
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
First-principles study of novel Cs2LuCoH6 and Cs2LuZnH6 double hydride perovskites for hydrogen storage applications
Rasmiah S. Almufarija,
Malik Shafqat Hayatb,
R. M. Arif Khalilc,
Shoug M. Alghamdid,
Elsammani Ali Shokrallaj,
Jack Arayrof,
Mohamed Abdelsabour Fahmygh,
Mohamed A. Siddige,
Ahmed Samiri and
Arslan Ashfaq *b
*b
aDepartment of Chemistry, College of Science, Princess Nourah Bint Abdulrahman University, P. O. Box 84428, Riyadh 11671, Saudi Arabia
bDepartment of Physics, Emerson University, Multan 60000, Pakistan. E-mail: arslan.ashfaq201@gmail.com
cInstitute of Physics, Bahauddin Zakariya University, Multan, 60800, Pakistan
dDepartment of Physics, College of Science in Yanbu, Taibah University, Yanbu Governorate, Saudi Arabia
eDepartment of Physics, Faculty of Science, Al-Baha University, Alaqiq 65779-7738, Saudi Arabia
fCollege of Engineering and Technology, American University of the Middle East, Egaila 54200, Kuwait
gDepartment of Mathematics, Adham University College, Umm Al-Qura University, Adham, Makkah 28653, Saudi Arabia
hDepartment of Basic Sciences, Faculty of Computers and Informatics, Suez Canal University, New Campus, Ismailia 41522, Egypt
iPhysics Department, Faculty of Science and Arts, King Khalid University, Muhayil Asir, Saudi Arabia
jDepartment of Physics, College of Science, University of Bahri, Sudan
First published on 28th May 2026
Abstract
This work presents a comprehensive evaluation of the structural, electronic, vibrational, magnetic, and mechanical characteristics of the double hydride perovskites Cs2LuCoH6 and Cs2LuZnH6, with the aim of determining their potential for hydrogen storage and related energy applications. The fully relaxed and optimized structures exhibit negative cohesive energies of −3.57 eV per atom for Cs2LuCoH6 and −2.65 eV per atom for Cs2LuZnH6, confirming their thermodynamic stability. Electronic band structure analysis reveals semiconductor behavior with an energy band gap of 0.917 eV for Cs2LuCoH6, while Cs2LuZnH6 displays metallic behavior with a zero band gap. Phonon dispersion calculations confirm the dynamic stability of Cs2LuCoH6, showing no imaginary modes, whereas Cs2LuZnH6 exhibits a few negative phonon frequencies, indicating partial instability. Magnetic analysis demonstrates a ferromagnetic phase with an overall magnetic moment of 1.25 µB for Cs2LuCoH6 and nonmagnetic behavior for Cs2LuZnH6. The computed hydrogen storage capacities (by weight) are 4.87 wt% for Cs2LuCoH6 and 4.46 wt% for Cs2LuZnH6. The tolerance factors (0.92 for Cs2LuCoH6 and 0.84 for Cs2LuZnH6) further confirm the structural symmetry and mechanical robustness of these compounds. These DFT-based results suggest that Cs2LuCoH6 and Cs2LuZnH6 are promising and novel candidates for use in next-generation hydrogen storage devices and energy-related applications.
Introduction
In the current time, the ultimate challenge is to achieve progress in the field of the development of energy systems that are environmentally friendly, using renewable, sustainable energy sources to overcome our dependency on fossil fuels. Due to the burning of fossil fuels, nitrogen oxides and carbon dioxide, along with other harmful particles, are released. Due to these factors, climate change and local extreme air pollution are observed globally.1,2Attaining the Sustainable Development Goals (SDGs) affects food conservation, the enhancement of human well-being and the management of waste in several forms. According to the seventh global goal (SDG7), energy is essential for widespread access to reliable, sustainable, contemporary and economical energy sources.3,4 Three key benchmarks for achieving SDG 7 (Affordable and Clean Energy) at the international level are: (i) increasing the share of renewable and green energy in the global energy mix; (ii) improving energy efficiency by maximizing the effective use of all generated energy; and (iii) ensuring universal access to reliable, affordable, and modern energy services for all. Nowadays, research is focused both theoretically and experimentally on producing clean energy and creating efficient carriers to accomplish sustainable civilization. Regarding the energy transition, technologies for energy generation (acquisition), storage, and transportation have attracted significant global attention.5,6
Hydrogen, in fact, has been observed as an excellent alternative energy carrier compared to fossil fuels such as natural gas, coal and oil. However, the development of hydrogen as an energy carrier presents significant hurdles, especially concerning its storage. Hydrogen can be effectively and safely stored using hydride perovskites.7–9 Materials for hydrogen storage purposes must satisfy some common requirements, such as large volumetric and gravimetric hydrogen storage capacities, ambient conditions for hydrogen release, good kinetics, etc. Perovskites are referred to as materials with these unique chemical and physical characteristics. Due to the vast variety of material structures, a wide range of physicochemical properties has been observed for the perovskite family.10–12 Perovskite-based structures deliver a large surface area for hydrogen adsorption and can be adapted for maximum hydrogen adsorption. In such materials, a large variety of physical characteristics has been observed. As concerns hydrogen storage, perovskite hydrides are ideal. The simulation of solid-state hydrogen storage compounds, particularly ABH3 hydride perovskites, has exposed various compounds for hydrogen storage.13 It is crucial to highlight that simulations of various compounds have been carried out, and many of them have gained popularity as hydrogen storage materials, such as (Ca/Sr)CuH3, NaMgH3, (Rb/Cs)InH3, and (Rb/Cs)BH3, along with the unique perovskite hydrides (Rb/Cs)2CaH4 (X = Ba, Sr, Cs) and (Sr/Ba)NiH3. Furthermore, first-principles simulations offer an economical and valuable technique to study and locate ideal H2 storage compounds.14 According to current knowledge, double-hydride perovskites have hardly been investigated experimentally or theoretically. The structure of double-hydride perovskites is a three-dimensional system with positive cations surrounded by octahedral structures. In the A2BXH6 structure, hydrogen (H) acts as an anion (hydride). The B-site is typically occupied by a transition metal, while the X-site is an alkali metal. The A-site is filled by a positively charged cation, which helps stabilize the overall crystal structure.15 Our research group has simulated a large number of hydride perovskite materials for hydrogen storage applications. The XCuH3 (X = Ni, Co, Zn) series represents a class of hydride perovskites with promising hydrogen storage capability. The gravimetric hydrogen storage capacities are calculated to be 3.0 wt% for NiCuH3, 2.8 wt% for CoCuH3, and 2.7 wt% for ZnCuH3, indicating slight variations depending on the transition metal substitution at the X-site. Moreover, metallic character is observed for all these materials.16 A series of LiXH3 (X = Cr, Co, Fe, Zn) materials was simulated by our research group. These are unique materials for hydrogen storage and related applications.17 In addition, Cs2CaTlH6, Cs2SrTlH6, and Cs2BaTlH6 are another class of hydride materials with novel and significant physicochemical properties that make these materials favorable for hydrogen (H) energy storage systems.18 Importantly, recent advances have demonstrated a growing interest in hydride-based and double perovskite systems;19,20 however, most reported studies focus on conventional transition-metal or alkaline-earth systems, while rare-earth (lanthanide)-based double hydride perovskites remain largely unexplored. In particular, the role of Lu-based double hydride perovskites combined with transition metals has not been systematically investigated for hydrogen storage applications, despite their potential to exhibit tunable electronic structures, magnetic behavior, and enhanced stability. Motivated by this research gap, we propose two novel double hydride perovskites, Cs2LuCoH6 and Cs2LuZnH6, for a comprehensive first-principles investigation.
In this article, we examine the properties of Cs2LuCoH6 and Cs2LuZnH6 double hydride compounds for H energy storage and related applications. DFT-based simulations are calculated using the CASTEP simulation code and by utilizing the hybrid complex HSE06 functional. The first section of the present article consists of an introduction to hydride perovskites, the second section illustrates the DFT-based methodology, and finally, the third section provides the results and discussion about the Cs2LuCoH6 and Cs2LuZnH6 double hydride perovskites. From these DFT-based calculations, it has been illustrated that these Cs2LuCoH6 and Cs2LuZnH6 double hydride compounds have the potential to revolutionize the field and motivate research into synthesizing these compounds for hydrogen energy storage systems. The present study highlights that these novel double hydride perovskites exhibit promising multifunctional properties, demonstrating superior stability and potential for hydrogen storage applications.
Research methodology
Density functional theory (DFT) is a powerful first-principles approach widely used to investigate the structural, electronic, vibrational, and mechanical properties of materials. In this study, all calculations involving the double hydride perovskites Cs2LuCoH6 and Cs2LuZnH6 were performed using the CASTEP code within the framework of plane-wave pseudopotential DFT. The electronic exchange–correlation effects were treated using the generalized gradient approximation (GGA) in the form of Perdew–Burke–Ernzerhof (PBE) for structural optimization, while the HSE06 hybrid functional was employed to obtain more accurate electronic band structures. The Kohn–Sham equations were solved self-consistently using a plane-wave basis set. Structural optimizations were carried out using the Broyden–Fletcher–Goldfarb–Shanno (BFGS) minimization algorithm until convergence was achieved.21 The convergence criteria were set to a total energy tolerance of 5 × 10−6 eV per atom, maximum force of 0.01 eV Å−1, maximum stress of 0.02 GPa, and maximum atomic displacement of 5 × 10−4 Å, ensuring highly accurate geometry optimization. A plane-wave cutoff energy of 520 eV was used for all calculations.22,23 The Brillouin zone was sampled using a Monkhorst–Pack k-point grid of appropriate density (optimized for convergence tests) to ensure numerical accuracy in total energy and electronic structure calculations. The phonon dispersion and vibrational stability were evaluated using the finite displacement (supercell) method, as implemented in CASTEP. A 1 × 2 × 1 supercell was constructed to calculate interatomic force constants and phonon spectra, which were used to assess the dynamical stability of the investigated materials.24 The dielectric and optical properties were analyzed using the Kramers–Kronig relations, which relate to the real and imaginary parts of the dielectric function and describe the interaction between electromagnetic radiation and the material.25–27 All structural relaxations were performed using the conjugate gradient method, and self-consistency was achieved when energy changes between successive iterations were less than the defined convergence threshold.28 These computational parameters ensure reliable and reproducible predictions of the structural, electronic, vibrational, and mechanical properties of Cs2LuCoH6 and Cs2LuZnH6 for hydrogen storage applications.Results and discussion
Fig. 1 presents the relaxed, stable and energy-minimized crystal structure of Cs2LuCoH6 and Cs2LuZnH6 double hydride perovskites with a cubic crystal structure. In the equilibrium structure obtained after relaxation, X = Co, Zn atoms are positioned in face-centered (1/2, ½, ½) positions, Cs atoms are positioned in body-centered positions, Lu atoms are located at corner sites, and H is fixed at face-centered positions with coordinates (1/4, ¼, ¼) in the optimized crystal structure of the materials under consideration. The geometries of these compounds were optimized and structurally relaxed by utilizing the DFT-based hybrid complex HSE06 hybrid functional, as implemented in the CASTEP simulation package. The point group P1 (32: oh, m![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, 4/m–32/m) and space group C1 (225: Fmm, −F 423) are found for the Cs2LuCoH6 and Cs2LuZnH6 double hydride perovskites. The supercell structures of Cs2LuCoH6 and Cs2LuZnH6 consist of 10 atoms associated with 6 hydride (H−) ions per formula unit, forming the extended crystal lattice of these materials. The relaxed structure contains a total of four atomic species, and the optimized supercell uses a maximum of four k-points in the Brillouin zone sampling. The geometrical optimization and relaxation of the single-point crystal structure are simulated by the variational principle. The calculated total energies of the perovskites show that Cs2LuCoH6 has a lower energy value (−7167.43 eV) compared to Cs2LuZnH6 (−7067.53 eV), indicating that Cs2LuCoH6 is energetically more stable than Cs2LuZnH6. The negative values of the total energy predict the chemical, thermodynamic, and structural stability and reliability of the Cs2LuCoH6 and Cs2LuZnH6 materials.
m, 4/m–32/m) and space group C1 (225: Fmm, −F 423) are found for the Cs2LuCoH6 and Cs2LuZnH6 double hydride perovskites. The supercell structures of Cs2LuCoH6 and Cs2LuZnH6 consist of 10 atoms associated with 6 hydride (H−) ions per formula unit, forming the extended crystal lattice of these materials. The relaxed structure contains a total of four atomic species, and the optimized supercell uses a maximum of four k-points in the Brillouin zone sampling. The geometrical optimization and relaxation of the single-point crystal structure are simulated by the variational principle. The calculated total energies of the perovskites show that Cs2LuCoH6 has a lower energy value (−7167.43 eV) compared to Cs2LuZnH6 (−7067.53 eV), indicating that Cs2LuCoH6 is energetically more stable than Cs2LuZnH6. The negative values of the total energy predict the chemical, thermodynamic, and structural stability and reliability of the Cs2LuCoH6 and Cs2LuZnH6 materials.
 | ||
| Fig. 1 The relaxed and energy-minimized crystal structure of Cs2Lu(Co, Zn)H6 double hydride perovskite. | ||
Another novel structural parameter is the cohesive energy that illustrates the properties of the Cs2LuCoH6 and Cs2LuZnH6 double hydride perovskite materials. The binding energy is actually the energy needed to separate the constituent atoms from the corresponding material and is given by the following expression:29
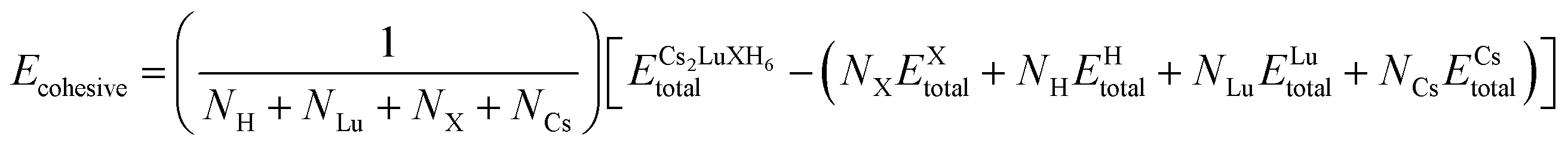 | (1) |
The electronic properties, including the band structure, DOS, and PDOS, play a key role in determining the material behavior and its potential applications across various fields.30 The electronic properties actually determine the material's response to electrical influence, affecting its behavior in various ways, such as its conductivity, optical properties, device performance, energy applications and use in materials science. The band structure illustrates the range of energy levels that an electron can occupy in the solid material. By calculating the energy band gap, we can predict the type of material: conductor, semiconductor or insulator.31,32 The calculated band structure, DOS, and PDOS of the double hydride systems Cs2LuCoH6 and Cs2LuZnH6 are calculated by applying the complex hybrid HSE06 functional, and the results are illustrated in Fig. 2(a–d) and in Fig. 3(a and b). From Fig. 2(a) and (c) it is observed that in Cs2LuCoH6, a very narrow energy band gap of 0.917 eV exists, indicating the semiconductor behavior of the material. However, in Cs2LuZnH6, the conduction and valence energy bands in the band structure overlap with zero band gap, illustrating the metallic behavior of the material. The shape of the band structure is different in each material due to the different cations, i.e., Co or Zn, in Cs2LuCoH6 and Cs2LuZnH6.
 | ||
| Fig. 2 The computed plots for the energy bands (a and c) and DOS (b and d) for hydrides Cs2LuCoH6 and Cs2LuZnH6, using the hybrid HSE06 functional. | ||
 | ||
| Fig. 3 The calculated PDOS plots (a and b) for the double hydrides Cs2LuCoH6 and Cs2LuZnH6, obtained using the hybrid HSE06 functional. | ||
Furthermore, the DOS illustrates the number of available electronic states per unit energy interval in a given compound. The DOS plots give valuable insights into material behavior and properties.33 Fig. 2(b) and (d) represent the DOS plots for Cs2LuCoH6 and Cs2LuZnH6 double hydride materials, predicting the behavior of these compounds. The maximum peaks of the DOS have been observed in the conduction energy bands as 34.52 states per eV at 1.97 eV and 33.52 states per eV at 3.86 eV, along with peaks in the valence energy bands of 57.58 states per eV at −0.57 eV and 43.31 states per eV at −2.76 eV in the Cs2LuCoH6 material, as shown in Fig. 2(b). Similarly, the maximum values of DOS have been noted in the valence energy bands as 30.52 states per eV at 0.98 eV and 36.97 states per eV at 2.65 eV, along with peaks in the valence energy bands of 17.98 states per eV at −3.28 eV and 26.87 states per eV at −4.35 eV in the Cs2LuZnH6 material, as shown in Fig. 2(d).
The concept of PDOS in solid-state physics explains the contribution of each specific atomic state to the overall electronic DOS in the compound under consideration. A PDOS examination highlights how individual atomic orbitals contribute to the electronic properties.34,35 The PDOS for the Cs2LuCoH6 and Cs2LuZnH6 double hydride perovskite materials are calculated, and the results for the PDOS are illustrated in Fig. 3(a and b). The electronic configurations of Cs, Lu, Co, Zn, and H in Cs2LuCoH6 and Cs2LuZnH6 perovskites are listed as 5s2 5p6 6s1, 5s2 5p6 5d1 6s2, 3s2 3p6 3d7 4s2, 3p6 4s2 3d10, and 1s1, respectively. This means that 6s1 of Cs, 5d1 of Lu, 3d7 of Co, and 1s1 of H make major contributions to the PDOS plots for Cs2LuCoH6. The peak values are listed for s-states as 28.68 states per eV at −2.34 eV, for p-states as 8.91 states per eV at −2.42 eV, for d-states as 48.73 states per eV at −0.19 eV, and for sum-states as 58.78 states per eV at −0.17 eV for the Cs2LuCoH6 material, as represented in Fig. 3(a). 6s1 of Cs, 5d1 of Lu, and 1s1 of H make major contributions to the PDOS plots for Cs2LuZnH6. The maximum values for Cs2LuZnH6 material are 13.83 states per eV at 0.74 eV for s-states,10.72 states per eV at 1.08 eV for p-states, and 21.52 states per eV at 0.88 eV for sum-states, as represented in Fig. 3(b).
Phonons act as the primary carriers of vibrational energy in the materials under investigation. The allowed frequency modes in the primitive lattice cell of the crystal of the material for the propagation of vibrational waves can be separated into two portions. The lower and upper branches are identified as acoustic and optical sections, respectively. At low frequency, the wavelength of acoustic phonons becomes large, and they behave as a sound wave in the primitive cell of the material crystal.36 The longitudinal and transverse phonons in the structural arrangement of the studied materials are abbreviated as LA and TA, respectively. Few non-zero minimum frequencies for optical phonons, even at very large wavelengths, still exist. In this regard, there is an interaction between the incident radiation and optical phonons of the material, termed as the infra-active region in the phonon dispersion curves.37 Raman scattering between incident radiation and optical phonons is termed as the Raman active region. The longitudinal and transverse phonons here are abbreviated as LO and TO, respectively. The dynamics, vibrations, stability and compatibility of a considered material are verified based on the phonon dispersion curves. Phonon dispersion with real (positive) modes of phonons confirms the vibrational and dynamic stability of the considered material.38 Phonon dispersion graphs and DOS for Cs2LuCoH6 and Cs2LuZnH6 double hydride perovskites are plotted by constructing a 1 × 2 × 1 supercell. Fig. 4(a–d) predicts the results for the phonon dispersion relations and the DOS for these double hydride perovskites. Fig. 4(a) and (c) illustrate that real (positive) modes of phonons are observed for Cs2LuCoH6 with no negative (imaginary) modes of phonons. However, in the case of the Cs2LuZnH6 perovskite, as shown in Fig. 4(b), a few imaginary phonons are observed, but a majority of real (positive) phonons are observed in the phonon dispersion relationship. The results obtained from the phonon dispersion curves illustrate the dynamic and vibrational compatibility, reliability, and stability of these Cs2LuCoH6 and Cs2LuZnH6 hydride perovskites for use in hydrogen storage devices. Fig. 4(b) and (d) illustrates the DOS for the phonons of Cs2LuCoH6 and Cs2LuZnH6 hydride perovskites. It is observed that no phonon states are observed in the negative (imaginary) region around the Fermi level of the DOS for the Cs2LuCoH6 perovskite material. The highest DOS peak for Cs2LuCoH6 of 0.36 states per eV at 19.27 eV is represented in Fig. 4(b). However, there are some states per eV in the imaginary (negative) region of the DOS for Cs2LuZnH6, but the maximum peaks are observed in the positive (real) region of the DOS, with maximum peaks listed at 0.009 states per eV at 61.33 eV, 0.008 states per eV at 81.33 eV, and 0.007 states per eV at 113.33 eV, as shown in Fig. 4(d). Finally, the results of DOS for the phonons again confirmed that Cs2LuCoH6 and Cs2LuZnH6 have vibrational and dynamical stability, rendering these materials suitable for hydrogen storage technologies and associated applications.
 | ||
| Fig. 4 Simulated plots showing the dispersion curves (a and c) and DOS (b and d) for Cs2LuCoH6 and Cs2LuZnH6, respectively, using the HSE06 hybrid functional. | ||
The magnetic behavior of a material describes its response to an external magnetic field and is fundamentally governed by the spin-dependent electronic structure. In functional materials for hydrogen energy storage and related applications, magnetic properties are particularly important as they provide insight into electronic exchange interactions, spin polarization, and orbital contributions, which can significantly influence the overall electronic stability and transport behavior.39
In the present study, the magnetic properties of Cs2LuCoH6 and Cs2LuZnH6 were investigated through DOS and PDOS calculations using the HSE06 hybrid functional, as shown in Fig. 5a, b and 6a, b, respectively.
 | ||
| Fig. 5 The computed spin-resolved DOS spectra for (a) Cs2LuCoH6 and (b) Cs2LuZnH6, obtained by utilizing the HSE06 functional. | ||
 | ||
| Fig. 6 The calculated graphs for spin-polarized PDOS for (a) Cs2LuCoH6 and (b) Cs2LuZnH6, utilizing the HSE06 functional. | ||
For Cs2LuCoH6, the spin-resolved DOS exhibits clear asymmetry between spin-up (↑) and spin-down (↓) channels near the Fermi level (set at 0 eV). This spin polarization arises primarily from the partially filled Co-3d orbitals, which undergo exchange splitting and contribute unequally to the two spin channels. As a result, a net magnetic moment of 1.25 µB is obtained, confirming a ferromagnetic ground state. The PDOS further reveals strong hybridization between Co-3d and H-1s states, which contributes to magnetic ordering through indirect exchange interactions.40
In contrast, Cs2LuZnH6 exhibits a completely symmetric spin-up and spin-down DOS, indicating no spin polarization and a non-magnetic ground state. This behavior originates from the fully filled Zn-3d10 electronic configuration, where the Zn-3d states lie deep below the Fermi level and do not contribute to exchange splitting. Consequently, both spin channels are identical, leading to a zero net magnetic moment (0 µB), as confirmed by DOS and PDOS analysis.
These results determine that the magnetic behavior in Cs2LuCoH6 is driven by partially filled transition-metal Co-3d states, whereas the absence of unpaired d-electrons in Zn leads to non-magnetic behavior in Cs2LuZnH6.
In addition, the spin-polarized PDOS profiles as a function of frequency for Cs2LuCoH6 and Cs2LuZnH6 double hydride perovskites have also been explored to verify the magnetic properties as predicted by DOS for these materials. The results showing the PDOS for Cs2LuCoH6 and Cs2LuZnH6 are presented in Fig. 6(a and b). From Fig. 6(a), it is observed that the spin-down ↓ and spin-up ↑ states of s-states in the valence band and the d-state located at the Fermi energy make contributions to the magnetic behavior of the Cs2LuCoH6 material. Also, there is an exact mirror reflection of the spin-down ↓ and spin-up ↑ states of p-states and f-states in the PDOS plots for the Cs2LuCoH6 material. The PDOS plots confirmed the magnetic behavior of the Cs2LuCoH6 material with a magnetic dipole moment of 1.25 Bohr. Furthermore, the PDOS plots of the Cs2LuZnH6 material have also been calculated, and the results are presented in Fig. 6(b). It is observed that there is an exact mirror reflection of the spin-down ↓ and spin-up ↑ states for the s-states, p-states, d-states and f-states in the PDOS plots for the Cs2LuZnH6 material. From the exact replicas of the spin-down ↓ and spin-up ↑ states in Fig. 4(b), it is confirmed that the Cs2LuZnH6 material has nonmagnetic behavior with a zero magnetic dipole moment.
The mechanical parameters of a compound illustrate its ability to withstand applied mechanical forces and deformations under various loading conditions. These properties provide insight into the compound's integrity, defect behavior, microstructural integrity and internal bonding strength. Physically, these properties arise from crystal structure, microstructural, and atomic bonding features, such as dislocations, phase compositions and grain boundaries.41 The most commonly evaluated properties include ductility, fracture toughness, strength, hardness, and impact resistance. In this study, the mechanical properties of Cs2LuCoH6 and Cs2LuZnH6 double hydride perovskite compounds were calculated via the HSE06 hybrid functional. The intrinsic elastic stiffness parameters C12, C11 and C44 were thus calculated for these hydride perovskites under an applied mechanical load. Born defined the elastic stability criteria as follows:42
| C44 > 0, C11 > C12 and C11 + 2C12 > 0 | (2) |
Stiffness mechanical parameters such as bulk modulus (B), Voigt shear modulus (Gv), Reuss isotropic shear modulus (Gr), shear modulus (G), and Young's modulus (Y) are calculated via Voigt–Reuss–Hill and are represented by the given relationships:43
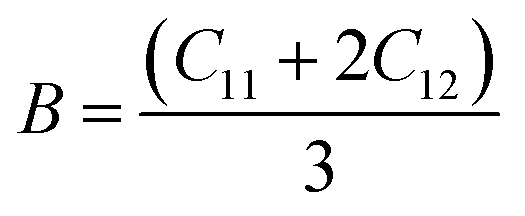 | (3) |
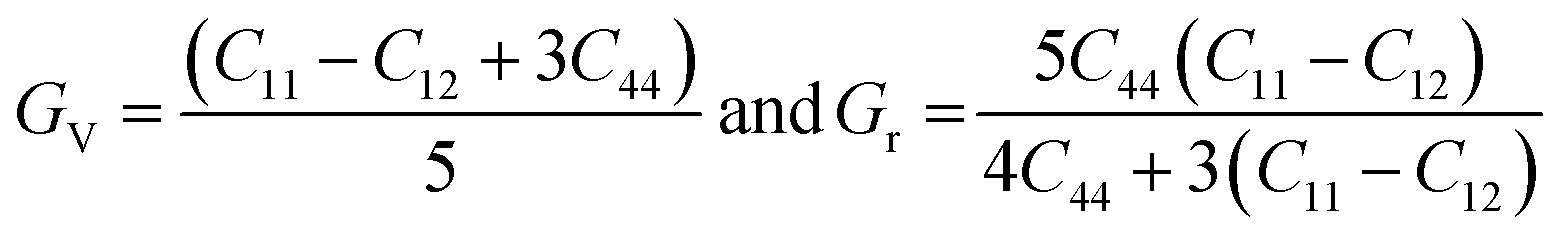 | (4) |
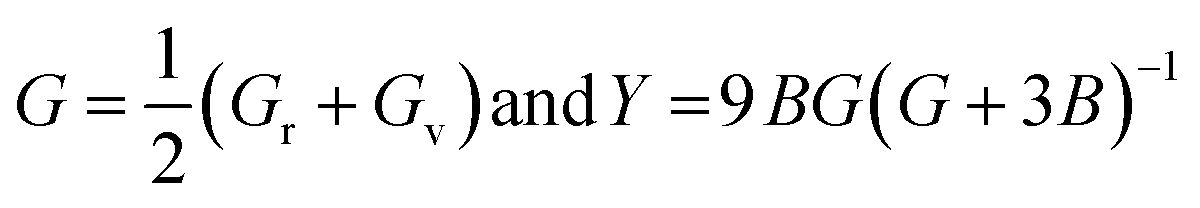 | (5) |
Poisson's ratio (ν) and the anisotropy factor (A) are dimensionless parameters used to describe the elastic and mechanical properties of a material, such as plasticity, anisotropy, and stiffness, and are defined by the following relationships:44
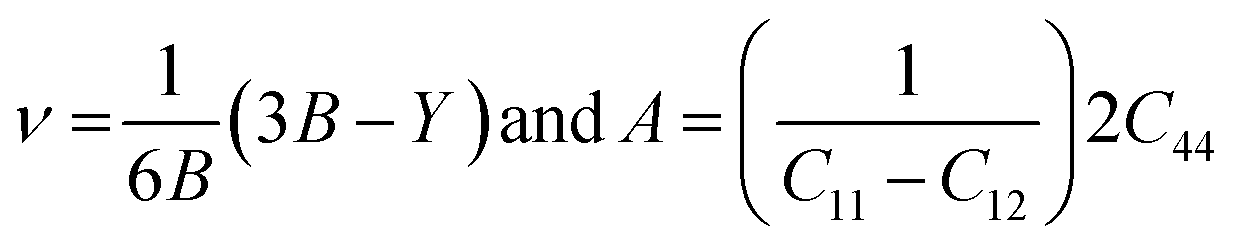 | (6) |
For a given material, the melting temperature in terms of elastic mechanical constants is expressed by the following relationship:
| Tmelt(K) = (553 + C12 5.922) ± 300 K | (7) |
Table 1 shows the calculated mechanical parameters for the hydride perovskites Cs2LuCoH6 and Cs2LuZnH6. The listed values include the elastic stiffness constants C11, C12, and C44 (in GPa), Poisson's ratio (ν), Cauchy pressure (Cp) in GPa, and Pugh's ratio (B/G). Additionally, the table summarizes the bulk modulus (B), compressibility (β) in GPa, Young's modulus (Y), anisotropy factor (A), shear modulus (G), and the estimated melting temperature (Tmelt) in K, providing a comprehensive overview of the mechanical stability and elastic behavior of these hydride perovskite materials. The bulk modulus of a material quantifies its resistance to uniform compressive stress, reflecting the strength of the interatomic bonds. The trend of the calculated bulk modulus values is given as Cs2LuCoH6 (3.03 GPa) > Cs2LuZnH6 (0.52 GPa). The deviation in the calculated values of Young's modulus is listed as Cs2LuCoH6 (35.26 GPa) > Cs2LuZnH6 (6.67 GPa). The determined values of Young's modulus illustrate that Cs2LuCoH6 and Cs2LuZnH6 are softer materials, as their calculated values are less than 40 GPa, which predicts their value for use in hydrogen storage systems and associated applications. Furthermore, the Poisson's ratio (ν) is an important parameter in terms of mechanical properties to distinguish between ductile and brittle studied materials. A material exhibits predominantly brittle nature when ν is less than 0.26, whereas for other values of ν, ductile behavior is prominent. The calculated values for the Poisson ratio (ν) are listed as Cs2LuCoH6 (2.44) > Cs2LuZnH6 (1.64). The determined values for ν illustrate that Cs2LuCoH6 and Cs2LuZnH6 possess ductile behavior, as ν > 0.26.45 The additional mechanical property of Pugh's ratio (B/G) is also used to identify the ductile or brittle behavior of a studied material. The critical threshold for the B/G ratio is 1.75. A ductile nature is dominant for a material when B/G is greater than 1.75, whereas for other values, brittle behavior is prominent. The computed values for B/G follow the trend Cs2LuZnH6 (1.82) > Cs2LuCoH6 (4.00). These values of B/G confirm the ductile nature of the Cs2LuCoH6 and Cs2LuZnH6 materials. The inherent ductility of Cs2LuCoH6 and Cs2LuZnH6 enhances their suitability for hydrogen storage by improving resistance to cracking, accommodating defects, and enabling practical use in storage tanks and pipelines. The anisotropy factor is a crucial parameter for judging the isotropic or anisotropic behavior of a studied material. Isotropic behavior is dominant for A ≥ 1, whereas for other values, anisotropic behavior is prominent in a material.46–48 The calculated trend for the anisotropy factor is listed as Cs2LuCoH6 (−7.73) > Cs2LuZnH6 (−9.76). The determined values predict an anisotropic nature for the Cs2LuCoH6 and Cs2LuZnH6 materials. In conclusion, these mechanical properties predict that these materials are novel and transformative and should motivate experimental researchers to synthesize these compounds for hydrogen storage and related applications. Fig. 7(a–d) shows the 3D visualizations of the mechanical properties of Young's modulus, linear compressibility, shear modulus, and Poisson's ratio.
| Parameter | Cs2LuCoH6 | Cs2LuZnH6 |
|---|---|---|
| C44 | 10.167 | 9.68 |
| C11 | −6.26 | 2.68 |
| C12 | −1.42 | 0.56 |
| ν | 2.43 | 1.64 |
| B/G | 1.82 | 4.00 |
| B | 3.04 | 0.52 |
| β | 0.0000 | 0.0000 |
| Y | 35.26 | 6.67 |
| A | −7.73 | −9.76 |
| GR | 9.14 | 5.42 |
| GV | 5.13 | −5.16 |
| G | 1.67 | 0.13 |
| Tmelt | 858.92 | 837.75 |
 | ||
| Fig. 7 3D visualizations of mechanical properties: (a) Young's modulus, (b) linear compressibility, (c) shear modulus, and (d) Poisson's ratio. | ||
The gravimetric H storage capacity, also known as the hydrogen weight percent or gravimetric hydrogen density, is defined as the fraction of hydrogen mass that can be released or stored by a material under consideration. The gravimetric H storage capacity represents the fraction of hydrogen mass as a percentage of the total material mass. As hydrogen is a low-cost and lightweight element, materials with large gravimetric ratios are considered the best materials, as these materials can store large amounts of hydrogen without much extra mass.49 Materials with a large gravimetric ratio have more energy per unit mass, making these materials crucial for many mobile applications like drones, fuel cells for vehicles, and portable devices. The gravimetric ratio for hydrogen storage materials is calculated by the given relationship:50
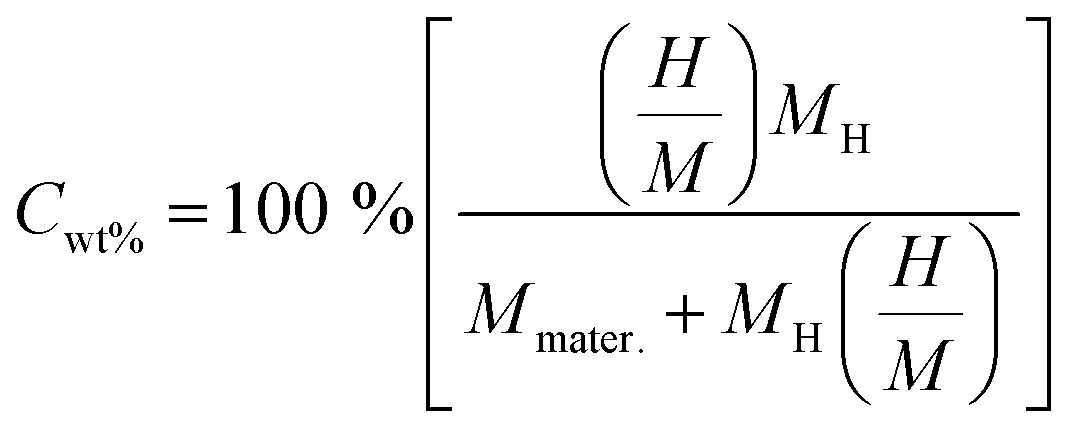 | (8) |
The sufficiently large gravimetric ratios illustrate that these double hydride perovskites possess capable hydrogen storage capabilities. Therefore, Cs2LuCoH6 and Cs2LuZnH6 can be considered as potential candidates for the design and computational modeling of compounds for hydrogen-based energy storage and fuel cell applications.
Tolerance factor
The tolerance factor (τ), commonly referred to as Goldschmidt's tolerance factor, is an important dimensionless parameter used to assess the structural stability and physical characteristics of perovskite materials.52,53 It helps predict whether compounds with compositions such as A2BB′X6 or ABX3 are likely to crystallize in cubic, distorted, symmetric, stable, or non-perovskite phases before performing computational optimization or experimental synthesis. The expression for τ is given as follows:54,55
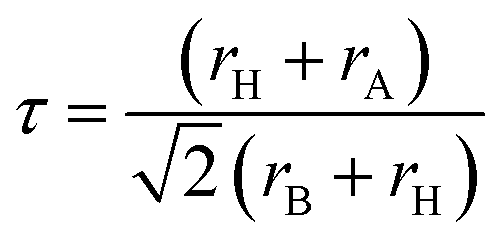 | (9) |
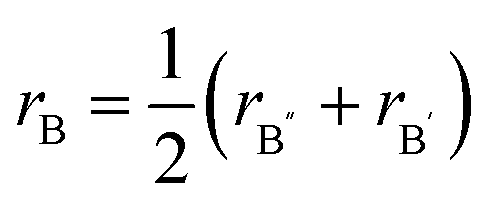 for a double hydride A2B′B″H6 perovskite material.
for a double hydride A2B′B″H6 perovskite material.
For perovskite-type hydride materials, the tolerance factor (τ) typically exists within the range of 0.8 ≤ τ ≤ 1.0. Moreover, a τ value between 0.9 and 1.0 matches an ideal and highly symmetric cubic hydride perovskite crystal structure.56–59 The determined τ values for the considered hydride double compounds illustrate the trend Cs2LuCoH6 (0.92) > Cs2LuZnH6 (0.84). These results predict that Cs2LuCoH6 possesses nearly perfect perovskite hydride geometry, while Cs2LuZnH6 shows slight crystal structural distortion. The confirmed suitable tolerance factors verified the structural feasibility and stability of these compounds within the framework of perovskite structures. These results illustrate that Cs2LuCoH6 and Cs2LuZnH6 are promising and game-changing materials for simulations and for the synthesis of hydrogen energy fuel cells and many other clean hydrogen storage systems.
Conclusions
Large calculated gravimetric ratios of 4.87 wt% (Cs2LuCoH6) and 4.46 wt% (Cs2LuCoH6) are observed for these materials. The calculated mechanical parameters include bulk modulus, where Cs2LuCoH6 (3.03 GPa) > Cs2LuZnH6 (0.52 GPa); Young's modulus, where Cs2LuCoH6 (35.26 GPa) > Cs2LuZnH6 (6.67 GPa); Poisson's coefficient (ν), where Cs2LuCoH6 (2.44) > Cs2LuZnH6 (1.64); and anisotropy factor, where Cs2LuCoH6 (−7.73) > Cs2LuZnH6 (−9.76). The metallic character of Cs2LuZnH6 and the semiconductor character of Cs2LuCoH6 are observed from the electronic properties. The non-magnetic behavior of Cs2LuZnH6 and magnetic behavior of Cs2LuCoH6 with a magnetic moment of 1.25 Bohr are predicted from the magnetic properties of these materials. The positive modes of phonons for Cs2LuCoH6, with no negative modes, but with a few negative modes for Cs2LuZnH6, are observed in the phonon dispersion graphs. Finally, the results of physical property analysis, such as electronic, vibrational, magnetic and mechanical properties, predict that Cs2LuCoH6 and Cs2LuZnH6 are potential and game-changing materials for hydrogen storage devices and related applications.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Data availability
All data that support the findings of this study are included with the article.Acknowledgements
The authors would like to thank Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2026R316), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. The authors extend their appreciation to the Deanship of Research and Graduate Studies at King Khalid University for funding this work through a Large Research Project under grant number RGP2/359/46.References
- P. Jain, C. Lal and A. Jain, Hydrogen storage in Mg: A most promising material, Int. J. Hydrogen Energy, 2010, 35(10), 5133–5144 CrossRef.
- S. Singh, S. Jain, A. K. Tiwari, M. R. Nouni and J. K. Pandey, Hydrogen: A sustainable fuel for future of transport sector, Renew. Sustain. Energy Rev., 2015, 51, 623–633 CrossRef CAS.
- B. Ahmed, M. B. Tahir, S. Nazir, M. Alzaid, A. Ali, M. Sagir and H. Alrobei, AN Ab-initio simulation of boron-based hydride perovskites XBH3(X = Cs and Rb) for advance hydrogen storage system, Comput. Theor. Chem., 2023, 1225, 114173 CrossRef CAS.
- A. I. Bashir, H. Arif, S. Azam and M. Irfan, First-principles quantum computations to investigate prospects of Mg2FeH6 for optoelectronics and hydrogen-storage applications, Int. J. Hydrogen Energy, 2023, 48(62), 23930–23942 CrossRef CAS.
- A. Hassan, H. S. Ramadan, M. A. Saleh and D. Hissel, Hydrogen storage technologies for stationary and mobile applications: Review, analysis and perspectives, Renew. Sustain. Energy Rev., 2021, 149, 111311 CrossRef.
- S. Al, M. Yortanlı and E. Mete, Lithium metal hydrides (Li2CaH4 and Li2SrH4) for hydrogen storage; mechanical, electronic and optical properties, Int. J. Hydrogen Energy, 2020, 24, 1–13 Search PubMed.
- A. Chibani, C. Bougriou and S. Merouani, Simulation of hydrogen absorption/desorption on metal hydride LaNi5-H2: Mass and heat transfer, Appl. Therm. Eng., 2018, 142, 110–117 CrossRef CAS.
- A. H. Reshak, NaMgH3 a perovskite-type hydride as advanced hydrogen storage systems: electronic structure features, Int. J. Hydrogen Energy, 2015, 40(46), 16383–16390 Search PubMed.
- H. H. Raza, G. Murtaza and R. M. A. Khalil, Optoelectronic and thermal properties of LiXH3 (X = Ba, Sr and Cs) for hydrogen storage materials: a first principle study, Solid State Commun., 2019, 299, 113659 CrossRef CAS.
- K. Komiya, N. Morisaku, R. Rong, Y. Takahashi, Y. Shinzato, H. Yukawa and M. Morinaga, Synthesis and decomposition of perovskite-type hydrides, MMgH3 (M = Na, K, Rb), J. Alloys Compd., 2008, 453(1–2), 157–160 CrossRef CAS.
- Z. Tan, Y. Chu, J. Chen, J. Li, G. Ji, G. Niu, L. Gao, Z. Xiao and J. Tang, Lead-free perovskite variant solid solutions Cs2Sn1-xTexCl6: bright luminescence and high anti-water stability, Adv. Mater., 2020, 32(32), 2002443–2002455 Search PubMed.
- M. Nabi and D. C. Gupta, Small-band gap halide perovskite for optoelectronic properties, Int. J. Energy Res., 2020, 233, 1–13 Search PubMed.
- Z. Xiao, Z. Song and Y. Yan, From lead halide perovskites to lead-free metal halide perovskites and perovskite derivatives, Adv. Mater., 2019, 31(47), 1803792–1804804 CrossRef CAS PubMed.
- D. Y. Hu, X. H. Zhao, T. Y. Tang, M. Lu, L. Li, L. K. Gao and Y. L. Tang, First-principles calculations to investigate the structural, electronic and optical properties of lead-free double perovskites Rb2SeI6 and K2SeI6, Sol. Energy, 2022, 231, 236–242 Search PubMed.
- W. Shi, T. Cai, Z. Wang and O. Chen, The effects of monovalent metal cations on the crystal and electronic structures of Cs2MBiCl6 (M = Ag, Cu, Na, K, Rb, and Cs) perovskites, J. Chem. Phys., 2020, 153(14), 141101–141114 CrossRef CAS PubMed.
- N. A. Noor, W. Iqbal, T. Zelai, A. Mahmood, H. M. Shaikh, S. M. Ramay and W. A. Masry, Analysis of direct band gap A2ScInI6 (A = Rb, Cs) double perovskite halides using DFT approach for renewable energy devices, J. Mater. Res. Technol., 2021, 13, 2491–2500 Search PubMed.
- E. T. McClure, M. R. Ball, W. Windl and P. M. Woodward, Cs2AgBiX6 (X= Br, Cl): new visible light absorbing, lead-free halide perovskite semiconductors, Chem. Mater., 2016, 28, 1348–1354 Search PubMed.
- E. T. McClure, M. R. Ball and W. P. M. Windl, Woodward Cs2AgBiX6 (X=Br, Cl): new visible light absorbing, lead-free halide perovskite semiconductors, Chem. Mater., 2016, 28, 1348–1354 CrossRef CAS.
- L. Wang, P. Yao, F. Wang, S. Li, Y. Chen, T. Xia, E. Guo, K. Wang, B. Zou and H. Guo, Pressure-Induced Structural Evolution and Bandgap Optimization of Lead-Free Halide Double Perovskite (NH4)2SeBr 6, Adv. Sci., 2020, 7(6), 1–13 CAS.
- R. Fletcher and M. C. Reeves, Function minimization by conjugate gradients, J. Comput., 1964, 7(2), 149–154 CrossRef.
- R. Fletcher and M. C. Reeves, Function minimization by conjugate gradients, J. Comput., 1964, 7(2), 149–154 CrossRef.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, Hybrid Functionals based on a screened Coulomb potential, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- P. P. Bruesch, Theory and Experiment I, Springer Series in Solid-State Sciences, 1982, ISBN 978-3-642-81781-6 Search PubMed.
- D. Alfè, PHON: a program to calculate phonons using the small displacement method, Comput. Phys. Commun., 2009, 180(12), 2622–2633 CrossRef.
- B. G. Pfrommer, M. Cote, S. G. Louie and M. L. Cohen, Relaxation of crystals with the Quasi-Newton Method, J. Comput. Phys., 1997, 131, 233–240 CrossRef CAS.
- R. de L. Kronig, On the Theory of Dispersion of X-Rays, J. Opt. Soc. Am., 1926, 12, 547–557 CrossRef CAS.
- H. B. Ozisik, K. Colakoglu, G. Surucu and H. Ozisik, Structural and lattice dynamical properties of Zintl Naln and NaTl compounds, Comput. Mater. Sci., 2011, 50, 1070–1076 CrossRef CAS.
- Y. Aierken, D. Çakır, C. Sevik and F. M. Peeters, Thermal properties of black and blue phosphorenes from a first-principles quasi-harmonic approach, Phys. Rev. B:Condens. Matter Mater. Phys.c, 2015, 92(8), 081408–0181412 CrossRef.
- M. S. H. Suleiman and D. P. Joubert, Elastic moduli of advanced orthorhombic binary and ternary metal (V, Ta, Nb) nitrides with the U2S3 structure, Phys. Status Solidi B, 2015, 252, 1–13 CrossRef.
- M. S. H. Suleiman and D. P. Joubert, Elastic moduli of advanced orthorhombic binary and ternary metal (V, Ta, Nb) nitrides with the U2S3 structure, Phys. Status Solidi B, 2015, 252, 1–13 CrossRef.
- B. R. Sahu, Electronic structure and bonding of ultralight LiMg, Mater. Sci. Eng., B, 1997, 49(1), 1–12 CrossRef.
- P. G. Collins, K. Bradley, M. Ishigami and A. Zettl, Extreme oxygen sensitivity of electronic properties of carbon nanotubes, sensitivity of electronic properties of carbon nanotubes, Science, 2000, 287, 1801–1804 CrossRef CAS PubMed.
- C. Robert, J. Bowman and B. Fultz, Metallic hydrides: Hydrogen storage and other gas- phase applications, MRS Bull., 2002, 27(9), 688–693 CrossRef.
- Y. Hai-yan, W. Qun, C. Shao-mei and G. Ping, A frst-principle calculation of structural, calculation of structural, mechanical and electronic properties of titanium borides, Trans. Nonferrous Met. Soc. China, 2011, 21, 1627–1633 CrossRef.
- M. K. Yaakob, N. H. Hussin, M. F. M. Taib, T. I. T. Kudin, O. H. Hassan, A. M. M. Ali and M. Z. A. Yahya, First Principles LDA+U Calculations for ZnO Materials, Integr, Integr. Ferroelectr., 2014, 155, 15–22 CrossRef CAS.
- M. S. Hayat and R. M. A. Khalil, Computational predictions of optoelectronic energy materials Cs2SiBr6, Cs2GeBr6 & Cs2SnBr6 for phenomenal photovoltaic applications; a first principles study, Comput. Theor. Chem., 2024, 24, 114532 CrossRef.
- M. S. Hayat, R. M. A. Khalil, S. Almufarij, I. Ragab, E. A. Shokralla, M. A. Fahmy, A. Rehman and A. Ashfaq, Exploring Cs-based double halide perovskites for energy applications: a study of thermoelectric, optical, and spin-polarized magnetic properties, J. Mater. Sci.: Mater. Electron., 2025, 36, 1372 CrossRef CAS.
- M. S. Hayat, R. M. A. Khalil, R. U. Hassan, A. U. Rehman, Y. M. Alanazi and J. Yuennan, The Ab-Initio Investigation of Structural, Electronic, Mechanical, Dynamical and Thermodynamic Properties of Double Halide Cs2SrTaCl6, Cs2SrTaBr6, and Cs2BaTaBr6 Perovskites: A Dispersion Corrected DFT Study. Journal of Inorganic and Organometallic Polymers and Materials, J. Mater. Sci.: Mater. Electron., 2025, 36, 1372 CrossRef CAS.
- Z.-Li Zhu, J. H. Gu, Yu Jia and X. Hu, Antiferromagnetism of perovskite EuZrO3 from first-principles study, Physica B, 2011, 406, 3985–3987 CrossRef CAS.
- R. Sivasamy, P. Venugopal and R. Espinoza-Gonzalez, Structure, electronic structure, optical and magnetic studies of double perovskite Gd2MnFeO6 nanoparticles: First principle and experimental studies, Mater. Today Commun., 2020, 25, 101603 CrossRef CAS.
- K. Edalati, A. Yamamoto, Z. Horita and T. Ishihara, High-pressure torsion of pure magnesium: Evolution of mechanical properties, microstructures and hydrogen storage capacity with equivalent strain, Scr. Mater., 2011, 64(9), 880–883 CrossRef CAS.
- M. Hamal, S. J. Asadabadi, I. Ahmad and H. A. R. Aliabad, Elastic constants of cubic crystals, Comput. Mater. Sci., 2014, 95, 592–598 CrossRef.
- E. Guler and M. Guler, Elastic and Mechanical Properties of Cubic Diamond under Pressure, Chin. J. Phys., 2015, 53(2), 040807–040811 Search PubMed.
- M. I. Hussain, R. M. A. Khalil, F. Hussain, A. M. Rana and M. Imran, Ab-initio prediction of the mechanical, magnetic and thermoelectric behavior of perovskite oxidesXGaO3(X = Sc, Ti, Ag) using LDA+U functional: For optoelectronic devices, J. Mol. Graph. Model., 2020, 99, 107621 CrossRef CAS PubMed.
- D. F. Bahr and J. A. Reid, Mechanical properties of cubic zinc carboxylate IRMOF-1 metal-organic framework crystals, Phys. Rev. B, 2007, 76, 184106 CrossRef.
- R. Sivasamy, P. Venugopal and R. Espinoza-Gonzalez, Structure, electronic structure, optical and magnetic studies of double perovskite Gd2MnFeO6 nanoparticles: First principle and experimental studies, Mater. Today Commun., 2020, 25, 101603 CrossRef CAS.
- S. Hayat, R. M. A. Khalil, M. I. Hussain and A. M. Rana, A DFT study of perovskite type halides KBeBr3, RbBeBr3, and CsBeBr3 in triclinic phase for advanced optoelectronic devices, Solid State Commun., 2022, 344, 114674 CrossRef CAS.
- M. S. Hayat and R. M. A. Khalil, A DFT engineering of double halide type perovskites Cs2SiCl6, Cs2GeCl6, Cs2SnCl6 for optoelectronic applications, Solid State Commun., 2023, 361, 115064 CrossRef CAS.
- S. Hayat, R. M. A. Khalil, M. I. Hussain, A. M. Rana and F. Hussain, First-principles investigations of the structural, optoelectronic, magnetic and thermodynamic properties of hydride perovskites XCuH3 (X = Co, Ni, Zn) for hydrogen storage applications, Optik, 2020, 24, 1–13 Search PubMed.
- M. S. Hayat and R. M. A. Khalil, Ab-initio exploration of unique and substantial computational properties of double hydrides Cs2CaTlH6, Cs2SrTlH6, & Cs2BaTlH6, for the computational manufacturing of hydrogen fuel cell: A DFT study, J. Mol. Graph. Model., 2023, 125, 108600 CrossRef PubMed.
- S. Hayat, R. M. A. Khalil, M. I. Hussain, A. M. Rana and F. Hussain, Ab-initio study of the structural, optoelectronic, magnetic, hydrogen storage properties and mechanical behavior of novel combinations of hydride perovskites LiXH3 (X=Cr, Fe, Co, & Zn) for hydrogen storage applications, J. Comput. Electron., 2021, 20, 2284–2299 CrossRef CAS.
- B. Sakintuna, F. Lamari-Darkrim and M. Hirscher, Metal hydride materials for solid hydrogen storage: a review, Int. J. Hydrogen Energy, 2007, 32, 1121–1140 CrossRef CAS.
- U. Eberle, M. Felderhoff and F. Schuth, Chemical and physical solutions for hydrogen storage, Angew. Chem., Int. Ed., 2009, 48, 6608–6630 CrossRef CAS PubMed.
- N. S. Mustafa, F. A. Halim Yap, M. S. Yahya and M. Ismail, The hydrogen storage properties and reaction mechanism of the NaAlH4 +Ca(BH4)2 composite system, Int. J. Hydrogen Energy, 2018, 43, 11132–11140 CrossRef CAS.
- M. B. Baysal, G. Surucu, E. Deligoz and H. Ozisik, The effect of hydrogen on the electronic, mechanical and phonon properties of LaMgNi4 and its hydrides for hydrogen storage applications, Int. J. Hydrogen Energy, 2008, 43, 23397–23408 CrossRef.
- A. Gencer and G. Surucu, Investigation of structural, Electronic and lattice dynamical properties of XNiH3(X = Li, Na and K) perovskite type hydrides and their hydrogen storage applications, Int. J. Hydrogen Energy, 2019, 44, 15173–15282 CrossRef CAS.
- W. Travis, E. N. K. Glover, H. Bronstein, D. O. Scanlonbc and R. G. Palgrave, On the application of the tolerance factor to inorganic and hybrid halide perovskites: a revised system, Chem. Sci., 2016, 7(7), 4548–4556 RSC.
- C. Li, X. Lu, W. Ding, L. Feng, Y. Gao and Z. Guo, Formability of ABX3(X = F, Cl, Br, I) halide perovskites, Acta Crystallogr., Sect. B: Struct. Sci., 2008, 92008, 702–707 CrossRef PubMed.
- Y. P. Fu, M. P. Hautzinger, Z. Y. Luo, F. F. Wang, D. X. Pan, M. M. Aristov, I. A. Guzei, A. L. Pan, X. Y. Zhu and S. Jin, Incorporating large a cation into lead iodide perovskite cages: relaxed Goldschmidt tolerance factor and impact on exciton–phonon interaction, ACS Cent. Sci., 2019, 5(8), 1377–1386 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |