
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTotal syntheses of ent-aogacillin A and aogacillin B
Huixing
Gu†
a,
Ziyi
Li†
a,
Xinwei
Zhang
b,
Ruocheng
Sang
b,
Zhendong
Li
a,
Jinyi
Ren
a,
Xiaojing
Chen
a,
Boya
Ma
a,
Wenhao
Qiu
a,
Zebin
Yang
a,
Xiaoyan
Li
 *a,
Rongbiao
Tong
*b and
Wei
Zhang
*a
*a,
Rongbiao
Tong
*b and
Wei
Zhang
*a
aHebei Technology Innovation Center for Energy Conversion Materials and Devices, College of Chemistry and Material Science, Hebei Normal University, No. 20, East Road of Nan Er Huan, Shijiazhuang 050024, China. E-mail: zhangwei@hebtu.edu.cn
bDepartment of Chemistry, The Hong Knog University of Science and Technology, Clearwater Bay, Kowloon, Hong Kong, China. E-mail: rtong@ust.hk
First published on 26th November 2025
Abstract
Aogacillins A and B, circumventors of arbekacin resistance in MRSA, possess a densely oxidized spirolactone moiety and an electron deficient terminal exocyclic double bond. This represents a challenging target for total synthesis. Herein, the evolution of a successful strategy for the total synthesis of ent-aogacillin A and aogacillin B is described. Although the spirocyclic skeleton could be efficiently constructed by the Achmatowicz rearrangement-based strategy, the elaboration of the dihydropyranone moiety to the fully functionalized lactone of aogacillins was not successful. A new strategy is proposed and successfully implemented, which features allylic C–H oxidation and a one-pot sequential aldol reaction and lactonization. This enables us to achieve the first and asymmetric total synthesis of aogacillin B and ent-aogacillin A in 7 steps.
Introduction
Aogacillins A and B (1a and 1b, Scheme 1) were isolated by Shiomi and co-workers in 2013 from a culture broth of Simplicillium sp. FKI-5985 and have been found to be capable of overcoming arbekacin (ABK) resistance in methicillin-resistant Straphylococcus aureus (MRSA).1 Arbekacin, a clinically used, potent antibiotic for the treatment of infections caused primarily by multi-resistant bacteria such as MRSA, has recently begun to suffer from much resistance, possibly due to phosphorylation or acetylation by bacterial aminoglycoside-modifying enzymes.2 Aogacillins were found to inhibit the growth of MRSA with a MIC value of 2.0 µg mL−1 and considerably reduce the MIC value of arbekacin against arbekacin-resistant MRSA from 256 µg mL−1 to 8 µg mL−1, which suggested that aogacillins could be specific circumventors for ABK-resistant MRSA. It was also reported that aogacillins could be used to prepare drugs for treating renal insufficiency,3 renal carcinoma,4 breast cancer,5 Alzheimer's disease6 and type-2 diabetes.7 Therefore, they hold great potential for clinical applications. Structurally, aogacillins represent an unusual δ-lactone with a spiro-fused 2-ethyl-6-methylcyclohexane, and their highly dense functionalities (especially the continuous high oxidation states) on the δ-lactone pose a major challenge for their chemical synthesis. The terminal exo-cyclic alkene conjugated with the carbonyl, acting as a Michael acceptor, also makes these compounds unstable. Aogacillin A (1a) differs structurally from aogacillin B (1b) only at C3 stereochemistry, which makes their separation very difficult as evident from NMR spectra of 1b containing residues from those of 1a. The total syntheses of both aogacillins A and B have not been reported so far. Herein, we describe our efforts to achieve the first total synthesis of aogacillin B and the enantiomer of aogacillin A.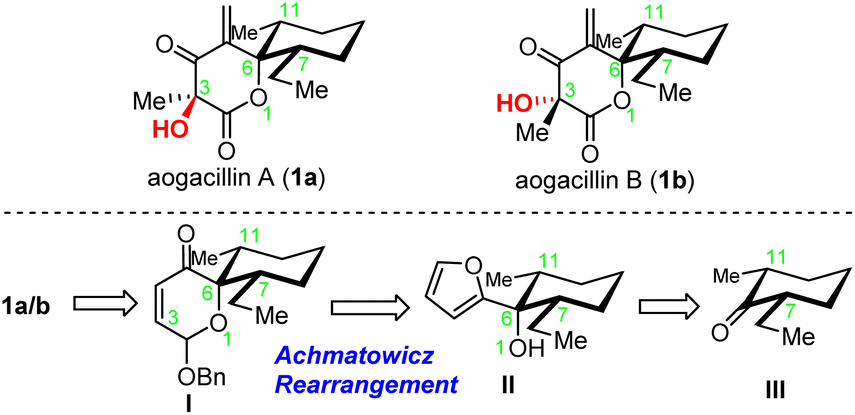 | ||
| Scheme 1 Aogacillins A and B and retrosynthetic analysis. | ||
Results and discussion
Our original retrosynthetic analysis was proposed in 2013 and hinged on the Achmatowicz rearrangement as depicted in Scheme 1. The fully functionalized δ-lactone of aogacillins A and B could be generated from the dihydropyranone acetal moiety of intermediate I, which was derived from the Achmatowicz rearrangement of furfuryl alcohol II. The addition of 2-lithiofuran to cis-2-ethyl-6-methyl-cyclohexanone delivered II as the substrate for the Achmatowicz rearrangement. It should be noted that the Achmatowicz rearrangement has been applied as the key strategic transformation in the total syntheses of various natural products containing tetrahydropyrans,8 spiroketals,9 and oxa-bridged bicycles10 and we have extensive experience in the exploitation of Achmatowicz rearrangement.As shown in Scheme 2, our synthesis commenced with the preparation of cis-2-ethyl-6-methyl-cyclohexanone 5. Claisen condensation of ethyl formate with commercially available 2-methyl cyclohexanone 2 (ref. 11) was followed by enamine formation and the Benary reaction12 to deliver E-ethylidene ketone 4.13 Hydrogenation with Pd/C in ethyl acetate gave the desired cis product with high yield and good diastereoselectivity. This scalable four-step-one-purification sequence provided cis-2-ethyl-6-methyl-cyclohexanone 5 (ref. 14) in multigram quantities in a single batch. Addition of lithiated furan to 5 afforded the single diastereomeric compound 6 as the substrate for the Achmatowicz rearrangement, which occurred smoothly with m-chloroperoxybenzoic acid (mCPBA) to provide the spiro-dihydropyranone acetal framework.15 Protection of the labile acetal as benzyl ether delivered 7 with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric ratio at the acetal carbon. The excellent scalability of the Achmatowicz rearrangement allowed us to prepare 7 on a 30-gram scale.
:1 diastereomeric ratio at the acetal carbon. The excellent scalability of the Achmatowicz rearrangement allowed us to prepare 7 on a 30-gram scale.
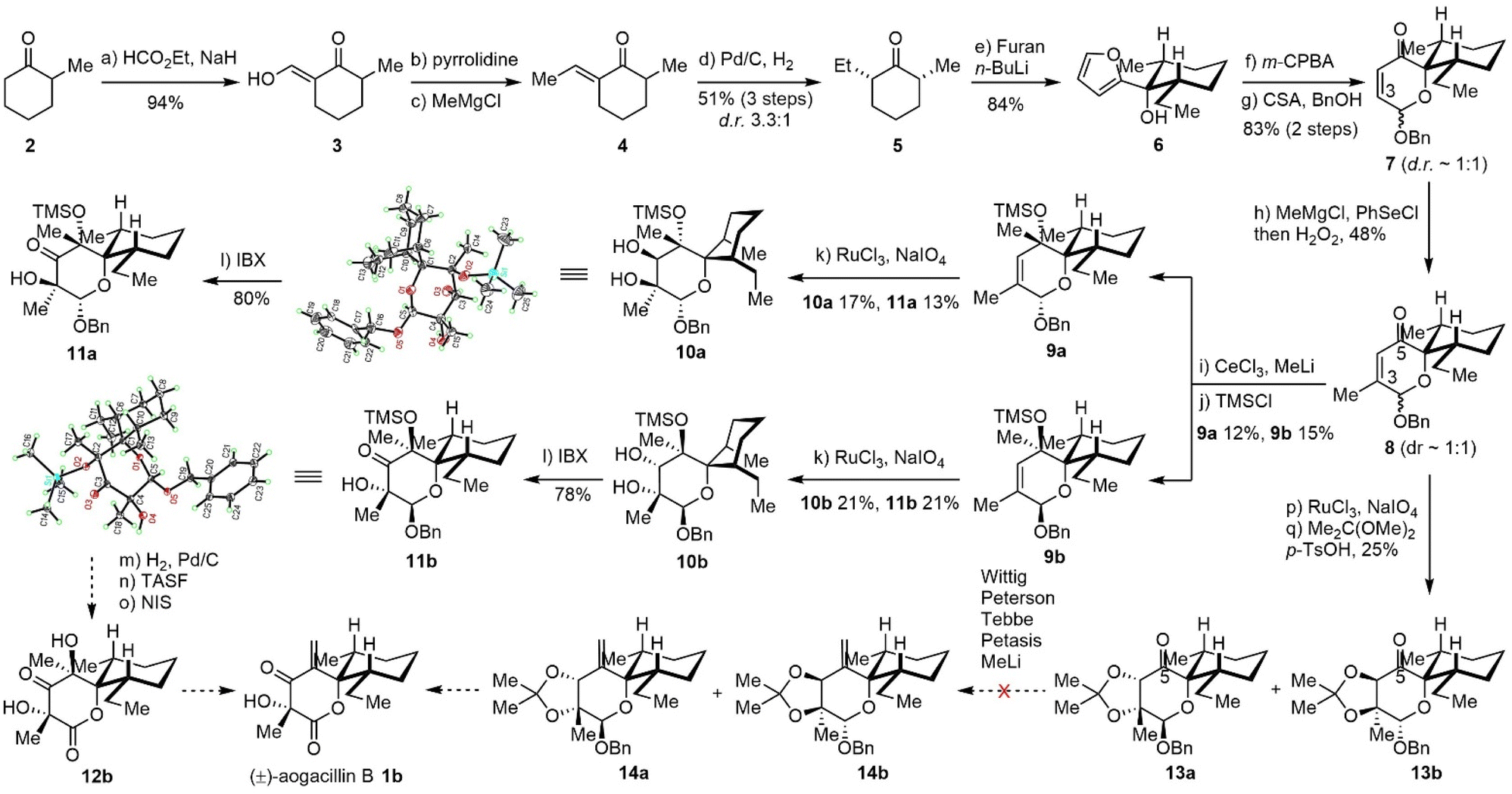 | ||
| Scheme 2 Synthetic efforts towards aogacillins A and B based on the Achmatowicz rearrangement strategy. | ||
With a reliable supply of spiro-framework 7 in hand, we set out to functionalize the dihydropyranone into the lactone corresponding to aogacillins. Installation of the methyl group at C3 was performed with a methyl Grignard reagent followed by PhSeCl/H2O2 (ref. 16) to afford compound 8 in 48% yield. CeCl3 mediated nucleophilic 1,2-addition17 of MeLi to 8 delivered inseparable products with poor yield. Improvement of the yield was attempted without success due to the extraordinary steric hindrance from the adjacent quaternary carbon (similar to the neopentyl effect). After TMS protection of the tertiary alcohol, the diastereoisomers 9a and 9b could be separated by flash column chromatography on silica gel and used individually in the subsequent reactions. The Upjohn dihydroxylation was found to proceed slowly with low conversion even after 5 days. Fortunately, ruthenium-catalyzed syn-dihydroxylation18 could afford the desired diols 10a and 10b as well as over oxidized hydroxyketones 11a and 11b in moderate yields. The diols 10a/10b could also be oxidized to hydroxyketones using IBX. The structures of 10a (CCDC 2464462) and 11b (CCDC 2464461) were further confirmed by X-ray crystallographic analysis. It is interesting to note that the methyl and ethyl groups on the cyclohexane moiety of 11b are in the equatorial positions based on the X-ray data, while the hydroxyketone 10a places the methyl and ethyl groups in the unfavorable axial positions. With 11a and 11b in hand, we tried to accomplish the total synthesis by transforming the benzyl acetal to lactone as well as dehydrating the C-5 tertiary alcohol to a terminal alkene. However, we encountered unexpected difficulty in dehydration under various conditions and suspected that the adjacent carbonyl might be responsible for the failure of dehydration. We also tried to perform the dihydroxylation from 8 and protection as acetonides 13a/13b. Then olefination was tested using the Wittig reagent, Peterson conditions (TMSCH2MgCl),19 Tebbe reagent,20 and Petasis reagent,21 but unfortunately, none of them could deliver the desired olefin. Methyl addition and dehydration did not work either due to decomposition. This failure forced us to re-design a new synthetic strategy that was not based on the Achmatowicz rearrangement.
We recognized that there was one major obstacle in the Achmatowicz rearrangement-based strategy: unsuccessful olefination of the C5 carbonyl due to the extraordinary steric hindrance and electronic effects of the neighboring functional groups. In order to avoid this problem, we proposed to install this terminal alkene at an early stage through vinyl metallic reagent addition to cis-2-ethyl-6-methyl-cyclohexanone (Scheme 3a, III + VII →VI). The δ-lactone could be constructed by aldol condensation of lactic acid derivative V and β-hydroxyl-α-methylene-aldehyde VI followed by lactonization.
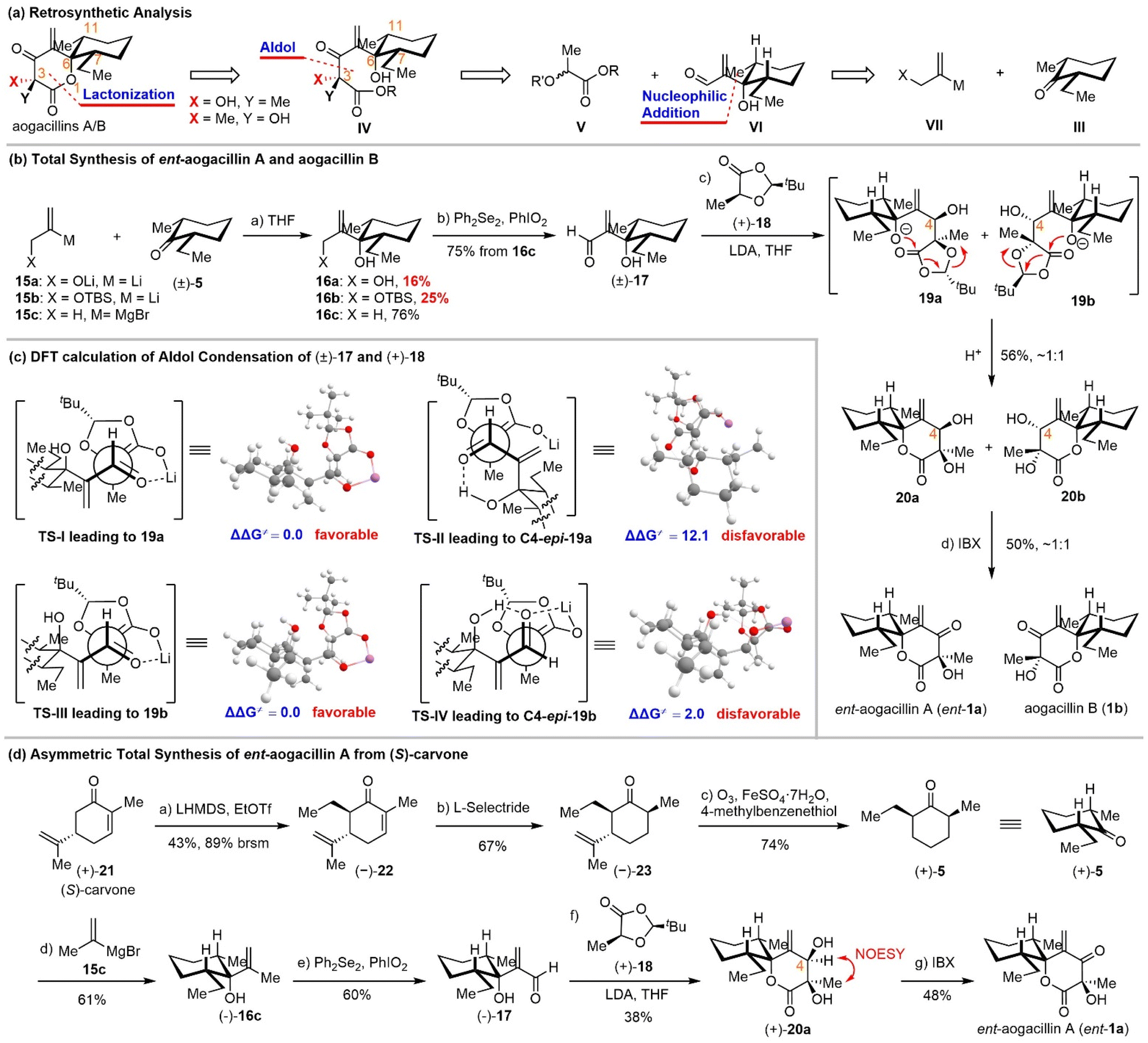 | ||
| Scheme 3 Total syntheses of aogacillin B and ent-aogacillin A. | ||
Initially, 2-bromoallyl alcohol and TBS protected 2-bromoallyl alcohol were employed for lithium–halogen exchange to generate in situ the requisite alkenyl lithium reagents 15a/15b, which were added to cis-2-ethyl-6-methyl-cyclohexanone 5 (Scheme 3b). Unfortunately, both reactions gave 16a/16b in poor yields (16% and 25%, respectively). We suspected that the lithium alkoxide might aggregate in the solution and disfavor the nucleophilic addition to the bulky enolizable ketone 5. If tert-butyldimethylsilyl (TBS) ether 15b was used as the protecting group, the propargyl addition product became the major byproduct. To improve this nucleophilic addition, we chose the isopropenyl Grignard reagent 15c for the addition to cis-2-ethyl-6-methyl-cyclohexanone 5 and performed an allylic oxidation with Ph2Se2/PhIO2 (ref. 22) to obtain the aldehyde 17 in 76% yield. It is noteworthy that the allylic oxidation with SeO2 under conventional conditions23 proceeded in only 15% yield. Next, aldol condensation of racemic aldehyde 17 and Seebach–Fráter chiral 1,3-dioxolan-4-one 18 (prepared through condensation of (S)-lactic acid and 2,2-dimethylpropanal)24 afforded an inseparable 1:1 mixture of lactones 20a and 20b, which might be derived from an intramolecular lactonization (19a/b → 20a/b). The subsequent IBX oxidation of 20a/20b furnished a mixture of aogacillin B and the enantiomer of aogacillin A, which were finally separated by reversed-phase preparative HPLC. The spectroscopic data (1H NMR, 13C NMR, and HRMS) and specific rotation of our synthetic materials were consistent with those reported for the natural products (Tables S1 and S2). It should be noted, however, that the HPLC separation was very inefficient. Only 5.8 mg of pure ent-aogacillin A and 4 mg of pure aogacillin B could be obtained from 40 mg of their mixture.
The accomplishment of the first total synthesis of aogacillins warranted some comments. First, the strategic use of Seebach–Fráter chiral 1,3-dioxolan-4-one 18 for the aldol reaction with racemic aldehyde was expected to deliver enantiomerically pure aogacillins if the diastereomeric products 20a/20b could be separated through column chromatography. This idea was achieved finally by chiral HPLC and thus our total synthesis is asymmetric. Second, β-hydroxyl-α-methylene aldehyde (i.e., 17) was employed for the first time in the aldol reaction with 1,3-dioxolanones and the concomitant lactonization, which offers a new venue for the synthesis of functionalized δ-lactones. Third, stereochemical outcomes from the sequential aldol reaction/lactonization are intriguing because only two diastereomers were isolated among eight possible stereoisomers (Scheme 3c). This suggested that the aldol reaction of 17a/b with 18 was highly diastereoselective. Density functional theory (DFT) calculations were performed to predict the configurations of the newly formed C-4 hydroxyl groups. DFT calculations showed that the energy barrier of transition state TS-II leading to C4-epi-19a is higher than that of TS-I leading to 19a; meanwhile, the Gibbs free energy of C4-epi-19a is also higher than that of 19a. This indicates that 19a is favored both kinetically and thermodynamically (see the SI). The energy barrier of transition state TS-IV leading to C4-epi-19b is higher than that of TS-III leading to 19b; however, the Gibbs free energy of C4-epi-19b is lower than that of 19b. (see the SI) This indicates that 19b is favored kinetically and C4-epi-19b is favored thermodynamically. As we conducted the reaction at −78 °C, the kinetically controlled product 19b should be the major product. Besides, it should be noted that this calculated stereochemical outcome is different from those reported by Seebach24 and Battaglia,25 but is consistent with those predicted by Zimmerman–Traxler models, although the β-hydroxyl aldehyde would form intramolecular hydrogen bonds.
Since a mixture of aogacillin B and ent-aogacillin A was obtained due to the use of racemic cis-2-ethyl-6-methyl-cyclohexanone (±)-5 and HPLC separation was required, we proposed to synthesize optically active (or pure) cis-2-ethyl-6-methyl-cyclohexanone 5 and expected to achieve the asymmetric synthesis of either aogacillin A or aogacillin B. To this end, we employed (S)-carvone as the chiral non-racemic starting material for the preparation of (+)-5 (Scheme 3d). α-Alkylation of (S)-carvone with ethyl triflate afforded the trans ethyl addition product (−)-22 with excellent diastereoselectivity. Although the conversion was low even using 5 equivalents of ethyl triflate, (S)-carvone could be recycled. The conjugate reduction with L-selectride could give (−)-23 in 67% yield with high diastereoselctivity. Under Kwon's hydrodealkenylation conditions,26 enantio-pure cis-2-ethyl-6-methyl-cyclohexanone (+)-5 could be prepared smoothly in 74% yield. Following the Grignard addition and allylic oxidation procedures as for the racemic substrate, enantiopure 17 was synthesized. The next aldol condensation with Seebach–Fráter chiral 1,3-dioxolan-4-one 18 afforded enantiopure lactone 20a. The absolute configuration of the C-4 hydroxyl group was confirmed by the NOESY spectrum. This was consistent with the DFT calculation results. Final oxidation with IBX accomplished the enantioselective synthesis of ent-aogacillin A.
Conclusions
In summary, we have explored two synthetic strategies for the total synthesis of the bioactive natural products aogacillins and achieved the first total synthesis of ent-aogacillin A and aogacillin B in 7 steps. The first strategy hinges on Achmatowicz rearrangement but fails to furnish aogacillins due to the unsuccessful olefination of C5 ketone which is sterically hindered and enolizable. To overcome this challenge, we redesigned our synthetic strategy which features the installation of the terminal alkene at an early stage and the construction of the spirocyclic δ-lactone scaffold through a newly developed diastereoselective one-pot aldol reaction and concomitant lactonization, which, as a new method, might be applicable to the synthesis of other fully functionalized δ-lactones and δ-lactone-containing natural products.Author contributions
H. G., Z. L., R. S., J. R., X. C., B. M., W. Q., Z. Y. and W. Z. performed the synthetic experiments. H. G. and Z. L. contributed equally to this work. X. Z. conducted the HPLC isolation. Z. L. performed the DFT calculations. W. Z., R. T. and X. L. supervised and provided guidance to the project. W. Z. drafted the manuscript and SI. R. T. revised the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
CCDC 2464461 (11b) and 2464462 (10a) contain the supplementary crystallographic data for this paper.27a,bExperimental procedures and characterization data are available within this article and its supplementary information (SI). Data are also available from the corresponding author on request. Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc04554a.
Acknowledgements
This research was financially supported by the Hebei Natural Science Foundation (Grant No. B2023205007), funding from the Hebei Provincial Department of Human Resources and Social Security (Grant No. C20230340), funding from Hebei Normal University (Grant No. L2023B17) and the HKUST and Research Grants Council of Hong Kong (16308922, 16304023, 16303524, and C6022-22W).Notes and references
- K. Takata, M. Iwatsuki, T. Yamamoto, T. Shirahata, K. Nonaka, R. Masuma, Y. Hayakawa, H. Hanaki, Y. Kobayashi, G. A. Peterson, S. Omura and K. Shiomi, Org. Lett., 2013, 15, 4678–4681 CrossRef CAS.
- (a) M. Tabata, M. Shimizu, M. Araake and H. Ogawa, Jpn. J. Antibiot., 2003, 56, 36–43 CAS; (b) K. Ishino, J. Ishikawa, Y. Ikeda and K. Hotta, J. Antibiot., 2004, 57, 679–686 CrossRef CAS.
- L. Tian, CN Pat., CN105326830A, 2016 Search PubMed.
- L. Tian, CN Pat., CN105380942A, 2016 Search PubMed.
- M. Zhao, CN Pat., CN105663121A, 2016 Search PubMed.
- L. Tian, CN Pat., CN105380943A, 2016 Search PubMed.
- L. Tian, CN Pat., CN105343057A, 2016 Search PubMed.
- (a) K. L. Jackson, J. A. Henderson, H. Motoyoshi and A. J. Phillips, Angew. Chem., Int. Ed., 2009, 48, 2346–2350 CrossRef CAS PubMed; (b) K. C. Nicolaou, R. J. Aversa, J. Jin and F. Rivas, J. Am. Chem. Soc., 2010, 132, 6855–6861 CrossRef CAS PubMed; (c) A. T. Herrmann, S. R. Martinez and A. Zakarian, Org. Lett., 2011, 13, 3636–3639 CrossRef CAS PubMed; (d) H. Takamura, K. Tsuda, Y. Kawakubo, I. Kadota and D. Uemura, Tetrahedron Lett., 2012, 53, 4317–4319 CrossRef CAS; (e) J. A. Gazaille, J. A. Abramite and T. Sammakia, Org. Lett., 2012, 14, 178–181 CrossRef CAS PubMed; (f) A. K. Ghosh and Z. Chen, Org. Lett., 2013, 15, 5088–5091 CrossRef CAS PubMed; (g) L. Zhu, Y. Liu, R. Ma and R. Tong, Angew. Chem., Int. Ed., 2015, 54, 627–632 CrossRef CAS PubMed; (h) L. Zhu and R. Tong, Org. Lett., 2015, 17, 1966–1969 CrossRef CAS PubMed.
- (a) J. Robertson, C. North and J. E. R. Sadig, Tetrahedron, 2011, 67, 5011–5023 CrossRef CAS; (b) L. Zhu, L. Song and R. Tong, Org. Lett., 2012, 14, 5892–5895 CrossRef CAS; (c) Y. Wang and G. A. O'Doherty, J. Org. Chem., 2022, 87, 6006–6013 CrossRef CAS PubMed.
- (a) J. Ren, J. Wang and R. Tong, Org. Lett., 2015, 17, 744–747 CrossRef CAS; (b) M. A. Márquez-Cadena, J. Ren, W. Ye, P. Qian and R. Tong, Org. Lett., 2019, 21, 9704–9708 CrossRef; (c) L.-D. Guo, Z. Xu and R. Tong, Angew. Chem., Int. Ed., 2021, 61, e202115384 CrossRef; (d) K. Takahashi, R. Harada, Y. Hoshino, T. Kusakabe, S. Hatakeyama and K. Kato, Tetrahedron, 2017, 73, 3548–3553 CrossRef CAS; (e) N. Hauser, M. A. Imhof, S. S. Eichenberger, T. Kundig and E. M. Carreira, Angew. Chem., Int. Ed., 2022, 134, e202112838 CrossRef.
- X. Yu, J. Hu, Z. Shen, H. Zhang, J. Gao and W. Xie, Angew. Chem., Int. Ed., 2017, 56, 350–353 CrossRef CAS PubMed.
- F. Näf and R. Decorzant, Helv. Chim. Acta, 1974, 57, 1309–1317 CrossRef.
- R. E. Ireland and P. W. Schiess, J. Org. Chem., 1963, 28, 6–16 CrossRef CAS.
- T. Miyoshi, T. Miyakawa, M. Ueda and O. Miyata, Angew. Chem., Int. Ed., 2011, 50, 928–931 CrossRef CAS PubMed.
- (a) C. U. Dinesh, P. Kumar, R. S. Reddy, B. Pandey and V. G. Puranik, Tetrahedron: Asymmetry, 1995, 6, 2961–2970 CrossRef CAS; (b) M. P. Georgiadis, A. Tsekouras, S. I. Kotretsou, S. A. Haroutounian and M. G. Polissiou, Synthesis, 1991, 929–932 CrossRef CAS; (c) S. D. Koulocheri, S. A. Haroutounian, C. D. Apostolopoulos, R. K. Chada and E. A. Couladouros, Eur. J. Org Chem., 1999, 1449–1453 CrossRef CAS; (d) J. Yu, H. Ma, H. Yao, H. Cheng and R. Tong, Org. Chem. Front., 2016, 3, 714–719 RSC.
- H. J. Reich and S. Wollowitz, Org. React., 2004, 44, 1–296 Search PubMed.
- (a) C. R. Johnson and B. D. Tait, J. Org. Chem., 1987, 52, 281–283 CrossRef CAS; (b) Y. Sun, R. Li, W. Zhang and A. Li, Angew. Chem., Int. Ed., 2013, 52, 9201–9204 CrossRef CAS PubMed; (c) M. Kurosu and Y. Kishi, Tetrahedron Lett., 1998, 39, 4793–4796 CrossRef CAS.
- B. Plietker and M. Niggemann, J. Org. Chem., 2005, 70, 2402–2405 CrossRef CAS.
- D. J. Ager, Org. React., 2004, 38, 1–223 Search PubMed.
- F. N. Tebbe, G. W. Parshall and G. S. Reddy, J. Am. Chem. Soc., 1978, 100, 3611–3613 CrossRef CAS.
- N. A. Petasis and E. I. Bzowej, J. Am. Chem. Soc., 1990, 112, 6392–6394 CrossRef CAS.
- D. H. R. Barton and D. Crich, Tetrahedron, 1985, 41, 4359–4364 CrossRef CAS.
- (a) G. M. Strunz, R. Bethell, G. Sampson and P. White, Can. J. Chem., 1995, 73, 1666–1674 CrossRef CAS; (b) D. R. Williams, P. T. Gladen and J. R. Pinchman, J. Org. Chem., 2015, 80, 5474–5493 CrossRef CAS PubMed.
- D. Seebach, R. Naef and G. Calderari, Tetrahedron, 1984, 40, 1313–1324 CrossRef.
- A. Battaglia, G. Barbaro, P. Giorgianni, A. Guerrini, C. Bertucci and S. Geremia, Chem.–Eur. J., 2000, 6, 3551–3557 CrossRef CAS.
- A. J. Smaligo, M. Swain, J. C. Quintana, M. F. Tan, D. A. Kim and O. Kwon, Science, 2019, 364, 681–685 CrossRef CAS PubMed.
- (a) CCDC 2464461: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2nqgrm; (b) CCDC 2464462: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2nqgsn.
Footnote |
| † Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |