
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective electrochemical oxidation of biomass and waste plastic at higher current densities for simultaneous hydrogen generation through hybrid water electrolysis
Snehanjali
Behera
 ,
Akanksha
Negi
,
Gayatri
Joshi
,
Chetansinh
Chauhan
,
Dipak Suresh
Kanthali
,
Saumyakanti
Khatua
* and
Biswajit
Mondal
*
,
Akanksha
Negi
,
Gayatri
Joshi
,
Chetansinh
Chauhan
,
Dipak Suresh
Kanthali
,
Saumyakanti
Khatua
* and
Biswajit
Mondal
*
Department of Chemistry, IIT GandhinagarPalaj, Gujarat-382055, India. E-mail: mondal.biswajit@iitgn.ac.in
First published on 5th December 2025
Abstract
The electrochemical oxidation of lignocellulose and plastic waste has been considered a clean and reliable strategy to produce feedstocks for various chemicals and fuels. In this study, we tackle the challenging selective oxidation of various lignocellulose and mixed biodegradable and bio-nondegradable plastics by targeting the oxidative cleavage of the specific C(OH)–C moiety. A monometallic Ni(O)O–H electrocatalyst was used for valorization of cellulose-based biomass, a series of lignin-based model complexes, lignin-derived secondary alcohols (KA oil), and mixed plastic waste based on the catalyst's O–H bond dissociation free energy. The catalyst performs remarkably well to selectively oxidize cellulose and lignin-based model complexes like HMF and PED with excellent yield at a higher current density of 100 mA cm−2. Mechanical insights into this reaction were obtained by in situ transmitted light spectroscopy and Raman measurements. The catalyst was also able to oxidize KA oil to adipic acid with 54% yield at a constant current electrolysis of 20 mA cm−2. Furthermore, plastic waste precursors having a C(OH)–C bond were selectively oxidized using this catalyst, which was further expanded to mixed plastic waste upgradation resulting in the generation of formate and acetate with faradaic efficiencies of 66% and 74%, respectively, and 100% yield in terephthalic acid accompanied by the co-production of hydrogen.
1. Introduction
Renewable carbon sources can act as substitutes for limited fossil-based sources to take the world a step towards carbon neutrality and sustainability in the chemical industry. While lignocellulose biomass is the most abundant carbon source in nature, waste plastic can be considered the most prevalent synthetic carbon source.1,2 Lignocellulose is composed of three types of biopolymers: cellulose, lignin, and hemicellulose. A major constituent of waste plastic is polyethylene terephthalate (PET). Polylactic acid (PLA) is also an ester-linked polymer like PET and is commonly used in households. Despite their apparent differences, plastics and lignocellulose have many fundamental structural and functional similarities. Both of them have polymeric structures with secondary interactions like hydrogen bonding and cross-linking.3 Again, the primary structures of both were revealed to have the elemental composition of C, O, N, and H with connectivities of C–C and C–O.4Cellulosic biomass (which makes up up to 50% of lignocellulose) depolymerization and subsequent dehydration result in 5-hydroxymethylfurfural (HMF) having C(OH)–C and C(O)–C linkages.5 Further oxidation of C(OH)–C and C(O)–C of HMF can result in the formation of 2, 5-furandicarboxylic acid (FDCA), a potential platform chemical for polyethylene furanoate, a plastic which could replace the petrochemically derived polyethylene terephthalate.6 Similarly, lignin obtained from wood or as industrial waste from the pulp and paper industry can be considered a major source of non-petroleum-based aromatic chemicals.7–11 In contrast to cellulose and other biopolymers, lignin does not have crystalline repetitive monomeric units; instead, the monolignol subunits are linked via various β-O-4, β-5, β–β, and β-1 linkages.12,13 β-O-4 dominates over all other linkages with 43–65%.14,15 This β-O-4 linkage generally holds C(OH)–C type of connectivity (Scheme 1); therefore, to selectively activate lignin through β-O-4 linkage for its valorization, oxidative cleavage of C(OH)–C can be considered as one of the most convenient ways.
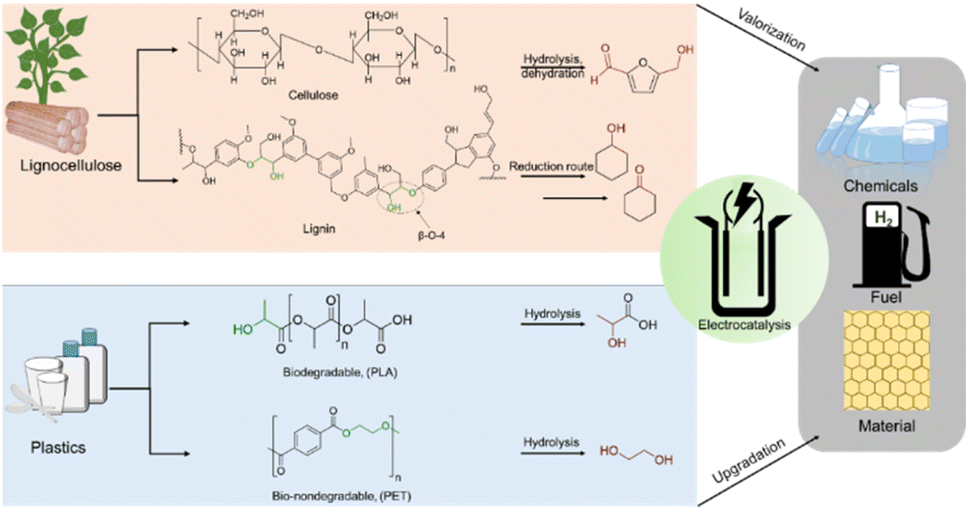 | ||
| Scheme 1 . Structural similarities between biomass and waste plastic and their electrochemical upgradation to valuable chemicals. | ||
Plastic pollution due to both biodegradable and non-biodegradable plastics has been a major environmental concern these days. For non-biodegradable plastics like polyethylene terephthalate (PET), the three end-of-life treatments are incineration, landfill/seawater disposal, and recycling; however, each option has severe restrictions because of the current population status.16 On the other hand, biodegradable plastics like polylactic acid (PLA) generate carbon footprints (CO2) whenever naturally degraded in soil and become non-degradable when disposed of into seawater.17–19 As the natural degradation kinetics is the slowest, it can cause severe accumulation in the environment, posing a serious global threat.20,21 These challenges motivate us to develop catalysts for the electrochemical valorization of plastics to promote plastic usage circularity.
As discussed earlier, a common linkage of C(OH)–C or C(O)–C is shared by the primary β-O-4 linkage of the lignin model complex, the cellulosic model complex 5-hydroxymethylfurfural (HMF) and monomeric units of biodegradable plastic (polylactic acid) and bio-nondegradable plastic (polyethylene terephthalate). It will be more effective to concentrate on this fundamental electrochemical oxidation unit for oxidative C–C bond cleavage to create value-added chemicals like carboxylates.
In this context, we present an exceptionally potent monometallic metal–organic framework Ni BDC/NF (Ni benzene dicarboxylic acid grown on Ni foam) as a pre-catalyst for different lignocellulose-based model complexes and plastic oxidation to value-added chemicals with the co-production of hydrogen. The correlation between the bond dissociation free energy (BDFE) of Ni(O)O–H vs. the reactive C–H bond of the substrate piqued our interest, leading us to evaluate the BDFE of activated Ni BDC/NF.22,23 We spectroscopically identified Ni(O)OH as a key catalytic intermediate for substrate oxidation. The high BDFE of Ni(O)O–H enables us to effectively screen a series of biomass and plastic-based substrates having C(OH)–C or C(O)–C linkages with C–H BDFE less than the BDFE of Ni(O)O–H. Using this principle, we have demonstrated biomass and waste plastic as substrates for oxidation to valuable carboxylates at a high current density and good recyclability with the co-production of hydrogen.
2. Results and discussion
2.1. Material characterization
The catalyst Ni BDC/NF was prepared via the hydrothermal method following the reported procedure. Briefly, a nickel salt, 1,4-benzenedicarboxylic acid, and nickel foam were placed in a hydrothermal chamber with water, ethanol, and DMF. It was heated for 3 h at 150 °C in an oven. It was washed thoroughly and subjected to electrochemical activation in 1 M KOH (Fig. 1a). The SEM of the as-prepared Ni BDC/NF shows a flake-like morphology, and it uniformly covers the nickel foam (Fig. 1b). The formation of MOF has been confirmed by its characteristic peak at ∼8° in the PXRD (Fig. 1c). The peaks at approximately 8° and 15° correspond to the 200 and 201 planes of the 2D NiBDC MOF, respectively. These match the data from CCDC number 985792.24 The FTIR data of the Ni BDC/NF also show two absorption peaks at 1580 and 1362 cm−1 due to the existence of carboxylate groups of the BDC ligand in the MOF structure.24 Upon electrochemical activation, although the flake-like morphology is retained, the peak at ∼8° as well as characteristic FTIR peaks diminished, suggesting destruction of the MOF network (Fig. 1b, c, S1 and S2). BDC ligands may remain on the surface, exhibiting a very weak interaction with the metal centre of Ni(O)OH of the activated Ni BDC/NF, as indicated by the less intense peaks for carboxylates in the FTIR spectra (Fig. S2). A report has recently been published in the context of HMF oxidation with TPA-modified Ni-foam electrodes, where the authors made similar observations.6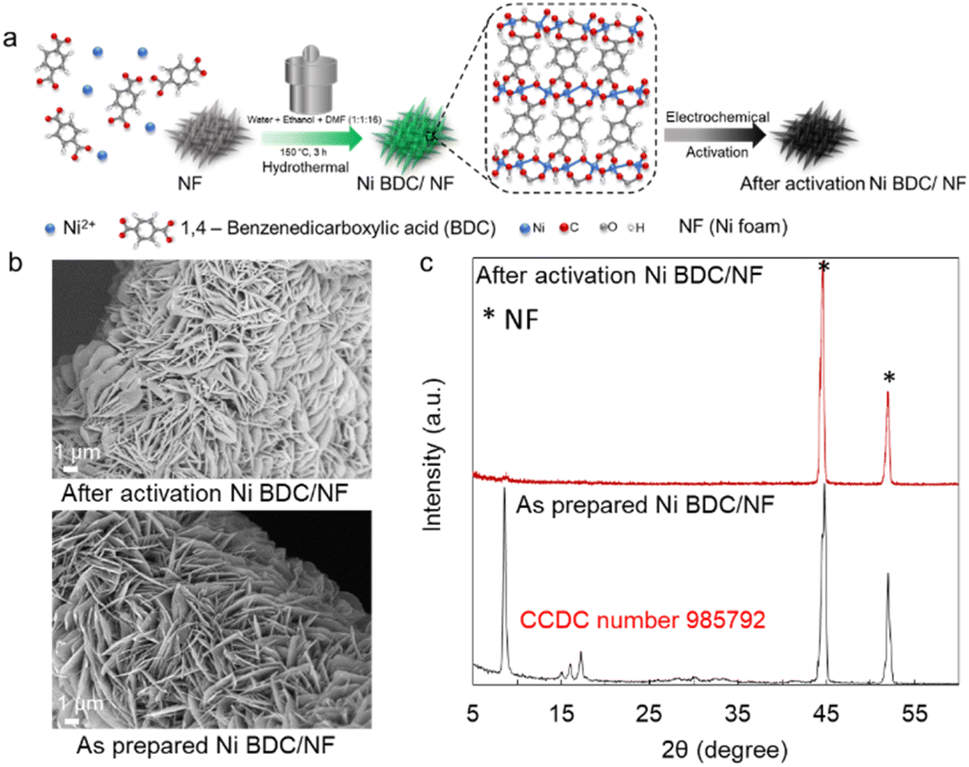 | ||
| Fig. 1 Preparation and physical characterization of electrochemically activated Ni BDC/NF: (a) schematic representation of the synthesis of Ni BDC/NF by a hydrothermal method followed by electrochemical activation; (b) SEM images of the as-prepared and activated Ni BDC/NF; (c) powder XRD pattern of the as-prepared Ni BDC/NF (grey line) and after its electrochemical activation (red line).28 | ||
The SEM data revealed a rough surface morphology, suggesting an increase in the surface area, which is favorable for catalysis. The Cdl value determined from the slope of the linear plot of non-faradaic current density vs. scan rate relates to the electrochemically active surface area, and is found to be at least 10 times higher than the Cdl value of the as-prepared Ni BDC/NF and 2.5 times higher than the Cdl value of Ni(O)OH prepared directly from activated Ni foam (Fig. 2a and S3). When the electrode is electrochemically activated by CV cycling around Ni(III/II) redox in 1 M KOH, a pair of featured bands of NiIII–O appeared at 474 and 558 cm−1 in the Raman spectra (Fig. 2b). This is also associated with a color change of the electrode from greenish-yellow to black, which remained stable for a few minutes, allowing us to ex situ characterize the electrode (Fig. S4). Thus, the electro-active species has been established to be Ni(O)OH (vide infra). This observation is further supported by the XPS spectra of Ni and O (Fig. S5a and b), which revealed major peaks at 855.4 eV, attributed to the 2p3/2 state of Ni2+, and at 856.8 eV, corresponding to the 2p3/2 state of Ni3+.25,26 Additionally, the O 1s spectrum shows peaks at 532.7 eV, 531.3 eV, and 530.5 eV, corresponding to surface adsorbed H2O, Ni–OH, and Ni–O species, respectively.27 These findings collectively confirm the formation of Ni(O)OH upon activation. The BDFEO–H of NiIIIO–H is crucial in determining its substrate oxidation efficiency. It is proposed that if the BDFEO–H is higher than the X–H (X = C, etc.) in the substrate, the X–H bond can be oxidatively cleaved (Fig. 2c). The BDFE of Ni(O)OH should ideally be determined by recording Ni(III/II)E1/2 at pH 0. However, the catalyst is not stable in acidic pH due to acid dissolution, and hence, the E1/2 of Ni(III/II) was measured at different pHs in the alkaline regime, where the catalyst is stable towards dissolution. A straight line is obtained from the plot of E1/2vs. pH, which is then extrapolated to pH 0 to get the E1/2 of the redox process at this pH (Fig. 2d and S6). The BDFEO–H for Ni(O)OH is determined to be 97.8 kcal mol−1 following eqn (1):
| BDFE = 23.06 E0 (pH) + 57.6 kcal mol−1 | (1) |
 | ||
| Fig. 2 Electrochemical characterization of electrochemically activated Ni BDC/NF: (a) Cdl measurement at different scan rates ranging from 20 to 90 mV s−1; (b) Raman signal showing the formation of Ni(O)OH after activation; (c) mechanism of X–H bond activation with the active Ni species; (d) E1/2vs. pH plot to determine the BDFE; (e) BDFE of some of the key substrates used in this study. | ||
As discussed earlier, one of the common linkages among biomass and waste plastic is C(OH)–C or C(O)–C, and the BDFEC–H of such linkage ranges from 75–87 kcal mol−1 (Fig. 2e), which is lower than the BDFEO–H for Ni(O)OH. This is intriguing as Ni(O)OH can be used as a common platform for the electro-oxidative upgradation of biomass and plastic. Further, the ipvs. scan rate plot suggests the redox process is surface-bound (Fig. S7).
2.2 Electrolysis experiments with various lignocellulose and plastic-based substrates
The activated NiBDC/NF sample was first tested for the oxidation of cellulose-based biomass 5-(hydroxymethyl) furfural (HMF). The catalyst shows a huge 240 mV gain in potential at 100 mA cm−2 following the addition of 100 mM HMF, and only requires 1.44 V vs. RHE to generate 100 mA cm−2 (Fig. 3a) compared to the OER. The electrochemical activity of the activated Ni BDC/NF for HMF oxidation was much higher in comparison to that of the as-synthesized Ni BDC/NF as well as Ni(O)OH derived directly from Ni foam (Fig. S8). The inset of Fig. 3a shows a Tafel slope of 80 mV dec−1 suggesting a facile electron transfer kinetics. A constant current electrolysis was performed at 100 mA cm−2 (Fig. S9) to assess the performance of the catalyst. The catalyst was able to selectively oxidize HMF into 2, 5-furfural dicarboxylic acid (FDCA), an important platform chemical for the polymer industry. 1H-NMR of the post-electrolysis electrolyte confirms the 100% conversion of HMF into FDCA with an 89% faradaic efficiency using 1,4-dioxane as an internal standard (Fig. 3b).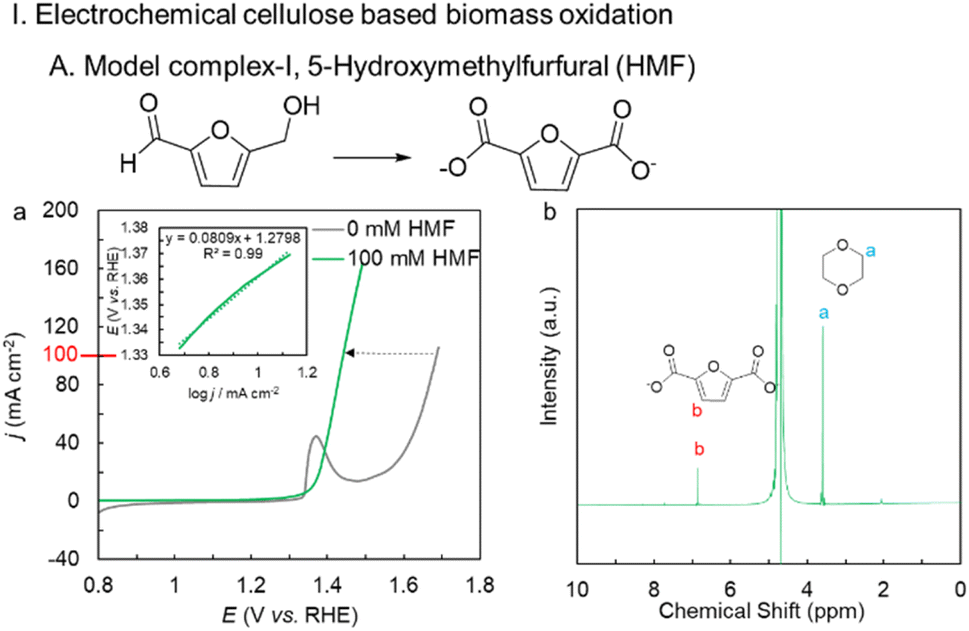 | ||
| Fig. 3 Electrochemical cellulose-based model complex oxidation by activated Ni BDC/NF: (a) LSV of 100 mM HMF oxidation (green line), 1 M KOH (grey line) (100% iR corrected), inset shows the Tafel plot of HMFOR measured in the kinetic region; (b) 1H-NMR spectrum of the post-electrolysis sample of HMFOR. | ||
Further, the electro-oxidation of lignin-based complexes derived from both oxidative as well as reductive routes after the depolymerization of lignin was explored. The lignin model complexes are so chosen that they contain the C(OH)–C motif, and BDFEC-H is less than the BDFEO–H of Ni(O)OH. One such model complex that is fully soluble in an aqueous medium is 1-phenylethane-1, 2-diol (PED) (lignin model complex I). The catalyst shows a significant reduction in the potential of 280 mV after the addition of 100 mM PED to 1 M KOH compared to the OER. The catalyst can efficiently oxidize PED by generating a current density of 100 mA cm−2 at only 1.41 V (Fig. 4a) with a facile electron transfer kinetics as evident from a low Tafel slope of 80 mV dec−1 in the kinetic region (Fig. 4a: inset). The decrease in charge transfer resistance in the EIS spectra suggests faster reaction kinetics between the electrode surface and electrolyte after the addition of 100 mM PED (Fig. S10). Also, it is interesting to note that the characteristic Raman bands for NiIII–O of Ni(O)OH vanish upon the addition of PED, suggesting NiIII–O as the key reactive intermediate of the process (Fig. S11). This is further confirmed by the in situ transmitted light spectroscopy study (Fig. S12). For the initial ∼20 seconds, both 1 M KOH and 1 M KOH + 100 mM PED solutions remained in an open circuit potential (OCP) state. After this period, a constant current electrolysis of 100 mA cm−2 is applied for ∼30 seconds. During the electrolysis, the activated Ni BDC/NF undergoes a color change from greenish to black in both cases, suggesting the formation of Ni(O)OH in both cases. Between 50 and 120 seconds, the black color remains stable in 1 M KOH where the alcohol substrate is absent. In contrast, in the presence of 100 mM PED, the electrode gradually reverts from black to its original color (Fig. S12). A constant current electrolysis at 100 mA cm−2 shows a steady potential vs. time curve until the theoretical charge passes for the completion of the reaction (Fig. S13). An aliquot of the post-electrolysis electrolyte was analyzed for the 1H- and 13C-NMR and it revealed that PED has undergone complete oxidative C–C bond cleavage to produce formate and benzoate with 98% and 100% chemical yield and faradaic efficiency (FE), respectively (Fig. 4a and S14).
 | ||
| Fig. 4 Electrochemical lignin-based model complex oxidation by activated Ni BDC/NF: (a) LSV of 100 mM PED oxidation (blue line) and 1 M KOH (grey line) (100% iR corrected), inset shows the Tafel plot of PEDOR measured in the kinetic region; (b) 1H-NMR spectrum of the post-electrolysis sample of PEDOR. | ||
1-Phenyl-ethanol (lignin model complex II) with C(OH)–C is a widely accepted lignin model complex resembling the β-O-4 unit. The electrochemical oxidation of the C(OH)–C unit is attempted with the catalyst. The catalyst shows a low onset potential of 1.35 V vs. RHE for its oxidation (70 mM; this is not fully miscible beyond this limit in 1 M KOH, Fig. S15). A constant current electrolysis at 20 mA cm−2 was carried out concurrently with time-dependent 1H-NMR to elucidate the reaction mechanism (Fig. 5a and b). The 2e− oxidation of model complex II into acetophenone as a major reaction is suggested by the stable electrolysis curve that remained unchanged until 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 seconds into the reaction at a lower potential of 1.4 V vs. RHE. It also shows strong agreement with the 1H-NMR data at 7500 and 12500 seconds. Peaks for benzoic acid (green) (62% FE), formate (blue) (35% FE), and acetophenone (30% FE) (maroon) can be found in the 1H-NMR spectra at 7500 seconds. The unreacted model complex II (yellow) makes up the remaining 36%.
000 seconds into the reaction at a lower potential of 1.4 V vs. RHE. It also shows strong agreement with the 1H-NMR data at 7500 and 12500 seconds. Peaks for benzoic acid (green) (62% FE), formate (blue) (35% FE), and acetophenone (30% FE) (maroon) can be found in the 1H-NMR spectra at 7500 seconds. The unreacted model complex II (yellow) makes up the remaining 36%.
 | ||
| Fig. 5 Electrochemical lignin-based model complex oxidation by activated Ni * sBDC/NF: (a) electrolysis curve of 1-phenyl ethanol oxidation at 20 mA cm−2, * shows the datapoints for NMR; (b) time-dependent 1H-NMR spectrum of 1-phenyl ethanol oxidation; (c) time-dependent 1H-NMR spectrum of 1-phenyl ethanol oxidation at 145 C, 175 C and 200 C at 1.40 V vs. RHE; (d) time-dependent 1H-NMR spectrum of 1-phenyl ethanol oxidation at 160 C and 200 C at 1.47 V vs. RHE. | ||
As the reaction continues, the 1H-NMR data at 12500 seconds reveal peaks for formate (blue) (36% FE), benzoic acid (green) (70% FE), and acetophenone (26% FE) (maroon), with just 9% of the model complex II (yellow) left. These observations lead to the conclusion that almost all of model complex II begins to convert to acetophenone at the beginning of the lower potential electrolysis curve. Concurrently, a portion of acetophenone undergoes oxidative C–C bond cleavage to produce formate and benzoate. In the electrolyte solution, the concentration of model complex II decreased, and the potential began to increase to generate 20 mA cm−2, thereby increasing the reaction kinetics of acetophenone oxidation into benzoate and formate. However, because of the possible potential increment, formic acid oxidation to carbonate also started taking place simultaneously. This is because, while acetophenone is less soluble in aqueous media, the kinetics of formic acid oxidation are comparable to those of acetophenone oxidation. This was further supported by 13C and 1H-NMR. The 1H-NMR at 34000 seconds suggests only 5% FE in acetophenone, 84% FE in benzoate, and only 1.6% FE in formate (Fig. S16a). A peak appears at 168 ppm in 13C-NMR as a result of the over-oxidation of formate into carbonate (Fig. S16b).
The overall yield for lignin model complex II electrooxidation at 34000 seconds by activated Ni BDC/NF is 13% for acetophenone and 66% for benzoate, along with carbonate, with 100% conversion. Also, constant potential electrochemical oxidation of 1-phenyl-ethanol was performed at 1.40 V vs. RHE and 1.47 V vs. RHE. The faradaic efficiency (FE) for 1.40 V vs. RHE was evaluated at three different charge-passing points (Fig. 5c and S17a). At 145 C, the FE values were 61.5% for benzoate, 7.65% for acetophenone, and 47% for formate, with no unreacted substrate remaining. Upon passing 170 C charge, the faradaic efficiencies were found to be 70% for benzoate, 6.36% for acetophenone, and 47% for formate, and at 200 C, 70% for benzoate, 3.4% for acetophenone and 43% for formate. The FE of electrochemical oxidation of 1-phenyl-ethanol at 1.47 V vs. RHE was evaluated at two different charge-passing points (Fig. 5d and S17b). At 160 C, the FE values were 77% for benzoate, 4% for acetophenone, and 7% for formate, with no unreacted substrate remaining. Upon passing an additional 40 C (total 200 C), the residual acetophenone was further oxidized to benzoate, resulting in an FE of 81% for benzoate and 2% for formate, with complete consumption of acetophenone. The overall yield of benzoate was determined to be 75% by using 1H-NMR. It is also evident that the overoxidation of formate is less at a milder potential. The catalytic performance of the activated Ni BDC/NF was also benchmarked against some recently reported literature for the electrooxidation of various lignin-model complexes (1-phenylethanol) and biomass (HMF), as summarized in Tables S1 and S4.
A lignin dimer model complex, 2-phenoxy-1-phenyl ethanol (lignin model complex III), was also tested for electrochemical oxidative valorization (Fig. 6a). 40 mM of the compound was dissolved in a solution of 1:1 acetonitrile to 0.2 M KOH aqueous solution since the compound is poorly soluble in aqueous media. Constant potential electrolysis at 1.5 V vs. RHE achieved a 90% conversion rate with a 78.33% yield of benzoic acid in just 16.5 hours (Fig. 6b and S18). Another substrate taken for screening was mesohydrobenzoin with C(OH)–C linkage. Owing to the lower solubility of the compound, only 9 mM was electrochemically oxidized with Ni BDC/NF, producing 79% benzoate with faradaic efficiency of 100% from constant current electrolysis of 10 mA cm−2 considering an 8 e-process (Fig. S19).
 | ||
| Fig. 6 . Electrochemical lignin-based model complex oxidation by activated Ni BDC/NF: (a) schematics of C–C bond cleavage for 2-phenoxy-1-phenyl ethanol; (b) 1H-NMR spectrum of the post-electrolysis sample of 2-phenoxy-1-phenyl ethanol. | ||
The reduction route after depolymerization of lignin affords KA oil, an important platform chemical (Fig. 7a). As KA oil also contains both C(OH)–C and C(O)–C functionalities, we expect our catalyst to be active for the electrooxidation of KA oil. The catalyst shows an onset of 1.34 V vs. RHE for 100 mM cyclohexanol oxidation in 1 M KOH (Fig. 7b). Constant current electrolysis at 20 mA cm−2 was performed to determine the product formation and selectivity (Fig. S20). For further understanding of the underlying reaction mechanism from cyclohexanol to adipate, a time-dependent 1H-NMR experiment was performed (Fig. 7c). It revealed that the cyclohexanol is converted into cyclohexanone first, where the cyclohexanone remains in its tautomeric form. Then this tautomer gets oxidized into adipate with 100% conversion and 54% yield. Moreover, this work provides an insightful method for the production of adipate, which is a monomer of Nylon-6,6.
 | ||
| Fig. 7 Electrochemical oxidation of KA oil obtained from reductive upcycling of lignin by activated Ni BDC/NF: (a) schematic illustration of lignin depolymerization to platform chemical generation; (b) LSV of 100 mM cyclohexanol oxidation (violet line), 1 M KOH (grey line) (no iR correction); (c) time-dependent 1H-NMR spectrum of cyclohexanol oxidation. | ||
Plastics have a negative carbon footprint regardless of whether they are biodegradable or not. In order to produce commodity chemicals, we examined the electrocatalytic activity of activated Ni BDC/NF for both the monomeric units of biodegradable and bio-nondegradable polymers, such as polylactic acid (PLA) and polyethylene terephthalate (PET), respectively. The hydrolysate of PLA contains lactate, and PET contains terephthalate and ethylene glycol. Terephthalate can be easily separated out as a solid terephthalic acid upon acid neutralization of the aqueous solution. However, separating ethylene glycol from the aqueous medium is a daunting task. Thus, electrocatalytic upcycling of aqueous EGOR to value-added formate is an impressive solution. Hence, the electrocatalytic activity of the monomeric structures of ethylene glycol and lactic acid was evaluated. Both monomers have C(OH)–C structures in them. The onset was moved to 1.36 V vs. RHE when 100 mM L-Lactic acid was introduced in 1 M KOH in the CV experiment. Only 1.56 V vs. RHE is needed by the catalyst to generate 100 mA cm−2 of current (Fig. 8a). Constant current electrolysis at 50 mA cm−2 was performed to obtain the product yield and selectivity of the lactic acid oxidation reaction, LAOR (Fig. 8b). The 1H-NMR spectra show that the post-electrolysis sample has a peak at 1.8 ppm, suggesting the production of acetic acid (Fig. 8c). Using 1, 4-dioxane as an internal standard, the FE and chemical yield of acetate are determined to be 100% and 91%, respectively. An additional peak attributed to carbonate at 168 ppm was detected during the analysis of the 13C-NMR of the analyte, indicating the oxidative C–C bond breaking (Fig. S21). We also tested the performance of the catalyst towards the electro-oxidation activity of ethylene glycol (EG), one of the monomers of PET plastic. For the EG oxidation reaction (EGOR), the catalyst only requires 1.35 V vs. RHE for the onset current, and to reach 100 mA cm−2 current density, it only requires 1.46 V vs. RHE (Fig. 8d), which exhibits significantly higher activity than the as-synthesized Ni BDC/NF and Ni(O)OH directly prepared from Ni foam (Fig. S22). For product quantification, constant current electrolysis was performed at 100 mA cm−2 (Fig. 8e). A steady curve was displayed by the catalyst throughout the electrolytic process. The formation of formate from the oxidation of ethylene glycol is responsible for the development of a substantial intensity peak at 8.3 ppm in the 1H-NMR of the electrolyte after electrolysis. When the product was quantified from the internal standard, the FE for formate was found to be 100% with a chemical yield of 77% (Fig. 8f). 13C-NMR provides additional confirmation of all 1H-NMR peaks (Fig. S23). Thus, we were able to oxidatively cleave the C(OH)–C link of the monomeric units of PLA and PET polymers in order to produce value-added chemicals. This was further employed for the electrooxidation of individual PLA and PET hydrolysate. For PLA hydrolysate oxidation (55 mg in 4 mL PLA in 1 M KOH, 0.84 mmol of LA), the catalyst shows 80% FE with 80% yield in 50 mA cm−2 chronopotentiometry for acetic acid formation. For PET hydrolysate oxidation (80 mg PET in 4 mL of 1 M KOH, 0.37 mmol of EG and TPA), the catalyst shows 75% FE with 74% yield in 100 mA cm−2 chronopotentiometry for formate production (Fig. S24). The electro-oxidation of a mixed plastic containing PLA and PET hydrolysate was also attempted. In this instance, 60 mg PET and 25 mg PLA were hydrolyzed in 1 M KOH to generate 0.35 mmol of EG and TPA, each with 0.372 mmol of LA, respectively. The catalyst shows an onset of 1.35 V vs. RHE, and to generate a 100 mA cm−2 current, it only requires 1.53 V vs. RHE. With a faradaic efficiency of 66% and 74%, respectively, formate and acetate were produced by constant current electrolysis at 100 mA cm−2. It was found that both had a chemical yield of over 50% within just 3 h. The acidification of the post-electrolysis sample afforded 100% terephthalic acid (Fig. S25).
 | ||
| Fig. 8 Electrochemical oxidation of a plastic-based model complex by activated Ni BDC/NF: (a) LSV of 100 mM lactic acid oxidation (maroon line) and 1M KOH (grey line) (no iR correction); (b) electrolysis curve of L-lactic acid oxidation reaction (LAOR) at 50 mA cm−2; (c) 1H-NMR spectrum of lactic acid oxidation before and after the completion of the reaction; (d) CV of 100 mM ethylene glycol oxidation (red line) and 1M KOH (grey line) (no iR corrected); (e) electrolysis curve of the ethylene glycol oxidation reaction (EGOR) at 100 mA cm−2; (f) 1H-NMR spectrum of ethylene glycol oxidation before and after the completion of the reaction. | ||
The post-electrolysis characterization of the activated Ni BDC/NF electrode was carried out using PXRD and SEM. The data revealed no apparent change in the morphology of the catalyst (Fig. S26). Also, the post-electrolysis sample was further examined by XPS, which displayed major peaks at 855.2 eV, attributed to the 2p3/2 state of Ni2+, and at 857.3 eV, corresponding to the 2p3/2 state of Ni3+ (Fig. S26a).29 The positive shift of binding energy for Ni3+ from 856.4 to 857.3 eV might be due to the formation of a more surface oxidized Ni(O)O–H phase during electrolysis. Additionally, the O 1s spectrum shows peaks at 532.9 eV, 531.2 eV, and 530.5 eV, corresponding to surface adsorbed H2O, Ni–OH, and Ni–O species, respectively (Fig. S27). The aNi BDC/NF shows two main peaks at 855.4 eV (Ni2+) and 856.8 eV (Ni3+), with a Ni3+/Ni2+ ratio of approximately 4.3:1. After electrolysis, the Ni 2p3/2 peaks appear at 855.2 eV (Ni2+) and 857.3 eV (Ni3+), and the Ni3+/Ni2+ ratio decreases markedly to 0.4:1, reflecting a much larger population of Ni2+ species in the post-catalysis sample. This indicates that the surface undergoes partial reduction and reconstruction during electrolysis, consistent with the formation of more stable Ni(OH)2-like phases under operating conditions. The recyclability of the catalyst is also tested, and it shows the retention of activity for at least 3 cycles (Fig. S28).
Dr H. Zhou et al. reported a bimetallic manganese-doped cobalt oxyhydroxide catalyst for a series of lignin-based model complexes and KA oil oxidation at constant potential electrolysis.30 J. Miao et al. also reported an Ir-doped Ir–NiFeO@NF catalyst for the valorization of some lignocellulose-based biomass upgradation with constant potential electrolysis.31 Xu J. et al. Reported a Mo@NiCoOOH nanorod catalyst to valorize 1 mM of 2-phenoxy-1-phenyl ethanol in an organic medium to produce benzaldehyde, methyl benzoate, and phenol.32 For plastic upcycling, Wang et al. designed a Cu-based CuO nanowire/Cu foam catalyst for the electro-upcycling of PET hydrolysate at constant potential electrolysis at 1.5 V vs. RHE to generate formate with 88% FE.33 Our group also reported a cobalt-based 1D coordination polymer for PET plastic upcycling to afford nearly 80% formate at 1.42 V vs. RHE.34 Another study by Liu. F reported the catalytic bifunctional activity of a multi-metallic catalyst, CuCo2O4/Ni foam, for constant potential electrolysis of the PETOR/HER.35 Multi-metallic nitrites, phosphides, and selenides are reported for PET and PLA electrooxidation.36–38 Some multi-metallic nitrides are also reported for bifunctional PET oxidation (Table S2).39,40 In this scenario, the present study provides a rationale based on the BDFE of the Ni(O)O–H for selectively oxidizing the structurally similar C(OH)–C or C(O)–C motifs present in both biomass and waste-plastic. However, there are limitations involved with the Ni(O)OH anode material as follows: it can undergo passivation due to the absorption of reaction intermediates, which often leads to an overoxidized product, carbonate, owing to C–C bond cleavage. From Pourbaix analysis, it is also observed that the Ni(O)OH phase is not stable at a higher potential, which is required for operation at a higher current density. Further, halides or other impurities present in complex feedstock environments may hamper the performance of these electrode materials.
We further intend to integrate (EG + LA) OR/HER and PEDOR/HER to create a bifunctional electrolytic platform because of the exceptional performance of activated Ni BDC/NF for various substrate electro-oxidation due to its good activity for the HER in 1 M KOH. During the HER, in 1 M KOH, the pristine Ni BDC/NF catalysts only require overpotentials of 170 mV and 440 mV to generate 10 mA cm−2 and 100 mA cm−2, respectively (Fig. S29). The bifunctionality of the system was explored using Ni BDC/NF as both the anode and cathode, where the anodic reaction is substrate oxidation and the cathodic reaction is hydrogen evolution. For the case study, 50 mM EG + 50 mM LA in 1 M KOH was taken as an analyte. 1H-NMR of the post-electrolysis sample revealed FE of 96.48% formate and nearly 100% acetate, along with simultaneous 100% FE of H2 gas generation (Fig. 9 and S30). Similarly, 100 mM PED in 1 M KOH was taken as an analyte in another case. Electrolysis at 100 mA cm−2 was performed for product quantification. 1H-NMR of the post-electrolysis analyte indicates the formation of benzoate and formate with 93.3% and 51% faradaic efficiency, respectively (Fig. S31). H2 gas production with 93% FE was observed during time-dependent gas chromatography analysis of the gaseous product (Fig. S31).
 | ||
| Fig. 9 Electrochemical oxidation of mixed plastic precursors with activated Ni BDC/NF as anode and cathode: (a) schematic of Ni BDC/NF as anode and cathode for (EG + LA) OR and HER, respectively; (b) 1H-NMR spectrum of (EG + LA) OR after the completion of the reaction; (c) time-dependent H2 quantification from (EG + LA) OR at 100 mA cm−2. | ||
Owing to the superior activity observed during the bifunctionality test with PEDOR/HER and (EG + LA) OR/HER with a 3-electrode system, a two-electrode configuration was subsequently employed, taking an activated Ni BDC/NF as anode and pristine Ni BDC/NF as cathode to evaluate the same transformation. For the lignin-model complex, PEDOR/HER was performed in 1 M KOH under the same two-electrode configuration. To achieve 100 mA cm−2, the system required an applied voltage of 2.0–2.4 V (Fig. S32a). The faradaic efficiencies were calculated to be 96% for benzoate and 82% for formate (Fig. S32b). The hydrogen evolution at the cathode also increased progressively with electrolysis time, with an overall faradaic efficiency of 89% (inset of Fig. S32a), with an energy efficiency of 62–74% (Higher Heating Value, HHV) and 51–61.5% (Lower Heating Value, LHV). Similarly, for the mixed plastic-based model complex oxidation (EG + LA) OR/HER at a current density of 100 mA cm−2 (Fig. S33a), the system required an applied voltage of 2.0–2.2 V. The faradaic efficiencies, determined from 1H-NMR analysis (Fig. S33b), were approximately 100% for formate and 85% for acetate. Concurrently, hydrogen evolution occurred at the cathode with a comparable faradaic efficiency of 95% (inset of Fig. S33a) with energy efficiencies of 67–74% (HHV) and 56–61.5% (LHV). The overall activity of the system was further benchmarked against similar reported systems, summarized in Table S3.
3. Experimental section
The electrode surface characterization was performed by Scanning Electron Microscopy (SEM) with a Jeol JSM7600F instrument, Powder X-ray Diffraction (PXRD) with a Bruker D8 DISCOVER instrument, and TEM with a Titan Themis d3898. SERS was done with 785 nm laser diode excitation with 10 mW power with 10 s laser exposure using a Raman probe (Kaisar Raman rxn System). The oxidation state of Ni was determined by X-ray photoelectron spectroscopy (XPS) with model AXIS SUPRA C332905/01.All the measurements were performed in a three-electrode system using a Metrohm Autolab PGSTAT 101 potentiostat. Ag/AgCl (saturated KCl) and a Pt wire were used as the reference and counter electrodes, respectively. All the potentials were converted to the normal hydrogen electrode (NHE) scale using the equation:
| ENHE (V) = EAg/AgCl + 0.197 |
or to a reversible hydrogen electrode (RHE) scale using the Nernst equation at room temperature:
| ERHE (V) = EAg/AgCl + 0.0592 × pH + 0.197 |
The scan rate was kept at 5 mV s−1 for cyclic voltammetry (CV) and linear sweep voltammetry (LSV) measurements. Constant current electrolysis was performed at room temperature and constant stirring in an undivided cell with a 0.5 × 0.5 cm2 Ni BDC/NF as the working electrode. During the mixed plastic and PED oxidation bifunctionality test for both the cathode and anode sides, 0.5 × 0.5 cm2 Ni BDC/NF was used and Ag/AgCl (saturated KCl) was used as a reference electrode.
0.1 M phosphate buffer adjusted to pH 7–12 was used as an electrolyte and for pH 14 and pH 13, 1 M KOH and 0.1 M KOH were used, respectively.
% Faradaic yield (FY) was determined from the following equations
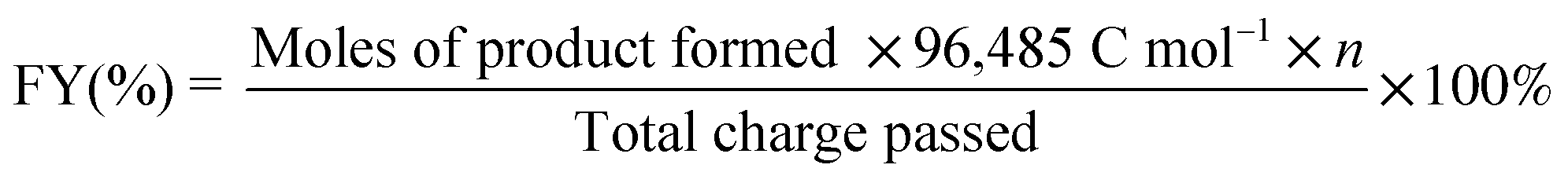
where n is the number of moles of electrons transferred per mole of product.
Electrochemical active surface area (ECSA) was determined from the following equation.
| ECSA = (Cdl/Cs) × Ageo |
During the coupled electrolysis experiments using a two-electrode system, activated Ni BDC/NF and pristine Ni BDC/NF were employed as the anode and cathode, respectively, in an undivided cell. The system was evaluated for both PED oxidation (PEDOR) and mixed plastic-based model compound oxidation. For PEDOR, the electrolyte consisted of 100 mM PED dissolved in 4 mL of 1 M KOH. For the model plastic oxidation, a mixture containing 50 mM ethylene glycol (EG) and 50 mM lactic acid (LA) in 4 mL of 1 M KOH was used as the electrolyte.
The energy efficiency of the cell was calculated using the following equation.41
| η = Ecell/Etheoretical × 100% |
The theoretical Ecell values were calculated from alkaline water electrolysers, as the standard electrode potentials for the substrates used in the two-electrode systems have not yet been reported.
The product yields were obtained through NMR (Bruker Avance III, 500 MHz), having dioxane as an internal standard in D2O.
The amount of produced hydrogen in the reactor headspace was analyzed using a gas chromatograph (CIC-Dhruva) equipped with a thermal conductivity detector (TCD).
4. Conclusions
In summary, this work presents a unified electrochemical approach for the simultaneous selective conversion of biomass and waste plastic into valuable chemicals and hydrogen by electrochemical oxidative C(OH)–C bond cleavage at high current densities with a Ni(O)O–H catalyst derived from Ni BDC/NF. This study explores various lignocellulose-based model complex substrates, including HMF, PED, 1-phenyl ethanol, meso-hydrobenzoin, 2-phenoxy-1-phenyl ethanol, and the lignin-derived substrate KA oil. Notably, for HMF and PED, our system achieved a high current density of 100 mA cm−2 at a remarkably low onset potential of approximately 1.4 V vs. RHE. Furthermore, time-dependent 1H-NMR studies provided insights into the possible reaction mechanisms for 1-phenyl ethanol and KA oil oxidation. Similarly, in the upgradation of mixed (biodegradable + non-biodegradable) waste plastics, we first evaluated the catalyst's performance using individual monomeric units before extending its application to real-life plastic waste. The correlation between the BDFEO–H of Ni(O)O–H and the BDFEC–H of the substrates, which generally get cleaved during the rate-limiting step, allowed us to facilitate a series of lignocellulose-based biomass and mixed plastic oxidation reactions. Using Ni(O)O–H as a bifunctional catalyst integrates biomass and plastic waste oxidation with H2 production in a single electrochemical system with a large current density of 100 mA cm−2, which was further extended to a two-electrode configuration to perform the same transformations at higher current densities (100 mA cm−2), enabling enhanced faradaic efficiencies for value-added product formation along with concurrent hydrogen generation. This not only addresses important environmental issues related to waste management, but it also provides a viable path for the production of valuable chemical products and clean energy. This unified approach represents a major step forward in the pursuit of sustainable and efficient resource utilization, paving the way for future developments in electrochemical upgradation and valorization techniques.Author contributions
B. M conceived and planned the experiments. S. B, A. N, and C. C performed the electrochemical experiments. G. J, S. B, C. C, D. S. K, and S. K. K performed the spectroscopic data collection and analysis. B. M, S. B and S. K. K analyzed the data. B. M and S. B wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Supplementary information (SI): additional CV data, electrolysis curves, and NMR data. See DOI: https://doi.org/10.1039/d5sc04651c.
Acknowledgements
B. M acknowledges an IIT Gandhinagar internal grant and SERB-SRG (SRG/2022/001736) for funding; S. B acknowledges IIT Gandhinagar; A. N and C. C acknowledge UGC-CSIR for fellowship. We acknowledge the CRTDH lab and Fire and Safety lab at IIT Gandhinagar for granting us access to the gas chromatography equipment and hydrothermal synthesis facilities, respectively.References
- P. Lanzafame, G. Centi and S. Perathoner, Chem. Soc. Rev., 2014, 43, 7562–7580 RSC.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS.
- S. K. Ritter, Lignocellulose: A Complex Biomaterial, Plant Biochem., 2008, 86, 15 Search PubMed.
- K. Lee, Y. Jing, Y. Wang and N. Yan, Nat. Rev. Chem., 2022, 6, 635–652 CrossRef PubMed.
- M. E. Zakrzewska, E. Bogel-Łukasik and R. Bogel-Łukasik, Chem. Rev., 2011, 111, 397–417 Search PubMed.
- D. Chen, W. Li, J. Liu and L. Sun, Energy Environ. Sci., 2025, 18, 3120–3128 Search PubMed.
- I. F. Demuner, J. L. Colodette, A. J. Demuner and C. M. Jardim, BioResources, 2019, 14, 7543–7581 Search PubMed.
- O. P. Verma, G. Manik and S. K. Sethi, Sustain. Energy, 2019, 100, 90–109 CrossRef.
- S. H. Choi, J. H. Lee, J. Yoo, J. H. Park, J. S. Bae and C. Y. Park, J. Ind. Eng. Chem., 2024, 129, 38–52 Search PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 Search PubMed.
- Z. Sun, G. Bottari, A. Afranasenko, M. C. A. Sturt, P. J. Deuss, B. Fridrich and K. Barta, Nat. Catal., 2018, 1, 82–92 CrossRef CAS.
- L. L. Landucci, S. Luque and S. Ralph, J. Wood Chem. Technol., 1995, 15, 493–513 CrossRef CAS.
- R. Vanholme, B. Demedts, K. Morreel, J. Ralph and W. Boerjan, Plant Physiol., 2010, 153, 895–905 CrossRef CAS PubMed.
- D. V. Evtuguin, C. P. Neto, A. M. Silva, P. M. Domingues, F. M. Amado, D. Robert and O. Faix Wood, J. Agric. Food Chem., 2001, 49, 4252–4261 CrossRef CAS PubMed.
- P. C. Rodrigues Pinto, E. A. Borges da Silva and A. E. Rodrigues, Ind. Eng. Chem. Res., 2011, 50, 741–748 CrossRef CAS.
- J. Li, Y. Song and Y. Cai, Environ. Pollut., 2020, 257, 113570 Search PubMed.
- R. T. Martin, L. P. Camargo and S. A. Miller, Green Chem., 2014, 16, 1768–1773 RSC.
- A. R. Bagheri, C. Laforsch, A. Greiner and S. Agarwal, Chall, 2017, 1, 1700048 Search PubMed.
- T. P. Haider, C. Völker, J. Kramm, K. Landfester and F. R. Wurm, Angew. Chem., Int. Ed., 2019, 58, 50–62 CrossRef CAS PubMed.
- G. X. Wang, D. Huang, J. H. Ji, C. Völker and F. R. Wurm, Adv. Sci., 2021, 8, 2001121 CrossRef CAS PubMed.
- C. Gioia, G. Giacobazzi, M. Vannini, G. Totaro, L. Sisti, M. Colonna, P. Marchese and A. Celli, ChemSusChem, 2021, 14, 4167–4175 CrossRef CAS.
- C. F. Wise and J. M. Mayer, J. Am. Chem. Soc., 2019, 141, 14971 CrossRef CAS.
- B. Mondal, N. Karjule, C. Singh, R. Shimoni, M. Volokh, I. Hod and M. Shalom, Adv. Energy Mater., 2021, 11, 2101858 CrossRef CAS.
- F. Cheng, X. Peng, L. Hu, B. Yang, Z. Li, C. L. Dong, J. L. Chen, L. C. Hsu, L. Lei, Q. Zheng, M. Qui, L. Dai and Y. Hou, Nat. Commun., 2022, 13, 6486 CrossRef CAS PubMed.
- E. Sadeghi, N. S. Peighambardoust, S. Chamani and U. Aydemir, ACS Mater. Au, 2022, 3, 143–163 CrossRef PubMed.
- D. Zheng, H. Wen, X. Sun, X. Guan, J. Zhang, W. Tian, H. Feng, H. Wang and Y. Yao, Chem.–Eur. J., 2020, 26, 17149–17155 CrossRef CAS.
- X. M. Zhu and Y. S. Zhang, Appl. Surf. Sci., 1998, 125, 11–16 Search PubMed.
- C. Chauhan, T. Gupta and B. Mondal, Small, 2025, 2410228 CrossRef CAS PubMed.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- H. Zhou, Z. Li, S. M. Xu, L. Lu, M. Xu, K. Ji, R. Ge, Y. Yan, L. Ma, X. Kong, L. Xheng and H. Duan, Angew. Chem., 2021, 133, 9058–9064 Search PubMed.
- J. Miao, Y. Ma, X. Wang, Y. Li, H. Wang, L. Zhang, J. Zhang, Y. Qin and J. Gao, Appl. Catal., B, 2023, 336, 122937 CrossRef CAS.
- J. Xu, J. Meng, Y. Hu, Y. Liu, Y. Lou, W. Bai, S. Dou, H. Yu and S. Wang, Research, 2023, 6, 0288 Search PubMed.
- J. Wang, X. Li, T. Zhang, Y. Chen, T. Wang and Y. Zhao, J. Phys. Chem. Lett., 2022, 13, 622–627 Search PubMed.
- S. Behera, S. Dinda, R. Saha and B. Mondal, ACS Catal., 2022, 13, 469–474 CrossRef.
- F. Liu, X. Gao, R. Shi, E. C. M. Tse and Y. Chen, Green Chem., 2022, 24, 6571–6577 Search PubMed.
- Y. Li, Y. Zhao, H. Zhao, Z. Wang, H. Li and P. Gao, J. Mater. Chem. A, 2022, 10, 20446–20452 RSC.
- H. Zhou, Y. Ren, Z. Li, M. Xu, Y. Wang, R. Ge, X. Kong, L. Zheng and H. Z. Duan, Nat. Commun., 2021, 12, 4679 CrossRef CAS.
- Z. Chen, N. Han, R. Zheng, Z. Ren, W. Wei and B. J. Ni, Advances and perspectives, SusMat, 2023, 3, 290–319 CrossRef CAS.
- X. Liu, Z. Fang, X. Teng, Y. Niu, S. Gong, W. Chen, T. J. Meyer and Z. Chen, J. Energy Chem., 2022, 72, 432–441 CrossRef CAS.
- X. Liu, Z. Fang, D. Xiong, S. Gong, Y. Niu, W. Chen and Z. Chen, Nano Res., 2023, 16, 4625–4633 CrossRef CAS.
- A. L. Santos, M. J. Cebola and D. M. Santos, Energies, 2021, 14, 3193 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |