduA and 2.68 eV for PduBB′ (Fig. 2c and 4c) respectively, indicating a greater propensity for electron emission in the latter. Valence band maxima (VBM) were extracted by linear extrapolation of the leading edge of the UPS spectra near the Fermi level, where the photoemission intensity begins to increase. The intersection of this extrapolated line with the baseline was taken as the VBM position. The VBM values were found to be 1.05 eV for PduA and 1.40 eV for PduBB′ with respect to the Fermi level (Fig. 5, S6aiii and biii). When combined with the optical band gaps, 3.84 eV for PduA and 3.63 eV for PduBB′, these values allowed estimation of conduction band minima (CBM) and full electronic band structures.
The VBM positions further indicate a substantial number of unoccupied states in the conduction band, facilitating electron mobility and higher conductivity, particularly for PduBB′. The deeper VBM position of PduBB′ relative to its Fermi level indicates a smaller energy gap between the filled valence band and the empty conduction band. This gap minimizes electron trapping in the valence band, allowing electrons to efficiently transition into the conduction band, contributing to increased electron flow and enhanced current conduction explaining the superior photocurrent and corresponding performance metrics observed in the self-assembled PduBB′ compared to PduA. The band gaps of the shell proteins PduA (3.84 eV) and PduBB′ (3.63 eV) position them within the range of well-known wide band gap semiconductors, such as gallium nitride (GaN, ∼3.4 eV) and silicon carbide (SiC, ∼3.2 eV).30,31
However, conventional wide band gap semiconductors require costly and complex fabrication methods, such as epitaxial growth, and are limited by rigidity and poor biocompatibility. In contrast, shell proteins self-assemble spontaneously into nanostructures like sheets and tubes, offering a bottom–up, energy-efficient synthesis route. Their biological origin grants them mechanical flexibility, environmental sustainability, and inherent biocompatibility critical for emerging applications in soft electronics, implantable devices, and bio-optoelectronic interfaces. Moreover, the electronic properties of shell proteins can be rationally tuned via amino acid substitutions, enabling customized band alignment and functional optimization. These advantages highlight the potential of protein-based materials as a new class of programmable, sustainable semiconductors.
Shell proteins as high-performance bio-photodetectors
The semiconductive and photoresponsive properties of the shell proteins make them ideal candidates for photodetector applications. A crucial factor in assessing a photodetector is its sensitivity towards varying powers of incident UV light. We measured the photoresponse of both PduA and PduBB′ shell proteins at varying UV light power (0.11 mW, 0.15 mW, 0.25 mW, and 0.31 mW), as shown in (Fig. 6a and b). We observed a corresponding increase in photocurrent generation with increasing UV light power. | ||
| Fig. 6 Photodetector performance metrics of PduA and PduBB′ shell proteins: (a and b) power-dependent photoresponse of PduA and PduBB′, illustrating increased photocurrent generation with rising UV light power, highlighting the sensitivity of both proteins to UV illumination; (c) responsivity of PduA and PduBB′ to incident UV light, demonstrating rapid current generation upon illumination, indicating their potential for fast photoresponse applications; (d) detection limits of PduA and PduBB′ at varying UV light power (0.11 mW, 0.15 mW, 0.25 mW, and 0.31 mW), as depicted in figures (a) and (b), showcasing the capability of these shell proteins to detect low-power light and function efficiently across a range of illumination conditions. | ||
The PduA shell protein exhibited an increase in photocurrent from 20 nA to 50 nA, while PduBB′ showed a surge from 50 nA to 90 nA, underscoring its enhanced photoresponsivity. This performance enhancement is consistent with its lower work function and higher density of redox-active tyrosine residues. To further evaluate the photodetector performance of the shell proteins, we estimated key figures of merit, including responsivity (R), detectivity (D*), and external quantum efficiency (EQE)32 as shown in Fig. 6c, d, and Table 1, using eqn (1)–(3) described in the Experimental section.
| UV light power | PduA (EQE%) | PduBB′ (EQE%) |
|---|---|---|
| 0.11 mW | 0.1 | 0.25 |
| 0.15 mW | 0.11 | 0.28 |
| 0.25 mW | 0.15 | 0.35 |
| 0.31 mW | 0.24 | 0.43 |
Across all power levels, PduBB′ outperformed PduA in every figure of merit, achieving higher responsivity, detectivity, and quantum efficiency. This superior performance likely stems from its optimized energy band alignment and higher density of tyrosine residues, which facilitate efficient photoinduced charge separation through a tyrosine mediated electron transfer mechanism. Taken together, these results establish that shell proteins, particularly PduBB′, can serve as scaffold-free, genetically encodable photodetectors with competitive sensitivity metrics. Compared to existing protein-based optoelectronic materials, which often rely on synthetic chromophores or rigid scaffolds, our system demonstrates higher stability, responsiveness, and biocompatibility, opening avenues for next-generation bio-optoelectronic technologies in wearable sensors, medical diagnostics, and environmentally adaptive devices.
Discussion
This study establishes the first report of semiconducting and photoresponsive behavior in bacterial microcompartment (BMC) shell proteins, specifically PduA and PduBB′. This marks a fundamental shift from their conventionally recognized roles as static structural elements. While previous research has extensively detailed their self-assembly into ordered 2D lattices and polyhedral architectures,33 the electronic functionalities of these protein materials have remained uncharacterized. Here, we demonstrate that these proteins can be drop-cast into thin films that exhibit stable photocurrent responses under UV illumination, driven by a biologically encoded tyrosine based electron transfer mechanism. This mechanism is facilitated by spatially organized tyrosine residues, which act as conduits for long-range electron transport in the absence of cofactors or external chromophores.Among the proteins tested, PduBB′ displayed the highest performance, generating a photocurrent of ∼90 nA under 0.31 mW UV light, approximately three times greater than that of PduA. It also exhibited faster response kinetics (τrise ∼0.35 s) and excellent durability over repeated on–off cycles. Mutational disruption of tyrosine residues resulted in >70% reduction in photocurrent, highlighting the essential role of aromatic amino acid side chains in facilitating efficient charge conduction. Furthermore, the non-linear current–voltage behavior and sub-bandgap work function values (as measured by ultraviolet photoelectron spectroscopy) place these materials within the category of wide-bandgap semiconductors. It is comparable to materials like GaN or SiC with distinct advantages in aqueous processability and biocompatibility.34
Importantly, this is the first direct electrical characterization of sheet-like BMC shell protein architectures, expanding their utility beyond compartmentalization and into bioelectronic domains. Previous studies have explored protein-based films such as BSA, silk fibroin, and photosystem complexes, but these often require external chromophores, lipids, or synthetic modifications to support charge transfer or light harvesting.36 For instance, in a recent study by Suresh et al., UV detection relied on coupling engineered photosynthetic proteins with external UV enhancer molecules to achieve sensitivity at low light intensities35 (Table 2). In contrast, our work shows that native shell proteins alone without any chemical coupling or enhancer molecules can facilitate robust photocurrent generation, purely due to their intrinsic sequence-encoded architecture and self-assembly properties. Notably, the external quantum efficiency (EQE) of our PduBB′ shell protein reaches 0.43%, substantially higher than the 0.048% EQE of the reported photosynthetic protein system, underscoring its superior photon-to-current conversion efficiency.
| Protein system | EQE% |
|---|---|
| Photosynthetic protein coupled with UV enhancer molecules35 | 0.048% |
| Shell protein PduBB′ | 0.43% |
To further evaluate the contribution of structural organization to the observed photoresponse, we compared the shell proteins with BSA, a globular control protein that does not form ordered 2D assemblies. BSA was selected because it represents a well-studied, non-assembling protein system, enabling a clear comparison for assessing how morphology governs charge transport. Unlike BSA, which forms disordered and discontinuous films, the BMC shell proteins self-assemble into continuous 2D sheets that provide extended pathways for electron delocalization and transport. This comparison highlights that the inherent sheet-like topology and ordered lattice symmetry of PduA and PduBB′ are key determinants of their enhanced conductivity and photonic response. It is also worth noting that, to date, there are very few studies in which a single, standalone protein has been used as an active photodetecting material. Most protein-based photodetectors rely on hybrid systems, such as photosynthetic proteins coupled with UV enhancers or semiconducting nanostructures (e.g., Suresh et al., Chem, 2019 (ref. 35)), where the protein primarily acts as a sensitizer. Thus, the intrinsic semiconducting and photoresponsive behavior observed here in native shell proteins represents a significant advancement in the development of purely biological materials for optoelectronic applications.
Furthermore, the shell proteins naturally assemble into stable 2D sheets that, as previously reported, are inherently resistant to denaturation under a wide range of physicochemical conditions.37 This structural robustness, combined with their semiconducting properties, opens up new avenues for integrating them into solid-state bio-optoelectronic platforms. Taken together, our findings position PduBB′ and PduA as genetically encodable, scalable, and sustainable components for UV photodetectors and bioelectronic circuits. Their self-assembling nature, high degree of symmetry, and aqueous stability render them uniquely suited for future development of soft photonic devices and wearable electronics.
Conclusion
This work identifies bacterial microcompartment shell proteins, particularly PduBB′, as a novel class of genetically encodable, intrinsically semiconducting and photoresponsive biomaterials. Unlike previously reported protein scaffolds such as those incorporating photosystem complexes, engineered cytochromes, or chromophore-conjugated proteins, shell proteins operate differently. These earlier systems rely on extrinsic cofactors, synthetic dyes, or metal clusters to induce optoelectronic functionality.35,38,39 PduBB′ exhibits native photocurrent generation without any chemical modification. The observed UV-induced electron flow is driven by the intrinsic electronic structure of the protein and electron transfer facilitated by aromatic amino acid residues like tyrosine. This sets shell proteins apart as the first example of structurally ordered protein assemblies that inherently exhibit semiconducting behavior.Shell proteins offer several unique advantages that position them as next-generation bioelectronic materials. Their genetic tunability allows precise modification of amino acid composition, surface properties, and charge distribution, enabling fine control over their electronic behavior.40 In contrast to inorganic semiconductors, they are biocompatible, biodegradable, and processable under mild aqueous conditions, making them ideal candidates for soft, implantable, or environmentally sustainable electronics.41 Furthermore, their intrinsic ability to self-assemble into higher-order architectures, such as nanosheets or polyhedral shells, provides built-in nanostructuring critical for device fabrication without external templates.42,43 These inherent features make shell proteins particularly attractive for developing bio-optoelectronic applications. Potential uses include flexible UV photodetectors, wearable biosensors, implantable diagnostic tools, and even protein-based logic devices. Importantly, these materials also open new avenues to explore fundamental electron transport mechanisms in biological systems without the need for non-native modifications. The scalability of bacterial expression systems, coupled with the structural and functional programmability of shell proteins, supports their potential for real-world deployment in cost-effective, sustainable electronic platforms. Looking forward, future efforts will focus on expanding the spectral sensitivity of these proteins through site-directed mutagenesis or chromophore integration. These efforts will also include integrating them with hybrid soft-matter systems and fabricating prototype devices to evaluate performance under application-relevant conditions. This study not only demonstrates the first native protein scaffold with semiconducting and photoresponsive properties but also establishes a foundational platform for building the next generation of functional, genetically encoded bioelectronics.
Experimental section
Unless otherwise mentioned, all the chemicals were procured from Sigma-Aldrich (India). Ultrapure water was used throughout the experiments. The shell protein construct was a kind gift from Prof. T. A. Bobik, Iowa State University, U. S. A.Cloning, expression, and purification of PduBMC shell proteins
To mutate tyrosine to alanine (PduA Y35A) and tryptophan (PduA Y35W) of PduA shell protein, we performed overlap extension PCR using primers mentioned in Tables S1 and S2. The point mutation was confirmed by Sanger sequencing. The mutant PCR product with BglII and HindIII restriction sites was cloned into a pET41a vector having a kanamycin-selectable marker and transformed into chemically competent E. coli DH5α cells. The positive colonies were selected on kanamycin plates. The recombinant plasmids were purified and transformed into BL21(DE3) cells for protein expression.To purify the shell proteins, 1% of an overnight grown culture was inoculated in bulk secondary media and incubated for 1.5 to 2 hours until reaching an OD600 of 0.5. The expression was induced by adding 0.5 mM IPTG to the secondary culture, followed by incubation at 28 °C for 12 hours. The cells were harvested by centrifugation and lysed in lysis buffer (50 mM Tris-base pH 7.5, 200 mM NaCl, and 5 mM imidazole) along with lysozyme (2.3 mg g−1 of cell pellet) and PMSF (0.5 mM). Furthermore, cells were sonicated and centrifuged at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 minutes. The supernatant was loaded onto a Ni-NTA affinity column, pre-equilibrated with column buffer, and washed with wash buffer (50 mM Tris base, 200 mM NaCl, and 50 mM imidazole; pH 7.5). The bound protein was eluted using elution buffer (50 mM Tris base, 200 mM NaCl, and 200 mM imidazole; pH 7.5). The eluted fractions were checked for purity on SDS-PAGE. The concentration of the protein was estimated using Bradford assay.
000 rpm for 30 minutes. The supernatant was loaded onto a Ni-NTA affinity column, pre-equilibrated with column buffer, and washed with wash buffer (50 mM Tris base, 200 mM NaCl, and 50 mM imidazole; pH 7.5). The bound protein was eluted using elution buffer (50 mM Tris base, 200 mM NaCl, and 200 mM imidazole; pH 7.5). The eluted fractions were checked for purity on SDS-PAGE. The concentration of the protein was estimated using Bradford assay.
TEM imaging
The individual protein samples of the shell proteins PduA and PduBB′ were prepared by drop casting 10 µl (5 µM) of the protein on the formvar-coated 300-carbon mesh grid followed by adding 10 µl of 1% uranyl acetate for negative staining. Samples were incubated for 30 s in the dark, followed by washing with water and air drying. TEM imaging of the protein samples was performed by using JEM 2100 TEM (JEOL, USA) operated at 120 kV.Field-emission scanning electron microscopy (FESEM) imaging
The morphology of PduA and PduBB′ was further confirmed by performing FESEM imaging. Silicon wafers were cleaned with distilled water followed by isopropyl alcohol. A 10 µl aliquot of 5 µM protein solution of both the shell proteins was dropcast on the cleaned and dried silicon wafer. The sample was allowed to incubate on the wafer for 3 minutes followed by wicking off with Whatman filter paper. The sample was allowed to dry in air and under vacuum in a desiccator. The imaging of the sample was performed using a FESEM instrument (JEOL JSMIT 300).Confocal laser scanning microscopy
To check the sheet like morphology of shell proteins in solution, we labelled PduA, PduBB′ and BSA with FITC by incubating each protein with the dye at a 1:10 molar ratio at 4 °C for 8 hours. After incubation, we dialyzed the samples in phosphate buffer (10 mM, pH 7.4) using a 3 KDa dialysis membrane in order to remove unreacted dyes. 10 µl of 5 µM labelled proteins were then imaged with an Olympus FV 3000 CLSM using oil-immersion objectives and post imaging processing was then performed with image J.
Ultraviolet photoelectron spectroscopy
Shell proteins were transferred onto a cleaned ITO substrate ensuring the formation of a uniform film and were allowed to dry under ambient conditions overnight. The ITO substrate was thoroughly cleaned following established protocols before drop-casting the protein solution onto it. The UPS spectra were recorded using a pass energy of 2 eV and a step size of 0.05 eV, with a He I source (hν = 21.22 eV). The analyzer lens axis was set at 90° relative to the sample surface, and the take-off angle was fixed at ∼30° to focus on a smaller solid angle. The instrument was calibrated using a gold target sample in an ultrahigh vacuum chamber, with a spectrometer resolution of 0.12 eV, to ensure accuracy.I–V characterization
We used an ITO-coated glass substrate (1 × 1 cm2) for I–V characterization. First, we cleaned the substrate with IPA and acetone solution followed by bath sonication for 5 minutes. The substrate was dried and a channel a few microns wide was created on the ITO substrate. Two external copper wires were attached on both sides of the electrode for electrical measurement. Subsequently, the sample solution was transferred to the channel to make a proper interface between two sides of the ITO electrodes. Furthermore, voltage bias was applied to the sample to measure the change in the current using a source meter.Microscopic imaging of the ITO substrate
The channel cast on the ITO substrate was imaged under a 10× objective lens using an Olympus lX73 microscope. Images were further analyzed using ImageJ software to calculate the width and length of the cast channel.Photocurrent experiment
To check the photo response of the material, the sample was illuminated with short-wavelength UV light (254 nm, periodically switched on and off). Correspondingly the change in current concerning to the light was recorded under non-biased conditions. We calculated the τrise when the light source was turned on and it reached the maximum photocurrent. Similarly, for τdecay, the difference between the maximum current and the lower optimum photocurrent was taken on switching off the light. The area used to calculate both times is represented in from the zoomed regions shown in Fig. 2f, g, 4f and g. Furthermore, to interpret the photodetector's performance, we estimated various figures of merit such as responsivity (R), detectivity (D*), and external quantum efficiency (EQE), summarized in Table 1, using the following standard equations32 | (1) |
 | (2) |
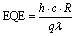 | (3) |
Detectivity represents the sensitivity of the photodetector to weak light signals, while EQE reflects the efficiency of converting incident photons into electrons, defined as the number of charge carriers generated per photon.32
pH-dependent photocurrent measurements
To investigate the involvement of tyrosine based electron transfer mechanisms in the photoresponse of the PduA shell protein, photocurrent measurements were performed under varying pH conditions. The pH of the PduA shell protein solution was adjusted prior to measurement to allow equilibration. Photocurrent responses were recorded under non-biased conditions with UV illumination at 254 nm in ON/OFF cycles. The change in current was measured at each pH, and the resulting trends were analyzed to evaluate the role of pH in modulating photo-induced electron transfer.Effect of catalase on photocurrent generation
To investigate the role of electron transfer dynamics in the photoresponsive behavior of the PduBB′ shell protein, catalase was used as an external electron quencher. Catalase was prepared in phosphate buffer and added to the shell at final concentrations ranging from 0.1 µM to 1 µM. For each measurement, the catalase solution was incubated with PduBB′ for 10 minutes to ensure sufficient interaction. Photocurrent measurements were carried out under non-biased conditions using a shortwave UV light source (254 nm) with periodic ON/OFF illumination cycles. Current responses were recorded to assess the effect of catalase on photo-induced electron transfer.Author contributions
SB & SMR: conceptualization, experimental design, execution, methodology, data analysis, investigation, manuscript drafting, and editing. SS: Supervision, project investigation, conceptualization, design, analysis, writing, reviewing, and editing.Conflicts of interest
There is no conflict of interest to declare.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Supplementary information: all data supporting the findings of this study including electrical measurements, photocurrent data, UV-vis spectra, TEM/FESEM images, band-gap calculations, protein sequences, and primer information are available. See DOI: https://doi.org/10.1039/d5sc05716g.
Acknowledgements
SB acknowledges DST-INSPIRE for the fellowship and INST for research facilities. SMR acknowledges INST for the fellowship and facilities. The authors also acknowledge all the SS Lab members for insightful discussions.References
- M. Torculas, J. Medina, W. Xue and X. Hu, ACS Biomater. Sci. Eng., 2016, 2, 1211–1223 CrossRef CAS.
- C. D. Bostick, S. Mukhopadhyay, I. Pecht, M. Sheves, D. Cahen and D. Lederman, Rep. Prog. Phys., 2018, 81, 026601 CrossRef PubMed.
- R. Montoya, P. Deckerman and M. O. Guler, BBA Adv., 2025, 7, 100149 CrossRef CAS PubMed.
- F. Greco, A. J. Bandodkar and A. Menciassi, APL Bioeng., 2023, 7, 020401 CrossRef PubMed.
- Q. Wu, J. Liu, X. Wang, L. Feng, J. Wu, X. Zhu, W. Wen and X. Gong, Biomed. Eng. Online, 2020, 19, 9 CrossRef.
- B. Mu, X. Hao, X. Luo, Z. Yang, H. Lu and W. Tian, Nat. Commun., 2024, 15, 903 CrossRef CAS PubMed.
- L. Zhang, J. R. Lu and T. A. Waigh, Adv. Colloid Interface Sci., 2021, 287, 102319 CrossRef CAS.
- L. A. Baker and S. Habershon, Proc. R. Soc. A, 2017, 473, 20170112 CrossRef PubMed.
- N. Sekar and R. P. Ramasamy, J. Photochem. Photobiol., C, 2015, 22, 19–33 CrossRef CAS.
- S. Cogliati, J. L. Cabrera-Alarcón and J. A. Enriquez, Biochem. Soc. Trans., 2021, 49, 2655–2668 CrossRef CAS PubMed.
- A. J. Bischoff, L. M. Hamerlynck, A. J. Li, T. D. Roberts, N. S. Ginsberg and M. B. Francis, J. Am. Chem. Soc., 2023, 145, 15827–15837 CrossRef CAS PubMed.
- M. Cordes and B. Giese, Chem. Soc. Rev., 2009, 38, 892–901 RSC.
- S. D. Glover, C. Jorge, L. Liang, K. G. Valentine, L. Hammarström and C. Tommos, J. Am. Chem. Soc., 2014, 136, 14039–14051 CrossRef CAS PubMed.
- A. Nilsen-Moe, C. R. Reinhardt, S. D. Glover, L. Liang, S. Hammes-Schiffer, L. Hammarström and C. Tommos, J. Am. Chem. Soc., 2020, 142, 11550–11559 CrossRef CAS PubMed.
- L. Hammarström and S. Styring, Energy Environ. Sci., 2011, 4, 2379–2388 RSC.
- D. G. Nocera, J. Am. Chem. Soc., 2022, 144, 1069–1081 CrossRef CAS PubMed.
- J. Tong, P. Zhang, L. Zhang, D. Zhang, D. N. Beratan, H. Song, Y. Wang and T. Li, Sens. Actuators, B, 2020, 310, 127838 CrossRef CAS PubMed.
- C. A. Kerfeld, C. Aussignargues, J. Zarzycki, F. Cai and M. Sutter, Nat. Rev. Microbiol., 2018, 16, 277–290 CrossRef CAS.
- E. Y. Kim, M. F. Slininger and D. Tullman-Ercek, Protein Sci., 2014, 23, 1434–1441 CrossRef CAS PubMed.
- J. M. Ochoa, O. Mijares, A. A. Acosta, X. Escoto, N. Leon-Rivera, J. D. Marshall, M. R. Sawaya and T. O. Yeates, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2021, 77, 275–285 CrossRef CAS.
- T. Esposito, P. H. Dinolfo and K. M. Lewis, Org. Electron., 2018, 63, 58–64 CrossRef CAS.
- N. Nelson and C. F. Yocum, Annu. Rev. Plant Biol., 2006, 57, 521–565 CrossRef CAS PubMed.
- T. Biskup, B. Paulus, A. Okafuji, K. Hitomi, E. D. Getzoff, S. Weber and E. Schleicher, J. Biol. Chem., 2013, 288, 9249–9260 CrossRef CAS.
- M. Kasanmascheff, W. Lee, T. U. Nick, J. Stubbe and M. Bennati, Chem. Sci., 2016, 7, 2170–2178 RSC.
- V. Sobolev, A. Sorokine, J. Prilusky, E. E. Abola and M. Edelman, Bioinformatics, 1999, 15, 327–332 CrossRef CAS.
- M.-T. Zhang and L. Hammarström, J. Am. Chem. Soc., 2011, 133, 8806–8809 CrossRef CAS PubMed.
- T. G. McCaslin, C. V. Pagba, S.-H. Chi, H. J. Hwang, J. C. Gumbart, J. W. Perry, C. Olivieri, F. Porcelli, G. Veglia, Z. Guo, M. McDaniel and B. A. Barry, J. Phys. Chem. B, 2019, 123, 2780–2791 CrossRef CAS PubMed.
- B. P. Lehman, C. Chowdhury and T. A. Bobik, J. Bacteriol., 2017, 199(8), e00785 CrossRef CAS.
- P. A. Prakash, U. Yogeswaran and S.-M. Chen, Sensors, 2009, 9, 1821–1844 CrossRef CAS.
- M. Dalla Vecchia, S. Ravyts, G. Van den Broeck and J. Driesen, Energies, 2019, 12, 2663 CrossRef CAS.
- R. Singh, Microelectron. Reliab., 2006, 46, 713–730 CrossRef CAS.
- J. M. Wu and W. E. Chang, ACS Appl. Mater. Interfaces, 2014, 6, 14286–14292 CrossRef CAS.
- K. A. Dryden, C. S. Crowley, S. Tanaka, T. O. Yeates and M. Yeager, Protein Sci., 2009, 18, 2629–2635 CrossRef CAS PubMed.
- M. Mishra, A. Gundimeda, S. Krishna, N. Aggarwal, L. Goswami, B. Gahtori, B. Bhattacharyya, S. Husale and G. Gupta, ACS Omega, 2018, 3, 2304–2311 CrossRef CAS PubMed.
- L. Suresh, J. V. Vaghasiya, D. K. Nandakumar, T. Wu, M. R. Jones and S. C. Tan, Chem, 2019, 5, 1847–1860 CAS.
- A. Nawade, K. B. Busi, K. Ramya, G. K. Dalapati, S. Mukhopadhyay and S. Chakrabortty, ACS Omega, 2022, 7, 20906–20913 CrossRef CAS PubMed.
- M. Faulkner, L.-S. Zhao, S. Barrett and L.-N. Liu, Nanoscale Res. Lett., 2019, 14, 54 CrossRef.
- A. Mershin, K. Matsumoto, L. Kaiser, D. Yu, M. Vaughn, M. K. Nazeeruddin, B. D. Bruce, M. Graetzel and S. Zhang, Sci. Rep., 2012, 2, 234 CrossRef.
- Y. Tan, R. Y. Adhikari, N. S. Malvankar, S. Pi, J. E. Ward, T. L. Woodard, K. P. Nevin, Q. Xia, M. T. Tuominen and D. R. Lovley, Small, 2016, 12, 4481–4485 CrossRef CAS.
- B. Ferlez, M. Sutter and C. A. Kerfeld, Metab. Eng., 2019, 54, 286–291 CrossRef CAS PubMed.
- Q. Luo, C. Hou, Y. Bai, R. Wang and J. Liu, Chem. Rev., 2016, 116, 13571–13632 CrossRef CAS PubMed.
- H. Liu, R. Jian, H. Chen, X. Tian, C. Sun, J. Zhu, Z. Yang, J. Sun and C. Wang, Nanomaterials, 2019, 9, 950 CrossRef CAS PubMed.
- M. Irimia-Vladu, Chem. Soc. Rev., 2014, 43, 588–610 RSC.
| This journal is © The Royal Society of Chemistry 2026 |