
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceD-Leucine modified Sansalvamide A analogs: photo-induced synthesis, target prediction, and antitumor potential study
Yujun Bao†
 ac,
Fujing Guan†d,
Fengmei Shic,
Su Wangc,
Liguo Weib,
Pengfei Li*c,
Zhanjiang Pei*c,
Lishuang Zhao*b and
Chuncheng Hao*a
ac,
Fujing Guan†d,
Fengmei Shic,
Su Wangc,
Liguo Weib,
Pengfei Li*c,
Zhanjiang Pei*c,
Lishuang Zhao*b and
Chuncheng Hao*a
aDepartment of Head and Neck Radiation Oncology, Harbin Medical University Cancer Hospital, Harbin, 150081, China
bCollege of Environmental and Chemical Engineering, Heilongjiang University of Science and Technology, Harbin, 150025, China
cKey Laboratory of Energy Utilization of Main Crop Stalk Resources, Heilongjiang Academy of Black Soil Conservation and Utilization, Harbin, 150086, China
dPhysical and Chemical Inspection Department, Harbin Center for Disease Control and Prevention, Harbin, 150056, China
First published on 22nd May 2026
Abstract
Cyclic peptides show promise in cancer therapy but are hindered by low natural yields and poor metabolic stability. This study synthesized Sansalvamide A analogs (Compound 1–5) via Leucine configuration modification and green intramolecular photoinduced single-electron-transfer cyclization, which introduces antitumor isoindolinone into the skeleton to enhance the structural rigidity. In vitro MTT assay showed that cyclic peptides exhibit a certain degree of tumor cell inhibitory activity, among which Compound 5 (with multiple D-leucine modifications) exhibits relatively stronger activity. Target prediction and protein–protein interaction analysis identified BCL2 as a key hub target, a finding further confirmed by molecular docking, which demonstrated that Compound 5 exhibits high binding affinity to BCL2 through stable hydrogen bonds and hydrophobic interactions (alkyl, π-cation and π-sigma interactions). Meanwhile, JC-1 assay verified that Compound 5 can effectively reduce mitochondrial membrane potential, which in turn endows it with the potential to induce tumor cell apoptosis. In silico ADMET predictions further supported the practical application potential of Compound 5. This research has provided a useful guide for peptide-based drug-target design.
1 Introduction
In recent years, polypeptide drugs have developed rapidly in the fields of disease treatment and healthcare.1–3 Among them, cyclic peptide compounds, owing to their stable conformations, hold significant potential in cancer therapy.4–6 Currently, research on the artificial synthesis of cyclic peptides (to address the yield issues of natural cyclic peptides) and their structural modification (to further improve physicochemical properties) is attracting increasing attention.7–9 For instance, introducing rigid structural fragments to enhance the structural stability of cyclic peptides; appropriately altering amino acid configurations to strengthen the hydrophobic interactions between cyclic peptides and proteins.10,11As a crucial step in cyclic peptide synthesis, cyclization methods severely limit the efficient synthesis and structural modification of cyclic peptides. Compared with traditional cyclization methods, the intramolecular photoinduced single-electron-transfer (SET) cyclization reaction, developed by Yoon and colleagues, offers rapid reaction rates while aligning with green chemistry concepts.12 This reaction constructs an intramolecular electron “donor–acceptor” system by introducing phthalimide as the electron acceptor and peptide chains with terminal trimethylsilyl (TMS) groups as the electron donor. Under photoexcitation, the TMS group departs rapidly, with intramolecular double terminal free radicals forming and coupling simultaneously, which in turn completes cyclization, introduces isoindolinone (with antitumor activity) into the cyclic peptide skeleton, and significantly enhances its structural rigidity (Scheme 1).
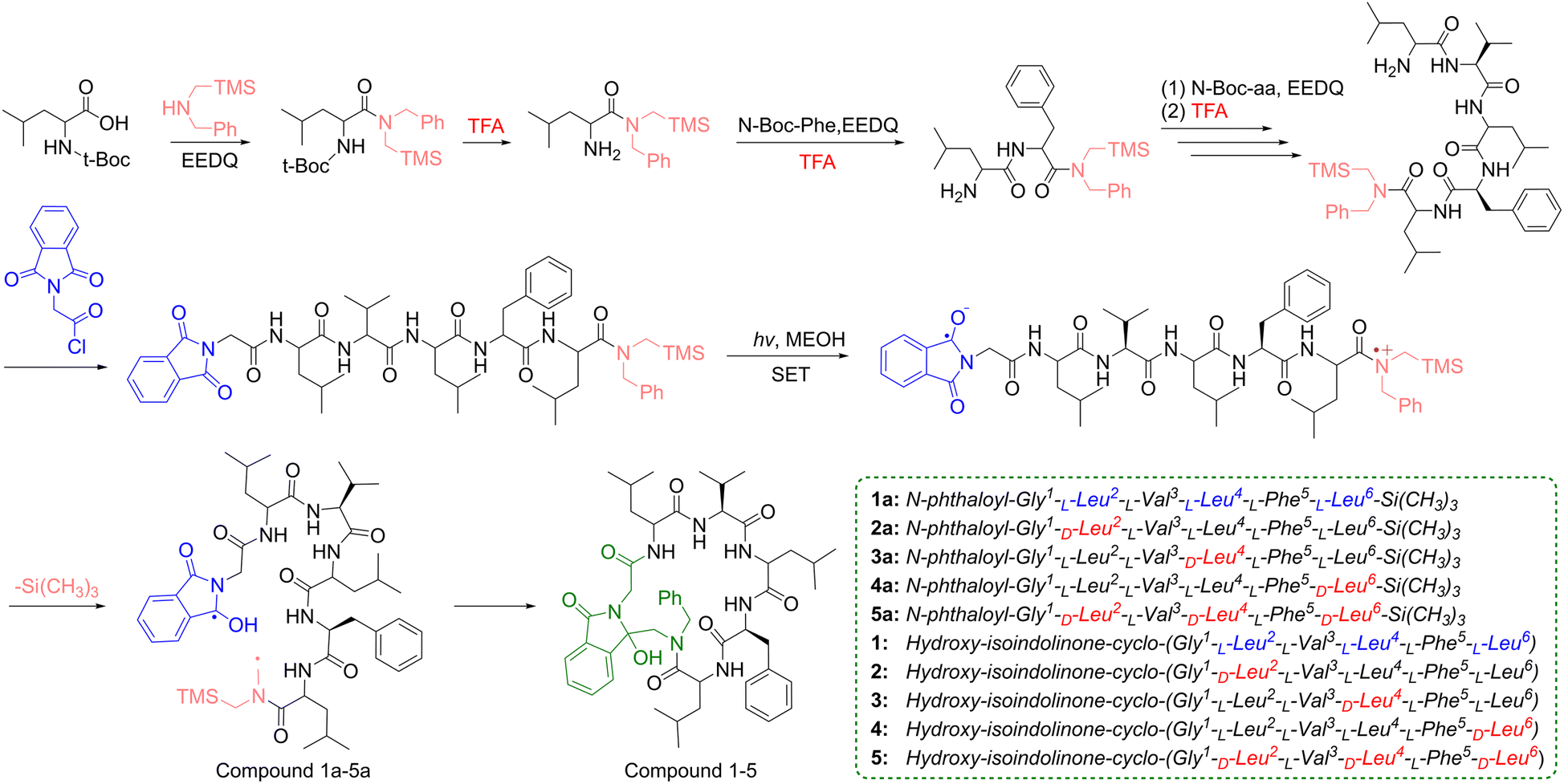 | ||
| Scheme 1 The synthesis of target linear peptides 1a–5a and cyclic peptides 1–5. | ||
Sansalvamide A (San A) is a natural cyclic penta-depsipeptide with multiple bioactivities containing two L-Leu-residues, one L-Phe residue, one L-Val-residue and one S–O-Leu-residue, which was isolated from the fermentation product of a marine fungus of the genus Fusarium.13–15 It has been reported that San A analogs modified with D- and N-methyl amino acids can exhibit enhanced bioactivity, and some of these derivatives have demonstrated inhibitory efficacy against colon cancer cell line (HT-29) comparable to that of 5-fluorouracil.16 This finding has attracted our attention in the research on the structural modification of San A to enhance the antitumor activity.
The tumor microenvironment is abundant in peptidases and proteases that target natural L-amino acid residues. These enzymes can accurately recognize and rapidly cleave peptide bonds, thereby accelerating the hydrolytic inactivation of cyclic peptides in vivo. In contrast, D-amino acids can evade hydrolysis and may also indirectly optimize the binding of cyclic peptides to the hydrophobic active pockets of tumor targets.17–19 Therefore, altering the configuration of amino acid residues to enhance the metabolic stability and target-binding specificity of cyclic peptide compounds is an important modification strategy. In our previous work, we have successfully synthesized the San A derivative (Compound 1) via the photoinduced SET reaction, which exhibits potential tumor inhibitory activity (Fig. 1).20 In this study, the configuration of Leucine (Leu) residues was further altered, and a series of San A analogs were designed and prepared via the SET cyclization reaction. The antitumor activity of the San A analogs was preliminarily evaluated and screened by MTT colorimetric tetrazole dye assay. In addition, the structure of the prepared cyclic peptides was further optimized, and the tumor-related active biomolecules potential targets were predicted. Meanwhile, the binding energies and interaction sites with target proteins were further calculated via molecular docking, and the association between D-amino acids and targets, as well as the impact on in silico ADMET (Absorption, Distribution, Metabolism, Excretion and Toxicity), was analyzed in detail.
 | ||
| Fig. 1 The five designed Sansalvamide A analogs. | ||
2 Results and discussion
2.1 Synthesis
A key step in the photoinduced SET synthesis of cyclic peptides is constructing linear peptides with an intramolecular electron “donor–acceptor” system. Briefly, 2-ethoxy-1-ethoxycarbonyl-1,2-dihydroquinoline (EEDQ) was used to activate the carboxyl group of N-Boc-Leu, after which an equivalent of N-benzyl-1-(trimethylsilyl)-methylamine (BnTMSA) was added directly. After detecting the reaction via thin-layer chromatography (TLC), the target product (N-Boc-Leu-TMS) was obtained through purification by column chromatography. Subsequently, the N-Boc group of N-Boc-Leu-TMS, which served as a protecting group, was removed using trifluoroacetic acid (TFA). The resulting Leu-TMS then functioned as a new reactive building block and was coupled with the next amino acid in the designed sequence until the linkage of all amino acid residues was completed. Finally, phthaloylglycine was introduced as an electron acceptor group at the N-terminal amino group of the peptide chain to construct the precursor linear peptides (Compound 1a–5a) for the photoinduced SET reaction.The photochemical reaction was carried out in a home-built photoreactor, and the reaction system was operated in methanol solution under N2 protection. A 450 W medium-pressure mercury lamp (Hanovia) equipped with a Pyrex glass filter tube was used as the light source to provide 300–400 nm UV radiation. The reaction temperature was maintained by circulating water, and the progress of the photochemical reaction was monitored by TLC. The target photochemical reaction products (Compound 1–5) were purified by column chromatography. The synthesis of the peptides is presented in Scheme 1. All synthetic target products in this study (Compound 1a–5a and 1–5) were characterized by 1H NMR, 13C NMR, and mass spectrometry, with detailed data provided in the SI. Among these products, Compound 1a and 1 were reported in previous work.20
2.2 In vitro antitumor activities
To systematically evaluate the tumor cell inhibitory activity of the target compounds, the MTT assay was used to detect the proliferation inhibitory effects on the hepatocellular carcinoma cell line (HepG-2), breast cancer cell line (4T1), colon cancer cell line (CT26), and normal fibroblast cell line (L929), all of which were obtained from Harbin Medical University (Harbin, China). As shown in Fig. 2A and B, linear peptides exhibited only weak inhibitory activity against the above three tumor cell lines, while cyclic peptide compounds demonstrated significant advantages in tumor growth inhibition. This activity difference is closely related to the structural characteristics of the two types of compounds. Specifically, the enhanced activity of cyclic peptides is attributed to their unique rigid structure, which can also be demonstrated by the fused isoindolinone ring system that effectively provides additional hydrogen bonds during the binding process and further forms more stable non-covalent binding with target amino acid residues through stronger hydrophobic interactions and π–π stacking interactions. This interaction mode effectively overcomes the defect of low target binding specificity of linear peptides caused by conformational flexibility, which has been verified in our previous studies. In contrast, Compound 5 showed the most prominent inhibitory activity against the three tumor cell lines, a result that clearly indicates altering the configuration of the Leu residue can optimize biological activity through a dual pathway which comprises one that regulates the overall spatial conformation of the compound, thereby allowing it to more accurately fit the binding pocket of the target protein, and the other that adjusts the hydrophobic orientation of the Leu residue side chain to further enhance hydrophobic interactions with target amino acid residues, which in turn significantly improves target binding efficiency and specificity. When coupled with the lack of obvious toxicity to normal L929 cells, this characteristic indicates that Compound 5 may achieve selective recognition of tumor cell targets through structural modification, holding potential clinical application value. | ||
| Fig. 2 IC50 values of (A) linear peptides 1a–5a and (B) cyclic peptides 1–5 against HepG-2, 4T1, CT26 and L929 cells. (C) The potential target protein networks of Compound 1–5 constructed using the STRING database. | ||
2.3 PPI network analysis
To further investigate the potential impact of structural modification of compounds on tumor inhibitory activity, protein–protein interaction (PPI) network analysis reveals the connections between compounds and target proteins from a holistic perspective, clarifies the molecular mechanisms of diseases, and thereby facilitates the prediction of novel drug targets.21–24 In this study, after energy minimization, Compound 1–5 were subjected to target prediction using the SWISS database (https://www.swisstargetprediction.ch/). The overlapping targets with the total list of cancer-related genes (obtained from https://www.genecards.org/) amounted to 60. The PPI network of Compound 1–5 was constructed using the STRING database (https://cn.string-db.org/), while topological analysis was conducted via Cytoscape software, followed by ranking based on Degree values (Fig. 2C and Table S1).25–27 Among these targets, the six hub targets with the highest Degree values were identified as BCL2, MMP9, CTSB, CTSS, BCL2L1, and MMP2.2.4 GO and KEGG pathway enrichment analysis of predicted targets
To investigate the biological pathways of the predicted targets of Compound 1–5, Gene Ontology (GO) analysis was performed on the aforementioned 60 targets using the DAVID database.28,29 As shown in Fig. 3A, GO annotations showed that the related genes were enriched in pathways related to tumor apoptosis, such as apoptotic process, Notch receptor processing, BAD-BCL-2 complex, cysteine-type endopeptidase activator activity involved in apoptotic process and Bcl-2 family protein complex. Moreover, these genes were also enriched in the angiogenesis, extracellular matrix, cell cycle, mitochondria and lysosome. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis suggested that related genes were enriched in Apoptosis, Notch signaling pathway, Pathways in cancer, p53 signaling pathway, PI3K-Akt signaling pathway, and EGFR tyrosine kinase inhibitor resistance, all of which are pathways related to cancer (Fig. 3B). This also indicates that Compound 1–5 exhibit potential for anti-tumor therapy. | ||
| Fig. 3 (A) GO analysis (cellular component, biological processes, and molecular functions) and (B) KEGG analysis of the sixty proteins. | ||
2.5 Molecular docking analysis
To identify suitable targets, molecular docking was performed for cyclic peptides 1–5 and linear peptides 1a–5a with the six major hub targets obtained from PPI interaction analysis. The docking scores of binding energy derived from molecular docking, which directly reflect the interaction strength between molecules, indicate that a lower value corresponds to greater stability of the system under isothermal and isobaric conditions after the receptor binds to the ligand (binding energy <−7 kcal mol−1 signifying very tight binding). Molecular docking results are shown in Fig. 4A and B. Compared with the relevant targets (CTSL, CTSB, MMP2, MMP9, ACE, and EGFR) of linear peptides, the important relevant targets (BCL2, BCL2L1, CTSS, CTSB, MMP2, and MMP9) of cyclic peptides are more inclined to induce cell apoptosis. This can be primarily attributed to the distinctive three-dimensional structural features of the BCL2 family, which are distinct from those of traditional enzymes in that they lack the canonical “active pocket” for small-molecule drugs to dock into, while possessing a single “large and flat” binding interface that specifically interacts with pro-apoptotic proteins (e.g., BIM). Notably, these flat binding interfaces present a significant challenge for linear peptides to achieve stable and effective occupancy, whereas cyclic peptides exhibit substantially greater adaptability to such interfaces owing to their inherent structural properties that confer upon them a relatively larger binding surface area, thereby enabling more robust intermolecular interactions. Furthermore, through the visual analysis of docking results, it can be observed that the isoindolinone structure of the cyclic peptide further exerts new interactions on the BCL2 protein, which further enhances the binding ability. | ||
| Fig. 4 Docking scores (kcal mol−1) for molecular docking of (A) linear peptides 1a-5a and (B) cyclic peptides 1–5 with each of six hub target proteins. (C) The residue interaction types combined with Compound 1–5 in the BCL2 protein. (D) 2D and (E) 3D interaction diagrams of Compound 5 interacting with the active site of BCL2 (PDB ID: 8VWZ). | ||
Docking results of cyclic peptide 1–5 reveal that these compounds exhibit relatively superior binding affinity predominantly for BCL2, a core anti-apoptotic protein within the cellular apoptotic regulatory network that interacts with BH3-only proteins (e.g., BIM, BAD) or pro-apoptotic counterparts (BAX, BAK) via its conserved BH3 hydrophobic pocket. Specifically, the binding of BCL2 to these cognate proteins not only preserves mitochondrial membrane homeostasis but also abrogates the initiation of the intrinsic apoptotic cascade, thus conferring a pivotal regulatory function in the suppression of programmed cell death.30–33 As shown in the molecular docking results (Fig. 4B), Compound 5 exhibits a docking score of −10.8 with BCL2, indicating the excellent binding affinity for BCL2. Further analysis of the interactions (Fig. 4C) revealed that Compound 5 forms numerous interaction modes with BCL2, particularly high-strength conventional hydrogen bonds and π-cation interactions.
2D and 3D interaction diagrams were generated by PyMOL and Discovery Studio 2019 Visualizer (Fig. 4Dand E). The interaction analysis demonstrated that Compound 5 exhibited significant binding affinity toward the BCL2 protein. Specifically, Compound 5 formed van der Waals interactions with Tyr22, Leu175, Tpr176, Glu179, Phe207, Glu210, Gly294, Asp298, Glu318, Lys321 residues.
Furthermore, hydrogen bonding exerted a pivotal role in stabilizing the Compound 5-BCL2 complex. Compound 5 established hydrogen bonds with Ser26 and Arg297 residues of BCL2, while additionally undergoing hydrophobic interactions (including alkyl interactions, π-sigma and π-cation interactions) with a suite of residues including Arg,19 Trp,23 Met,27 Pro,29 Arg127, Arg183, Tyr208, Pro209 and Arg297. These cumulative interactions collectively validate that Compound 5 holds significant potential for targeting BCL2.
2.6 Detection of MMP by JC-1
BCL2, a core anti-apoptotic protein in the intrinsic apoptotic pathway, exerts a pivotal anti-apoptotic effect by maintaining mitochondrial membrane potential (MMP) homeostasis.34–37 Notably, a significant decrease in MMP serves as a key hallmark for the initiation of tumor cell apoptosis. In this study, the JC-1 probe was utilized to assess MMP changes in 4T1 cells, and the resultant fluorescence signals were visualized using fluorescence microscopy. As clearly depicted in Fig. 5, healthy mitochondria in the control group emitted intense red fluorescence. In contrast, after treatment with Compound 5, the intensity of red fluorescence in cells was significantly reduced, accompanied by a marked enhancement of green fluorescence signals. Furthermore, this trend became increasingly pronounced with the elevation of Compound 5 concentration. Collectively, these results indicate that Compound 5 may possess the capacity to specifically target and bind BCL2, thereby eliciting mitochondrial damage and ultimately inducing tumor cell apoptosis. | ||
| Fig. 5 Detection of MMP changes in 4T1 cells after different treatment via the JC-1 probe, scale bars: 20 µm. | ||
2.7 In silico ADMET study
ADMET prediction is a crucial method to preliminarily clarify the fundamental properties of drugs from multiple dimensions, providing a key basis for druggability evaluation in drug research and development. In this study, ADMET prediction was performed on linear peptides 1a–5a and cyclic peptides 1–5 using the ADMETlab 3.0 platform (https://admetlab3.scbdd.com/), with the results detailed in Table 1 and Table S15.38–41 From the perspective of physicochemical properties, Compound 5 contains 15 hydrogen bond acceptors (HA) and 6 hydrogen bond donors (HD), and the molecular weight (MW) exceeds 500, violating Lipinski's rule, which indicates limited feasibility for oral administration. However, its octanol–water partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) and water solubility (logS) both fall within the optimal ranges, and it complies with Pfizer's druggability criteria, demonstrating a solid druggability foundation. Compound 5 exhibits low blood–brain barrier (BBB) penetration, implying a relatively controllable risk of central nervous system (CNS) side effects. Enzymes such as CYP2C9 and CYP3A4 belong to the cytochrome P450 enzyme system (CYP450), the most prominent drug-metabolizing enzyme family in the human liver, involved in the metabolism of exogenous substances. ADME prediction reveals that Compound 5 possesses both CYP2C19 substrate and CYP3A4 inhibitor properties. This not only establishes a molecular basis for individualized and precise anti-tumor therapy but also demonstrates remarkable potential in synergizing combination therapies, directly inhibiting tumor metabolic pathways, and reversing drug resistance. In addition, its moderate half-life (T1/2 = 1.208 h) ensures sustained release of efficacy to maintain the inhibitory effect on tumor cells while allowing flexibility in adjusting the administration regimen.
P) and water solubility (logS) both fall within the optimal ranges, and it complies with Pfizer's druggability criteria, demonstrating a solid druggability foundation. Compound 5 exhibits low blood–brain barrier (BBB) penetration, implying a relatively controllable risk of central nervous system (CNS) side effects. Enzymes such as CYP2C9 and CYP3A4 belong to the cytochrome P450 enzyme system (CYP450), the most prominent drug-metabolizing enzyme family in the human liver, involved in the metabolism of exogenous substances. ADME prediction reveals that Compound 5 possesses both CYP2C19 substrate and CYP3A4 inhibitor properties. This not only establishes a molecular basis for individualized and precise anti-tumor therapy but also demonstrates remarkable potential in synergizing combination therapies, directly inhibiting tumor metabolic pathways, and reversing drug resistance. In addition, its moderate half-life (T1/2 = 1.208 h) ensures sustained release of efficacy to maintain the inhibitory effect on tumor cells while allowing flexibility in adjusting the administration regimen.
| Descriptors | Calculated | Empirical decision |
|---|---|---|
| a MW ≤ 500; logP ≤ 5; nHA ≤ 10; nHD ≤ 5. If two properties are out of range, a poor absorption or permeability is possible.b Compounds with a high logP (>3) and low TPSA (<75) are likely to be toxic.c low permeability: <2 × 10−6 cm s−1, medium permeability: 2–20 × 10−6 cm s−1, high passive permeability: >20 × 10−6 cm s−1.d For the classification endpoints, the prediction probability values are transformed into six symbols: 0–0.1 (—), 0.1–0.3 (—), 0.3–0.5 (—), 0.5–0.7 (+), 0.7–0.9 (++), and 0.9–1.0 (+++).e The output value is the probability of being toxic, within the range of 0 to 1.f The output value is the probability of being active. |
||
| MW | 893.51 | Optimal: 100–600 |
| nHA | 15 | Optimal: 0–12 |
| nHD | 6 | Optimal: 0–7 |
| nRot | 11 | Optimal: 0–11 |
| nRing | 5 | Optimal: 0–6 |
| nRig | 49 | Optimal: 0–30 |
| LogS |
−3.837 | Optimal: −4∼0.5 (log mol L−1) |
| LogP |
3.528 | Optimal: 0–3 (log mol L−1) |
| TPSA | 206.35 | Optimal: 0–140 |
| Flexibility | 0.224 | nRot/nRig |
| Lipinski | Rejected | a |
| Pfizer | Accepted | b |
| MDCK Permeability | −4.712 | c |
| CYP1A2 inhibitor | — | d |
| CYP1A2 substrate | — | d |
| CYP2C19 inhibitor | — | d |
| CYP2C19 substrate | ++ | d |
| CYP2C9 inhibitor | — | d |
| CYP2C9 substrate | — | d |
| CYP2D6 inhibitor | — | d |
| CYP2D6 substrate | — | d |
| CYP3A4 inhibitor | +++ | d |
| CYP3A4 substrate | — | d |
| CYP2B6 inhibitor | — | d |
| CYP2B6 substrate | — | d |
| CYP2C8 inhibitor | — | d |
| BBB | — | d |
| T1/2 | 1.208 | The unit is hours |
| DILI | 0.283 | e |
| Carcinogenicity | 0.010 | e |
| Hematotoxicity | 0.046 | e |
| Respiratory | 0.251 | e |
| SR-p53 | 0.493 | f |
Compounds with a topological polar surface area (TPSA) exceeding 140 Å2 often show high lipophilicity and strong protein-binding capacity. Compound 5 has a TPSA of 206.35, suggesting strong protein-binding potential. Meanwhile, its cyclic peptide structure with a high number of rigid bonds (nRig = 49) helps maintain conformational stability, enhances binding specificity to tumor-specific targets (such as kinases and receptors), and lays a structural foundation for improving the targeting of anti-tumor effects. In terms of toxicological safety, the carcinogenicity, hematotoxicity, and drug-induced liver injury (DILI) risk of this compound are all at extremely low levels. This builds a critical foundation for reducing adverse reactions and improving treatment compliance in long-term anti-tumor therapy, making it particularly suitable for solid tumor or hematological tumor treatment scenarios requiring prolonged administration. Therefore, overall, the outstanding performance of this small-molecule cyclic peptide in terms of toxicological safety, metabolic regulation, structural modifiability, and druggability basis endows it with significant potential as an anti-cancer candidate drug.
3 Conclusions
This study successfully synthesized Sansalvamide A analogs (Compound 1–5) by integrating Leu configuration modification with green intramolecular photoinduced SET cyclization, which introduces antitumor-active isoindolinone into the cyclic skeleton and thereby improves the stability and target-binding specificity. Compound 5 (with multiple D-leucine modifications) stood out among the analogs for its relatively stronger in vitro tumor inhibitory activity, while also exhibiting negligible toxicity to normal L929 cells. Target prediction, along with GO and KEGG enrichment analyses, revealed that the cyclic peptide analogs possess the potential to induce tumor cell apoptosis, with BCL2 identified as the key target. Further molecular docking results demonstrated that Compound 5 binds BCL2 with high affinity through hydrogen bonds and diverse hydrophobic interactions (including alkyl, π-cation, and π-sigma interactions). Additionally, results from JC-1 assays demonstrated that Compound 5 can induce a decrease in MMP, which in turn may confer the potential to induce tumor cell apoptosis. In silico ADMET predictions further corroborated that Compound 5 possesses both a foundation for druggability and a certain degree of toxicological safety. Overall, this study successfully designed and synthesized a promising cyclic peptide candidate (Compound 5) for cancer therapy, while simultaneously offering valuable insights to advance cyclopeptide-based drug design.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Yujun Bao: resources, funding acquisition, writing – original draft and writing – review & editing. Fujing Guan: visualization and resources. Fengmei Shi: project administration and software. Su Wang: funding acquisition and resources. Liguo Wei: formal analysis and investigation. Pengfei Li: funding acquisition and writing – original draft. Lishuang Zhao: conceptualization, data curation, methodology, supervision and validation. Chuncheng Hao: funding acquisition and supervision. Zhanjiang Pei: funding acquisition and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental details and supplementary data. 1H-NMR, 13C-NMR and MS of compounds (Fig. S1–S28), potential target protein networks (Fig. S29), GO analysis (Fig. S30), KEGG analysis (Fig. S31), Hub gene prediction results of compounds (Tables S1 and S2), Interaction of compounds with potential target proteins and amino acid residue statistics. (Tables S3–S14), 2D and 3D interaction maps of compounds docking with potential target proteins (Fig. S32–S90), Predicted ADME results of studied compounds (Tables S15). See DOI: https://doi.org/10.1039/d6ra02497a.Acknowledgements
Financial support for this research was funded by the Beijing Medical Award Foundation (No. YXTL-2023-726-0158), the Shanghai Zhenghao Charity Foundation (No. ZH-YXJK-34), the Key Research and Development Projects of Heilongjiang Province (2023ZX02B03), Doctoral Startup Fund Project of Heilongjiang Academy of Agricultural Sciences, Heilongjiang Provincial Research Institutes Special Business Expenses (CZKYF2026-1-C017, CZKYF2025-1-A002) and Innovation Project of Heilongjiang Academy of Agricultural Sciences (CX23JQ03, CX22YQ35).Notes and references
- K. Han, J. Zhang, X. Miao, Y. Yu, L. Zhang, J. Zhao, X. Wang, Q. Wei, Y. Li, Y. Ke, B. Ma and W. Wang, PSMA-Triggered Phase Separation of Peptide-Drug Conjugates Enables Zinc-Dependent Tumor Inhibition, Angew. Chem., Int. Ed., 2025, e18980, DOI:10.1002/anie.202518980.
- Y. Zhang, J. Kæstel-Hansen, D. Teze, Ge Huang, S. Schoffelen, M. Lisby, M. Zhang, N. S Hatzakis and M. Meldal, Click-Cyclized Cell Penetrating Peptides Containing Hydrophobic Proline Derivatives for Efficient Intracellular Delivery, Angew. Chem., Int. Ed., 2025, e202504862, DOI:10.1002/anie.202504862.
- J. Wang, Y. Du, G. Su, W. Tang, X. Long, Y. He and L. Feng, Dual-Functional pH-Sensitive Nanoliposomes Modified with CD47 Mimicry Peptide Enhance Icariin Delivery to Attenuate Neuroinflammation and Oxidative Stress in Epilepsy, Mater. Today Bio, 2025, 35, 102528, DOI:10.1016/j.mtbio.2025.102528.
- M. Hasan, N. Vodnala, Y. Glagovsky, Y. Saed, J. Kriegesmann, H. Suga and A. Brik, Direct Cellular Screening of Pd-Mediated Arylation of Cyclic Peptide Binders Targeting Ubiquitin Chains: Toward Modulating NEMO Liquid-Liquid Phase Separation, J. Am. Chem. Soc., 2025, 147, 28303–28312, DOI:10.1021/jacs.5c09059.
- M. Kong, Y. Peng, Y. Miao and L. Qiu, PDGFR-targeted nanovesicles for restraining breast cancer hepatic metastasis via hepatic stellate cell regression and NK cell activation, Mater. Today, 2024, 79, 1–15, DOI:10.1016/j.mattod.2024.07.005.
- A. H. Benfield, F. Vernen, R. S. Young, F. Nadal-Bufí, H. Lamb, H. Hammerlindl, D. J. Craik, H. Schaider, N. Lawrence, S. J. Blanksby and S. T. Henriques, Cyclic tachyplesin I kills proliferative, non-proliferative and drug-resistant melanoma cells without inducing resistance, Pharmacol. Res., 2024, 207, 107298, DOI:10.1016/j.phrs.2024.107298.
- F. Xiang, J. Deng, X. Bu, Y. Zhou, J. Zhang, Y. Weng and M. Gao, Electro-induced C–H/S–H cross-coupling for the functionalization/macrocyclization of cysteine-containing peptides, Nat. Commun., 2025, 16, 9617, DOI:10.1038/s41467-025-64615-4.
- H. Ogawa, Y. Nagata, T. Ki Chan, Y. Matsuda and H. Nakamura, Rapid Construction of a Tyr C6-Trp C5’ Linkage: Application in the Total Synthesis of Micitide 982, a Noncanonical Cyclic Peptide, Angew. Chem., Int. Ed., 2025, 64, e202516053, DOI:10.1002/anie.202516053.
- X. Y. Liu and J. Waser, Peptide/Protein Functionalization and Macrocyclization via Alkyne Umpolung with Hypervalent Iodine Reagents, Acc. Chem. Res., 2025, 58, 2852–2861, DOI:10.1021/acs.accounts.5c00451.
- R. Nordström, L. Nyström, H. Ilyas, H. S. Atreya, B. C. Borro, A. Bhunia and M. Malmsten, Microgels as carriers of antimicrobial peptides–effects of peptide PEGylation, Colloid. Surface., B, 2019, 565, 8–15, DOI:10.1016/j.colsurfa.2018.12.049.
- S. S. Denisov, J. H. Ippel, B. J. Mans, I. Dijkgraaf and T. M. Hackeng, SecScan: a general approach for mapping disulfide bonds in synthetic and recombinant peptides and proteins, Chem. Commun., 2019, 55, 1374–1377, 10.1039/C8CC08777F.
- U. C. Yoon, Y. X. Jin, S. W. Oh, C. H. Park, J. H. Park, C. F. Campana, X. Cai, E. N. Duesler and P. S. Mariano, A synthetic strategy for the preparation of cyclic peptide mimetics based on SET-promoted photocyclization processes, J. Am. Chem. Soc., 2003, 125, 10664–10671, DOI:10.1021/ja030297b.
- M. Cueto, P. R. Jensen and W. Fenical, N-Methylsansalvamide, a cytotoxic cyclic depsipeptide from a marine fungus of the genus Fusarium, Phytochemistry, 2000, 55, 223–226, DOI:10.1016/S0031-9422(00)00280-6.
- Y. Hwang, D. Rowley, D. Rhodes, J. Gertsch, W. Fenical and F. Bushman, Mechanism of Inhibition of a Poxvirus Topoisomerase by the Marine Natural Product Sansalvamide A, Mol. Pharmacol., 1999, 55, 1049–1053, DOI:10.1016/S0026-895X(24)23256-9.
- G. N. Belofsky, P. R. Jensen and W. Fenical, Sansalvamide: A new cytotoxic cyclic depsipeptide produced by a marine fungus of the genus Fusarium, Tetrahedron Lett., 1999, 40, 2913–2916, DOI:10.1016/S0040-4039(99)00393-7.
- T. J. Styers, A. Kekec, R. Rodriguez, J. D. Brown, J. Cajica, P. S. Pan, E. Parry, C. L. Carroll, I. Medina, R. Corral, S. Lapera, K. Otrubova, C. Pan, K. L. McGuire and S. R. McAlpine, Synthesis of Sansalvamide A derivatives and their cytotoxicity in the MSS colon cancer cell line HT-29, Bioorgan. Med. Chem., 2006, 14, 5625–5631, DOI:10.1016/j.bmc.2006.04.031.
- J. Liu, J. Liu, L. Chu, Y. Zhang, H. Xu, D. Kong, Z. Yang, C. Yang and D. Ding, Self-Assembling Peptide of d-Amino Acids Boosts Selectivity and Antitumor Efficacy of 10-Hydroxycamptothecin, ACS Appl. Mater. Interfaces, 2014, 6, 5558–5565, DOI:10.1021/am406007g.
- J. Wang, J. Song, Z. Yang, S. He, Y. Yang, X. Feng, X. Dou and A. Shan, Antimicrobial Peptides with High Proteolytic Resistance for Combating Gram-Negative Bacteria, J. Med. Chem., 2019, 62, 2286–2304, DOI:10.1021/acs.jmedchem.8b01348.
- S. Zhao, X. Qiao, M. Chen, Y. Li, X. Wang, Z. Xu, Y. Wu and X. Luo, D-Amino Acid-Based Antifouling Peptides for the Construction of Electrochemical Biosensors Capable of Assaying Proteins in Serum with Enhanced Stability, ACS Sens., 2022, 7, 1740–1746, DOI:10.1021/acssensors.2c00518.
- L. Zhao, H. Zhang, G. Tan, Z. Wang and Y. Jin, Photo-induced synthesis and in vitro biological activity of a Sansalvamide A analog, Tetrahedron Lett., 2017, 58, 1669–1672, DOI:10.1016/j.tetlet.2017.03.042.
- Z. He, L. Liu, D. Han, K. Gao, L. Dong, X. Qi, D. Bu, P. Huo, Z. Wang, X. Fan, J. Sun, S. Lei, W. Deng, J. Liu, H. He, Y. Zhao, J. Guo and Y. Wu, POINT: a web-based platform for pharmacological investigation enhanced by multi-omics networks and knowledge graphs, Sci. Bull., 2025, 71(3), 495–499, DOI:10.1016/j.scib.2025.10.002.
- Y. Wang, T. Zhao, C. Huang, F. Liu, Y. Zhang, D. Kong and Z. Fan, Effect and mechanism of Banxia Xiexin decoction in colorectal cancer: A network pharmacology approach, Phytomedicine, 2024, 123, 155174, DOI:10.1016/j.phymed.2023.155174.
- Y. Han, S. Guo, Y. Li, J. Li, L. Zhu, Y. Liu, Y. Lv, D. Yu, L. Zheng, C. Huang, C. Li, J. Hu and Z. Liu, Berberine ameliorate inflammation and apoptosis via modulating PI3K/AKT/NFκB and MAPK pathway on dry eye, Phytomedicine, 2023, 121, 155081, DOI:10.1016/j.phymed.2023.155081.
- B. Ge, R. Sang, W. Wang, K. Yan, Y. Yu, L. Kong, M. Yu, X. Liu and X. Zhang, Protection of taraxasterol against acetaminophen-induced liver injury elucidated through network pharmacology and in vitro and in vivo experiments, Phytomedicine, 2023, 116, 154872, DOI:10.1016/j.phymed.2023.154872.
- A. Daina, O. Michielin and V. Zoete, SwissTargetPrediction: updated data and new features for efficient prediction of protein targets of small molecules, Nucleic Acids Res., 2019, 47, W357–W364, DOI:10.1093/nar/gkz382.
- A. Daina and V. Zoete, Testing the predictive power of reverse screening to infer drug targets, with the help of machine learning, Commun. Chem., 2024, 7, 105, DOI:10.1038/s42004-024-01179-2.
- D. Gfeller, O. Michielin and V. Zoete, Shaping the interaction landscape of bioactive molecules, Bioinformatics, 2013, 29, 3073–3079, DOI:10.1093/bioinformatics/btt540.
- B. T. Sherman, M. Hao, J. Qiu, X. Jiao, M. W. Baseler, H. C. Lane, T. Imamichi and W. Chang, DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update), Nucleic Acids Res., 2022, 50, W216–W221, DOI:10.1093/nar/gkac194.
- D. W. Huang, B. T. Sherman and R. A. Lempicki, Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources, Nature Protoc., 2009, 4, 44–57, DOI:10.1038/nprot.2008.211.
- A. W. Roberts, A. H. Wei and D. C. Huang, BCL2 and MCL1 inhibitors for hematologic malignancies, Blood, 2021, 138, 1120–1136, DOI:10.1182/blood.2020006785.
- K. Ye, W. X. Meng, H. Sun, B. Wu, M. Chen, Y. P. Pang, J. Gao, H. Wang, J. Wang, S. H. Kaufmann and H. Dai, Characterization of an alternative BAK-binding site for BH3 peptides, Nat. Commun., 2020, 11, 3301, DOI:10.1038/s41467-020-17074-y.
- J. M. Byun, C. Chun, Y. Koh and T. Y. Yoon, Prediction of Venetoclax Sensitivity Based on BCL2 Protein–Protein Interaction for Acute Myeloid Leukemia Patients, Blood, 2021, 138, 3473, DOI:10.1182/blood-2021-150925.
- J. Deng, D. Park, M. Wang, Q. Deng, S. Matulis, L. H. Boise and X. Deng, Small Molecule Bda-366 As a Bcl2-BH4 Antagonist for Multiple Myeloma Therapy, Blood, 2015, 126, 2049, DOI:10.1182/blood.V126.23.2049.2049.
- M. G. Vander Heiden, N. S. Chandel, E. K. Williamson, P. T. Schumacker and C. B. Thompson, Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria, Cell, 1997, 91, 627–637, DOI:10.1016/S0092-8674(00)80450-X.
- S. Shimizu, M. Narita, Y. Tsujimoto and Y. Tsujimoto, Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC, Nature, 1999, 399, 483–487, DOI:10.1038/20959.
- H. Y. C. Emily, M. C. Wei, S. Weiler, R. A. Flavell, T. W. Mak, T. Lindsten and S. J. Korsmeyer, BCL-2, BCL-XL sequester BH3 domain-only molecules preventing BAX-and BAK-mediated mitochondrial apoptosis, Molecular cell, 2001, 8, 705–711, DOI:10.1016/S1097-2765(01)00320-3.
- M. C. Wei, W. X. Zong, E. H. Y. Cheng, T. Lindsten, V. Panoutsakopoulou, A. J. Ross, K. A. Roth, G. R. MacGregor, C. B. Thompson and S. J. Korsmeyer, Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death, Science, 2001, 292, 727–730, DOI:10.1126/science.1059108.
- Y. J. Duan, L. Fu, X. C. Zhang, T. Z. Long, Y. H. He, Z. Q. Liu, A. P. Lu, Y. F. Deng, C. Y. Hsieh, T. J. Hou and D. S. Cao, Improved GNNs for Log D7.4 Prediction by Transferring Knowledge from Low-Fidelity Data, J. Chem. Inf. Model., 2023, 63, 2345–2359, DOI:10.1021/acs.jcim.2c01564.
- G. Xiong, Z. Wu, J. Yi, L. Fu, Z. Yang, C. Hsieh, M. Yin, X. Zeng, C. Wu, X. Chen, T. Hou and D. Cao, ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties, Nucleic Acids Res., 2021, 49, W5–W14, DOI:10.1093/nar/gkab255.
- J. Dong, N. Wang, Z. Yao, L. Zhang, Y. Cheng, D. Ouyang, A. Lu and D. Cao, ADMETlab: a platform for systematic ADMET evaluation based on a comprehensively collected ADMET database, J. Cheminformatics, 2018, 10, 29, DOI:10.1186/s13321-018-0283-x.
- D. Jiang, T. Lei, Z. Wang, C. Shen, D. Cao and T. Hou, ADMET Evaluation in Drug Discovery. 20. Prediction of Breast Cancer Resistance Protein Inhibition through Machine Learning, J. Cheminformatics, 2020, 12, 1–26, DOI:10.1186/s13321-020-00421-y.
Footnote |
| † Yujun Bao and Fujing Guan contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |