DOI:
10.1039/D6RA02580C
(Paper)
RSC Adv., 2026,
16, 30272-30280
High-energy ball milling-driven La/Zr doping of magnetic Na-A zeolite derived from coal gangue for efficient phosphate removal
Received
28th March 2026
, Accepted 19th May 2026
First published on 3rd June 2026
Abstract
To overcome the cage-energy barriers of zeolites that limit the incorporation of large functional cations, coal gangue (CG) was calcined and alkali-activated to synthesize Na-A zeolite. High-energy ball milling (HEBM) of Fe3O4 with Na-A produced a magnetic zeolite (MZ), and a mechanochemically driven in situ ion-exchange process enabled rapid La3+ incorporation to form lanthanum magnetic zeolite (LMZ) for phosphate removal. Fe3O4 and Na-A were tightly interlocked at the nanoscale, forming 150–800 nm aggregates composed of 10–20 nm nanocrystallites. The disappearance of NaCl diffraction peaks after washing confirmed in situ Na+/La3+ exchange during milling. LMZ showed a specific surface area of 91.32 m2 g−1, over four times that of pristine Na-A zeolite, and a saturation magnetization of 25.6 emu g−1. At pH 3.0, the maximum phosphate uptake reached 44.47 mg P g−1 and remained stable over five adsorption–regeneration cycles. Under identical conditions, Zr4+-doped zirconium magnetic zeolite (ZMZ) displayed a similarly favorable composite structure, highlighting the generality of this strategy. This work offers a scalable mechanochemical route for introducing large functional cations into solid-waste-derived zeolites and developing recyclable phosphate sorbents.
Phosphorus (P) is an essential nutrient for aquatic ecosystems and agricultural production, yet its excessive discharge into water bodies has become a major environmental concern.1,2 Elevated phosphate concentrations can accelerate eutrophication, triggering algal blooms, dissolved oxygen depletion, deterioration of water quality, and the loss of aquatic biodiversity, thereby posing long-term risks to ecosystem stability and human water use.3 At the same time, phosphorus is a finite and strategically important resource, and its recovery from wastewater is increasingly regarded not only as a pollution-control measure but also as a promising route toward resource conservation and circular utilization.4,5 Against this background, the development of efficient materials capable of both phosphate capture and potential phosphorus recovery has attracted growing attention.6–8
Zeolites have been widely used in adsorption, catalysis, and ion exchange because of their ordered microporous structures, tunable surface chemistry,9 and tailorable framework compositions, and they have shown considerable promise in water pollution control.10 Beyond natural mineral precursors, solid wastes such as fly ash, red mud, and coal gangue can also serve as sustainable feedstocks for zeolite synthesis, offering a viable route for waste valorization and cleaner production.11–13 Zeolites consist of negatively charged frameworks and charge-compensating cations, and their silicon-to-aluminum ratio (Si/Al) governs key properties such as ion-exchange capacity and surface acidity/basicity; in general, lower Si/Al ratios favor cation exchange.14–16 These intrinsic characteristics make zeolites particularly attractive as host matrices for the incorporation of functional metal species aimed at enhancing phosphate affinity and selectivity.
Recent efforts have focused on introducing multivalent cations, particularly rare-earth ions, to enhance the affinity and selectivity of zeolites toward anionic pollutants.17–22 Among them, La3+ shows strong affinity for phosphate, while Zr-based sites can selectively capture phosphate through strong Lewis acidity and stable inner-sphere coordination.23–25 However, conventional ion-exchange and impregnation methods often suffer from low efficiency, long processing times, and the formation of aggregated surface phases, making it difficult to achieve effective dispersion of rare-earth species at zeolitic change sites.19,26 In particular, the large hydrated radii of rare-earth ions and their sluggish diffusion kinetics impose substantial steric and mass-transfer limitations on their entry into zeolitic cages and exchange domains, which restricts the efficient construction of highly accessible active sites.10,16,27,28 Therefore, strategies capable of overcoming these kinetic barriers while simultaneously promoting structural activation of the zeolite framework are highly desirable.
High-energy ball milling, as a representative mechanochemical approach, can promote ion exchange by inducing solid-state activation, defect formation, and short-range diffusion through intense shear and impact.14–17,29–32 In addition to promoting cation incorporation, ball-milling-induced structural reconstruction may alter pore accessibility and expose additional reactive sites, thereby influencing adsorption behavior. Meanwhile, the co-introduction of magnetic nanoparticles provides a practical route for the rapid magnetic separation and recovery of adsorbents after use.33–35 On this basis, this work employs CG-derived Na-A zeolite as the substrate and uses wet high-energy ball milling to construct magnetic composites, followed by mechanochemically enabled in situ ion exchange to introduce metal active sites into the zeolite phase. Particular attention is given to ball-milling-induced structural reconstruction, ion-exchange behavior, magnetic response, and phosphate removal performance, with the aim of evaluating this strategy for the development of efficient and recyclable zeolite-based phosphate adsorbents.
1 Experimental
1.1 Materials and chemicals
Coal gangue from the Huainan coal mine was used as the raw material for zeolite synthesis; its main components were Al2O3, SiO2, TiO2, and MgO, Table 1 and Table. S1 (SI). The chemicals used were NaOH (96.0%), Na2SiO3·9H2O, NaAlO2, Fe3O4, ZrOCl2·8H2O, C2H6O, HCl (1 mol L−1), LaCl3·7H2O, and KH2PO4. All reagents were of analytical grade and were used as received without further purification.
Table 1 Chemical composition of CG
| SPL |
N2O |
MgO |
Al2O3 |
SiO2 |
P2O5 |
SO3 |
K2O |
CaO |
TiO2 |
Fe2O3 |
| CG (%) |
0.59 |
0.63 |
27.37 |
53.59 |
0.06 |
0.89 |
1.07 |
2.19 |
0.86 |
2.98 |
1.2 Synthesis of materials
1.2.1 Preparation of Na-A zeolite. This procedure was adapted from previous studies on the synthesis of zeolites from solid wastes.5,6,9,17 Fig. 1, CG was crushed and sieved through a 200-mesh screen (75 µm). Part of the CG was calcined at 800 °C for 2 h in a nickel crucible using a muffle furnace (FO610C, JPN). After cooling, the sample was activated in 0.1 mol L−1 HCl at a solid-to-liquid ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 to remove oxide impurities. The solid was washed with deionized water to near neutral pH, dried, mixed and ground with an equal mass of NaOH, and recalcined at 400 °C for 2 h to obtain pretreated calcine coal gangue (CCG). Then, 4 g of CCG was mixed with an appropriate amount of Na2SiO3·9H2O, 0.5 g of NaAlO2, and 9 g of NaOH. The mixture was stirred and aged at 150 rpm for 2 h at a liquid-to-solid ratio of 8 mL g−1 on a digital heating magnetic stirrer (ZNCL-DL15, CHN) to form a gel. The gel was transferred to a 100 mL polytetrafluoroethylene reactor and crystallized at 80–110 °C for 6–24 h in a vacuum drying oven (DZF-6050, CHN). The product was collected by vacuum filtration through a 0.45 µm membrane (AP-01P, CHN), washed with deionized water until the filtrate reached pH 9, and dried overnight at 40–60 °C. The final product was denoted as Na-A zeolite.
:2 to remove oxide impurities. The solid was washed with deionized water to near neutral pH, dried, mixed and ground with an equal mass of NaOH, and recalcined at 400 °C for 2 h to obtain pretreated calcine coal gangue (CCG). Then, 4 g of CCG was mixed with an appropriate amount of Na2SiO3·9H2O, 0.5 g of NaAlO2, and 9 g of NaOH. The mixture was stirred and aged at 150 rpm for 2 h at a liquid-to-solid ratio of 8 mL g−1 on a digital heating magnetic stirrer (ZNCL-DL15, CHN) to form a gel. The gel was transferred to a 100 mL polytetrafluoroethylene reactor and crystallized at 80–110 °C for 6–24 h in a vacuum drying oven (DZF-6050, CHN). The product was collected by vacuum filtration through a 0.45 µm membrane (AP-01P, CHN), washed with deionized water until the filtrate reached pH 9, and dried overnight at 40–60 °C. The final product was denoted as Na-A zeolite.
 |
| | Fig. 1 High-energy ball milling-metal ion exchange process. | |
1.2.2 Preparation of magnetic zeolite. Na-A zeolite was acid-activated in 1.0 mol L−1 HCl at a solid-to-liquid ratio of 1:10 at 60 °C for 4 h, then filtered, washed to neutral pH, and dried at 105 °C for 12 h. Fe3O4 and Na-A zeolite were mixed at mass ratios of 1:1, 3:2, 2:1, 3:1, and 4:1, dispersed in 30 mL ethanol, and subjected to wet high-energy ball milling in a planetary mill (NP-100, JPN).14,17 Milling was performed in a 100 mL SUS304 stainless-steel jar with steel balls (large: medium: small = 4:10:102), a ball-to-powder ratio of 10:1, a filling volume of 60–70%, and a rotation speed of 300 r min−1 using a 15 min on/5 min off cycle for a net milling time of about 4 h, with water cooling.34,35 The product was magnetically separated, washed three times with ethanol, dried overnight at 40–60 °C, and denoted as MZ-1 to MZ-5.
1.2.3 Preparation of metal-ion-doped magnetic zeolites. MZ and LaCl3·7H2O were mixed in ethanol at a mass ratio of 4:3, ultrasonically dispersed for 30 min, and wet-ball-milled at 300 rpm for 60 h. The product was collected by magnetic separation, washed three times with ethanol, and vacuum-dried at 60 °C to obtain lanthanum magnetic zeolite (LMZ).16,19 Zirconium magnetic zeolite (ZMZ) was prepared analogously by replacing LaCl3·7H2O with ZrOCl2·8H2O.20,23
1.3 Characterizations
The morphology and microstructure of Na-A zeolite, MZ, ZMZ, and LMZ were characterized by scanning electron microscopy (SEM, JSM-7500F, JPN) and transmission electron microscopy (TEM, Tecnai F30, NL). The crystal phases of coal gangue and the synthesized zeolite products were analyzed by X-ray diffraction (XRD, Smartlab SE, JPN), while functional groups were identified by Fourier transform infrared spectroscopy (FT-IR, Nicolet iS50, USA). The surface composition and chemical states of the magnetic zeolites were examined by X-ray photoelectron spectroscopy (XPS, Escalab 250Xi, USA). Specific surface area and pore size distribution were determined by Brunauer–Emmett–Teller (BET, V-sorb2800p, CHN) analysis. The magnetic properties were measured using a vibrating sample magnetometer (VSM, HH-20, CHN). The concentration of solution was measured by ICP-AES techniques using an ICP-AES_OPTIMA7000DV equipment.
1.4 Batch study
To evaluate phosphate removal performance, 0.1 g of Na-A zeolite, MZ, LMZ, or ZMZ was added to 100 mL of phosphate solution (50 mg L−1) and shaken at 200 r min−1 for 12 h at 30 °C in a thermostatic shaker (ZHWY-2102C, CHN). Adsorption experiments were conducted over a pH range of 2–9.36 After adsorption, 5 mL of the supernatant was collected and filtered through a 0.22 µm syringe filter to eliminate interference from residual particles during absorbance measurement. During solid–liquid separation, the magnetic adsorbents could be readily collected on the inner wall of the vessel using an external magnet, demonstrating their good separability and recovery potential.37 LMZ reusability was evaluated by desorption in 0.5 M NaOH (1 g L−1, 200 rpm, 8 h, 25 ± 1 °C), followed by magnetic separation and phosphate analysis of the desorption solution. The recovered solid was washed to near neutral pH and vacuum-dried at 60 °C for 12 h to obtain regenerated LMZ. Re-adsorption was then carried out in freshly prepared phosphate solution (50 mg P L−1, pH 3.0, 1 g L−1, 200 rpm, 3 h, 25 ± 1 °C). The adsorption–desorption cycle was repeated five times. Fresh LMZ and LMZ after five cycles were further compared over pH 2–9, and the capacity retention was calculated.19,33 The residual phosphate concentration was determined by the ascorbic acid–ammonium molybdate method (GB/T 9727-2007 and GB/T 11893-89; UV, TU-1901, China, λ = 700 nm).35,38 Phosphate adsorption was calculated by eqn (1); model parameters are listed in Table 2.| |
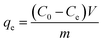 | (1) |
where qe is the equilibrium adsorption capacity (mg L−1; C0 and Ce are the initial and equilibrium phosphate concentrations (mg L−1), respectively; V is the solution volume (L); and m is the mass of adsorbent (g).
Table 2 Model equations and fitting parameters
| Model |
Model name |
Equation |
Model parameters |
| Kinetic |
Pseudo-first order (PFO) |
qt = qe(1 − e−K1t) |
qe: Equilibrium adsorption capacity (mg g−1); qt: adsorption capacity at time t (mg g−1); K1: PFO adsorption rate constant (min). |
| Pseudo-second order (PSO) |
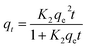 |
K2: PSO adsorption rate constant (g mg−1 min−1). |
| Isotherm |
Langmuir (L) |
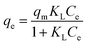 |
qm: Maximum monolayer adsorption capacity (mg g−1); KL: Langmuir adsorption affinity-related constant (L mg−1); RL: Shows that the isotherm is unfavorable (RL > 1), favorable (0 < RL < 1), linear (RL = 1), or irreversible (RL = 0). |
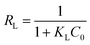 |
| Freundlich (F) |
qe = KFCe1/nF |
KF: Freundlich adsorption affinity-related constant (mg g−1) (mg L−1)−n; nF−1: adsorption strength. |
| Thermodynamics |
Adsorption thermodynamics |
ΔG0 = −RTln(K0e) |
ΔG0: Free energy change, ΔH0: Enthalpy change, and ΔS0: entropy change |
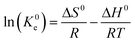 |
R: Gas constant (8.314 kJ mol−1), K0e: Langmuir equilibrium constant. |
2 Results and discussion
2.1 Characterization and instrumentation
SEM images (Fig. S4a (SI)) show that the coal gangue (Fig. S1 (SI))-derived Na-A zeolite possesses a well-defined cubic morphology with a particle size of about 2 µm (Fig. S2 (SI)). After wet high-energy ball milling (Fig.S4b–d (SI)), the sharp crystal facets and original long-range morphology disappeared, giving irregular nanoaggregates with secondary sizes of ca. 150–800 nm, indicative of fragmentation, re-agglomeration, and partial di sordering.15,17 The TEM images provide a clearer characterization of the structure of the LMZ sample and the distribution of its constituent elements (Fig. 2). Fe3O4 nanoparticles are embedded and intergrown with the zeolite matrix, with particle sizes of approximately 10–30 nm. This indicates that high-energy wet ball milling effectively reduced both the magnetic Fe3O4 and zeolite particles to the nanoscale, while simultaneously promoting their physical/chemical integration, resulting in the LMZ sample exhibiting uniform and strong magnetic properties.39–41 ICP-OES analysis (Table S3, (SI)).
 |
| | Fig. 2 TEM images and elemental mapping of LMZ (a). Elemental mapping of (b) Si, (c) Al, (d) O, (e) Fe and (f) La. | |
Interestingly, the spatial distribution of La atoms coincides with that of Si and Al, while being complementary to that of Fe. This observation suggests that La ions are uniformly distributed within the zeolite molecular cage framework.16 EDS mapping (Fig. S4e–m (SI)) shows uniform distributions of Na, Al, Si, O, and Fe in MZ. In LMZ, La is strongly co-localized with Si/Al but relatively distinct from Fe, indicating that La preferentially resides in the zeolite phase rather than the Fe3O4 domain. This supports the occurrence of in situ Na+/La3+ exchange during ball milling.19,22 ZMZ displays similar surface characteristics, confirming the versatility of the synthetic route.20,23
2.2 XRD and magnetic characteristics
XRD patterns (Fig. 3a, S3 and S5b (SI)) confirm that the hydrothermal product is well-crystallized Na-A zeolite (PDF# 39-0222), with characteristic reflections at (200), (220), (222), (420), (600), and (622).4 After wet high-energy ball milling, these reflections become weaker and broader in MZ, LMZ, and ZMZ, indicating decreased crystallinity and particle nanocrystallization, consistent with the SEM and TEM (Fig. 2) results.14,23 MZ retains the diffraction features of both Fe3O4 (PDF#85-1436) and Na-A zeolite, confirming a biphasic composite structure. LMZ and ZMZ show similar patterns, with no detectable crystalline La- or Zr-containing secondary phases. Together with the EDS results showing La co-localized with Si/Al but relatively separated from Fe, this indicates that no long-range crystalline La- or Zr-containing secondary phases were detected within the XRD detection limit. Together with ICP-OES, EDS mapping, and XPS results, this supports the incorporation of La/Zr species into the magnetic zeolite composites, although the possibility of poorly crystalline or amorphous La/Zr species cannot be excluded.22,23 Comparison of the unwashed and washed samples provides supporting evidence for Na+ release during mechanochemically assisted ion exchange. In unwashed LMZ-NaCl and ZMZ-NaCl, additional peaks at 2θ = 31.7° and 45.5° match the (200) and (220) reflections of NaCl (PDF# 99-0059), but disappear after washing. This indicates that Na+/La3+ and Na+/Zr4+ exchange occurred during milling, with released Na+ reacting with Cl− to form removable NaCl.38,42,43
 |
| | Fig. 3 XRD patterns of (a) Fe3O4, LMZ, LMZ-NaCl, ZMZ, ZMZ-NaCl, MZ and Na-A -zeolite; VSM magnetization curves of (b) Fe3O4, ZMZ, LMZ and MZ. | |
Fig. 3b and S5b (SI), shows that MZ, LMZ, and ZMZ exhibited saturation magnetizations of 20.3, 25.6, and 27.1 emu per g, respectively, indicating that the composites retained sufficient magnetic response for rapid magnetic separation. The slightly higher apparent Ms values of LMZ and ZMZ should be interpreted cautiously and may be related to changes in composite composition, particle dispersion, and removal of loosely bound nonmagnetic residues during washing rather than to a direct magnetic contribution from La/Zr species. Because Fe3O4 and γ-Fe2O3 have similar spinel structures with overlapping XRD reflections, the magnetic component is described here as a Fe3O4-derived spinel magnetic iron oxide phase.17
2.3 Adsorption–desorption analysis
Fig. 4a and Table S2 (SI), the specific surface area of the zeolite increased with decreasing crystal size. Most pores were distributed in the range of 2–50 nm, with relatively few outside this range, indicating that the increased hydroxide concentration promoted pore-volume development.15,31 Detailed crystal structure analysis is provided in Fig. S2 and S3 (SI) and Fig. 4b, the sample exhibited an adsorption–desorption isotherm with an H3-type hysteresis loop and no obvious saturation plateau. Fig. 4c and d and Table 1, MZ shows little change in surface area or micropore volume relative to Na-A zeolite, but its average pore size and total pore volume increase by more than twofold, indicating that co-ball milling with Fe3O4 mainly generates mesoporosity with limited disruption of the original micropores.18 By contrast, LMZ exhibits a much higher surface area (91.31 m2 g−1), nearly four times that of Na-A zeolite, and a total pore volume of 0.2152 cm3 g−1. Its micropore volume is about three times that of MZ, while the average pore size decreases toward that of the parent zeolite, suggesting that La incorporation facilitates micropore reconstruction.
 |
| | Fig. 4 N2 adsorption–desorption isotherms (a and c) and pore size distribution curves (b and d) of Na-A zeolite, MZ, LMZ, and ZMZ. | |
Both MZ and LMZ display type IV isotherms with H3 hysteresis loops at P/P0 0.4–0.9, indicating the formation of irregular hierarchical pore structures after milling.19,32 Zr incorporation gives a similar effect: ZMZ reaches a surface area of 82.31 m2 g−1 and a total pore volume of 0.2395 cm3 g−1, with a broad pore-size distribution of 2–185 nm (Table 3).17,20 Thus, the pore-structure evolution should be interpreted as the combined result of particle fragmentation, re-aggregation, interparticle mesoporosity, and partial micropore reconstruction induced by high-energy ball milling and La/Zr incorporation.32 The increased surface area and pore volume are discussed as improved site accessibility and mass-transfer pathways rather than as definitive evidence of complete zeolitic framework preservation.
Table 3 BET surface properties of Na-A zeolite and MZ, LMZ and ZMZ
| Adsorbents |
BET surface area (m2 g−1) |
Total pore volume (cm3 g−1) |
Micropore volume(cm3 g−1) |
Average pore diameter (nm) |
| Zeolite |
24.31 |
0.0598 |
0.0067 |
12.89 |
| MZ |
21.03 |
0.1872 |
0.0056 |
45.57 |
| LMZ |
91.31 |
0.2152 |
0.0239 |
12.62 |
| ZMZ |
82.31 |
0.2395 |
0.0209 |
15.54 |
2.4 Phosphate adsorption performance
Fig. 5a, pristine Na-A zeolite and MZ show only limited phosphate uptake, with maximum adsorption capacities of 6.58 and 11.36 mg g−1, respectively, whereas both LMZ and ZMZ exhibit markedly enhanced performance. In particular, LMZ reaches 34.95 mg g−1, indicating that the improvement mainly arises from the phosphate-affinitive La/Zr sites introduced by doping rather than from simple Fe3O4 incorporation.19,24,42 LMZ performs best under acidic conditions, reaching 44.47 mg g−1 at pH 3 (Table 4), whereas ZMZ shows a maximum uptake of 33.2 mg g−1 at pH 2 and is more sensitive to pH variation (Fig. 5b).
 |
| | Fig. 5 (a) Phosphate adsorption capacities of Na-A zeolite, MZ, LMZ, and ZMZ; (b) effect of pH on phosphate adsorption; (c) pH-drift curves; (d) pseudo-first-order kinetic fitting; (g) pseudo-second-order kinetic fitting; (e and f) Langmuir and Freundlich isotherm fitting for LMZ; (h and i) Langmuir and Freundlich isotherm fitting for ZMZ. | |
Table 4 Comparison of phosphate adsorption capacities among various zeolite-based adsorbents
| Adsorbent |
SBET (m2 g−1) |
Pore diameter (nm) |
T (K) |
Dosage (g L−1) |
pH |
Qe(P) (mg g−1) |
Ref. |
| LMZ |
21.725 |
89.673 |
298 |
0.5 |
4–9 |
9.14 |
29 |
| La-Z |
52.75 |
5.78 |
303 |
2.0 |
5 |
17.2 |
32 |
| La-A |
58.0 |
11.45 |
333 |
1.0 |
6 |
44.0 |
41 |
| BMS@ZrO2 |
146.57 |
3.55 |
298 |
0.5 |
2 |
16.47 |
44 |
| Zr–GO |
— |
2–50 |
298 |
— |
5 |
27.71 |
43 |
| Zr-FeBC |
25.51 |
— |
295 |
2.0 |
5 |
33.33 |
45 |
| ZMZ |
82.31 |
15.54 |
303 |
1.0 |
3 |
33.2 |
This work |
| LMZ |
91.31 |
12.62 |
303 |
1.0 |
3 |
44.47 |
This work |
At low pH, La/Zr sites are more readily protonated, which promotes ligand exchange with H2PO4− and the formation of inner-sphere La/Zr–O–P complexes, thereby leading to higher phosphate uptake. As the pH increases, site deprotonation and stronger OH− competition cause a marked decline in adsorption.35,36,46 The pH-drift results in Fig. 5c further support this mechan-ism: when the initial pH was 2–3, the final pH increased to about 5–6, indicating that adsorption was accompanied by surface coordination/ligand exchange and proton transfer; at higher initial pH, the final pH decreased slightly, suggesting the involvement of surface deprotonation and H+ release41,43 (eqn (2)). Overall, these results indicate that phosphate removal by LMZ/ZMZ is dominated by surface chemisorption rather than simple electrostatic attraction.
Fig. 5d and g, the adsorption kinetics of both LMZ and ZMZ are better described by the pseudo-second-order (PSO) model, with R2 values of 0.9956 and 0.9908, respectively, whereas the pseudo-first-order (PFO) model shows a more pro-nounced deviation (Table 5).15,16,47 This indicates that the rate-controlling step is not governed solely by physical diffusion, but is more consistent with surface active-site reactions and ligand exchange, together with a contribution from intraparticle diffusion. This interpretation agrees well with the stronger specific chemical interactions introduced by La/Zr doping and is further supported by the XPS results (Fig. 6f and i), including the positive shifts of La 3d (+0.5 eV) and Zr 3d (+1.0–1.2 eV), as well as the appearance of P 2p and O 1 s (P–O) signals.19,20,23,42
Table 5 Equilibrium parameters of Langmuir and Freundlich models for P adsorption onto LMZ and ZMZ
| Model |
Langmuir |
| SMP |
LMZ |
ZMZ |
LMZ |
ZMZ |
LMZ |
ZMZ |
| T (K) |
KF |
1/n |
R2 |
| 293 |
4.837 |
12.87 |
0.5088 |
4.969 |
0.9241 |
0.8677 |
| 303 |
4.961 |
14.17 |
0.4962 |
5.269 |
0.9193 |
0.8711 |
| 313 |
4.487 |
14.09 |
0.2108 |
4.839 |
0.9903 |
0.8909 |
| Model |
Freundlich |
| SMP |
LMZ |
ZMZ |
LMZ |
ZMZ |
LMZ |
ZMZ |
| T (K) |
qm (mg g−1) |
KL (L mg−1) |
R2 |
| 293 |
43.688 |
36.72 |
0.0048 |
0.0706 |
0.9918 |
0.9974 |
| 303 |
49.542 |
38.04 |
0.0057 |
0.0759 |
0.9975 |
0.9972 |
| 313 |
51.616 |
41.38 |
0.0056 |
0.0681 |
0.9971 |
0.9973 |
 |
| | Fig. 6 XPS images of (a), (h): LMZ-Cl, LMZ, LMZ-P, ZMZ-Cl and ZMZ;(b–g) Elemental mapping of P 2p, Al 2p, Si 2p, La 3d and Fe 2p for LMZ-P and LMZ, (i): Zr 3d for ZMZ-P, ZMZ and ZMZ-Cl. | |
The adsorption isotherms further show that LMZ is best fitted by the Langmuir model at 303 K (Fig. 5e), with an R2 of 0.9974 and a theoretical monolayer adsorption capacity of 49.542 mg g−1, which is superior to the Freundlich (Fig. 5f, R2 = 0.9766) and Temkin (Fig. 5i, R2 = 0.8909) models.27,37 ZMZ shows the same trend, with the Langmuir model providing the best fit (Fig. 5h and i) and a theoretical maximum adsorption capacity of 38.04 mg g−1 (Table 6).17,48,49 These results indicate that phosphate adsorption on both LMZ and ZMZ is consistent with a monolayer-like adsorption behavior involving surface coordination and ligand exchange.
Table 6 Kinetic fitting parameters for phosphate adsorption onto Na-A zeolite, MZ, ZMZ and LMZ
| Adsorbents |
qmax (mg g−1) |
PFO kinetic model |
PSO kinetic model |
| qe (mg g−1) |
K1 (min−1) |
R2 |
qe (mg g−1) |
K2 (g mg−1 min−1) |
R2 |
| Zeolite |
7.1 |
6.12 |
0.0256 |
0.9132 |
7.04 |
0.0045 |
0.9467 |
| MZ |
11.99 |
9.47 |
0.1499 |
0.8307 |
10.31 |
0.0209 |
0.9204 |
| ZMZ |
22.32 |
21.28 |
0.1618 |
0.9511 |
22.68 |
0.0121 |
0.9908 |
| LMZ |
42.15 |
32.79 |
0.2186 |
0.9723 |
34.49 |
0.0119 |
0.9956 |
To verify the adsorption performance of the adsorbents in the presence of interfering ions, coexisting-ion experiments were conducted, as detailed in Fig. S7 (SI). LMZ showed good recyclability over five adsorption–desorption cycles (Fig. S8 (SI)). The phosphate removal efficiency decreased gradually from about 98.5% in the first cycle to 80.0% in the fifth cycle, with values of 95.6%, 89.7%, and 82.8% in the second, third, and fourth cycles, respectively. Despite this decline, LMZ still retained appreciable phosphate removal ability after repeated regeneration, indicating good operational stability.The thermodynamic analysis is provided in Table. S4 (SI).
2.5 XPS and FTIR analysis
XPS results (Fig. 6a–i) provide further evidence for mechanochemical ion exchange and phosphate coordination. After calibration to C 1s at 284.8 eV, LMZ and LMZ-P show Na, La, Fe, O, Al, and Si signals in the survey spectra, whereas the unwashed sample LMZ-Cl additionally exhibits Na 1s (1071 eV) and Cl 2p (198 eV) peaks that disappear after washing. The ZMZ system shows the same evolution, consistent with the XRD results, confirming in situ Na+/La3+ or Na+/Zr4+ exchange during ball milling with removable NaCl as the by-product.15,16,41,45 After phosphate adsorption, LMZ-P shows a distinct P 2p doublet at 133.0–134.2 eV and an enhanced P–O component in O 1s at 531.3–531.8 eV, while Si 2p and Al 2p shift slightly to higher binding energy and broaden, indicating changes in the local framework environment induced by ion exchange and phosphate coordination.19,22,42,46
The La 3d spectrum shows La 3d5/2 and La 3d3/2 peaks at 835.6 and 852.4 eV, respectively, together with satellite peaks at 839.4 and 856.3 eV.24,30,45 Similarly, the Zr 3d doublet in ZMZ appears at 182.3 and 184.8 eV, in agreement with the concurrent changes in P 2p and O 1s. The slight positive shifts of La 3d and Zr 3d after adsorption suggest changes in the local chemical environment of La/Zr sites. As shown in Fig. S6 (SI), the band near 1630 cm−1 is assigned to the H–O–H bending vibration of adsorbed water. The broad band at 1000–1050 cm−1 is mainly associated with the overlap between zeolitic Si–O–Si/Si–O–Al framework vibrations and phosphate-related P–O stretching vibrations. After phosphate adsorption, LMZ-P and ZMZ-P showed noticeable changes in this region, indicating that phosphate species altered the surface vibration environment of the adsorbents. In addition, the low-wavenumber band around 580 cm−1, related to Fe–O and other metal–oxygen vibrations, changed after adsorption, suggesting the involvement of metal-containing sites in phosphate binding. In contrast, the Fe 2p peaks at 710.9 and 724.5 eV remain essentially unchanged, showing that Fe3O4 mainly provides magnetic separability rather than directly participating in phosphate binding.34,42
2.6 Adsorption mechanism analysis
As illustrated in the adsorption mechanism scheme (Fig. 7), Na-A zeolite, Fe3O4, and La/Zr precursors undergo continuous collision and shear in an ethanol medium during high-energy ball milling, leading to the formation of magnetic zeolite composites. The mechanochemical process promotes the intimate coupling between Fe3O4 and the zeolitic matrix, thereby enabling rapid magnetic separation. Meanwhile, it facilitates Na+ exchange with La3+/Zr4+, allowing La/Zr active sites to be introduced into or anchored within the zeolitic framework and pore environment. During phosphate adsorption, H2PO4−/HPO42− diffuses through the pore channels toward the La/Zr active sites and interacts with protonated La/Zr–OH groups under acidic conditions through ligand exchange and surface coordination, forming La/Zr–O–P-related binding structures eqn (3)–(6). The changes in P–O vibrations observed by FTIR (Fig.S5 (SI)), together with the variations in P 2p, O 1s, and La/Zr chemical environments revealed by XPS (Fig. 6), support this process. Overall, phosphate removal by LMZ/ZMZ is mainly governed by La/Zr-site-mediated surface coordination and ligand exchange, while the hierarchical pore structure facilitates mass transfer and Fe3O4 primarily provides magnetic recoverability.| | |
La-OH + H2PO4− → La (H2PO4) + OH−
| (3) |
| | |
2La-OH + HPO42− → La2(HPO4) + 2OH−
| (4) |
| | |
Zr-OH2+ + H2PO4− → (Zr–OH2+)(H2PO4−)
| (5) |
| | |
Zr-OH2+ + HPO42− → (Zr–OH2+)(HPO42−)
| (6) |
 |
| | Fig. 7 Adsorption mechanism diagram. | |
3 Conclusion
In conclusion, a wet high-energy ball milling/in situ ion-exchange strategy was developed to introduce large functional cations into Na-A zeolite while simultaneously achieving nanoscale coupling with Fe3O4 and magnetic separability. La3+ doping preserved the microporous framework of Na-A zeolite and promoted hierarchical pore reconstruction, improving site accessibility and mass transfer. As a result, LMZ exhibited a maximum phosphate uptake of 44.47 mg g−1 at pH 3, while ZMZ reached 33.2 mg g−1. Kinetic and isotherm analyses indicated that phosphate adsorption was dominated by monolayer chemisorption, and multiple lines of evidence supported a mechanism involving in situ Na+/La3+ or Na+/Zr4+ exchange and subsequent inner-sphere La/Zr–O–P coordination. The successful preparation of zirconium magnetic zeolite through the same route further highlights the versatility of this strategy for other large functional cations.
Author contributions
Xiangwu Meng: writing – original draft, formal analysis; Jianjun Li: investigation, funding acquisition; Liangji Xu: conceptualization, approved and corrected; Changguo Xue: supervision; Xuekai Wang: methodology, reviewed; Linlin Song: data curation, visualization.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental details, and extended characterization. See DOI: https://doi.org/10.1039/d6ra02580c.
Acknowledgements
This work was financially supported by Natural Science Foundation of Universities in Anhui Province (2023AH010025), National Natural Science Foundation of China (No. 11872001).
References
- M. Delkash, B. E. Bakhshayesh and H. Kazemian, Microporous Mesoporous Mater., 2015, 214, 224–241, DOI:10.1016/j.micromeso.2015.04.039.
- H. Figueiredo and C. Quintelas, J. Hazard. Mater., 2014, 274, 287–299, DOI:10.1016/j.jhazmat.2014.04.012.
- N. Widiastuti, H. Wu, H. M. Ang and D. K. Zhang, Desalination, 2008, 218, 271–280, DOI:10.1016/j.desal.2007.02.022.
- J. Čejka, R. Millini, M. Opanasenko, D. P. Serrano and W. J. Roth, Catal. Today, 2020, 345, 2–13, DOI:10.1016/j.cattod.2019.10.021.
- C. Belviso, E. Agostinelli, S. Belviso, F. Cavalcante, S. Pascucci and D. Peddis, Microporous Mesoporous Mater., 2015, 202, 208–216, DOI:10.1016/j.micromeso.2014.09.059.
- J. Cao, Q. Sun, P. Wang, J. Shen and X. Dai, Z. Anorg. Allg. Chem., 2020, 646, 1666–1670, DOI:10.1002/zaac.202000215.
- P. Wang, Q. Sun, Y. Zhang and J. Cao, Mater. Res. Express, 2020, 7, 016104, DOI:10.1088/2053-1591/ab609c.
- C. Wang, H. Gui, C. Li, J. Chen and C. Chen, Water Sci. Technol., 2021, 84, 941–953, DOI:10.2166/wst.2021.271.
- X. Querol, N. Moreno, J. C. Umaña, A. Alastuey, E. Hernández, A. López-Soler and F. Plana, Int. J. Coal Geol., 2002, 50, 413–423, DOI:10.1016/S0166-5162(02)00124-6.
- P. Ning, H.-J. Bart, B. Li, X. Lu and Y. Zhang, J. Environ. Sci., 2008, 20, 670–674, DOI:10.1016/S1001-0742(08)62111-7.
- A. Ateş, J. Colloid Interface Sci., 2018, 523, 266–281, DOI:10.1016/j.jcis.2018.03.115.
- A. Ateş, Powder Technol., 2019, 344, 199–207, DOI:10.1016/j.powtec.2018.12.018.
- X. Du, X. Gao, H. Zhang, X. Li and P. Liu, Catal. Commun., 2013, 35, 17–22, DOI:10.1016/j.catcom.2013.02.010.
- X. He, X. Huang, Z. Wang and Y. Yan, Microporous Mesoporous Mater., 2011, 142, 398–403, DOI:10.1016/j.micromeso.2010.12.029.
- X. H. Vu, M. S. Marschall, V. T. Tran, T. P. Ngo, T. T. Dang, D. M. Dinh, T. K. T. Dao, O. Busse and J. J. Weigand, J. Phys. Chem. Solids, 2021, 152, 109962, DOI:10.1016/j.jpcs.2021.109962.
- Y. Ji, Y. Wang, B. Xie and F.-S. Xiao, Comments Inorg. Chem., 2016, 36, 1–16, DOI:10.1080/02603594.2015.1031375.
- L. L. Song, J. Li, Z. Zhang, Z. Li, T. Hu, F. Li, Y. Zhao and X. Peng, Mater. Lett., 2021, 303, 130542, DOI:10.1016/j.matlet.2021.130542.
- R. Xu, M. Zhang, R. J. G. Mortimer and G. Pan, Environ. Sci. Technol., 2017, 51, 3418–3425, DOI:10.1021/acs.est.6b05623.
- W. Sang, L. Mei, S. Hao, D. Li, X. Li, Q. Zhang, X. Jin and C. Li, Water Sci. Technol., 2020, 82, 2975–2989, DOI:10.2166/wst.2020.541.
- D. Guaya, C. Valderrama, A. Farran, A. Armijos and J. L. Cortina, Chem. Eng. J., 2015, 271, 204–213, DOI:10.1016/j.cej.2015.03.003.
- A. Jayaseelan Arun, S. S. Vigneshwar and A. Swetha, J. Water Process Eng., 2020, 38, 101567, DOI:10.1016/j.jwpe.2020.101567.
- Q. Xu, Z. Chen, Z. Wu, X. Xu, D. Li, L. Li and Y. Liu, Bioresour. Technol., 2019, 289, 121600, DOI:10.1016/j.biortech.2019.121600.
- Y. Zou, R. Zhang, L. Wang, Y. Xue, J. Chen and X. Zhang, Appl. Clay Sci., 2020, 192, 105638, DOI:10.1016/j.clay.2020.105638.
- L. Chen, X. Zhao, B. Pan, W. Zhang, M. Hua, L. Lv and W. Zhang, J. Hazard. Mater., 2015, 284, 35–42, DOI:10.1016/j.jhazmat.2014.10.048.
- D. Cordell, J.-O. Drangert and S. White, Glob. Environ. Change, 2009, 19, 292–305, DOI:10.1016/j.gloenvcha.2008.10.009.
- H. Liu, X. Sun, Y. Yin and C. Hu, J. Hazard. Mater., 2008, 151, 616–622, DOI:10.1016/j.jhazmat.2007.06.033.
- M. T. Vu, H. C. Duong, Q. Wang, A. Ansari, Z. Cai, N. B. Hoang and L. D. Nghiem, Environ. Technol. Innov., 2023, 30, 103114, DOI:10.1016/j.eti.2023.103114.
- S. J. Ng, B. Li, Z. He, J.-C. Han, M. T. Munir, X. Wu and Y. Huang, Resour. Policy, 2023, 85, 103781, DOI:10.1016/j.resourpol.2023.103781.
- S. Zhang, W. Wu, X. Hu, G. Tang, Y. Liu and B. Zhou, J. Environ. Chem. Eng., 2025, 13, 116779, DOI:10.1016/j.jece.2025.116779.
- H. Zeng, S. Li, J. Li, Z. Li, D. Lin, J. Zhang and D. Li, J. Water Process Eng., 2025, 77, 108597, DOI:10.1016/j.jwpe.2025.108597.
- C. Sun, H. Cao, C. Huang, P. Wang, J. Yin, H. Liu, H. Tian, H. Xu, J. Zhu and Z. Liu, Bioresour. Technol., 2022, 362, 127851, DOI:10.1016/j.biortech.2022.127851.
- Y. He, H. Lin, Y. Dong and L. Wang, Appl. Surf. Sci., 2017, 426, 995–1004, DOI:10.1016/j.apsusc.2017.07.272.
- Z. Wu, Q. Zheng and H. D. Peng, J. Clean. Prod., 2023, 392, 136294, DOI:10.1016/j.jclepro.2023.136294.
- B. B. Qiu and F. Duan, Colloids Surf., Ac, 2019, 571, 86–93, DOI:10.1016/j.colsurfa.2019.03.041.
- M. El Bouraie and A. A. Masoud, Appl. Clay Sci., 2017, 140, 157–164, DOI:10.1016/j.clay.2017.01.021.
- F. Lin, Y. Zhou, X. Wang, H. Wang and B. Xue, J. Ind. Eng. Chem., 2026, 156, 464–475, DOI:10.1016/j.jiec.2025.08.052.
- Y. Zhang and T. C. Ling, Constr. Build. Mater., 2020, 234, 117424, DOI:10.1016/j.conbuildmat.2019.117424.
- S. M. Muscarella, L. Badalucco, B. Cano, V. A. Laudicina and G. Mannina, Bioresour. Technol., 2021, 341, 125812, DOI:10.1016/j.biortech.2021.125812.
- F. Pan, H. Wei, Y. Huang, J. Song, M. Gao, Z. Zhang, R. Teng and S. Jing, J. Clean. Prod., 2024, 444, 141233, DOI:10.1016/j.jclepro.2024.141233.
- W. Ding, S. Bai, H. Mu and G. Naren, Water Sci. Technol., 2017, 76, 785–792, DOI:10.2166/wst.2017.241.
- J. Goscianska, M. Ptaszkowska-Koniarz, M. Frankowski, M. Franus, R. Panek and W. Franus, J. Colloid Interface Sci., 2018, 513, 72–81, DOI:10.1016/j.jcis.2017.11.003.
- B. Paul, J. J. Dynes and W. Chang, Desalination, 2017, 419, 141–151, DOI:10.1016/j.desal.2017.06.009.
- X. Luo, X. Wang, S. Bao, X. Liu, W. Zhang and T. Fang, Sci. Rep., 2016, 6, 39108, DOI:10.1038/srep39108.
- L. Zhang, H. Dan, O. T. Bukasa, L. Song, Y. Liu, L. Wang and J. Li, ACS Omega, 2020, 5, 25326–25333, DOI:10.1021/acsomega.0c03657.
- M. A. Rahman, D. Lamb, A. Kunhikrishnan and M. M. Rahman, Water, 2021, 13, 3320, DOI:10.3390/w13233320.
- X. Lu, Z. Zhong, R. Yan, F. Zan, X. Zhang, Y. Wu, Y. Liu and X. Li, J. Clean. Prod., 2022, 373, 133746, DOI:10.1016/j.jclepro.2022.133746.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069, DOI:10.1515/pac-2014-1117.
- Y. Zhan, H. Zhang, J. Lin, Z. Zhang and J. Gao, J. Mol. Liq., 2017, 243, 624–637, DOI:10.1016/j.molliq.2017.08.091.
- H. S. Rangappa, I. Herath, C. Lin and S. Ch, Environ. Pollut., 2024, 343, 123140, DOI:10.1016/j.envpol.2023.123140.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *d,
Liangji Xuc,
Changguo Xue
*d,
Liangji Xuc,
Changguo Xue










