DOI:
10.1039/D6RA04227A
(Paper)
RSC Adv., 2026,
16, 29981-29995
Decoding the hydrogen storage and functional properties of MgBH3 (B = Mo and In) via first-principles simulations
Received
15th May 2026
, Accepted 22nd May 2026
First published on 2nd June 2026
Abstract
This present study presents a complete first-principles investigation of the structural, hydrogen storage, optoelectronic, mechanical, and thermodynamic properties of MgBH3 (B = Mo, In). Cubic phase structural stability was determined by formation energies, Goldschmidt tolerance factors, and octahedral factors. Hydrogen storage capacities were calculated as 2.45 wt% for MgMoH3 and 2.13 wt% for MgInH3, indicating reasonable desorption suitable for real-world energy storage. Electronic structure calculations (GGA-PBE) determine metallic conductivity as a result of valence and conduction band overlap. Optical analyses show that the high refractive index and strong absorption of MgBH3, combined with their metallic nature, ensure efficient charge transport and lattice stability. Mechanical stability is confirmed by the elastic constants satisfying Born's criteria. This stability, coupled with their distinct ductile nature, ensures robust structural integrity and prevents microcracking, making repeated hydrogen cycling highly stable. Strong elastic anisotropy is indicative of the directional dependence of hydride perovskites. Thermodynamic assessment: Debye temperature, lattice and minimum thermal conductivities, and Grüneisen parameter offer insight into phonon transport and heat capacity, confirming their utility as thermal barrier coatings at high temperatures. In short, MgBH3 (B = Mo, In) hydrides are multifunctional materials exhibiting moderate hydrogen-storage capability, robust mechanical stability, favorable thermal-management characteristics, and distinct dielectric and metallic optical responses.
1. Introduction
As the global population continues to rise, so too has the quantity of energy used worldwide. This increase stems from the increased energy needs caused by technical progress and advances in science aimed at providing modern amenities for society. Fossil fuels, which are non-renewable resources that take a long time to replenish or return to their original state, provide a sizable portion of the world's energy.1 Since carbon-based combustion and the reliance on inefficient fossil fuels have been linked to nearly a 35% increase in CO2 emissions, hydrogen is gaining attention as a promising alternative source of energy due to its abundance, non-toxicity, high efficiency, and long-term sustainability.2 Hydrogen-based energy systems were launched after the global oil crisis of the 1970s, and they have the potential to revolutionize the transportation and power engineering industries.3 As the most abundant element in the universe and the lightest fuel available, hydrogen is recognized as a highly effective energy carrier,4 with certain metal hydride perovskites demonstrating efficiency improvements from around 3.8% to above 20%.5 One of the first uses of hydrogen as a reserve fuel was the first human voyage to the moon, and several nations are now using it for transportation.6 Materials that can store hydrogen at room temperature and pressure while attaining high volumetric and gravimetric hydrogen densities are the subject of the current study.7 The 2019 launch of Hyundai's Nexo fuel cell vehicle, which sold over 1000 units in South Korea and demonstrated the growing market acceptability of hydrogen-based transportation options, is a concrete example of this expanding trend toward hydrogen utilization commercially.8 High volumetric and gravimetric ratios, quick reaction kinetics, robust mechanical stability, and the capacity to release hydrogen under normal operating conditions are all expected to be characteristics of ideal energy storage materials.9 To industrially implement hydrogen as a sustainable energy source, a range of technical challenges must be addressed and overcome.10 Hydrogen has been increasingly adopted worldwide as an alternative clean fuel because it generates energy without producing carbon dioxide, with water being its only by-product, free from pollutants, ozone-depleting agents, or harmful synthetic chemicals.11 However, storing hydrogen efficiently continues to be a significant challenge compared to conventional methods. Compression increases safety concerns and restricts the amount of hydrogen that can be contained under high pressure,12 whereas liquefaction, despite saving storage space, raises the risks of evaporation and emission losses,13 making the search for safer and more cost-effective storage materials crucial.14 Extensive experimental and theoretical studies in recent years have evaluated their potential for efficient hydrogen storage.15 For these materials to be useful in the long run, they must fulfill a number of important criteria.16 ABH3 compounds can generally be classified according to the elements they contain: an A-site element from group I or II of the periodic table and a B-site element from the transition metal group, which was first discovered by German scientist Gustav Rose in the Ural Mountains in 1839.17 In the general ABX3 formula, A denotes an alkali metal, B a transition metal, and X represents oxygen or a halogen element commonly forming a cubic lattice within the Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m crystallographic space group.18
m crystallographic space group.18
According to research, oxides rather than hydrides are frequently formed as a result of hydrogen's poor stability and cation restrictions. Nonetheless, lightweight metal hydrides have garnered interest because of their high gravimetric densities, reversible absorption/desorption behavior, and exceptional thermal stability, which make them attractive options for real-world hydrogen storage applications.19 However, some inherent characteristics, such as high formation enthalpy, strong thermodynamic stability, sluggish kinetics of hydrogen absorption and desorption, and vulnerability to oxidation in air, restrict the practical application of magnesium hydrides.20
CaXH3 (X = Mn, Fe, and Co) hydride-type perovskites have been studied in the past to assess their feasibility for storing hydrogen.21 Theoretical investigations point out that MgBH3 and MgNH3 perovskite materials can gain hydrogen storage levels well above the Department of Energy's (DOE) target of 5.5 wt% for onboard applications, achieving roughly 15.4 wt% and 14.9 wt% respectively.22 Magnesium-based perovskite hydrides can also be prepared experimentally using a variety of synthesis approaches, including solution-based techniques, ball milling, and conventional solid-state methods. Siddique et al.23 also reported that AeVH3 (Ae = Be, Mg, Ca, Sr) perovskite hydrides exhibit ionic bonding with no bandgap, possess gravimetric hydrogen storage capacities of 3.45, 3.52, 3.60, and 3.65 wt% for BeVH3, MgVH3, CaVH3, and SrVH3, respectively.
The growing need for effective hydrogen storage and sophisticated optoelectronic materials underscores the need to explore additional members of this material class, even in spite of significant progress in the study of magnesium-based perovskites. The current study offers a first-principles density functional theory (DFT) investigation of the structural, elastic, thermodynamic, optoelectronic, and hydrogen storage features of MgBH3 (B = Mo, In), motivated by its unique physical properties and exciting potential uses. The goal of this theoretical investigation is to establish a thorough understanding of these materials and evaluate their applicability for real-world energy storage and electronic device applications.
2. Computational details
In this investigation, the CASTEP package was utilized, which is draws on a plane-wave pseudopotential formalism for total energy calculations, making it suitable for reliable simulations of material attributes. Our study ensured that the generalized gradient approximation (GGA), specifically the Perdew Burke Ernzerhof (PBE) functional24 and its revised form PBEsol,25 reproduced the lattice constants and unit-cell volumes of MgBH3 with greater precision than the local density approximation (LDA), and were in closer agreement with experimental findings. Hence, the PBE scheme was chosen for all subsequent computations. The geometric optimization of the crystal structures was carried out using the Broyden Fletcher Goldfarb Shanno (BFGS) minimization algorithm,26 confirming that the most energetically stable configuration of MgBH3 (X = Mo, In) was acquired. The plane-wave basis set cut-off energy and Brillouin zone sampling were carefully optimized through convergence tests of total energy, atomic forces, and stress to ensure computational accuracy. A cut-off energy of 500 eV and a 6 × 6 × 6 Monkhorst–Pack k-point grid were found to provide well-converged results for both hydrides. The electronic minimization was performed using self-consistent field (SCF) iterations with a total energy convergence tolerance of 2.0 × 10−5 eV/atom. The eigenvalue and Fermi energy tolerances were set to 6.25 × 10−6 eV and 2.72 × 10−13 eV, respectively, and a smearing width of 0.1 eV was applied to facilitate convergence of the metallic states. The convergence criteria for structural relaxation were set to 0.05 eV Å−1 for the maximum force, 0.002 Å for atomic displacement, and 0.1 GPa for the maximum stress, ensuring reliable optimization of the equilibrium structures. The optimized structures were then used to evaluate their electronic band dispersions, TDOS, and PDOS. To probe the optical behavior, the complex dielectric function was computed, which represents the interaction of photons with the electronic system across a wide energy range. Specifically, the real part, ε1(ω), was derived utilising the Kramers–Kronig transformation, while the imaginary part, ε2(ω), was obtained from the transition probabilities between occupied and unoccupied electronic states. Once ε(ω) was evaluated, additional optical characteristics such as the absorption coefficient, refractive index, optical conductivity, reflectivity, and energy loss function were systematically extracted.
3. Results and discussions
3.1 Structural properties and energetics of MgBH3 (B = Mo, In)
The cubic perovskite hydrides MgMoH3 and MgInH3 crystallize in the ABX3 structure, with Mg atoms occupying the A-site, the transition metal B (Mo, In) occupying the B-site, and hydrogen atoms forming corner-sharing BH6 octahedra (Fig. 1). Table 1 summarizes the optimized lattice parameters (a, V), formation energies (ΔEf), total energies (E0), and geometric stability factors including the Goldschmidt tolerance factor (t), octahedral factor (µ), and physically based tolerance factor (τ) for MgMoH3 and MgInH3. The optimized lattice constants were determined through energy-volume optimization. MgMoH3 exhibits a lattice parameter of 3.55 Å, corresponding to a unit cell volume of 44.72 Å3, whereas MgInH3 shows an expanded lattice with a lattice constant of 4.11 Å and a volume of 69.41 Å3. The larger lattice of MgInH3 is attributed to the greater ionic radius of In relative to Mo, leading to an expanded cubic framework.
 |
| | Fig. 1 Compounds unit cell of MgBH3 (B = Mo and In). | |
Table 1 Structural, Thermodynamic, and Geometric Stability of Cubic MgBH3 (B = Mo and In) Hydrides
| Materials |
a (Å) |
V (Å3) |
ΔHf (eV per Atom) |
E0 (Ry) |
t |
µ |
τ |
| Stability criteria |
— |
— |
ΔHf < 0 |
E0 < 0 |
0.813 < t < 1.107 |
0.37< µ < 0.859 |
τ < 4.18 |
| MgMoH3 |
3.55 |
44.72 |
−0.769 |
−217.08 |
0.997 |
0.448 |
3.756 |
| MgInH3 |
4.11 |
69.41 |
−0.475 |
−189.30 |
0.949 |
0.519 |
3.693 |
To examine the stability of MgMoH3 and MgInH3 relative to competing phases, we computed their formation energies using the following equation:
| | |
ΔHf = Eprpducts − Ereactance
| (1) |
For the decomposition reaction of MgBH3, we used the following equation:
The corresponding formation enthalpy is calculated as:
| |
 | (2) |
where
EMgBH3 represents the total energy of the MgBH
3 unit cell,
EH2 is the energy of the hydrogen molecule,
EMgH2 is the energy of magnesium hydride, and
EB is the energy of B (B = Mo, In). Using this approach, the calculated formation enthalpies for MgMoH
3 and MgInH
3 are −0.769 eV/atom and −0.475 eV/atom, respectively. These values suggest that both hydrides are thermodynamically stable with respect to their competing phases. These highly negative values indicate that the formation of these hydrides from their elemental precursors is strongly exothermic, confirming their intrinsic thermodynamic stability. The total energies, −217.08 Ry for MgMoH
3 and −189.30 Ry for MgInH
3, further support the strong cohesive interactions within the lattice, reflecting robust structural integrity. For comparison, previously reported perovskite hydrides such as MgAlH
3 (−0.499 eV/atom), MgScH
3 (−0.958 eV/atom), and MgZrH
3 (−0.871 eV/atom)
27 show less negative formation energies, highlighting the greater intrinsic stability of MgMoH
3 and MgInH
3. Since a complete convex-hull calculation was not carried out in this study, the calculated formation enthalpies should be regarded as indicative of relative stability against the considered decomposition pathway rather than proof of absolute thermodynamic stability.
3.2 Geometric stability
The geometric stability of the cubic perovskite structure was further evaluated using the Goldschmidt tolerance factor t, octahedral factor µ, and physically based tolerance factor τ, calculated as:28| |
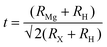 | (3) |
| |
 | (4) |
| |
 | (5) |
For cubic perovskite stability, t should fall within 0.813 to 1.107, µ within 0.37 to 0.859, and τ below 4.18. For MgMoH3, the tolerance factor of 0.997 indicates near-ideal cubic packing, while the octahedral factor of 0.448 confirms that the BH6 octahedra are appropriately coordinated. MgInH3, with a slightly smaller tolerance factor of 0.949 and a higher octahedral factor of 0.519, remains within the stability range, reflecting minor lattice adjustments due to the larger B-site cation. The physically based tolerance factors for both compounds, 3.756 for MgMoH3 and 3.693 for MgInH3, are below the critical threshold, further confirming that the cubic perovskite structure is energetically and geometrically favorable. The calculated values are listed in Table 1.
Collectively, the combination of strongly negative formation energies, appropriate ionic packing, and favorable tolerance factors indicates that both MgMoH3 and MgInH3 are stable cubic hydrides. Although their gravimetric hydrogen-storage capacities remain below the DOE onboard targets, their structural robustness and moderate volumetric storage characteristics may still be relevant for stationary or niche hydrogen-storage applications.
3.3 Hydrogen storage performance
One of the most efficient energy carriers for stationary and transportation uses is hydrogen. Particularly in nations with limited natural resources, it provides a renewable substitute that can reduce dependency on imported petroleum.29
The two main processes by which hydrogen is stored in solid-state materials are physisorption and chemisorption. In chemisorption, hydrogen and the solid (usually metals) undergo a chemical process that produces hydride phases, whereas in physisorption, hydrogen molecules interact weakly with the host material through van der Waals forces.20 In addition to the well-researched metal hydrides, recently produced perovskite-hydrides with increased hydrogen storage capacity (gravimetric density) have garnered a lot of attention.30 The conversion of excess electrical energy into hydrogen provides a practical means of controlling power consumption and guaranteeing stability between output and consumption. Nowadays, a primary area of technical research in this area is the hunt for cutting-edge hydrogen storage materials.31 A dependable energy management technique that allows for improved control over supply and demand fluctuations is the conversion of excess electrical output into hydrogen. Because of its technological significance, the creation of novel hydrogen storage devices is currently garnering a lot of scientific interest. One of the main obstacles to using hydrogen as fuel is the lack of materials with a sufficient gravimetric density. An integrated strategy targeted at creating and developing materials capable of achieving increased storage capacities with improved density is necessary to address this restriction.
This section focuses on assessing the hydrogen storage capacity of Mg-based perovskite hydrides, namely MgBH3 (X = Mo, In). Because it measures the quantity of hydrogen that may be stored per unit mass, the gravimetric storage capacity is an important requirement. This statistic provides an exact figure that makes it easier to assess the material's overall appropriateness and efficiency for hydrogen storage applications. The gravimetric storage capacity (Cwt%) of a hydrogen-absorbing material is generally determined by calculating the proportion between the molar mass of the absorbed hydrogen and the molar mass of the host compound.32
| |
 | (6) |
In this equation,
n is the hydrogen-to-host atomic ratio (
H/
M),
mH is the molar mass of hydrogen, and
mhost is the molar mass of the host lattice. For MgBH
3 (B = Mo, In), calculated gravimetric hydrogen storage capacities are 2.45 wt% and 2.13 wt%, respectively. The lower capacity of MgInH
3 stems from the greater atomic mass of In. In general, ABH
3-type perovskite hydrides exhibit gravimetric hydrogen densities in the range of 1.2–6.0 wt%.
33 Although the hydrogen storage capacities of MgMoH
3 and MgInH
3 are moderate, they are comparable to or higher than several reported perovskite hydrides, including RbCaH
3,
34 CsPtH
3,
35 FrPtH
3,
35 CsSnH
3,
36 FrSnH
3,
36 KSrH
3,
32 InMgH
3,
37 RbGaH
3,
38 CsGaH
3,
38 FrGaH
3,
38 CaCuH
3,
39 SrCuH
3,
39 CoCuH
3,
40 NiCuH
3,
40 ZnCuH
3,
40 LiNiH
3,
41 NaNiH
3,
41 KNiH
3,
41 CsBH
3,
42 and RbBH
3.
42 These results indicate that MgMoH
3 and MgInH
3 possess competitive hydrogen-storage characteristics within the broader class of perovskite hydrides, particularly for moderate-capacity or stationary storage applications.
The thermodynamic stability of the studied hydrides was evaluated through their dehydrogenation reactions, in which molecular hydrogen (H2) is taken as the reference state. Dehydrogenation corresponds to the release of hydrogen from the hydride upon heating and can be expressed as:
Following the approach proposed by Ikeda,43 the hydrogen decomposition enthalpy (ΔH) is calculated as:  , where H represents the total enthalpy of each system. The enthalpy is determined using: H = Eele + EZPE. Here, Eele and EZPE denote the electronic total energy and the zero-point energy, respectively. The zero-point energy is obtained from the phonon density of states as:
, where H represents the total enthalpy of each system. The enthalpy is determined using: H = Eele + EZPE. Here, Eele and EZPE denote the electronic total energy and the zero-point energy, respectively. The zero-point energy is obtained from the phonon density of states as:  , where h, ω, and g(ω) are Planck's constant, phonon frequency, and phonon density of states, respectively. The hydrogen desorption temperature (Tdes) is estimated using:44
, where h, ω, and g(ω) are Planck's constant, phonon frequency, and phonon density of states, respectively. The hydrogen desorption temperature (Tdes) is estimated using:44  , where, ΔS is the entropy change of the reaction. In this work, ΔS is approximated by the entropy of hydrogen gas, taken as 130.7 J mol−1 K−1. Using the calculated formation energies as an approximation to the decomposition enthalpy, the estimated desorption temperatures are 567 K for MgMoH3 and 350 K for MgInH3. These values indicate moderate thermal stability, suggesting that hydrogen release can occur at relatively accessible temperatures. Such moderate desorption temperatures are beneficial for practical hydrogen storage, as they reduce thermal stress during cycling while maintaining sufficient structural stability to prevent degradation or phase separation.
, where, ΔS is the entropy change of the reaction. In this work, ΔS is approximated by the entropy of hydrogen gas, taken as 130.7 J mol−1 K−1. Using the calculated formation energies as an approximation to the decomposition enthalpy, the estimated desorption temperatures are 567 K for MgMoH3 and 350 K for MgInH3. These values indicate moderate thermal stability, suggesting that hydrogen release can occur at relatively accessible temperatures. Such moderate desorption temperatures are beneficial for practical hydrogen storage, as they reduce thermal stress during cycling while maintaining sufficient structural stability to prevent degradation or phase separation.
To further assess storage performance, the volumetric hydrogen storage capacity was evaluated using the relation:  , where, NH is the number of hydrogen atoms per formula unit, mH is the mass of a hydrogen atom, V is the unit cell volume, and NA is Avogadro's number. The calculated volumetric hydrogen storage capacities are 112 kg m−3 for MgMoH3 and 72 kg m−3 for MgInH3. The higher volumetric capacity of MgMoH3 arises from its smaller lattice parameter and more compact crystal structure, which allows denser hydrogen packing. In contrast, the larger ionic radius of In leads to lattice expansion in MgInH3, resulting in reduced volumetric storage density. These results highlight a clear trade-off between atomic size and hydrogen packing efficiency, where smaller B-site elements favour enhanced volumetric storage performance.
, where, NH is the number of hydrogen atoms per formula unit, mH is the mass of a hydrogen atom, V is the unit cell volume, and NA is Avogadro's number. The calculated volumetric hydrogen storage capacities are 112 kg m−3 for MgMoH3 and 72 kg m−3 for MgInH3. The higher volumetric capacity of MgMoH3 arises from its smaller lattice parameter and more compact crystal structure, which allows denser hydrogen packing. In contrast, the larger ionic radius of In leads to lattice expansion in MgInH3, resulting in reduced volumetric storage density. These results highlight a clear trade-off between atomic size and hydrogen packing efficiency, where smaller B-site elements favour enhanced volumetric storage performance.
Although MgMoH3 and MgInH3 exhibit moderate volumetric hydrogen-storage capacities and favorable structural stability, their gravimetric storage capacities remain below the U.S. Department of Energy (DOE) onboard hydrogen-storage target. In addition, practical hydrogen-storage performance may also be influenced by factors such as hydrogen desorption kinetics, reversibility, and equilibrium hydrogen pressure, which were not explicitly investigated in the present study.45–47 Therefore, the current work should be regarded as a theoretical assessment of the structural stability, hydrogen-storage behavior, and physical properties of MgBH3 (B = Mo, In) hydrides. Experimental validation is encouraged to confirm the predicted stability and hydrogen-storage characteristics of these compounds. Furthermore, the hydrogen-storage performance may potentially be improved through elemental doping, defect engineering, or nanostructuring approaches, which could enhance surface activity and hydrogen adsorption/desorption kinetics compared with bulk materials.48–51
3.4 Mechanical properties
Revealing the mechanical stability of materials is crucial for harnessing their full potential in real-time applications. The elastic constants C11, C12, and C44 for MgMoH3 and MgInH3 are reported in Table 2, revealing a robust and stable material that meets the stringent Born stability criteria for cubic crystals: C11 − C12 > 0, C11 + 2C12 > 0, C44 > 0, confirming that both compounds are mechanically stable.45 Specifically, MgMoH3 shows C11 − C12 = 212.52 GPa and C44 = 36.72 GPa, while MgInH3 exhibits higher longitudinal stiffness (C11 − C12 = 73.02 GPa) but lower shear resistance (C44 = 9.22 GPa).
Table 2 Elastic Constants and moduli of compound MgBH3 (B = Mo and In) hydrides
| Compounds |
C11 (GPa) |
C12 (GPa) |
C44 (GPa) |
B (GPa) |
G (GPa) |
Y (GPa) |
| MgMoH3 |
258.77 |
46.25 |
36.72 |
117.09 |
57.14 |
147.44 |
| MgInH3 |
96.22 |
23.20 |
9.22 |
47.54 |
16.65 |
44.73 |
The mechanical response of a material is characterized by its stiffness coefficients, adding longitudinal stiffness (C11), transverse stiffness (C12), and shear stiffness (C44). Table 2 represents the computed properties, consisting of bulk moduli and shear moduli, derived through the Voigt, Reuss, and Hill methods. The values present in Table 2 are derived using the Hill method, offering a reliable estimate of the material's mechanical behavior. The mechanical properties of MgBH3 (B = Mo, In) have been thoroughly investigated by computing the Voigt and Reuss bulk moduli (BV and BR), shear moduli (GV and GR), Young's modulus (E), Poisson's ratio (ν), and elastic anisotropy (A). The computed values are presented in Table 3 and are determined using the following formulas:44
| |
 | (7) |
| |
 | (8) |
| |
 | (9) |
| |
 | (10) |
Table 3 Mechanical indicators of MgBH3 (B = Mo, In) hydrides
| Compounds |
ν |
B/G |
CP (GPa) |
A |
µm |
HV (GPa) |
Mechanical nature |
| MgMoH3 |
0.29 |
2.05 |
9.53 |
0.40 |
3.19 |
6.12 |
Ductile, metallic bonding, moderate anisotropy, good hardness |
| MgInH3 |
0.34 |
2.85 |
13.98 |
0.79 |
5.16 |
2.89 |
Softer, highly ductile, metallic bonding, strong anisotropy |
The overall bulk modulus (B) and shear modulus (G) are then calculated as the arithmetic mean of the Voigt and Reuss values, in accordance with Hill's theory:52
| |
 | (11) |
| |
 | (12) |
In addition, Young's modulus (
E) and Poisson's ratio (
ν) are determined using:
53| |
 | (13) |
| |
 | (14) |
The bulk moduli of the two hydrides differ significantly: MgMoH3 exhibits B = 117.09 GPa, whereas MgInH3 has B = 47.54 GPa, showing a disparity of about 59%. This indicates that MgMoH3 is considerably more resistant to uniform compression than MgInH3, meaning the two compounds will respond differently to pressure variations during hydrogenation cycles. In contrast, their resistance to shape deformation also shows notable variations. The shear modulus decreases from G = 57.14 GPa for MgMoH3 to G = 16.65 GPa for MgInH3, a reduction of nearly 71%, while the Young's modulus drops from 147.44 GPa to 44.73 GPa (around 70%). Thus, although MgMoH3 maintains greater rigidity, MgInH3 is mechanically softer with lower shear and tensile stiffness.
Poisson's ratio (v) is an important mechanical parameter for identifying both the bonding type and the ductile or brittle nature of a material. Typically, ν ≈ 0.10 corresponds to covalent bonding, ν ≈ 0.25 to ionic, and ν ≈ 0.33 to metallic bonding.43 In addition, ν = 0.26 is often taken as the dividing line between brittle (ν < 0.26) and ductile (ν > 0.26) behavior.44 For MgMoH3 (ν = 0.29) and MgInH3 (ν = 0.34), the values are above the ductility threshold and fall within or above the metallic regime, suggesting significant metallic bonding contributions and ductile characteristics. Another useful indicator is Pugh's ratio (B/G), where values greater than 1.75 denote ductility, while values below 1.75 correspond to brittleness.52 Both hydrides satisfy this criterion, confirming their ductile nature. The inverse ratio (G/B), with values of 0.34 for MgMoH3 and 0.26 for MgInH3, lies well below the typical covalent (∼1.1) and ionic (∼0.6) benchmarks, further supporting their metallic-like bonding behavior.53 Cauchy pressure (CP = C12 − C44) provides additional insight: positive values indicate ionic/metallic bonding and ductility, whereas negative values point to covalent and brittle characteristics. Both MgMoH3 (9.58 GPa) and MgInH3 (13.98 GPa) exhibit positive CP, again consistent with ductile metallic bonding.
The elastic anisotropy factor, defined as  , is a critical parameter for distinguishing isotropic (A = 1) from anisotropic (A ≠ 1) materials. The computed value is A = 0.40 for MgMoH3 and A = 0.79 for MgInH3. These hydride perovskites are mechanically anisotropic, as evidenced by the large deviations of both values from unity. A more unequal distribution of bonding stiffness across crystallographic orientations is reflected in the higher deviation in MgInH3, which suggests a greater directional dependency of its elastic response.
, is a critical parameter for distinguishing isotropic (A = 1) from anisotropic (A ≠ 1) materials. The computed value is A = 0.40 for MgMoH3 and A = 0.79 for MgInH3. These hydride perovskites are mechanically anisotropic, as evidenced by the large deviations of both values from unity. A more unequal distribution of bonding stiffness across crystallographic orientations is reflected in the higher deviation in MgInH3, which suggests a greater directional dependency of its elastic response.
The machinability index µM = B/C44, gauges intrinsic plasticity and workability (higher µM ⇒ easier machining (values ≳1 are considered workable)).54,55 High workability is indicated by MgMoH3's µM = 3.19 and MgInH3's µM = 5.16, while lower shear resistance is reflected by MgInH3's greater value. In contrast to MgMoH3, which strikes a compromise between moderate shear rigidity and increased tensile strength for better structural integrity, MgInH3 is softer and more compliant due to its higher shear rigidity but lower tensile resistance. Both machinability index values of both investigated materials are higher than those of LiPH3 (1.29), NaPH3 (1.62), and KPH3 (1.63),56 and AClH3 (A = Rb, Cs, K: 1.45, 1.32, 1.56),57 verifying their excellent flexibility and fabrication capabilities. This improved machinability is particularly beneficial for hydrogen storage, as it reduces microcrack formation.
The Vickers hardness, estimated using the relation  , measures a material's resistance to plastic deformation and serves as an indicator of the mechanical durability of hydrogen storage compounds. The calculated hardness for MgMoH3 is 6.12 GPa, whereas MgInH3 has 2.89 GPa. The soft-to-moderately hard domain (HV < 10 GPa)41 is where these hydride perovskites fall for both values, which is advantageous for preserving structural integrity and preventing excessive brittleness during hydrogen cycling. When compared with other reported hydride perovskites, such as MgCuH3 (0.77 GPa), RbNiH3 (0.64 GPa), BeGaH3 (3.02 GPa), and BeInH3 (3.17 GPa),58 both MgGaH3 and MgTlH3 show significantly higher hardness values, underscoring their enhanced mechanical stability. In hydrogen storage applications, this enhanced hardness and ductility (shown by B/G > 1.75) imply that these compounds can withstand microcrack development and structural deterioration, guaranteeing dependable cycle performance.
, measures a material's resistance to plastic deformation and serves as an indicator of the mechanical durability of hydrogen storage compounds. The calculated hardness for MgMoH3 is 6.12 GPa, whereas MgInH3 has 2.89 GPa. The soft-to-moderately hard domain (HV < 10 GPa)41 is where these hydride perovskites fall for both values, which is advantageous for preserving structural integrity and preventing excessive brittleness during hydrogen cycling. When compared with other reported hydride perovskites, such as MgCuH3 (0.77 GPa), RbNiH3 (0.64 GPa), BeGaH3 (3.02 GPa), and BeInH3 (3.17 GPa),58 both MgGaH3 and MgTlH3 show significantly higher hardness values, underscoring their enhanced mechanical stability. In hydrogen storage applications, this enhanced hardness and ductility (shown by B/G > 1.75) imply that these compounds can withstand microcrack development and structural deterioration, guaranteeing dependable cycle performance.
3.5 Electronic properties
To comprehend a material's electronic properties, it is essential to look at its electronic band structure and density of states. The vertical axis of these figures represents the energy levels, while the horizontal axis traces the wave vector (k) along specific high-symmetry areas in the Brillouin zone (BZ). Fig. 2, 3 displays the total density of states (DOS) and calculated electronic band structures for the cubic phases along the high-symmetry path X–M–R–Γ–R. The reference between the valence and conduction bands is the Fermi level, which is represented by a dashed line at 0 eV. The energy range in which the electronic band structures are shown is −4 to +4 eV. After the structural optimization, the electrical characteristics of MgBH3 (B = Mo, In) were investigated using first-principles calculations. The band structures of the studied compounds are depicted in Fig. 2 and 3, and the lack of a band gap verifies that the lowest unoccupied and highest occupied electronic states are not separated in energy. According to this, electrons can easily move from the valence band to the conduction band, confirming the metallic nature of these perovskite hydrides and boosting to their excellent electrical conductivity.59 Comparable electronic configurations have been observed in other magnesium-based perovskites, with previous studies consistently reporting metallic behavior.60 In agreement with these findings, our investigation of MgBH3 (B = Mo, In) also demonstrates a comparable electronic structure, further confirming their metallic nature.
 |
| | Fig. 2 Band diagram of MgBH3 (a) MgMoH3, (b) MgInH3 hydrides. | |
 |
| | Fig. 3 Compounds partial density of states (a) MgMoH3, (b) MgInH3 hydrides. | |
The TDOS and PDOS for MgBH3 (B = Mo, In) at zero temperature and pressure are displayed in Fig. 3. Mg-p, Mo-p/d, and H-p orbitals primarily control the conduction and valence bands in MgMoH3, whereas Mg-s/p, In-p/d, and H-p orbitals dominate the contributions in MgInH3, as seen in Fig. 3. These states play a crucial role in determining the electrical characteristics of these hydrides because most of them are found in the energy range of −3 eV to +5 eV. The PDOS analysis reveals pronounced p-d hybridization between the H-p orbitals and the Mo/In-p, d or Mg-s, p orbitals in both MgMoH3 and MgInH3. This strong orbital interaction signifies substantial covalent bonding, contributing to the electronic stability of these perovskite hydrides. Additionally, MgInH3 exhibits a slightly higher density of states near the Fermi level compared to MgMoH3, suggesting enhanced electronic conductivity and more efficient charge carrier mobility.
The electronic band structures of MgMoH3 and MgInH3 were further evaluated using the hybrid HSE06 functional to improve the accuracy of exchange-correlation effects, particularly near the Fermi level. The calculated HSE06 band structures, shown in Fig. 4a and b, reveal that both compounds retain their metallic character, as evidenced by the absence of a band gap and the clear overlap between valence and conduction bands at the Fermi level (EF = 0 eV). For MgMoH3, multiple bands cross the Fermi level along the high-symmetry directions (X–R–M–Γ–R), indicating a high density of itinerant charge carriers and strong electronic conductivity. The dispersion of bands near EF suggests significant hybridization between Mo-d and H-p states, which facilitates electron mobility. Similarly, MgInH3 exhibits band crossings at the Fermi level, confirming its metallic nature. However, compared to MgMoH3, the bands near EF appear relatively less dispersive in certain regions, implying slightly different carrier dynamics and orbital contributions, likely arising from the In-p/d states interacting with H-p orbitals.
 |
| | Fig. 4 Electronic band structures of (a) MgMoH3 and (b) MgInH3 calculated using the HSE06 hybrid functional along the high-symmetry directions (X–R–M–Γ–R). | |
Importantly, the use of the HSE06 functional provides more reliable electronic structure predictions than standard GGA. The results confirm that the metallic behavior is intrinsic and not an artifact of the exchange–correlation approximation. This metallic nature is beneficial for hydrogen storage, as it promotes efficient charge transfer. It may also enhance hydrogen adsorption and desorption kinetics.
3.6 Optical properties
In this study, the optical properties of MgBH3 (B = Mo, In) were systematically investigated over a photon energy range up to 12 eV. The analysis focused on several key parameters, including the absorption coefficient (α(ω)), photoconductivity (σ(ω)), reflectivity (R(ω)), real and imaginary components of the dielectric function (ε(ω)), energy-loss function (L(ω)), extinction coefficient (k(ω)), and refractive index (n(ω)). Examining these frequency-dependent optical properties provides a comprehensive understanding of the electronic transitions and light-matter interactions in these compounds.
The optical behavior of a material is governed by its interaction with incident electromagnetic radiation and provides important insight into its dielectric and metallic response characteristics. This is described by the complex dielectric function, ε(ω) = ε1(ω) + iε2(ω), following Ehrenreich and Cohen. Here, ω is the angular frequency, ε1(ω) represents the real part obtained via the Kramers–Kronig relation, reflecting the induced polarization, while ε2(ω), derived from Kohn–Sham electronic transitions,9 indicates the material's absorption of incoming radiation.
| |
 | (15) |
| |
 | (16) |
In the first expression,
p denotes the primary parameter. Conversely, the second relation introduces the terms
v,
e,
h,
p,
kn, and
k'n which correspond to the crystal cell volume, elementary electric charge, reduced Planck's constant, momentum transition operator, valence-band wave function, and conduction-band wave function, respectively. Accurate evaluation of these optical parameters is vital for improving the performance of electronic and optoelectronic devices. In the low-energy region, the imaginary part
ε2(
ω) of the dielectric function decreases sharply with increasing photon energy (
Fig. 5b), indicating a reduced probability of interband electronic transitions, as
ε2(
ω) directly reflects photon absorption
via electronic excitations.
61 Conversely, the real part
ε1(
ω) of the dielectric function rises notably in this energy range, reflecting increased polarization and charge displacement within the lattice under an external electric field. Both MgMoH
3 and MgInH
3 exhibit high static dielectric constants, indicating strong interaction with incident electromagnetic radiation and pronounced dielectric response characteristics associated with their metallic electronic structure. In the UV region, the imaginary part
ε2(
ω) approaches zero, indicating negligible absorption at higher energies. This suggests minimal dielectric loss, consistent with the metallic behavior observed in the band structure and DOS analyses, thus supporting the theoretical predictions. Another important feature occurs in the high-energy (UV) region, where the
ε1(
ω) and
ε2(
ω) curves intersect and the dielectric constant approaches unity. This marks the critical transition at which the material becomes transparent to incident electromagnetic radiation, allowing photons above this energy to pass through with minimal attenuation. This high-energy transparency reflects reduced photon absorption in the ultraviolet region and highlights the distinct dielectric and optical response characteristics of these metallic hydrides at higher photon energies. Furthermore, these optical characteristics underline the importance of Mg-based hydrides as candidate materials for multifunctional applications. The high dielectric response at low photon energies reflects strong polarization behavior, while the reduced absorption at higher photon energies highlights the characteristic metallic and dielectric optical response of these hydrides across a broad energy range.
 |
| | Fig. 5 Compounds MgBH3 (B = Mo, In) dielectric function (a) ε1(ω) and (b) ε2(ω). | |
From the calculated values of ε1(ω) and ε2(ω) a range of optical responses was determined, such as the complex refractive index, optical conductivity (real part), absorption spectrum, reflectivity, and the electron energy loss spectrum. The refractive index was expressed in its complex form as n(ω) − ik(ω), where n(ω) denotes the real component associated with light propagation, and k(ω) refers to the extinction coefficient describing attenuation within the medium. These quantities were systematically obtained using standard relations and widely adopted theoretical expressions.62,63
| |
 | (17) |
| |
 | (18) |
| |
 | (19) |
| |
 | (20) |
The refractive index n(ω) is an important optical parameter that governs the propagation of electromagnetic radiation through a material and provides valuable insight into its dielectric and optical response characteristics. Since the real component of Δ(ω) characterizes the degree of light refraction at the interface between various media, and it is associated with the transparency of the material. As shown in Fig. 6a, the refractive index at 0 eV exceeds 10 for both MgMoH3 and MgInH3. In the infrared region, the refractive index first reaches a maximum, then slightly decreases, and rises again in the visible region. Across the visible spectrum, the values gradually decline from about 2.62 to 1.50 for MgInH3 and 1.29 to 1.96 for MgMoH3. In the ultraviolet range, Δ(ω) continues to fall, approaching nearly zero, which is lower than silicon nitride (∼1.9).64
 |
| | Fig. 6 Optical parameters of MgBH3 (B = Mo, In): (a) refractive index and (b) extinction coefficient. | |
One important metric that reacts to the amount of light absorption and scattering in a material is the extinction coefficient k(ω); higher values indicate stronger absorption of the incoming radiation. As shown in Fig. 6b, both MgMoH3 and MgInH3 exhibit their maximum extinction in the infrared region. In the visible spectrum, the extinction coefficient decreases, ranging from 4.00 to 2.54 for MgInH3 and 1.60 to 1.30 for MgMoH3, indicating a moderate reduction in absorption strength. In the ultraviolet region, the extinction coefficient approaches zero, reflecting reduced absorption at higher photon energies and the characteristic energy-dependent optical response of these metallic hydrides.
Optical conductivity reflects a material's ability to absorb photons and generate charge transport under electromagnetic excitation, offering key insights into free-electron contribution and its relation to reflectivity. At zero energy, the conductivity is high, but it decreases with increasing photon energy in the infrared region. For MgInH3, conductivity rises again from the infrared to the visible range, showing prominent peaks above 4.36 Ω−1 cm−1 in the infrared. In contrast, MgMoH3 exhibits a steady increase from the infrared to the ultraviolet, with its strongest peaks in the UV region, reaching values above 8.58 Ω−1 cm−1. At higher photon energies, the conductivity continues to decline and eventually approaches zero, consistent with the weakening photon absorption. Interestingly, the spectra also exhibit wave-like oscillations (rise-fall features) as a function of energy, reflecting the interplay of interband transitions and the availability of electronic states. This behavior, presented in Fig. 7a, confirms the strong optical response of these hydrides in the lower-energy domain while indicating reduced conductivity in the ultraviolet regime.
 |
| | Fig. 7 The optical response of MgBH3 (X = Mo, In) showing (a) optical conductivity and (b) reflectance. | |
Optical reflectivity, R(ω), represents the fraction of incoming photons reflected from the surface of the crystal. As shown in Fig. 7b, its spectral trend is inversely related to the absorption curve, displaying clear variations with photon energy. At zero photon energy, both MgMoH3 and MgInH3 exhibit high static reflectivity values of approximately 0.98. With increasing photon energy, the reflectivity decreases from the infrared to visible region before increasing again in the ultraviolet range. This oscillatory behavior originates from interband electronic transitions and variations in the dielectric response of the materials. The combination of high refractive index and characteristic reflectivity behavior further reflects the distinct metallic and dielectric optical response of these hydrides.
The mathematical expression for absorption coefficient α(ω) is obtained by combining the real and imaginary contributions of the dielectric response, which allows accurate estimation of a material's photon absorption capability across the studied energy spectrum.65
| |
 | (21) |
Visible-range absorption (1.6 to 3.2 eV, 780 to 380 nm) is especially important to solar and light-driven devices. For the compounds studied, the initial absorption peak appears in the infrared region, with MgInH3 showing about 10.55 × 104 cm−1 at 1.50 eV and MgMoH3 about 9.22 × 104 cm−1 at 0.50 eV. With increasing photon energy, absorption rises again within the IR domain. In the visible range, MgMoH3 exhibits values of 4.00 × 104 to 6.00 × 104 cm−1, while MgInH3 ranges between 11.22 × 104 and 13.50 × 104 cm−1. A continuous increase in the absorption coefficient is observed as the photon energy shifts toward the ultraviolet (UV) region, where both compounds attain their maximum absorption strengths. MgMoH3 reaches a peak value of 28.85 × 104 cm−1 at 9.50 eV, while MgInH3 exhibits a maximum value of 18.65 × 104 cm−1 at 4.66 eV, as shown in Fig. 8a. These high absorption coefficients indicate strong photon-electron interactions and efficient electromagnetic energy absorption over a broad energy range. Their strong electronic response may still be relevant for understanding energy-transfer processes that could influence hydrogen adsorption/desorption kinetics under external thermal or electromagnetic stimulation.
 |
| | Fig. 8 Optical response of MgBH3 (B = Mo, In): (a) absorption coefficient (eV) and (b) absorption coefficient (nm). | |
3.7 Thermodynamic properties
Debye temperature (ΘD) is the temperature at which wavelength of phonons approaches the lattice parameter of the unit cell, and the limit between low- and high-temperature behavior of solids. Debye temperature is a characteristic parameter as the materials with higher ΘD will have stronger atomic bonding, higher melting points, increased hardness, increased velocity of propagation of acoustic waves, and lower mean atomic weights. Debye temperature is estimated from the average sound velocity (vm), which may be calculated using both shear modulus and bulk modulus.| |
 | (22) |
In formula 22, vt corresponds to the transverse acoustic velocity, while vl represents the longitudinal acoustic velocity. These sound velocities can be obtained using the following expressions.| |
 | (23) |
The Debye temperature (ΘD)66 can be determined using the following mathematical relation:
| |
 | (24) |
The expression is here h being Planck's constant, kB the Boltzmann constant, V0 is the unit-cell volume, n the number of atoms in the unit cell, and vm the average sound velocity. The Debye temperatures were calculated as 565.32 K and 307.91 K for MgMoH3 and MgInH3, respectively (Table 4). These compounds possess lower ΘD and therefore expand more gradually on being heated and endure thermal stress, thereby becoming potential candidates for thermal barrier coatings (TBCs) on various substrates.
Table 4 Calculated thermodynamic and thermal transport parameters
| Compound |
ρ (g cm−3) |
vt (km s−1) |
vl (km s−1) |
vm (km s−1) |
θD (K) |
Tm (K) |
kph (W m−1 K−1) |
γ |
kmin (W m−1 K−1) |
| MgMoH3 |
4.578 |
3.533 |
6.498 |
3.941 |
565.32 |
2082.33 |
11.14 |
1.71 |
1.26 |
| MgInH3 |
3.401 |
2.213 |
4.529 |
2.486 |
307.91 |
1121.66 |
1.68 |
2.05 |
0.59 |
Thermal conduction in crystalline solids is mainly conducted through vibrational motion of the lattice ions, known as lattice thermal conductivity (κph). For the estimation of such a property, Slack's empirical relation67 was employed, a usual method to estimate phonon-conducted thermal conductivity in semiconductors and perovskite complex compounds.
| |
 | (25) |
Based on this model, lattice thermal conductivities (κph) of the studied perovskites hydrides are 11.14 W m−1 K−1 for MgMoH3 and 1.68 W m−1 K−1 for MgInH3, as shown in Table 4. MgInH3's smaller κph indicates more phonon scattering, which could be due to the heavier atomic mass and larger unit-cell distortions of In, while MgMoH3's larger value offers more efficient phonon transport. These results are consistent with other perovskites, e.g., lead-halide MAPbI3 possessing ultralow κph (0.5 to 1.0 W m−1 K−1) at RT due to anharmonicity and lattice softness,67 highlighting the varied thermal behavior in perovskite families.
The Grüneisen parameter (γ) is a well-known thermal descriptor that quantifies lattice anharmonicity and is extremely widely used in solid-state physics to evaluate how the lattice vibrations respond to stress or volumetric changes. Higher values of γ are indicators of greater anharmonicity, affecting thermal expansion, phonon scattering, and heat transport. For MgBH3 (B = Mo, In), γ is calculated by applying Poisson's ratio,66 providing a quantitative measure of anharmonic behavior and phonon scattering in such hydride perovskites [eq. (26)].
| |
 | (26) |
The Grüneisen parameter (γ) quantifies lattice anharmonicity and its influence on thermal properties. For the studied hydride perovskites, γ values calculated are 1.71 for MgMoH3 and 2.05 for MgInH3 (Table 4). The calculated values are in line with typical solids, which usually show γ ≈ 2.0,66 indicating average anharmonic influences in the lattice.
Measurement of minimum thermal conductivity (Kmin) is most important for applications at high temperatures because Kmin is the most easily accessible thermal conductivity at high temperatures. Vacancies or dislocations in the structure have no effect on Kmin, because phonon wavelengths at high temperatures approach the interatomic distance. Average sound velocity and Debye temperature are intrinsic properties which determine mainly Kmin and influence temperature-dependent thermal properties and electrical conductivity. For the studied compounds, Kmin was calculated using Clarke's model65 eqn (27), which considers the average sound velocity, molecular weight, density, and atomic packing.
| |
 | (27) |
The calculated minimum thermal conductivity (kmin) values for MgMoH3 and MgInH3 are 1.26 W m−1 K−1 and 0.59 W m−1 K−1, respectively, as summarized in Table 4. Materials with higher Debye temperatures and greater average sound velocities tend to exhibit increased kmin values. Therefore, an enhancement in either Debye temperature or sound velocity is generally associated with a rise in the minimum thermal conductivity of the material, highlighting its thermal transport efficiency under high-temperature conditions.
4. Conclusion
In the current work, structural, electronic, optical, mechanical, and thermodynamic properties of double hydride perovskites MgBH3 (B = Mo, In) have been investigated using density functional theory in detail. The two compounds have been found to be stable thermodynamically with negative formation energies and positive tolerance and octahedral factors. Theoretical hydrogen-storage analysis indicates moderate gravimetric and competitive volumetric storage capacities together with moderate desorption temperatures, suggesting potential relevance for stationary or niche hydrogen-storage applications. Electronic structure analysis confirms the metallic nature of these hydrides, while the calculated high optical absorption (∼104 cm−1) reflects strong photon interaction and dielectric/metallic optical response characteristics. From the mechanical viewpoint, the hydrides are tough with ductile character and high elastic anisotropy, suggesting robustness under operating conditions. Thermodynamic analysis: Debye temperature, melting point, lattice and minimum thermal conductivity, and Grüneisen parameter reflect efficient conduction of heat, suggesting applicability in high-temperature technologies such as thermal barrier coatings. The calculated results suggest that MgBH3 (B = Mo, In) hydrides possess stable structural frameworks, ductile mechanical behavior, moderate hydrogen-storage capacity, and distinct dielectric/metallic optical responses. This work expands the current theoretical understanding of Mg-based hydride perovskites and may guide future studies on multifunctional hydride materials.
Author contributions
Md. Shahazan Parves: writing manuscript-reviewing and editing. Md. Hasan Mia: conceptualization; methodology; writing manuscript-reviewing and editing; data curation; validation; supervision Omar Alsalmi: formal analysis, data curation, review-editing Md. Zahid Hasan: formal analysis; validation; review-editing; and supervision.
Conflicts of interest
The authors declare that they have no known conflicting financial interests or personal ties that may have seemed to affect the work presented in this study.
Data availability
Relevant data from this study are available from the corresponding author upon a reasonable request.
Acknowledgements
The authors extend their appreciation to Umm Al-Qura University, Saudi Arabia, for funding this research work through grant number: 26UQU4300099GSSR02. Funding statement: this research work was funded by Umm Al-Qura University, Saudi Arabia under grant number: 26UQU4300099GSSR02.
References
- I. A. Hassan, H. S. Ramadan, M. A. Saleh and D. Hissel, Hydrogen storage technologies for stationary and mobile applications: Review, analysis and perspectives, Renew. Sustain. Energy Rev., 2021, 149, 111311 CrossRef CAS.
- L. Zhou, Progress and problems in hydrogen storage methods, Renew. Sustain. Energy Rev., 2005, 9, 395–408 CrossRef CAS.
- M. R. Raupach, et al., Global and regional drivers of accelerating CO2 emissions, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(24), 10288–10293, DOI:10.1073/pnas.0700609104.
- A. Gencer, G. Surucu and S. Al, MgTiO3Hx and CaTiO3Hx perovskite compounds for hydrogen storage applications, Int. J. Hydrogen Energy, 2019, 44(23), 11930–11938 CrossRef CAS.
- W. Hao and J. Xie, Reducing diffusion-induced stress of bilayer electrode system by introducing pre-strain in lithium-ion battery, J. Electrochem. Energy Convers. Storage, 2021, 18(2), 020909 Search PubMed.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, Organometal halide perovskites as visible-light sensitizers for photovoltaic cells, J. Am. Chem. Soc., 2009, 131(17), 6050–6051 CrossRef CAS PubMed.
- H. Zhou, Q. Chen, G. Li, S. Luo, T.-B. Song, H.-S. Duan, Z. Hong, J. You, Y. Liu and Y. Yang, Interface engineering of highly efficient perovskite solar cells, Science, 2014, 345(6196), 542–546 CrossRef CAS PubMed.
- Z. X. Guo, C. Shang and K. F. Aguey-Zinsou, Materials challenges for hydrogen storage, J. Eur. Ceram. Soc., 2008, 28(7), 1467–1473 CrossRef CAS.
- J. Zheng, C.-G. Wang, H. Zhou, E. Ye, J. Xu, Z. Li and X. J. Loh, Current research trends and perspectives on solid-state nanomaterials in hydrogen storage, Research, 2021, 2021, 3750689 CrossRef CAS PubMed.
- N. A. Ali, M. S. Yahya, N. Sazelee, M. F. M. Din and M. Ismail, Influence of nanosized CoTiO3 synthesized via a solid-state method on the hydrogen storage behavior of MgH2, Nanomaterials, 2022, 12(17), 3043 CrossRef CAS PubMed.
- D. P. Broom, Potential storage materials, in Hydrogen Storage Materials: The Characterisation of Their Storage Properties, Springer, London, 2011, pp. 19–59 Search PubMed.
- L. Kong and G. Liu, Synchrotron-based infrared microspectroscopy under high pressure: An introduction, Matter Radiat. Extremes, 2021, 6(6), 068202 CrossRef CAS.
- M. Kaur and K. Pal, Review on hydrogen storage materials and methods from an electrochemical viewpoint, J. Energy Storage, 2019, 23, 234–249 CrossRef.
- R. K. Ahluwalia, X. Wang, A. Rousseau and R. Kumar, Fuel economy of hydrogen fuel cell vehicles, J. Power Sources, 2004, 130(1–2), 192–201 CrossRef CAS.
- S.-H. Ri, U.-G. Jong, T.-S. Im and U.-R. Rim, Perovskite-type hydrides ACaH3 (A = Li, Na): computational investigation on materials properties for hydrogen storage applications, RSC Adv., 2025, 15, 19245–19253 Search PubMed.
- P. Ahmadi and E. Kjeang, Realistic simulation of fuel economy and life cycle metrics for hydrogen fuel cell vehicles, Int. J. Energy Res., 2017, 41(5), 714–727 CrossRef.
- Q. Sun, N. Wang, Q. Xu and J. Yu, Nanopore-supported metal nanocatalysts for efficient hydrogen generation from liquid-phase chemical hydrogen storage materials, Adv. Mater., 2020, 32(44), 2001818 CrossRef CAS PubMed.
- X. Zhang, Y. Tang, F. Zhang and C.-S. Lee, A novel aluminum–graphite dual-ion battery, Adv. Energy Mater., 2016, 6(11), 1502588 CrossRef.
- E. Rivard, M. Trudeau and K. Zaghib, Hydrogen storage for mobility: a review, MDPI, 2019, 12(12), 1973 CAS.
- H. Jiang, Z. Ding, Y. Li, G. Lin, S. Li, W. Du, Y. Chen, L. L. Shaw and F. Pan, Hierarchical interface engineering for advanced magnesium-based hydrogen storage: synergistic effects of structural design and compositional modification, Chem. Sci., 2025, 16(18), 7610–7636 RSC.
- G. Surucu, A. Gencer, A. Candan, H. H. Gullu and M. Isik, CaXH3 (X = Mn, Fe, Co) perovskite-type hydrides for hydrogen storage applications, Int. J. Energy Res., 2020, 44, 2345–2354 Search PubMed.
- M. K. Shahzad, S. Hussain, M. N. Khan, M. J. Aslam, R. M. Mohammed, V. Tirth, H. Alqahtani, A. Algahtani, T. Al-Mughanam and W. Azeem, Computational insights of double perovskite Na2CaCdH6 hydride alloy for hydrogen storage applications: a DFT investigation, Sci. Rep., 2024, 14(1), 25102 CrossRef CAS PubMed.
- A. Siddique, et al., Structures and hydrogen storage properties of AeVH3 (Ae= Be, Mg, Ca, Sr) perovskite hydrides by DFT calculations, Int. J. Hydrogen Energy, 2023, 48(63), 24401–24411 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868, DOI:10.1103/PhysRevLett.77.3865.
- J. P. Perdew, et al., Generalized gradient approximation for solids and their surfaces, Phys. Rev. Lett., 2008, 100(13), 136406, DOI:10.1103/PhysRevLett.100.136406.
- T. H. Fischer and J. Almlof, General methods for geometry and wave function optimization, J. Phys. Chem., 1992, 96(24), 9768–9774, DOI:10.1021/j100203a036.
- S. Bahhar, A. Tahiri, A. Jabar, M. Louzazni, M. Idiri and H. Bioud, Computational assessment of MgXH3 (X= Al, Sc and Zr) hydrides materials for hydrogen storage applications, Int. J. Hydrogen Energy, 2024, 58, 259–267 Search PubMed.
- C. Li, X. Lu, W. Ding, L. Feng, Y. Gao and Z. Guo, Formability of abx3 (x= f, cl, br,i) halide perovskites, Struct. Sci., 2008, 64(6), 702–707 Search PubMed.
- B. Sakintuna, F. Lamari-Darkrim and M. Hirscher, Metal hydride materials for solid hydrogen storage: a review, Int. J. Hydrogen Energy, 2007, 32(9), 1121–1140 CrossRef CAS.
- P. Schouwink, et al., Structure and properties of complex hydride perovskite materials, Nat. Commun., 2014, 5(1), 5706 CrossRef CAS PubMed.
- D. P. Broom, The accuracy of hydrogen sorption measurements on potential storage materials, Int. J. Hydrogen Energy, 2007, 32(18), 4871–4888 Search PubMed.
- H. H. Raza, G. Murtaza, U. Hani, N. Muhammad and S. M. Ramay, First-principle investigation of XSrH3 (X = K and Rb) perovskite-type hydrides for hydrogen storage, Int. J. Quantum Chem., 2020, 120(24), e26419 CrossRef CAS.
- K. Ikeda, T. Sato and S. Orimo, Perovskite-type hydrides–synthesis, structures and properties, Int. J. Mater. Res., 2008, 99(5), 471–479 CrossRef CAS.
- H. Murtaza, Q. Ain, A. Kumar, A. M. Ali, A. D. Oza and J. Munir, The Effect of Pressure in Tuning the Optoelectronic, Thermoelectric and Hydrogen Storage Properties of RbCaH3 for Renewable Energy Applications, J. Energy Storage, 2025, 134, 118183 Search PubMed.
- A. Bakar, H. Muhammad, M. Ahmed, M. S. Kiani, A. Quader, S. Batay and H. Elhosiny Ali, Investigations for hydrogen storage applications of XPtH3 (X= Cs, Fr) hydrides: A first principles study, Chem. Phys., 2025, 591, 112566 CrossRef CAS.
- M. K. Masood, W. Khan, K. Chaoui, Z. Ashraf, S. Bibi, A. Kanwal, A. A. Alothman and J. Rehman, Theoretical investigation of XSnH3 (X: Rb, Cs, and Fr) perovskite hydrides for hydrogen storage application, Int. J. Hydrogen Energy, 2024, 63, 1248–1257 Search PubMed.
- M. Hadhoud, O. Eddahmani, A. El Mekkaouy, A. Tahiri and R. Touti, Indium-Based Perovskite Hydrides InZH3 (Z = Be, Ca, Mg) as Promising Hydrogen Storage Materials: A Comprehensive First-Principles Study, Int. J. Hydrogen Energy, 2026, 231, 154842 CrossRef CAS.
- R. M. Khalil, S. Hayat, M. I. Hussain, A. M. Rana and F. Hussain, DFT based first principles study of novel combinations of perovskite-type hydrides XGaH3 (X= Rb, Cs, Fr) for hydrogen storage applications, AIP Adv., 2021, 11(2), 025032 CrossRef CAS.
- B. Ahmed, M. B. Tahir, A. Ali and M. Sagir, First-principles screening of structural, electronic, optical and elastic properties of Cu-based hydrides-perovskites XCuH3 (X= Ca and Sr) for hydrogen storage applications, Int. J. Hydrogen Energy, 2024, 54, 1001–1007 CrossRef CAS.
- S. Hayat, R. M. A. Khalil, M. I. Hussain, A. M. Rana and F. Hussain, First-principles investigations of the structural, optoelectronic, magnetic and thermodynamic properties of hydride perovskites XCuH3 (X= Co, Ni, Zn) for hydrogen storage applications, Optik, 2021, 228, 166187 CrossRef CAS.
- A. Gencer and G. Surucu, Investigation of structural, electronic and lattice dynamical properties of XNiH3 (X= Li, Na and K) perovskite type hydrides and their hydrogen storage applications, Int. J. Hydrogen Energy, 2019, 44(29), 15173–15182 CrossRef CAS.
- B. Ahmed, M. B. Tahir, S. Nazir, M. Alzaid, A. Ali, M. Sagir and A. Hussein, An Ab-initio simulation of boron-based hydride perovskites XBH3 (X= Cs and Rb) for advance hydrogen storage system, Comput. Theor. Chem., 2023, 1225, 114173 CrossRef CAS.
- M. Baaddi, R. Chami, O. Baalla, S. El Quaoubi, S. Ali, L. El H. Omari and M. Chafi, The effect of strain on hydrogen storage characteristics in K2NaAlH6 double perovskite hydride through first principle method, Environ. Sci. Pollut. Res., 2024, 31(53), 62056–62064 CrossRef CAS PubMed.
- Md Hasan Mia, Md Shahazan Parves, O. Alsalmi and Md Z. Hasan, Investigating Stability, Thermomechanical, Optoelectronic, and Hydrogen Storage Potential of MgXH3 (X= Ga, Tl) Perovskites via DFT and AIMD Simulations, ACS Appl. Eng. Mater., 2025, 3(11), 4218–4232 CrossRef CAS.
- L. Charpin and A. Ehrlacher, Estimating the Poroelastic Properties of Cracked Materials, Acta Mech., 2014, 225(9), 2501–2519 CrossRef.
- B. Ahmed, M. B. Tahir, M. Sagir, A. Parveen, Z. Abbas and A. A. Nassani, First-Principles Study of Ti-Based X2TiH5 (X= Mg, Ca, Sr) Hydrides for Advanced Hydrogen Storage Applications, Chem. Phys., 2025, 589, 112499 CrossRef CAS.
- Md H. Mia, Md Rasheduzzaman, O. Alsalmi and Md Z. Hasan, Theoretical Investigation of Gravimetric Capacity, Magnetic Behavior, and Stability of CsAH3 (A= Fe, Cu, and Tl) Hydrides via DFT and DFT-Based AIMD Simulations, Int. J. Hydrogen Energy, 2026, 209, 153607 CrossRef CAS.
- A. Manzoor, M. A. Ullah, M. W. Yasin, S. S. A. Gillani, M. Ikram and M. Rizwan, Investigation SrLiF3-xHx Perovskites through Ion Replacement Mechanism for Advance Solid Hydrogen Storage Systems: A Prediction through DFT-Based Calculations, J. Energy Storage, 2024, 88, 111639 CrossRef CAS.
- M. Rizwan, H. F. Arooj, F. Noor, K. Nawaz, M. A. Ullah, Z. Usman, A. Ali and T. Mahmood, Computational Study to Investigate Effectiveness of Titanium Substitution in CaFeH3 Perovskite-Type Hydride: An Approach towards Advanced Hydrogen Storage System, J. Mater. Res. Technol., 2024, 31, 2676–2684 CrossRef CAS.
- M. A. Ullah, K. N. Riaz and M. Rizwan, Computational Evaluation of KMgO3-xHx as an Efficient Hydrogen Storage Material, J. Energy Storage, 2023, 70, 108030 CrossRef.
- D. P. Broom, C. J. Webb, G. S. Fanourgakis, G. E. Froudakis, P. N. Trikalitis and M. Hirscher, Concepts for Improving Hydrogen Storage in Nanoporous Materials, Int. J. Hydrogen Energy, 2019, 44(15), 7768–7779 CrossRef CAS.
- F. Mouhat and F.-X. Coudert, Necessary and sufficient elastic stability conditions in various crystal systems, Phys. Rev. B, 2014, 90(22), 224104, DOI:10.1103/PhysRevB.90.224104.
- R. Hill, The elastic behaviour of a crystalline aggregate, Proc. Phys. Soc., London, Sect. A, 1952, 65(5), 349 CrossRef.
- I. R. Shein and A. L. Ivanovskii, Elastic properties of mono-and polycrystalline hexagonalAlB2-like diborides of s, p and d metals from first-principles calculations, J. Phys.: Condens. Matter, 2008, 20(41), 415218 CrossRef.
- I. N. Frantsevich, Elastic Constants and Elastic Moduli of Metals and Insulators, 1982 Search PubMed.
- Z. Sun, D. Music, R. Ahuja and J. M. Schneider, Theoretical investigation of the bonding and elastic properties of nanolayered ternary nitrides, Phys. Rev. B, 2005, 71(19), 193402, DOI:10.1103/PhysRevB.71.193402.
- D. V. Suetin, I. R. Shein and A. L. Ivanovskii, Elastic and electronic properties of hexagonal and cubic polymorphs of tungsten monocarbide WC and mononitride WN from first-principles calculations, Phys. Status Solidi B, 2008, 245(8), 1590–1597, DOI:10.1002/pssb.200844077.
- H. Murtaza, Q. Ain, S. A. Issa, H. M. Zakaly and J. Munir, A precise prediction for the hydrogen storage ability of perovskite XPH3 (X= Li, Na, K) hydrides: First-principles study, Int. J. Hydrogen Energy, 2024, 94, 1084–1093 CrossRef CAS.
- H. Murtaza, S. M. Qaid, H. M. Ghaithan, A. A. A. Ahmed and J. Munir, The excellent performance of AClH3 (A= Rb, Cs, K) perovskite hydrides for hydrogen storage applications, Renewable Energy, 2025, 252, 123491 CrossRef CAS.
- W. Khan and M. K. Masood, The investigation of rubidium-based hydrides for hydrogen storage application: density functional theory study, Mater. Sci. Semicond. Process., 2024, 173, 108149 CrossRef CAS.
- W. Kuang, H. Wang, X. Li, J. Zhang, Q. Zhou and Y. Zhao, Application of the thermodynamic extremal principle to diffusion-controlled phase transformations in Fe-CX alloys: Modeling and applications, Acta Mater., 2018, 159, 16–30 CrossRef CAS.
- H. Ehrenreich and H. R. Philipp, Optical Properties of Ag and Cu, Phys. Rev., 1962, 128(4), 1622–1629, DOI:10.1103/PhysRev.128.1622.
- D. N. Basov, R. D. Averitt and D. Hsieh, Towards properties on demand in quantum materials, Nat. Mater., 2017, 16(11), 1077–1088 CrossRef CAS PubMed.
- A. V. Zayats, I. I. Smolyaninov and A. A. Maradudin, Nano-optics of surface plasmon polaritons, Phys. Rep., 2005, 408(3–4), 131–314 CrossRef CAS.
- M. Irfan, E. M. Ahmed, S. A. M. Issa and H. M. H. Zakaly, New eco-friendly lead-free double perovskites Rb2FeCdO6 driven semiconducting material for radiation shielding and energy applications: DFT + U based nanoarchitectonics, Opt. Quant. Electron., 2024, 56(8), 1355, DOI:10.1007/s11082-024-07263-0.
- R. Sa, Y. Wei, W. Zha and D. Liu, A first-principle study of the structural, mechanical, electronic and optical properties of vacancy-ordered double perovskite Cs2TeX6 (X= Cl, Br, I), Chem. Phys. Lett., 2020, 754, 37538 CrossRef.
- M. Tarekuzzaman, M. S. Parves, M. Z. Rahman and S. S. Hasan, Cesium-Based Perovskite Hydrides: A Theoretical Insight into Hydrogen Storage and Optoelectronic Characteristics, Solid State Commun., 2025, 404, 116043 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Md. Hasan Mia
a,
Md. Hasan Mia





 , where H represents the total enthalpy of each system. The enthalpy is determined using: H = Eele + EZPE. Here, Eele and EZPE denote the electronic total energy and the zero-point energy, respectively. The zero-point energy is obtained from the phonon density of states as:
, where H represents the total enthalpy of each system. The enthalpy is determined using: H = Eele + EZPE. Here, Eele and EZPE denote the electronic total energy and the zero-point energy, respectively. The zero-point energy is obtained from the phonon density of states as:  , where h, ω, and g(ω) are Planck's constant, phonon frequency, and phonon density of states, respectively. The hydrogen desorption temperature (Tdes) is estimated using:44
, where h, ω, and g(ω) are Planck's constant, phonon frequency, and phonon density of states, respectively. The hydrogen desorption temperature (Tdes) is estimated using:44  , where, ΔS is the entropy change of the reaction. In this work, ΔS is approximated by the entropy of hydrogen gas, taken as 130.7 J mol−1 K−1. Using the calculated formation energies as an approximation to the decomposition enthalpy, the estimated desorption temperatures are 567 K for MgMoH3 and 350 K for MgInH3. These values indicate moderate thermal stability, suggesting that hydrogen release can occur at relatively accessible temperatures. Such moderate desorption temperatures are beneficial for practical hydrogen storage, as they reduce thermal stress during cycling while maintaining sufficient structural stability to prevent degradation or phase separation.
, where, ΔS is the entropy change of the reaction. In this work, ΔS is approximated by the entropy of hydrogen gas, taken as 130.7 J mol−1 K−1. Using the calculated formation energies as an approximation to the decomposition enthalpy, the estimated desorption temperatures are 567 K for MgMoH3 and 350 K for MgInH3. These values indicate moderate thermal stability, suggesting that hydrogen release can occur at relatively accessible temperatures. Such moderate desorption temperatures are beneficial for practical hydrogen storage, as they reduce thermal stress during cycling while maintaining sufficient structural stability to prevent degradation or phase separation. , where, NH is the number of hydrogen atoms per formula unit, mH is the mass of a hydrogen atom, V is the unit cell volume, and NA is Avogadro's number. The calculated volumetric hydrogen storage capacities are 112 kg m−3 for MgMoH3 and 72 kg m−3 for MgInH3. The higher volumetric capacity of MgMoH3 arises from its smaller lattice parameter and more compact crystal structure, which allows denser hydrogen packing. In contrast, the larger ionic radius of In leads to lattice expansion in MgInH3, resulting in reduced volumetric storage density. These results highlight a clear trade-off between atomic size and hydrogen packing efficiency, where smaller B-site elements favour enhanced volumetric storage performance.
, where, NH is the number of hydrogen atoms per formula unit, mH is the mass of a hydrogen atom, V is the unit cell volume, and NA is Avogadro's number. The calculated volumetric hydrogen storage capacities are 112 kg m−3 for MgMoH3 and 72 kg m−3 for MgInH3. The higher volumetric capacity of MgMoH3 arises from its smaller lattice parameter and more compact crystal structure, which allows denser hydrogen packing. In contrast, the larger ionic radius of In leads to lattice expansion in MgInH3, resulting in reduced volumetric storage density. These results highlight a clear trade-off between atomic size and hydrogen packing efficiency, where smaller B-site elements favour enhanced volumetric storage performance.







 , is a critical parameter for distinguishing isotropic (A = 1) from anisotropic (A ≠ 1) materials. The computed value is A = 0.40 for MgMoH3 and A = 0.79 for MgInH3. These hydride perovskites are mechanically anisotropic, as evidenced by the large deviations of both values from unity. A more unequal distribution of bonding stiffness across crystallographic orientations is reflected in the higher deviation in MgInH3, which suggests a greater directional dependency of its elastic response.
, is a critical parameter for distinguishing isotropic (A = 1) from anisotropic (A ≠ 1) materials. The computed value is A = 0.40 for MgMoH3 and A = 0.79 for MgInH3. These hydride perovskites are mechanically anisotropic, as evidenced by the large deviations of both values from unity. A more unequal distribution of bonding stiffness across crystallographic orientations is reflected in the higher deviation in MgInH3, which suggests a greater directional dependency of its elastic response. , measures a material's resistance to plastic deformation and serves as an indicator of the mechanical durability of hydrogen storage compounds. The calculated hardness for MgMoH3 is 6.12 GPa, whereas MgInH3 has 2.89 GPa. The soft-to-moderately hard domain (HV < 10 GPa)41 is where these hydride perovskites fall for both values, which is advantageous for preserving structural integrity and preventing excessive brittleness during hydrogen cycling. When compared with other reported hydride perovskites, such as MgCuH3 (0.77 GPa), RbNiH3 (0.64 GPa), BeGaH3 (3.02 GPa), and BeInH3 (3.17 GPa),58 both MgGaH3 and MgTlH3 show significantly higher hardness values, underscoring their enhanced mechanical stability. In hydrogen storage applications, this enhanced hardness and ductility (shown by B/G > 1.75) imply that these compounds can withstand microcrack development and structural deterioration, guaranteeing dependable cycle performance.
, measures a material's resistance to plastic deformation and serves as an indicator of the mechanical durability of hydrogen storage compounds. The calculated hardness for MgMoH3 is 6.12 GPa, whereas MgInH3 has 2.89 GPa. The soft-to-moderately hard domain (HV < 10 GPa)41 is where these hydride perovskites fall for both values, which is advantageous for preserving structural integrity and preventing excessive brittleness during hydrogen cycling. When compared with other reported hydride perovskites, such as MgCuH3 (0.77 GPa), RbNiH3 (0.64 GPa), BeGaH3 (3.02 GPa), and BeInH3 (3.17 GPa),58 both MgGaH3 and MgTlH3 show significantly higher hardness values, underscoring their enhanced mechanical stability. In hydrogen storage applications, this enhanced hardness and ductility (shown by B/G > 1.75) imply that these compounds can withstand microcrack development and structural deterioration, guaranteeing dependable cycle performance.


















