DOI:
10.1039/D6RA03016E
(Review Article)
RSC Adv., 2026,
16, 27593-27609
Advancements in the manufacturing routes for the synthesis of marketed PARP inhibitors
Received
10th April 2026
, Accepted 6th May 2026
First published on 21st May 2026
Abstract
The significant role of PARP inhibitors in the treatment of various cancers, especially ovarian and breast cancer, is very well established. To date, seven PARP inhibitors have received marketing approval, namely, olaparib, rucaparib, niraparib, talazoparib, fuzuloparib, pamiparib and senaparib, in various countries. This article reviews the manufacturing routes for the synthesis of these marketed drugs reported in the literature since 2017. Over the past decade, synthetic strategies for PARP inhibitors have transitioned from step-intensive medicinal chemistry synthesis routes to more streamlined, industrially viable processes. These modern approaches integrate key transformations, such as amide bond formation, C–H activation, carbopalladation, heterocycle construction, and stereocontrolled cyclisation, enabling improved efficiency and scalability. In parallel, the incorporation of green chemistry principles, particularly in one of the manufacturing routes of olaparib, has led to more sustainable process development.
 Dhurgam Rani | Dhurgam Rani completed her B. Pharmacy from RBVRR Women's College of Pharmacy, Hyderabad, in 2022, building a strong foundation in pharmaceutical sciences and research. She further pursued her M. Pharmacy in Medicinal Chemistry at NIPER Hyderabad, India, where she graduated with distinction and was awarded a Gold Medal for academic excellence in 2024. Currently, she is pursuing her PhD through the BITS–RMIT Joint PhD Program. Her research focuses on the design and development of novel PARP inhibitors for the treatment of ovarian cancer, integrating medicinal chemistry, molecular biology, and advanced drug discovery approaches for improved therapeutic outcomes. |
 Magdalena Plebanski | Magdalena Plebanski is an immunologist. Her group integrates immunology, nanotechnology, and translational science to develop innovative diagnostics, vaccines and therapies. She completed her PhD at the University of Bristol and postdoctoral studies at the University of Oxford, UK, where she pioneered work on immuno-suppression and vaccine design. She subsequently held senior research and leadership roles at the Burnet Institute and Monash University before joining RMIT University, where she is now a Distinguished Professor, and Director of the Biomedical & Health Innovation Enabling Impact Platform and the Accelerator for Translational Research and Clinical Trials (ATRACT) Centre. Her research currently focuses on immune suppression, cancer immunotherapy and diagnostic and prognostic biomarkers in ovarian cancer. She has published over 250 peer-reviewed papers, has an H-index of 70, and is a top 0.5% highly cited scholar. A number of her 60 patents across multiple patent families on vaccines and prognostic biomarker technologies progressed to clinical trials. She has received numerous international honours, for example, the Howard Hughes International Scholar (HHMI) Award, USA, and the Distinguished Mexican Award. |
 Tanmay Chatterjee | Tanmay Chatterjee obtained his MSc degree in Chemistry from IIT Delhi in 2009. He received a PhD degree in 2014 from Jadavpur University for his work on green synthesis under the supervision of Prof. B. C. Ranu at IACS, Kolkata, India. After a postdoctoral stint with Prof. E. J. Cho in South Korea, he started his independent career at BITS Pilani, Hyderabad Campus, India, in 2018. Currently, he is working as an Associate Professor, and his research interests include the development of green synthetic methodologies by iodine catalysis, visible-light photocatalysis, electrochemical organic transformations, and solvent-mediated or controlled, reagentless organic transformations. His group also focuses on the design and discovery of novel drug molecules for the treatment of breast cancer, ovarian cancer, and Huntington's disease. To date, he has published 68 papers in reputable international journals, with an H-index of 31, as well as 3 book chapters and 4 patents. He received the prestigious Thieme Chemistry Journals Award 2025 and was recognised as an Emerging Investigator 2024 by the journal Chemical Communications. |
1. Introduction
Poly ADP-ribose polymerase (PARP) is a family of nuclear enzymes that catalyse the poly(ADP-ribosyl)ation process, playing a central role in genomic stability by sensing DNA damage. The critical function of PARP is well established in both healthy and cancerous cells, orchestrating the DNA repair mechanism. However, compared to healthy cells, cancerous cells express high levels of PARP, enabling these cells to repair damaged DNA and thereby sustain their viability. Among the 17 known PARP isoforms, PARP1 and PARP2 are the best characterised and together account for the majority of cellular poly(ADP-ribosyl)ation activity.1 PARP1, in particular, functions as a rapid DNA damage sensor that, upon encountering DNA single-strand breaks, binds to the damaged DNA through its N-terminal zinc finger domain and activates the catalytic domain of the C-terminal. This activation triggers the transfer of ADP-ribose units from NAD+ to the target protein. This process produces long, branched PARP chains that further recruit DNA repair machinery to the site of damage. Through this mechanism, PARP1 acts as a key regulator of base excision repair (BER), chromatin remodelling, and the coordination of various DNA repair pathways.2 The therapeutic relevance of PARP emerged from the discovery of synthetic lethality between PARP inhibition and defects in homologous recombination repair (HRR), particularly mutations in BRCA1 and BRCA2. In HRR-deficient tumours, inhibition of PARP1/2 causes the accumulation of DNA damage, collapse of replication forks, and ultimately irreparable double-strand breaks, leading to cell death.3 This mechanistic vulnerability enabled the development of PARP inhibitors (PARPis) as a novel class of targeted anticancer agents. PARPis primarily act as reversible competitive inhibitors of the NAD+ binding site of the PARP enzyme, mimicking the nicotinamide moiety of NAD+. Clinically approved PARPis, such as olaparib, rucaparib, niraparib, talazoparib, fuzuloparib, pamiparib, and senaparib (Fig. 1), exhibit both catalytic inhibition and “PARP trapping,” wherein the inhibitor stabilises PARP–DNA complexes, converting them into cytotoxic DNA lesions.
 |
| | Fig. 1 Marketed PARP inhibitors developed for cancer treatment. | |
These marketed PARP inhibitors have transformed the therapeutic landscape for ovarian,4 breast,5,6 and prostate cancers,7,8 particularly in patients with BRCA1/2 mutations or broader HRR defects. Ongoing research is rapidly expanding its application to combination therapies with chemotherapy, radiotherapy, immune checkpoint inhibitors, and agents targeting DNA damage response pathways such as ATR, WEE1, and CDK12.9 Despite their clinical success, challenges, such as acquired resistance, limited biomarkers of response, and toxicity management, continue to shape current research efforts. Despite structural heterogeneity, marketed PARP inhibitors share key features that enable them to bind potently to the catalytic domain of PARP1/2. Most inhibitors emulate the nicotinamide mimicking amide/lactam core for NAD+ competitive binding and interact via π–π stacking, hydrogen bonding within the glycine-rich loop, and hydrophobic contacts within the donor site. Likewise, all the approved PARP inhibitors, while sharing the common pharmacophore, i.e., amide/lactam, differ significantly in their peripheral scaffolds, ranging from rigid fused heterocycles (talazoparib, rucaparib) to flexible benzamide derivatives (niraparib, senaparib), which modulate PARP trapping potency. Other heterocycles, such as piperazine and piperidine (olaparib, niraparib, and senaparib), and electron-withdrawing groups (EWG), such as –CF3 (fuzuloparib), control the pharmacokinetic and safety profiles. Medicinal chemists have explored diverse heterocyclic scaffolds, including phthalazinones, benzimidazoles, indazoles, dihydropyridazinones, and fused bicyclic systems, to achieve optimal target engagement and favourable drug-like properties.10 Importantly, not all clinical inhibitors rely equally on the classic catalytic inhibition mechanism. Some compounds, notably talazoparib, exhibit exceptionally strong PARP-trapping activity, stabilizing the PARP–DNA complex and thereby amplifying cytotoxicity. These mechanistic subtleties have guided substantial structural optimization and continue to influence the development of next-generation derivatives. As the clinical importance of PARP inhibitors continues to expand, understanding their synthetic evolution has become increasingly relevant. Synthesis is not merely a means to obtain the final drug molecule; it is central to optimizing structure–activity relationships, enabling late-stage functionalization, and improving selectivity across PARP isoforms. Furthermore, several PARP inhibitors have undergone extensive synthetic iterations even after clinical approval, leading to new derivatives with enhanced oral bioavailability, fewer off-target effects, improved CNS penetration, or activity in HRR-proficient tumours. The synthetic strategies developed for these inhibitors have also inspired chemists to design novel PARP-targeted conjugates, bifunctional degraders, radioligands for imaging, and dual-acting inhibitors capable of targeting complementary DNA-damage response pathways.
Given the rapid pace of innovation, a comprehensive understanding of the synthetic methodologies used to construct marketed PARP inhibitors is essential for medicinal chemists and chemical biologists aiming to design next-generation molecules. This review focuses exclusively on the recent advances in developing novel synthetic routes to access marketed PARP inhibitors, such as olaparib, rucaparib, and niraparib, from 2017 to 2025, as the literature on the same topic was reviewed by David, covering the developments before 2017.11 Soon after these molecules were developed, talazoparib was approved by the FDA in 2018, and fuzuloparib, pamiparib, and senaparib acquired marketing approval in China. Hence, this article also provides a comprehensive overview of the synthetic routes to talazoparib, fuzuloparib, pamiparib, and senaparib.
2. Olaparib
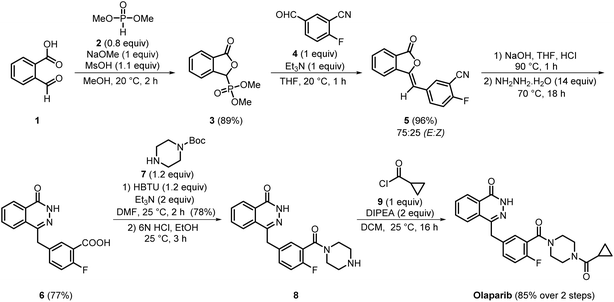
Olaparib is the first-in-class oral, small-molecule PARP1/2 inhibitor, demonstrating potent enzymatic inhibition with IC50 values of 5 and 1 nM, respectively.12 Owing to the therapeutic significance and distinctive structural features, the medicinal chemistry synthesis route devised for olaparib became a crucial component of its early development. The KUDOS team established an efficient and strategically modular pathway that enabled rapid access to the phthalazinone core and its essential substituents (Scheme 1). It commenced with readily available starting material, 2-carboxybenzaldehyde 1, which, upon reaction with dimethyl phosphite 2, sodium methoxide, and MsOH, afforded 3 in 89% yield. The intermediate 3 was further subjected to the Wittig reaction with 4, yielding compound 5 with an excellent yield of 96%, albeit with poor stereoselectivity (E/Z = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Subsequent hydrolysis of 5 using aq. NaOH and cyclisation with hydrazine hydrate afforded the key intermediate 6 in 77% yield. In the next step, acid amine coupling of 6 with Boc-protected piperazine 7, followed by Boc deprotection, produced penultimate intermediate 8 with 78% yield. Finally, the reaction of 8 with cyclopropyl carbonyl chloride 9 successfully produced olaparib in 85% yield.13,14
:1). Subsequent hydrolysis of 5 using aq. NaOH and cyclisation with hydrazine hydrate afforded the key intermediate 6 in 77% yield. In the next step, acid amine coupling of 6 with Boc-protected piperazine 7, followed by Boc deprotection, produced penultimate intermediate 8 with 78% yield. Finally, the reaction of 8 with cyclopropyl carbonyl chloride 9 successfully produced olaparib in 85% yield.13,14
 |
| | Scheme 1 Olaparib medicinal chemistry synthesis route developed by KUDOS. | |
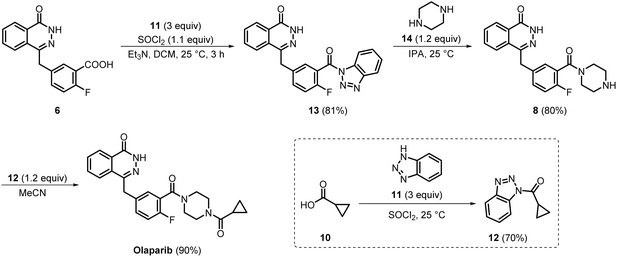
In 2017, Alembic Pharmaceuticals developed a high-yielding process and synthesised olaparib with 98% purity (Scheme 2). In this process, the carboxylic acid functionality of intermediate 6 was activated using benzotriazole 11 and a benzotriazolyl ester 13 was prepared, which significantly facilitated subsequent amide bond formation. Moreover, the recrystallisation of crude material with water and isopropanol enabled easy isolation of compound 13. This activation strategy significantly minimised competing side reactions and enhanced coupling efficiency, thereby improving the overall yield. Subsequently, the transamidation of intermediate 13 with piperazine 14, followed by recrystallisation from the same solvent system, i.e., water and isopropanol, afforded compound 8 with an isolated yield of 80%. In the final step, compound 8 was reacted with intermediate 12 (prepared earlier through an acid-amine coupling between 10 and 11 and recrystallised from methanol), giving olaparib in 90% yield. Finally, olaparib was effortlessly purified by performing simple filtration and washing with acetonitrile.15 Overall, this process offered several advantages over previously reported methods. Use of the benzotriazole activation strategy effectively minimised the formation of by-products, while reliance on crystallisation-based purification eliminated the need for the substantial amount of hazardous organic solvents required for column chromatography. Thus, this process could be considered greener and more cost-effective than the previously reported processes. Moreover, in this process, each step delivered products with high yield and excellent purity, making this route well-suited for the industrial-scale synthesis of olaparib.
 |
| | Scheme 2 Synthetic route to olaparib developed by Alembic Pharmaceuticals. | |
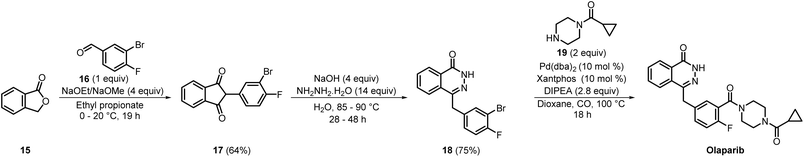
During the same period, Apotex developed a novel process for preparing intermediates 18 and 19, as well as olaparib (Scheme 3). In this process, phthalide 15 and compound 16 were initially reacted in ethyl propionate employing either sodium ethoxide or methoxide. Later, cooling the reaction mixture to 0 °C and acidifying to pH 1–2 with hydrochloric acid yielded a dark-red precipitate. This precipitate, upon further filtration and purification by column chromatography, yielded compound 17 in 64% yield. In the next step, compound 17 was refluxed with hydrazine hydrate in the presence of sodium hydroxide. Filtration of the reaction mixture upon completion furnished intermediate 18 in 75% yield. Finally, the Pd-catalysed amino carbonylation of intermediate 18 with compound 19 resulted in the production of olaparib.16 Overall, this process required only three steps for the synthesis of olaparib starting from commercially available compound 15, and thus it could be considered as a more step-economical process as compared to the medicinal chemistry synthesis route to olaparib. Although fewer chemicals were used in this process than in the medicinal chemistry synthesis route, it required a highly expensive, rare-earth-metal catalyst, Pd(dba)2, along with the xantphos ligand.
 |
| | Scheme 3 Preparation of intermediates 17 and 18 and the product olaparib. | |
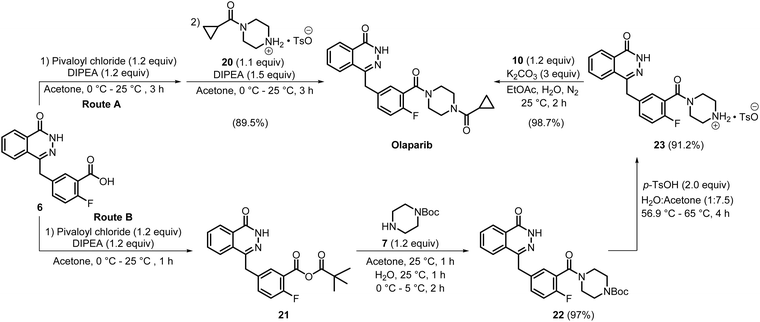
In the following year, two olaparib manufacturing routes, A and B (Scheme 4), were developed by ScinoPharm Singapore Pte, Ltd. In route A, acid amine coupling of compounds 6 and 20 was done using pivaloyl chloride as a coupling agent along with N,N-diisopropylethylamine (DIPEA). Pivaloyl chloride has excellent solubility in organic solvents, thereby facilitating a smooth and efficient coupling process and ultimately affording olaparib in 89.5% yield. In this route, olaparib was isolated as an off-white solid by performing a series of purification steps, including filtration after a basic workup, extraction using ethyl acetate, and recrystallisation from methanol and water. In route B, compounds 6 and 7 were subjected to acid–amine coupling under conditions similar to those in route A, enabling the formation of compound 22 in one pot via intermediate 21. The product was isolated by filtration followed by aqueous washing, affording 97% yield. Subsequent treatment of 22 with p-toluene sulfonic acid (p-TsOH), followed by filtration of the formed solid precipitate, and acetone washing, produced 23 in 91.2% yield. The final reaction of 23 with 10, using K2CO3 as the base in a biphasic solvent system, generated olaparib as a precipitate, which was isolated by filtration, with an excellent yield of 98.7%.17 The use of cost-effective and less hazardous reagents, such as pivaloyl chloride, instead of expensive coupling agents, such as HBTU, used in the medicinal chemistry synthesis route to olaparib, and the adoption of benign solvents such as acetone and water, along with the admirable yields of the products using simple filtration procedures without compromising purities, rendered these two routes industrially friendly.
 |
| | Scheme 4 Manufacturing process of olaparib developed by ScinoPharm Singapore Pte, Ltd. | |
Later, in 2022, a novel synthetic route (Scheme 5) for the key intermediate 6 was developed by Chen et al. Compound 27 was synthesised initially by brominating 26 using N-bromosuccinimide (NBS) and benzoyl peroxide (BPO), followed by Zn-incorporation, in 92% yield. Further, Negishi coupling of 27 and 25 (previously prepared from 24 using POCl3) afforded 28, which was isolated by filtration in 80% yield. Subsequent alkaline hydrolysis of 28 afforded the key intermediate 6 in 85% yield, which was isolated by filtration followed by aqueous washings.18 In this process, the main core of olaparib 6 was synthesised in 4 steps with good yields while leaving scope for further simplification of the process, including the elimination of expensive chemicals such as Pd catalysts and toxic chemicals such as POCl3 and BPO, as well as careful handling during the Negishi coupling reaction.
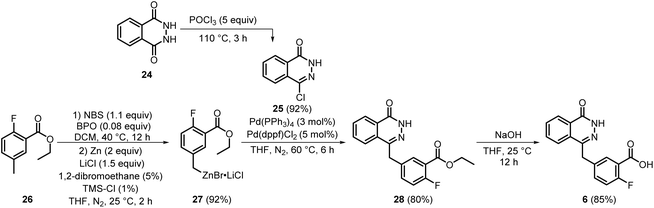 |
| | Scheme 5 Scalable manufacturing process for the synthesis of key intermediate 6 involved in olaparib synthesis. | |
In October 2023, an eco-friendly synthetic process to access olaparib using commercially inexpensive starting materials was developed by Chatterjee et al.,19 which involved four steps with an overall yield of 51% (Scheme 6). The first step of the synthetic protocol involved the nucleophilic substitution reaction of 9 with piperazine 15 in acetic acid, producing 19, selectively, in 88% yield. In the subsequent step, acid-amine coupling of 19 with 29 produced 30 in a moderate yield of 75%. Reaction of 30 with 2-acetyl benzoic acid 31 generated compound 32 in 85% yield, where the enolization of 31 using sodium tertiary butoxide took place initially, followed by regioselective radical-nucleophilic aromatic substitution (SRN1) reaction (where the solvent DMF acted as a source of radical initiator).
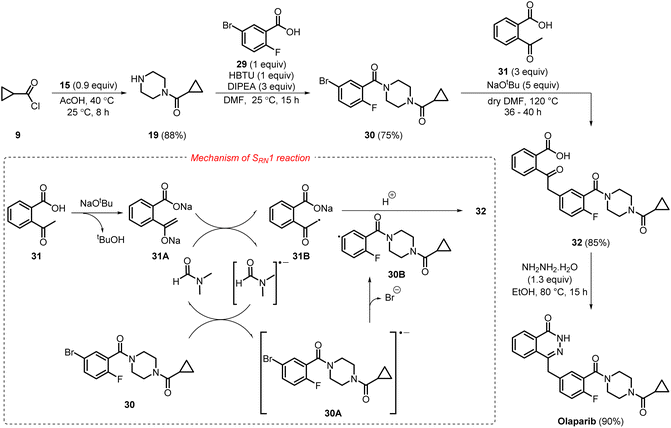 |
| | Scheme 6 Scalable and eco-friendly synthetic approach towards olaparib. | |
First, the 2-acetyl benzoic acid 31 underwent deprotonation and enolization to form the corresponding enolate 31A, which then transfers an electron (SET) to DMF, thus producing the enolate radical 31B and the corresponding radical anion of DMF. Then a single electron transfer (SET) from the radical anion of DMF to the substrate 30 generated the corresponding radical anion 30A, which immediately underwent bromide elimination to furnish the corresponding aryl radical 30B. Finally, radical–radical coupling between 30B and 31B furnished the desired product 32. The SRN1 reaction is highly regioselective, as bromide is a much better leaving group than fluoride. The final cyclisation of 32 with hydrazine hydrate furnished olaparib in 90% yield. This method addressed the various challenges associated with the medicinal chemistry synthesis route to olaparib. It successfully eliminated the use of transition metals and the formation of the by-product phosphine oxide, achieving a relatively high atom economy of 35.9% through a simple, environmentally benign process. Metrics observed include process mass intensity (PMI) of 18.2 g g−1, mass productivity of 5.5%, E-factor of 17.2 g g−1, and a reaction mass efficiency (RME) of 12.6% (Table 1). However, further optimisation of this process still requires eliminating the use of genotoxic hydrazine and achieving much better yields in large-scale synthesis.19
Table 1 Comparison of green metrics between the reported synthesis route19 and the medicinal chemistry synthesis route to olaparib
| S. no |
Parameters |
Medicinal chemistry synthesis route |
Reported synthesis route |
| 1 |
No of steps |
6 |
4 |
| 2 |
Atom economy |
26.0% |
35.9% |
| 3 |
Overall yield |
46.0% |
51.0% |
| 4 |
Atom efficiency |
12.0% |
18.3% |
| 5 |
Process mass intensity (PMI) |
51.8 g g−1 |
18.2 g g−1 |
| 6 |
Mass productivity |
1.9% |
5.5% |
| 7 |
E-factor |
50.8 g g−1 |
17.2 g g−1 |
| 8 |
Effective mass yield |
2.0% |
5.8% |
| 9 |
Reaction mass efficiency (RME) |
7.5% |
12.6% |
More recently, in 2024, Glenmark Life Sciences disclosed a novel synthetic process for the synthesis of olaparib (Scheme 7). This group attempted to synthesise intermediate 35 from the commercially available compound 4. By protecting the aldehyde group in compound 4 with trimethyl orthoformate (TMOF), the nitrile group was allowed to hydrolyse, producing 34 in 91% yield. In sequence, the acid amine coupling of compounds 34 and 19 produced 35 in 95% yield. Wittig reaction of 35 with 3 produced 36 in 90% yield. Compound 36 was cyclized with hydrazine hydrate in the final step, and olaparib was synthesised in 80% yield. Various impurities (Fig. 2) encountered during the olaparib synthesis process were reported to be less than 0.15%.20 The process consists of straightforward reactions that are easy to execute, yielding excellent results at each step. The isolation of products was primarily accomplished through filtration. Additionally, minimising impurity formation greatly improved the yield at each step.
 |
| | Scheme 7 Process for olaparib synthesis patented by Glenmark Life Sciences. | |
 |
| | Fig. 2 Impurities encountered during the synthesis of olaparib. | |
3. Rucaparib
Rucaparib is another small-molecule PARPi with good oral bioavailability. It selectively targets PARP1, 2, and 3 with an IC50 of 1 nM.21 The medicinal chemistry synthesis route for the synthesis of rucaparib, comprising 7 steps, was developed by Agouron and the Cancer Research Campaign Technology in 2002 (Scheme 8).22 The commercially available compound 43 was converted into its enamine form 44 by reacting with DMF–DMA in an excellent yield of 97%. Reductive cyclisation of 44 using Pd/C furnished 45 in 81% yield. In the subsequent step, 45 was reacted with 46 in the presence of a catalytic amount of t-butylcatechol, producing 47 in 89% yield. Zn-mediated reduction followed by cyclisation of 47 produced compound 48 in a moderate yield of 73%. Selective bromination at the 2-position of the indole ring of 48 gave 49, and the Suzuki coupling reaction of 49 with 4-formylphenylboronic acid 50 resulted in the formation of 51 in 77% yield. Final reductive amination of the aldehyde group of 51 using sodium cyanoborohydride delivered rucaparib in 68% yield.23
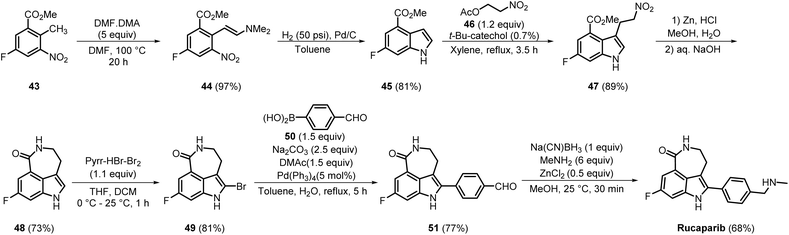 |
| | Scheme 8 Medicinal chemistry synthesis route for the synthesis of rucaparib. | |
In 2020, a concise synthetic method employing cascade carbopalladation followed by C–H amination using N,N-di-tert-butyldiaziridinone (Scheme 9) was developed by Cang Cheng et al. Sonogashira coupling of 52 and 53 produced 54 in a good yield 85%. Involving a substitution reaction, 54 was next converted into p-methoxybenzylamine (PMB) protected compound 55, which was isolated by filtration in 72% yield. Furthermore, compound 55 reacted with commercially available compound 56, yielding compound 57 in 82% yield. In the subsequent step, the key intermediate 57 was converted into 59 in 87% yield through a cascade carbopalladation followed by C–H amination. The final Boc deprotection of 59 using trifluoroacetic acid (TFA) yielded rucaparib in 88% purity.24 This approach allowed the successful synthesis of rucaparib using easily accessible starting materials and achieved good yields through a few straightforward steps, with flash chromatography utilised for purification. However, a significant drawback is the reliance on expensive catalysts such as Pd, which poses challenges for industrial scalability.
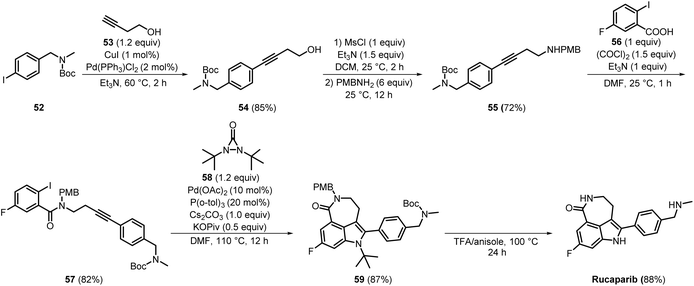 |
| | Scheme 9 Synthesis of rucaparib through cascade carbopalladation and C–H amination. | |
In 2022, rucaparib was synthesised by Beck et al., using a methodology that involved a streamlined, seven-step process suitable for industrial-scale synthesis via the production of novel intermediates (Scheme 10). In this approach, palladium/copper-catalysed Sonogashira coupling of compounds 62 with 63 resulted in the formation of 64 with an impressive yield of 86%. In the next step, acylation using trifluoroacetic anhydride (TFAA) in the presence of triethylamine produced 65 in 53% yield. Further treatment of 65 with TFA in a triflic acid/water solution (in a ratio of 3.6:1) gave intermediate 66 in a moderate yield of 66%. The Fisher indole reaction of 66 with 61 (previously generated via the diazotisation of 60 in 97% yield) produced 67 in 76% yield, involving a cyclic [3,3]-sigmatropic rearrangement as the key step. Compound 67 then underwent a transamidation reaction with hydrazine, followed by intramolecular cyclisation (acid–amine coupling) and TFA deprotection, affording 68 in 89% yield. Final debromination was facilitated using palladium/carbon, and rucaparib was delivered in an excellent yield of 96%.25 The method was used to successfully synthesise rucaparib on a 70 mmol scale with a good yield; however, the need for column chromatography for purification at every step, particularly re-purification by column chromatography during the preparation of compound 66, makes the process labour-intensive, with high organic solvent consumption, and reduces the overall yield.
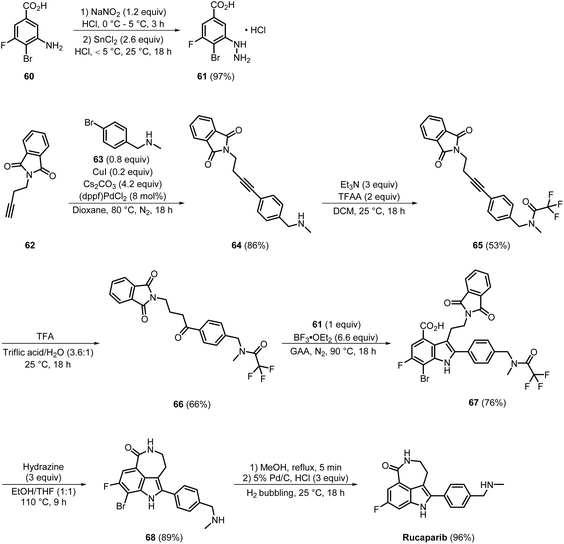 |
| | Scheme 10 Patented process for the synthesis of rucaparib. | |
4. Niraparib
Niraparib, a potent, orally active small-molecule inhibitor (IC50 = 4 nM), exhibited a 100-fold selectivity for PARP1 and PARP2 over other PARP family members.26 The medicinal chemistry synthesis route for the synthesis of niraparib was developed by Jones et al. in 2009 (Scheme 11). This process initially involved an independent synthesis of two key intermediates, 72 and (S)-77. For the synthesis of 72, compound 69 was initially converted into the ester, then brominated with NBS to produce 70 in 40% yield. This was followed by the reaction of compound 70 with 71, which provided 72 in 74% yield. Meanwhile, the second intermediate (S)-77 was synthesised from the commercially available compound 73, which was reacted with 74 in the presence of Pd (PPh3)4 to generate 75 in 71% yield. The nitro group in 75 was subsequently reduced to an amine 76, giving the product in 90% yield. Later, from 76, compound (S)-77 was resolved using L-tartaric acid, and the secondary amine was Boc-protected in a yield of 24.8% and an enantiomeric excess (ee) of 80–90%. In the subsequent step, both intermediates 72 and (S)-77 were reacted to form imine-containing compound 78. Compound 78 was then reacted with sodium azide, giving 79 in 46% yield. Following this, 79 was converted into 80 in 72% yield using ammonia. Finally, Boc deprotection of 80, followed by chiral separation, resulted in niraparib·HCl in 93% yield and an overall yield of 3–4%.27,28
 |
| | Scheme 11 Medicinal chemistry synthesis route to niraparib. | |
In 2021, Teva Pharmaceuticals reported a significant advancement in the synthesis of niraparib (Scheme 12). The procedure commenced with the transformation of the methyl group of 81 into aldehyde 72, giving the product in 57% yield. This conversion was facilitated through the formation of enaminone compound 82, utilising DMF/DMA and sodium periodate (NaIO4). Subsequently, 72 underwent aldehyde protection with methanesulfonic acid, giving compound 83 in a remarkable 95% yield. The ester functionality of 83 was then converted into amide 84 in 66% yield. Aldehyde deprotection produced 85 in 90% yield. Intermediate compound (S)-77 was synthesised from commercially available 86. Upon reacting 86 with 87, compound 88 was generated in a yield of 71%. Compound 88 was subsequently reacted with N-methylpyrrolidone (NMP) and methyl acrylate 89, affording 90 in 67% yield. Compound 76 was then synthesised from compound 91 via initial treatment with RANEY® nickel, producing compound 92 in 66% yield, followed by a reduction with NaBH4 and boron trifluoride etherate, which provided the product in 87% yield. Boc protection of 76 to 93 followed by further resolution yielded (S)-77. The two intermediates, 85 and (S)-77, were then reacted to produce compound 94 in 67% yield. Subsequently, 94 was cyclized using sodium azide in 2,6-lutidine, affording 80 in 66% yield. Finally, 80 was reacted with p-toluene sulfonic acid monohydrate, resulting in the desired niraparib tosylate in a yield of 65%.29 This innovative method notably reduced the use of hazardous chemicals and minimised the formation of regioisomers as impurities. By omitting the carbon–nitrogen cross-coupling step and converting the ester group into an amide early in the synthesis, the process demonstrated enhanced cost-effectiveness and was less labour-intensive, as compounds were isolated by simple extraction, filtration, and washing with different solvents at almost every step. While the use of hazardous chemicals has been reduced to some extent, the number of steps involved still presents an opportunity for improvement, as it impacts the overall yield of the process.
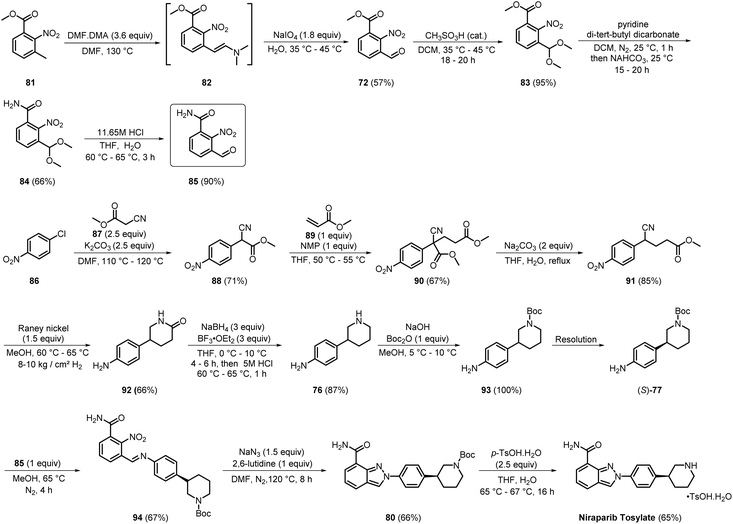 |
| | Scheme 12 Synthetic procedure developed by Teva Pharmaceuticals to access niraparib. | |
During the same period, the Zai Lab developed a synthetic process to access intermediate (S)-77 (Scheme 13). Initially, the acid amine coupling of 95 and 96, utilising CDI, was performed to produce 97 in 90% yield. Subsequently, the cyclised compound 98 was prepared in 30% yield from 97 by treatment with potassium carbonate and methyl tert-butyl ether (MTBE), and isolation by filtration followed by recrystallisation. Further reduction of the carbonyl group of 98 resulted in the formation of 99. In the next step, 99 was treated with 20% Pd (OH)2/C and HOAc to generate 100, which was isolated by simple extraction with ethyl acetate and purified by recrystallisation from a mixed solvent of ethyl acetate and n-heptane in a yield of 55% over 2 steps, achieving 98.5% ee. The final selective Boc protection of the cyclic amine of compound 100 produced the desired key intermediate (S)-77 in 85% yield, which was isolated by simple filtration and washings with water/isopropanol.30 This method successfully eliminated the use of precious metals, such as Pd, and chiral resolution via chromatography, which greatly reduced its associated cost and environmental burden. It also avoided steps such as carbon–nitrogen coupling, which often led to the formation of by-products. However, a multistep, complex procedure, along with a lower yield, especially in step 2, which involved the formation of compound 98, still requires further optimisation for better industrial suitability.
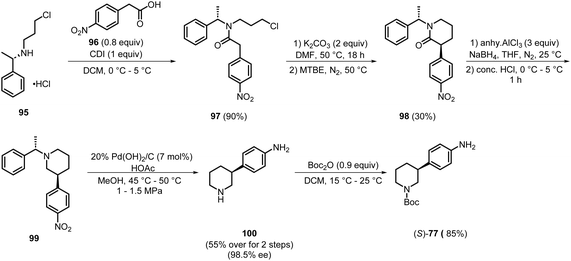 |
| | Scheme 13 Patented process for the synthesis of key intermediate (S)-77 by the Zai Lab. | |
In 2023, TESARO, Inc. disclosed a novel method for preparing niraparib (Scheme 14). Initially, intermediate 72 was prepared from the commercially available starting material 81. Compound 81 was converted into its enaminone form 82 using DMF·DMA, and a subsequent reaction with NaIO4 gave 72 in 80% yield. Concurrently, intermediate 112 was synthesised from 105. The amine group in 105 was acylated with benzoyl chloride, giving compound 106 in 97% yield. The borylation of compound 106 was then performed using n-butyllithium (n-BuLi) and triisopropoxyborane [B(O-iPr)3], delivering compound 107 in 92% yield. Afterwards, compound 107 was protected using pinacol (108), yielding 109 with full conversion. Next, compound 109 reacted with 103, which was separately synthesised from 101 in two steps: oxidation of 101 to form 102, followed by tosylation to generate 103 in 77% yield, along with 104. This reaction produced 110 in 91% yield. The double bond adjacent to the nitrogen in 110 was specifically reduced using a Rhodium catalyst and the chiral ligand Josiphos SL-J505-2, resulting in the formation of 111. Subsequent deprotection of the amine group afforded compound (S)-77 in 96% yield. Further reaction of compound (S)-77 with ethyl sulfonic acid produced 112 in 77.6% yield.
 |
| | Scheme 14 Patented process for the synthesis of niraparib by TESARO. | |
The two intermediates, 113 and (S)-77, were coupled to generate compound 114. The cyclisation of compound 114 was achieved using Cu(OTf)2, resulting in the production of 79. In the following steps, the ester group of 79 was converted into an amide. This was initiated by lithiation with LiOH, producing 115 in 86% yield, followed by coupling with ammonium hydroxide using CDI, yielding 80. Finally, tosylation of compound 80 followed by Boc deprotection gave niraparib tosylate in 87% yield.31 This convergent synthesis of niraparib employed simple reaction steps, and the products were mainly isolated by filtration, followed by washings with different solvents in each step. However, the use of expensive reagents such as TEMPO and Pd catalyst, together with the large number of steps, greatly affects the cost-effectiveness and overall yield of the process, respectively.
5. Talazoparib
Talazoparib, a potent oral PARP inhibitor (IC50 = 0.57 nM), was developed by Pfizer.32,33 The medicinal chemistry synthetic route to talazoparib was developed by BioMarin Pharmaceutical in 2016, and comprised six steps (Scheme 15). Knoevenagel condensation of commercially available 116 with 117 produced 118 in 78% yield. Subsequently, the cyclic intermediate 118 was cleaved to produce 119 in an excellent 99% yield. Reductive coupling of 119 and 120 generated 121 in 95% yield. Isolation of the compounds was done by filtration in all steps. The cyclisation of 121 to 122, followed by chiral separation, produced talazoparib with >99% ee.34,35
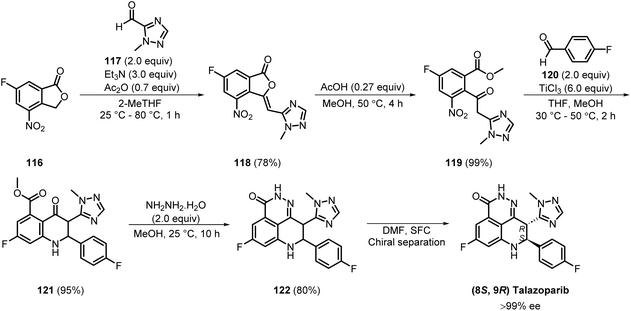 |
| | Scheme 15 Medicinal chemistry synthesis route for the synthesis of talazoparib developed by BioMarin Pharmaceutical Inc. | |
During the same period, a synthetic procedure for the triazole intermediate 124 was developed by Henderson et al. (Scheme 16). This triazole intermediate can serve as an alternative to compound 117 in talazoparib synthesis, as it involves simple filtration of the product 118 without much affecting the yield in the first step of Scheme 15. For this, compound 123 was reacted with lithium bis(trimethylsilyl)amide (LiHMDS) in DMF/2-methyl THF under cold conditions for about 1 hour, producing the desired triazole intermediate 124 in 72.5% yield.36
 |
| | Scheme 16 Synthesis of triazole intermediate 124. | |
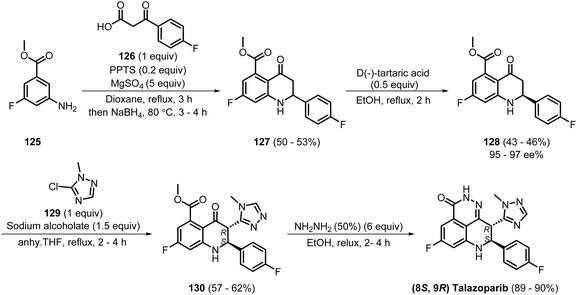
The following year, Xu et al. disclosed a concise synthetic route to talazoparib involving four steps (Scheme 17). In the first step, compound 125 was reacted with 126 in the presence of pyridinium p-toluene sulfonate (PPTS) and MgSO4, producing 127, which was purified using column chromatography in 50–53% yield. The transformation involved an imine formation via Schiff-base condensation, followed by intramolecular Friedel–Crafts acylation and reduction of the imine to the amine. Then, in the second step, compound 128 was separated from 127 using kinetic resolution with D(−)-tartaric acid, giving the product in 43–46% yield and 95–97% ee. Next, compound 128 was reacted with 129 using sodium alcoholate to produce 130 in a moderate yield of 57–62% via a nucleophilic heteroaromatic substitution reaction. The final cyclisation of compound 130 using hydrazine (NH2NH2) generated talazoparib in 89–90% yield. After the reaction was complete, talazoparib was isolated by evaporating the solvent, filtering, and then recrystallising as a white solid from ethanol.37 This method successfully reduced the number of steps required from six to four, leaving scope to further optimise the process and achieve better yields and industrial suitability.
 |
| | Scheme 17 Patented process for the synthesis of talazoparib. | |
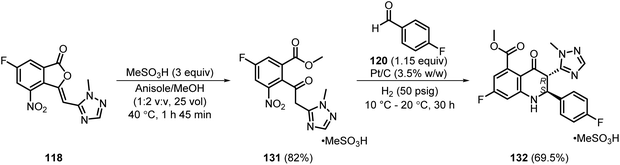
Later in 2021, Daniels et al. developed a step-economic process for the synthesis of the methanesulfonate salt form of 130; i.e., 132. A cascade methanolysis/nitro reduction/imine formation and cyclisation strategy was developed for the synthesis of 132 in 69.5% yield in just two steps, starting from 118 via the formation of 131 in 82% yield (Scheme 18).38
 |
| | Scheme 18 Cascade methanolysis/nitro reduction/imine formation/cyclisation strategy with 118 to synthesise 132. | |
6. Fuzuloparib
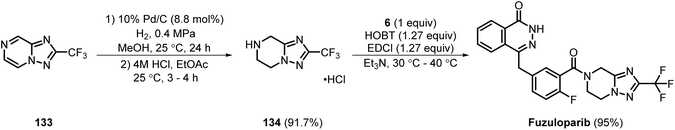
Fuzuloparib is a small molecule olaparib analogue and a potent PARP inhibitor (IC50 = 1.46 nM).39,40 Fuzuloparib was initially synthesised by Jiangsu Hengrui Pharmaceuticals. Acid amine coupling of compound 134 (which was prepared according to the patent procedure US 2021/0070760 A1 from 133) and the key intermediate of olaparib 6 using HOBt, EDCl coupling agents, delivered fuzuloparib in an excellent yield of 95% (Scheme 19).41,42
 |
| | Scheme 19 Synthetic process for fuzuloparib developed by Jiangsu Hengrui Pharmaceuticals Co., Ltd. | |
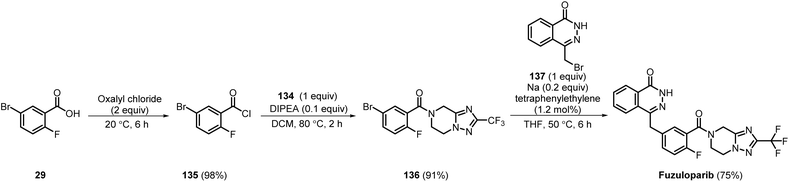
In 2022, Suzhou Fushilai Pharmaceutical disclosed a large-scale synthesis of fuzuloparib involving a 3-step procedure (Scheme 20). Commercially available 29 was first converted into its acid chloride 135 in 98% yield by treatment with oxalyl chloride. Next, amide coupling of 135 and 134 afforded 136 in 91% yield. Finally, the Wurtz–Fittig coupling of 136 with 137 using sodium metal and tetraphenyl ethylene resulted in the formation of fuzuloparib in 75% yield.43 Employing a few steps, fuzuloparib was synthesised and isolated by recrystallisation from ethyl acetate and petroleum ether with good yields in each step. By conversion into an acid chloride, the need for expensive coupling agents was eliminated. However, the generation of toxic or corrosive byproducts, such as HCl, during the acid chloride synthesis step requires proper ventilation, and the use of sodium metal requires inert conditions to be maintained.
 |
| | Scheme 20 Synthetic route of fuzuloparib developed by Suzhou Fushilai Pharmaceutical Co., Ltd. | |
7. Pamiparib
Pamiparib, a selective PARP1/2 inhibitor, exhibited IC50 values of 1.3 and 0.92 nM, respectively.44 The initial efficient route for the synthesis of pamiparib was developed by BeiGene Ltd. in 2016, and involved seven steps (Scheme 21). Initially, compound 138 was converted into 139 upon treatment with TFAA, and the latter was isolated by DCM extraction, giving the product in 98% yield. The reaction between 139 and 140, using tetramethylguanidine (TMG), Pd(PPh3)Cl2, and CuI, led to the formation of 141, which was purified by column chromatography to give pure 141 in 53% yield. Compound 141 was then treated with ZnBr2, giving 142 in 51% yield, which was isolated by column chromatography using an EtOAc/hexane solvent system. Further deprotection, followed by reaction with methyl 2-bromoacetate, produced 144, which was extracted using DCM in a moderate yield of 67%. The acid-mediated intramolecular cyclisation of 144 gave 145 in 33% yield, followed by condensation with hydrazine hydrate to afford pamiparib in 94% yield with >99% ee.45,46 The product was purified using preparative TLC employing DCM as the eluent. However, among the seven steps, many exhibited compromised yields, which significantly affected the overall yield of pamiparib, suggesting the need for novel synthetic process development with industrial suitability.
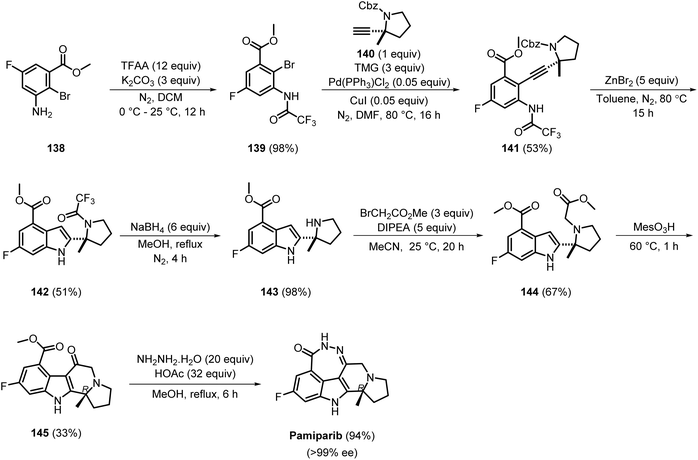 |
| | Scheme 21 Medicinal chemistry synthesis route for the synthesis of pamiparib. | |
Later in 2017, the same group developed a large-scale synthetic process for pamiparib (Scheme 22). N-Alkylation of 150 with 151 generated 152 in 83% yield. Further reaction of 152 with 153 in the presence of butyl lithium led to the formation of 154 in 54% yield. Desilylation of 154 using tetrabutylammonium fluoride (TBAF) gave 155 in 90% yield. Resolution of 155 using (−)-di-p-methylbenzoyl-L-tartaric acid separated the R-isomer 156 in a very low yield (27%), but with high optical purity (>96% ee). Cross-coupling of 156 with the previously synthesised intermediate 149 produced 157 in 66% yield. The intermediate 149 was initially prepared from the commercially available 146 by nitration to yield 147, followed by esterification of the acid group to produce 148. In both steps, the products were isolated by centrifugation and methanol and water washes. Subsequently, the nitro group was reduced to the amine 138, and the amine was extracted with ethyl acetate; tosylation of the amine produced intermediate 149. Compound 149 was isolated by first dissolving the crude precipitate in toluene and stirring for 20 minutes at 5–15 °C, followed by centrifugation and filtration. Similar to the medicinal chemistry synthesis route (Scheme 21), compound 157, upon intramolecular cyclisation and subsequent condensation, gave pamiparib in 95% yield.47 Compared to the medicinal chemistry synthesis route, this method employed simple unit operations and gave the products with high optical purity. However, the overall yield, the use of harsh chemicals such as n-BuLi, and the use of expensive, rare-earth metals such as Pd leave room for further improvement.
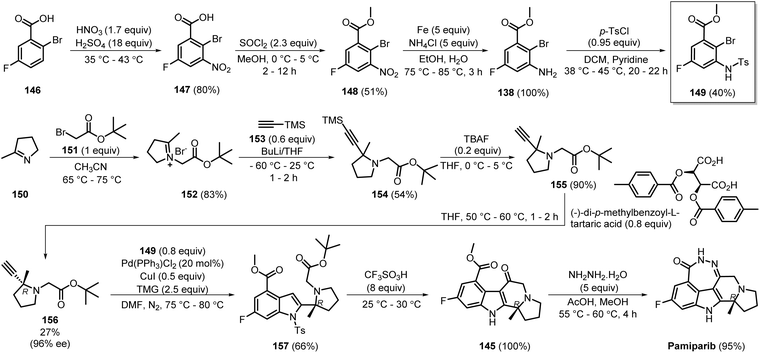 |
| | Scheme 22 Large-scale synthetic route of pamiparib. | |
8. Senaparib
Senaparib, a PARP 1/2 inhibitor, acts through a dual mechanism of catalytic inhibition and PARP-DNA trapping, with IC50 values of 6.27 and 1.57 nM, respectively.48,49 Being a very recently marketed PARP inhibitor, the synthetic pathways for senaparib have not been extensively explored. The synthetic pathway to senaparib developed by IMPACT Therapeutics involved a 7-step process (Scheme 23).50 First, the nucleophilic addition-elimination of compound 158 resulted in the formation of compound 159. Next, intramolecular cyclisation of 159 led to the generation of the core structure, 160, which then underwent rearrangement to produce 161 in 86.9% yield. Carbonyl protection of 161 using hexamethyldisilazane favours smooth nucleophilic displacement with 163 in subsequent steps, to deliver 164 in 74.6% yield. Ester hydrolysis gave 165, followed by acid–amine coupling of compounds 165 and 166, which delivered senaparib in 89.4% yield. In this process, the products were isolated by simple filtration followed by washing with various solvents such as methanol, water, and ethyl acetate at each step, minimising the use of organic solvents. However, the use of expensive reagents, such as compounds 163 and 166, and multi-step optimisation, shows the need for further optimisation to make the process industrially suitable.
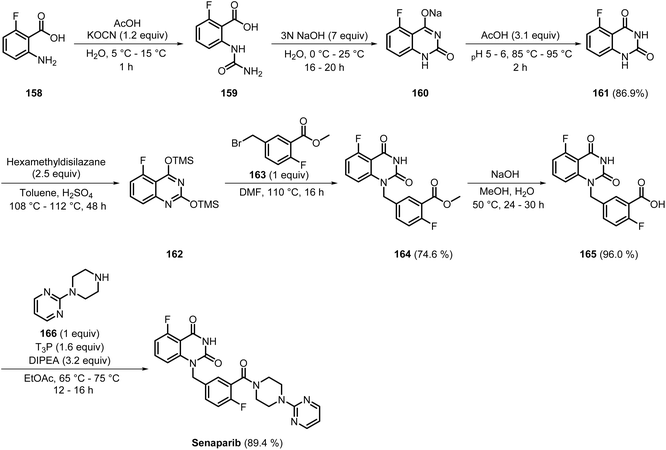 |
| | Scheme 23 Synthetic pathway of senaparib. | |
9. Conclusion and future scope
Despite PARP's well-established and validated role as an oncology target, only seven PARP inhibitors have been marketed worldwide. While significant advancements have been made in the synthesis and development of the earlier approved agents, there remains a pressing need to optimise the synthetic methodologies for the more recently approved PARP inhibitors, namely talazoparib, fuzuloparib, pamiparib, and senaparib. Future improvements in the industrial synthesis of these PARP inhibitors should focus on streamlining and greening their manufacturing routes. Key opportunities include telescoping multi-step sequences to reduce isolation and processing times, and replacing harsh reagents/solvents with greener, safer alternatives. Continuous-flow chemistry offers significant advantages for hazardous steps, such as reductive transformations and heterocycle formations, thereby improving safety and scalability. Biocatalysis presents a route to cleaner, more selective bond formation and chiral centre construction. Further gains lie in crystallisation to control impurities and improve downstream processing, along with cost optimisations through cheaper materials, convergent fragment assembly, and catalyst recycling. Overall, these advancements will make PARP inhibitor manufacturing more economical, sustainable, and industrially robust.
Author contributions
Dhurgam Rani: conceptualisation, writing, editing; Magdalena Plebanski: supervision, visualisation; and Tanmay Chatterjee: conceptualisation, supervision, visualisation, review & editing.
Conflicts of interest
There are no conflicts to declare.
Abbreviations
| NAD+ | Nicotinamide adenine dinucleotide |
| EU | European Union |
| IC50 | Half maximal inhibitory concentration |
| HR | Homologous recombination |
| ATR | Ataxia Telangiectasia and Rad3-related protein kinase |
| WEE1 | WEE1 G2 checkpoint kinase |
| CDK12 | Cyclin-dependent kinase 12 |
| MsOH | Methanesulfonic acid |
| K2CO3 | Potassium carbonate |
| HBTU | 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate |
| POCl3 | Phosphoryl chloride |
| DMF-DMA | N,N-Dimethylformamide dimethyl acetal |
| NaBH4 | Sodium borohydride |
| CDI | Carbonyl diimidazole |
| LiOH | Lithium hydroxide |
| DMF | N,N-Dimethylformamide |
| HOBt | 1-Hydroxybenzotriazole |
| EDCl | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| ee | Enantiomeric excess |
Data availability
No primary research results, software or codes have been included, and no new data were generated or analysed as part of this review.
Acknowledgements
Dr Tanmay Chatterjee thankfully acknowledges the financial support from the Anusandhan National Research Foundation (ANRF), Government of India (file no. CRG/2023/003045). Dhurgam Rani is grateful to BITS Pilani, Hyderabad Campus, and RMIT, Melbourne, for her fellowship.
References
- H. Juarez-Salinas, J. L. Sims and M. K. Jacobson, Nature, 1979, 282, 740–741 CrossRef CAS PubMed.
- W. L. Kraus, Mol. Cell, 2015, 58, 902–910 CrossRef CAS PubMed.
- P. K. Das, G. S. P. Matada, R. Pal, L. Maji, P. S. Dhiwar, B. V. Manjushree and M. P. Viji, Eur. J. Med. Chem., 2024, 274, 116535 CrossRef CAS PubMed.
- D. M. O'Malley, T. C. Krivak, N. Kabil, J. Munley and K. N. Moore, Targeted Oncol., 2023, 18, 471–503 CrossRef PubMed.
- G. R. Daly, M. M. AlRawashdeh, J. McGrath, G. P. Dowling, L. Cox, S. Naidoo, D. Vareslija, A. D. K. Hill and L. Young, Curr. Oncol. Rep., 2024, 26, 103–113 Search PubMed.
- T. Zhou and J. Zhang, Transl. Oncol., 2025, 57, 102410 CrossRef CAS PubMed.
- A. K. Taylor, D. Kosoff, H. Emamekhoo, J. M. Lang and C. E. Kyriakopoulos, Front. Oncol., 2023, 13, 1159557 CrossRef CAS PubMed.
- O. Longoria, N. Beije and J. S. de Bono, Semin. Oncol., 2024, 51, 25–35 CrossRef CAS PubMed.
- A. Jain, A. Barge and C. N. Parris, Oncogene, 2025, 44, 193–207 CrossRef CAS PubMed.
- P. G. Jain and B. D. Patel, Eur. J. Med. Chem., 2019, 165, 198–215 CrossRef CAS PubMed.
- D. L. Hughes, Org. Process Res. Dev., 2017, 21, 1227–1244 CrossRef CAS.
- E. D. Deeks, Drugs, 2015, 75, 231–240 CrossRef CAS PubMed.
- S. Bochum, S. Berger and U. M. Martens, Small Mol. Oncol., 2018, 211, 217–233 CAS.
- K. A. Menear, et al., J. Med. Chem., 2008, 51, 6581–6591 CrossRef CAS PubMed.
- S. R. Chennamaneni, M. Velisoju, S. P. Anegondi, Y. V. Joshi, A. Panwar, and M. Patel, WO2017/191562A1, 2017.
- S. S. Bratovanov and S. P. Green, US pat., US 2017/0233351 A1, 2017 Search PubMed.
- T.-Y. Hsiao and Y.-H. Chang, WO2018/038680A1, 2018.
- Z. Chen, S. Wang, K. Liu, R. Zhang, Q. Li, W. Bian, R. Qiao and C. Li, ACS Omega, 2022, 7, 6313–6321 CrossRef CAS PubMed.
- I. Chatterjee, D. Roy and G. Panda, Green Chem., 2023, 25, 9097–9102 RSC.
- V. R. C. Palle, P. R. Patel, N. Ali, S. H. Shelke, S. M. Lad, G. T. Patle, R. M. Shanmughasamy and P. R. Yadav, US pat., US 2024/0351989 A1, 2024.
- Y. Y. Syed, Drugs, 2017, 77, 585–592 CrossRef CAS PubMed.
- S. E. Webber, S. S. Canan-Koch, J. Tikhe and L. H. Thoresen, US pat., US 6,495,541 B1, 2002.
- A. C. Flick, H. X. Ding, C. A. Leverett, S. J. Fink and C. J. O'Donnell, J. Med. Chem., 2018, 61, 7004–7031 CrossRef CAS PubMed.
- C. Cheng, X. Zuo, D. Tu, B. Wan and Y. Zhang, Org. Lett., 2020, 22, 4985–4989 CrossRef CAS PubMed.
- B. C. Beck, J. B. Perales, J. D. Speake and I. Ferrando, US pat., US 11,220,507 B2, 2022.
- L. J. Scott, Drugs, 2017, 77, 1029–1034 CrossRef PubMed.
- P. Jones, J. Altamura, F. Boueres, H. Ferrigno, R. Fonsi, M. Giomini, S. Lamartina, M. Monteagudo, M. Ontoria, E. Orsale, G. Palumbi, F. Pesci, M. Roscilli, C. Scarpelli, M. Schultz-Fademrecht, F. Toniatti, C. Rowley, J. A. Narjes and P. Passacantilli, J. Med. Chem., 2009, 52, 7170–7185 CrossRef CAS PubMed.
- P. Jones, J. M. Ontoria, R. Scarpelli and C. Schultz-Fademrecht, US pat., US 8,071,623 B2, 2011.
- P. K. Luthra, S. L. Vasoya, B. T. Patil, A. K. Taneja, N. C. Srivastav and R. Singh, US pat., US 10,927,095 B2, 2021.
- T. Wang, US pat., US 10,927,077 B2, 2021.
- A. Stewart, A. J. Toto, F. X. Chen and G. Wu, US pat., US 11,629,137 B2, 2023.
- S. M. Hoy, Drugs, 2018, 78, 1939–1946 CrossRef PubMed.
- A. C. Flick, C. A. Leverett, H. X. Ding, E. McInturff, S. J. Fink, C. J. Helal, J. C. DeForest, P. D. Morse, S. Mahapatra and C. J. O'Donnell, J. Med. Chem., 2020, 63, 10652–10704 CrossRef CAS PubMed.
- B. Wang, J. Chu, D. K. C. Chow, J. G. Oplinger, J. F. Natarajan, J. A. Bardenhagen, J. R. S. Burke, M. A. D. Beckmann, D. S. Oleykowski, M. A. Hardwicke and J. M. Lindsley, et al., J. Med. Chem., 2016, 59, 335–357 CrossRef CAS PubMed.
- B. Wang, D. Chu, Y. Liu, Q. Jiang and L. Lu, US pat., US 9,926,303 B2, 2018.
- M. Henderson, US pat., US 2016/0280691 A1, 2016.
- Y. Xu, P. W. Yohi, M. Xu and D. Cruise, US pat., US 9,708,319 B1, 2017.
- D. S. Daniels, J. M. O'Brien, A. N. Calkins, M. J. McLaughlin, J. T. Reeves, M. A. Huffman and P. J. Strotman, Org. Process Res. Dev., 2021, 25, 2686–2692 CrossRef CAS.
- A. Lee, Drugs, 2021, 81, 1221–1226 CrossRef CAS PubMed.
- N. Li, Q. Liu, Y. Tian and L. Wu, Gynecol. Oncol., 2022, 33, e86 CrossRef CAS PubMed.
- X. Liu, Y. Zhang, H. Wang, J. Chen and L. Zhao, CN115650988A, 2023.
- W. Li and Y. Zhang, US pat., US 2021/0070760 A1, 2021.
- X. Zhang, X. Lü, J. Lu and H. Zhang, WO2022/036940A1, 2022.
- A. Markham, Drugs, 2021, 81, 1343–1348 CrossRef CAS PubMed.
- C. Zhou, B. Ren and H. Wang, US pat., US 9,260,440 B2, 2016.
- H. Wang, B. Ren, Y. Liu, B. Jiang, Y. Guo, M. Wei, L. Luo, X. Kuang, M. Qiu, L. Lv, H. Xu, R. Qi, H. Yan, D. Xu, Z. Wang, C.-X. Huo, Y. Zhu, Y. Zhao, Y. Wu, Z. Qin, D. Su, T. Tang, F. Wang, X. Sun, Y. Feng, H. Peng, X. Wang, Y. Gao, Y. Liu, W. Gong, F. Yu, X. Liu, L. Wang and C. Zhou, J. Med. Chem., 2020, 63, 15541–15563 CrossRef CAS PubMed.
- H. Wang, C. Zhou, B. Ren and X. Kuang, WO2017/032289A1, 2017.
- A. Lee, Drugs, 2025, 85, 1–5 Search PubMed.
- X. Man, Y. Zhang, J. He and P. Wang, Bioorg. Chem., 2025, 163, 108761 CrossRef CAS PubMed.
- S. Cai and P. Huang, US pat., US 10,562,889 B2, 2020.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a



























