DOI:
10.1039/D6RA02092E
(Review Article)
RSC Adv., 2026,
16, 19920-19968
Frontiers in manganese catalysis: a sustainable platform for bond construction and heterocycle synthesis
Received
11th March 2026
, Accepted 1st April 2026
First published on 17th April 2026
Abstract
The progress of earth-abundant transition metal-based catalytic methodologies has driven growing interest within the scientific community for the construction of bioactive organic molecules. Among the earth-abundant transition metals, manganese is an attractive alternative to precious metals in synthetic methodologies due to its availability, low toxicity, and variable oxidation states. Recently, there has been considerable progress in manganese catalysis for the selective formation of diverse chemical bonds. This review introduces recently developed techniques in manganese catalysis for various bond-forming strategies, especially C–N, C–C, C–O, and C–S bonds, indicating the importance of manganese-based methods in organic syntheses. Manganese-catalysed methodologies have also demonstrated considerable efficacy in the construction of heterocyclic moieties. The synthesis of heterocyclic compounds through manganese-based annulation, cyclization, and multicomponent techniques is briefly summarized in this review. We also discuss the main catalytic structures, reaction variables, ligand considerations, substrate scope, and functional group tolerance, and the importance of manganese catalysis in terms of both economic viability and sustainability. The need for eco-friendly manganese catalysts that can promote both diverse bond-forming approaches and the construction of heterocyclic moieties further accentuates their emerging importance in modern synthetic methodology.
 Asit Kumar Das | Dr Asit Kumar Das was born in Kakdwip, West Bengal, India. He obtained his BSc degree from the University of Calcutta and completed his MSc in Chemical Sciences at the Indian Institute of Technology Guwahati. He was awarded his PhD from Jadavpur University under the supervision of Professor Sanjay Bhar. Dr Das began his academic career as an Assistant Professor in the Department of Chemistry at Krishnath College in 2017. He is currently serving as an Assistant Professor of Chemistry at Murshidabad University. His research focuses on the development of innovative synthetic methodologies for constructing medicinally significant heterocyclic and carbocyclic frameworks, the advancement of heterogeneous catalysis, and the exploration of sustainable approaches in chemical transformations. |
 Kumares Sarkar | Dr Kumares Sarkar was born in the village of Kaligram in the district of Malda in the state of West Bengal, India. He completed his schooling from 2002 to 2010. He obtained his BSc (Chemistry Honours) from Krishnath College, University of Kalyani, West Bengal, and his MSc from the Indian Institute of Technology Guwahati, Assam. In 2024, he completed his PhD at the Indian Institute of Technology, Kharagpur, West Bengal, under the supervision of Prof. Tanmaya Pathak and Prof. Kumar Biradha. His research interests include the synthesis of functionalized carbohydrates and their transformation into various heterocyclic molecules. |
 Suchandra Bhattacharjee | Dr Suchandra Bhattacharjee completed her PhD at the Indian Institute of Technology (IIT) Guwahati, India, followed by postdoctoral research at the Fujian Institute of Research on the Structure of Matter (FJIRSM), Chinese Academy of Sciences (CAS), Fuzhou, Fujian, P. R. China. She is currently serving as an Assistant Professor at Chandigarh University, Mohali, Punjab. Her primary research interests include multicomponent reactions using novel catalytic systems and the synthesis of biologically active compounds through C–H activation and C–C coupling strategies. |
 Sushmita Gajurel | Dr Sushmita Gajurel, born in 1994 in Udalguri, Assam, is an Assistant Professor at the University Centre for Research and Development, Chandigarh University, Mohali, Punjab. She earned her PhD from North Eastern Hill University, Meghalaya, India, in 2024. She is experienced in developing green and sustainable heterogeneous catalysts, including graphitic carbon nitride, graphite oxide, metal–organic frameworks, and metal-grafted nanoparticles. Her research specializes in developing methodologies for organic reactions, green synthesis, and heterogeneous catalysis. |
 Subhendu Dhibar | Dr Subhendu Dhibar was born in 1991 in Purandarpur, Birbhum, West Bengal, India. He obtained his PhD in Chemistry from Visva-Bharati University, India, in 2020, specializing in supramolecular materials under the supervision of Prof. Biswajit Dey. In 2021, he was awarded the prestigious Dr D. S. Kothari Postdoctoral Fellowship and joined the research group of Prof. Bidyut Saha in the Department of Chemistry at the University of Burdwan, India. In 2025, he received the DST-CSRI Postdoctoral Fellowship and worked with Prof. Soumya Jyoti Ray at the Indian Institute of Technology (IIT) Patna, Bihar, India. Later in 2025, he was awarded the JSPS Postdoctoral Fellowship and is currently conducting research in the group of Prof. Koichi Mayumi at the Neutron Science Laboratory, Institute for Solid State Physics (ISSP), the University of Tokyo, Japan. Dr Dhibar's research is interdisciplinary, encompassing supramolecular chemistry, functional gel materials, and metal–organic frameworks, with applications in biomedicine, flexible electronics, and environmental science. His work particularly focuses on the design of tough hydrogels, self-healing metallogels, semiconducting soft materials, flexible microelectronic devices, and non-volatile resistive memory switching systems. |
 Sumit Kumar Panja | Dr Sumit Kumar Panja is currently serving as an Assistant Professor at the Tarsadia Institute of Chemical Science, Uka Tarsadia University, Surat-394350, Gujarat, India. He completed his PhD at Banaras Hindu University (2011–2016). Subsequently, he carried out postdoctoral research in the Department of Inorganic and Physical Chemistry at the Indian Institute of Science (2016–2018). His research interests include photophysical studies of organic materials, solvation dynamics, organic materials for solar cells, TADF materials, covalent organic frameworks (COFs), metal–organic frameworks (MOFs), and catalytic systems for H2O2 production, as well as hydrogen (H2) evolution. |
 Shubham Avinash Deshmukh | Dr Shubham Avinash Deshmukh is currently a faculty member at JSPM University, School of Basic and Applied Sciences, Faculty of Science and Technology, Department Chemistry, Wagholi, Pune. He obtained his PhD in bio-inspired porphyrin-based photocatalysis for organic transformation and biomass conversion. During his PhD tenure at Vellore Institute of Technology, from 2019 to 2023, he published 14 research articles in reputed international journals, with 267 citations. His focus is mainly on the synthesis of covalent organic frameworks for sunlight-driven photocatalytic organic transformation and biomass conversion to value-added products. Recently, he published 2 patents, review articles, 2 book chapters, and is currently serving his commitment to academic output. |
 Sujit Sarkar | Dr Sujit Sarkar is currently a faculty member at JSPM University, in the School of Basic and Applied Sciences, Faculty of Science and Technology Department Chemistry, Wagholi, Pune. He obtained his PhD in Synthetic Organic Chemistry and MSc in Chemistry from IIT Guwahati, Assam, India, in 2018 and 2013 under the supervision of Prof. A. K. Saikia and Prof. A. T. Khan, respectively. He has qualified for many national-level competitive examinations, such as IIT JAM (2011), CSIR-UGC NET JRF (AIR-63) in Dec 2012 and 2017 (AIR-74), and GATE in February 2012, 2013 and 2020. His research interests include synthetic methodology, C–H functionalization, multicomponent reactions, development of new reactions, synthesis and functionalization of heterocycles, asymmetric synthesis, total synthesis of natural products and bioactive molecules, synthesis and applications of metal–organic frameworks (MOFs) in gas/vapor/liquid adsorption, heterogeneous catalysis, and fluorescence sensing. He has also authored many textbooks as well as research papers in reputed journals. |
Introduction
The bond-forming strategies using transition-metal catalysts are of fundamental importance in the construction of structurally important organic frameworks and are extensively used in the fields of medicine, agriculture, and other chemical industries.1 Precious metals, such as palladium,2 iridium,3 rhodium,4 and ruthenium,5 have been utilized for diverse bond-forming reactions, but significant concerns have been raised owing to their toxicity, high cost, limited availability, and environmental impact. Chemists are constantly searching for simple construction strategies with a wider range of substrates, higher efficiency, and high selectivity for product generation.6 The introduction of sustainability in chemical manufacturing is highly essential to overcome the detrimental environmental effects of manufacturing from the viewpoint of green chemistry.7 Therefore, earth-abundant transition metals, including the first row, have been recognized as attractive substitutes for the existing precious metals.8 Manganese-mediated organic synthesis has attracted immense attention because of its natural abundance, eco-friendliness, economic viability, low toxicity, and unique redox chemistry.9 Manganese complexes have recently been found to be highly capable of constructing diverse bond-forming reactions, particularly carbon–nitrogen, carbon–carbon, carbon–oxygen, and carbon–sulfur bonds in organic synthesis, which establishes an essential strategy for assembling value-added organic molecules.10 In this regard, the manganese-catalyzed acceptorless dehydrogenative coupling (ADC) and borrowing hydrogen (BH) reactions have been extensively explored in the formation of C–X bonds (where X = C, N, O, and S) using alcohols or phenols as the substrates.11 Similarly, alkenes, alkynes, and carbonyl compounds have been employed as versatile starting materials in manganese-mediated bond-forming reactions, particularly annulation and coupling reactions.12 The manganese-catalyzed atom-economical bond-forming strategy through the C–H functionalization of allylic and benzylic substrates has also attracted great attention, which minimizes the necessity for pre-functionalized substrates.13 In parallel, the manganese-enabled C–H functionalization of heterocyclic molecules has opened new avenues for late-stage diversification of structurally significant organic molecules.14 Such functionalized heterocyclic moieties are essentially valuable in pharmaceutical, agrochemical, and industrial research. Moreover, the manganese-driven cleavage of strained rings for the construction of new bonds has been developed as a powerful tool for skeletal rearrangement and molecular complexity generation, which emphasizes the ability of non-toxic manganese catalysts in unusual activation modes.15
Functionalized and fused heterocyclic frameworks have been attracting considerable attention in recent times due to their widespread biological applications, including antitumour,16 anti-inflammatory,17 neuroprotective,18 anti-convulsant,19 antiepileptic,20 anti-microbial21 and anti-diabetic22 activities. The construction of such heterocyclic frameworks via manganese-driven annulation, cyclization, and multicomponent reactions has emerged as a powerful tool to access a wide variety of nitrogen-, oxygen-, and sulfur-containing heterocyclic units. For example, PGL-135,23 frentizole,24 riluzole,25 and R116010 (ref. 26) are extensively used against various chronic diseases. Despite impressive progress in biological fields, heterocyclic scaffolds remain indispensable in agrochemicals, including herbicides, insecticides, and fungicides (Fig. 1).27 Various heterocyclic skeletons play a crucial role in the efficient development of agrochemicals due to their biological compatibility, chemical flexibility, and optimized performance, enabling the advancement of benign and sustainable crop protection mediators. Therefore, the significant utility of earth-abundant manganese catalysts to assist both diverse bond-forming reactions and the construction of heterocyclic frameworks further underscores their emerging significance in modern synthetic methodology.
 |
| | Fig. 1 Biological and agrochemical applications of functionalized and fused heterocyclic molecules. | |
This review article provides a comprehensive overview of the recent progress in manganese-catalyzed organic synthesis, highlighting sustainable bond-forming strategies and the construction of heterocyclic frameworks. It covers the substrate classes and activation modes, along with C–X bond formation using various functional groups, such as alcohols and phenols, carbonyl compounds, and alkenes; allylic, benzylic, and heterocyclic C–H functionalization; strained-ring cleavage-driven bond construction; and manganese-catalyzed heterocycle synthesis (Fig. 2). Selective mechanistic pathways and corresponding examples are discussed to reveal the key developments, present challenges, and future scope in this rapidly growing field.
 |
| | Fig. 2 Mn-catalysed synthetic approaches for the formation of diverse bonds and heterocyclic frameworks. | |
Mn-catalyzed C–X bond formation using alcohols and phenols
Morrill et al. developed the Mn-catalyzed N-alkylation of sulfonamides. The yield of N-alkylated sulfonamides depends on the concentration of catalyst (Mn: 5 mol%) and the base (K2CO3: 10 mol%).28 Using the borrowing hydrogen strategy, a series of alkylated alkyl and aryl sulfonamides were synthesized (Scheme 1). Interestingly, electron-withdrawing groups containing sulfonamides (e.g., 4-NO2, 4-CN) afforded poor yields. The authors used benzyl alcohol as the alkylating agent in this reaction. A series of substituted benzylic alcohols was successfully tolerated and produced the corresponding mono-N-alkylated sulfonamides in good to excellent yields. The authors observed that thiophene-3-ylmethanol was smoothly transformed into its corresponding sulfonamide in 87% yield, whereas thiophene-2-ylmethanol remained unreacted, inhibiting the Mn catalysis via the weak coordination between substrate and catalyst. The survival of benzyl alcohol, bearing a reducible olefin moiety, demonstrated the chemoselectivity of the reaction.
 |
| | Scheme 1 Mn-catalyzed alcohol-mediated N-alkylation of sulfonamides. | |
The plausible catalytic cycle showed that the base-mediated initial activation of the Mn precatalyst resulted in the active manganese complex via the dehydrobromination reaction (Scheme 2).28 An alkoxo-type complex was generated after the coordination of the alcohol, and then dehydrogenation of the alcohol resulted in an aldehyde and manganese hydride species. The condensation between aldehyde and sulfonamide produced an N-sulfonylimine, which was then reduced by the resulting manganese hydride species and provided the N-alkylated product with the regeneration of the catalytically active manganese species.
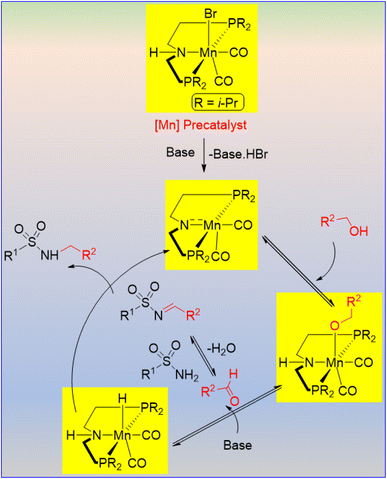 |
| | Scheme 2 Plausible mechanism for the Mn-catalyzed N-alkylation of sulfonamides via the borrowing-hydrogen strategy. | |
Azofra et al. reported a unique hydrogen auto-transfer energy pathway-based stereoselective amination of racemic alcohols.29 In order to produce optically pure products, the authors employed readily available substrates and a commercially available manganese complex as the catalyst. DFT studies indicate that using Mn as a precatalyst can result in optically pure products by following the existing protocol. The chiral imine intermediate can distinguish between the catalyst racemic mixture and its corresponding enantiomer. For example, if the catalyst's (S) enantiomer is present in the hydride transfer step, the imine intermediate can recognize it and furnish the expected product with that specific configuration of the catalyst's enantiomer. The catalyst serves as the chiral reagent and efficiently yields enantioselective products (Scheme 3). DFT analysis showed that the successive dehydrogenation of alcohols resulted in chiral manganese alkoxide intermediates. Only one among the four diastereomeric intermediates generated reacts at a time. The electronic nature of the groups attached to the alcohols has no influence on the reaction's stereochemistry.
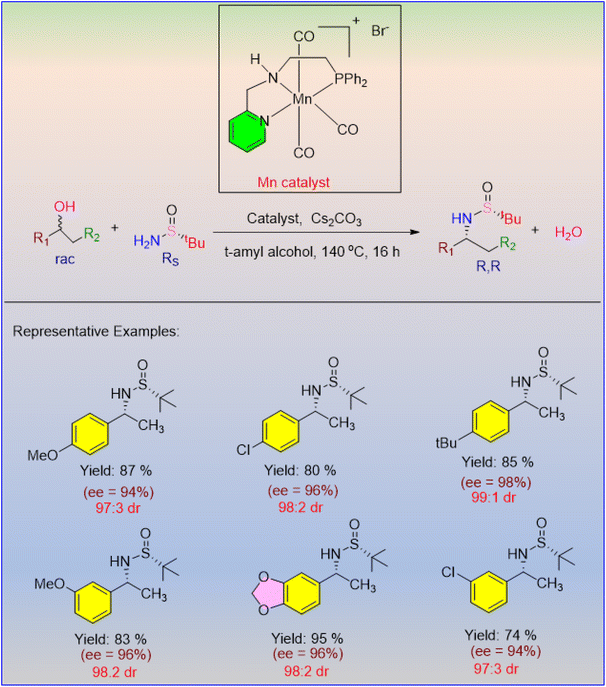 |
| | Scheme 3 Manganese-catalysed asymmetric amination of sec-alcohols. | |
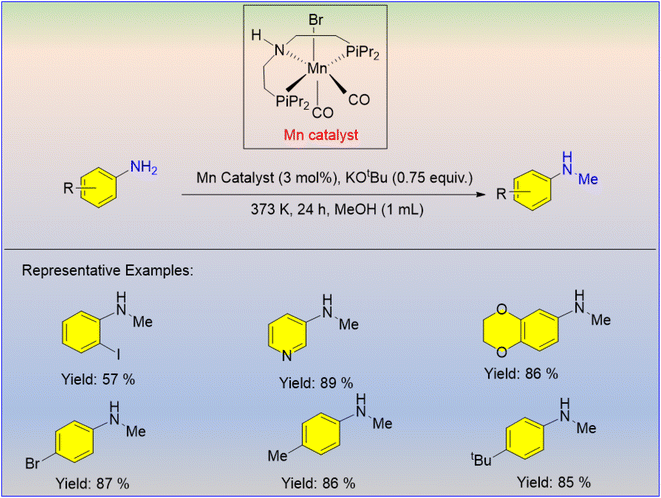
The methanol-driven N-methylation of aromatic amines using the second generation of manganese PNP pincer complexes was developed by Beller and his group.30 Good yields were obtained for various types of primary anilines by selective methylation at 100 °C for 16 hours with Mn (2 mol%) and potassium tert-butoxide (0.5 equiv.) as the base (Scheme 4). The utility of this methodology is that it allows for the use of various types of amines while requiring less catalyst and base loading.
 |
| | Scheme 4 Manganese-catalysed methanol-mediated N-methylation of amines. | |
The PN3-pincer ligand-supported Mn(I) complexes were designed by Hultzsch et al. for the alkylation of amines with primary and secondary alcohols.31 Most intriguingly, this transformation was carried out under mild reaction conditions (60–100 °C) and a low catalyst loading (0.5 mol%). Aromatic amines afforded better yields with benzyl alcohol at 60 °C as compared to aliphatic amines, which required 100 °C for good yields (Scheme 5). In contrast, the N-alkylation of aniline with secondary alcohols requires a high temperature (100 °C), which opposes substituted benzylic alcohols. This protocol has an advantage in synthesizing the drug cinacalcet, which involves the challenging alkylation of benzylamine under non-optimal conditions.
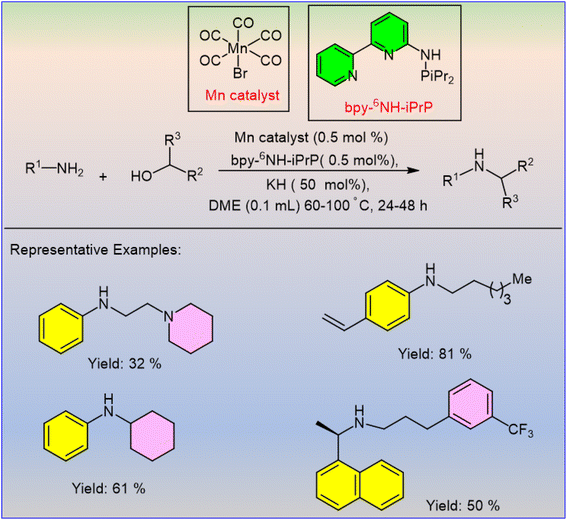 |
| | Scheme 5 Manganese-catalyzed primary and secondary alcohol-mediated N-alkylation of amines. | |
Peng et al. developed the alcohol-driven N-alkylation of aromatic amines using cheap and industrially available manganese salts like MnCl2 or Mn(CO)5Br, and triphenylphosphine (PPh3) as the ligand.32 Using this catalytic concentration (10 mol% Mn antecedent, 20 mol% PPh3, 1.2 equiv. KOtBu, 130 °C, 20 h), a series of (hetero) aromatic and aliphatic amines were specifically alkylated in moderate-to-high yields with aliphatic and aromatic alcohols (Scheme 6). By extension, this methodology allows the synthesis of indole through an intramolecular reaction and a resveratrol-derived amine. This catalytic method cannot be applied to some functional groups, such as nitro, ester, and hydroxy, and it does not require manganese complexes.
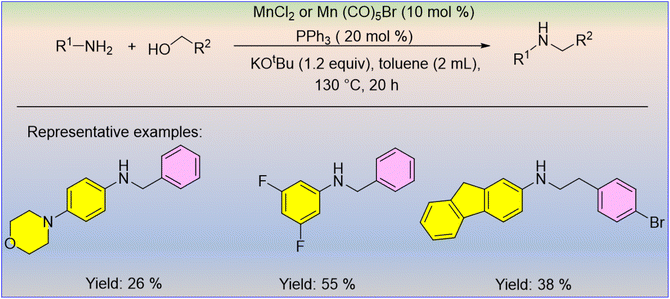 |
| | Scheme 6 Manganese-catalysed, alcohol and triphenylphosphine-mediated N-alkylation of amines. | |
Daw et al. published the first study on the construction of secondary amides using the dehydrogenative coupling of benzyl alcohol and ammonia.33 The amount of base (KH) used in this manganese-catalyzed technique was discovered to be crucial to the selectivity of amide formation, in which the electron-efficient benzyl alcohol showed high reactivity (Scheme 7). The research offered a potential mechanistic path whereby potassium hydride combines with alcohol to produce an alkoxide ion that may be in equilibrium with an excess quantity of NH3 in order to produce potassium amide (Scheme 8). The final product aldehydes were found through the dehydrogenation of the revived alcohol using an Mn catalyst. A hemiaminal intermediate is formed when ammonia or potassium amide attacks an aldehyde; this intermediate then undergoes deprotonation by potassium hydride to furnish an amino alkoxide intermediate (II). The final amide salt product (VI) is produced by N-alkylation of the resulting amide with the alcohol after a second manganese-catalyzed dehydrogenation of (II) results in the selective synthesis of primary amide salts (IV or V).
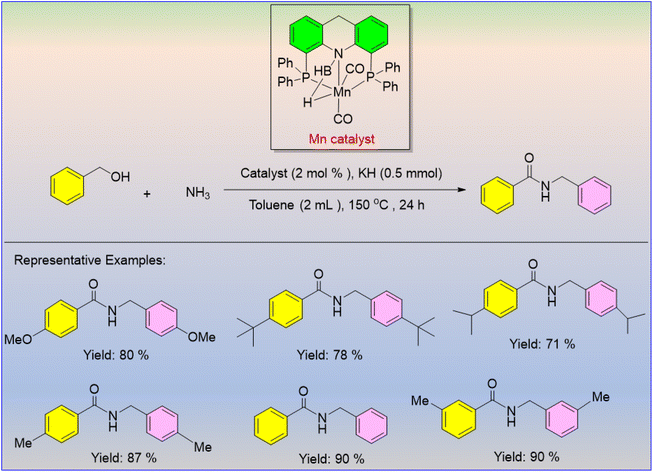 |
| | Scheme 7 Manganese-catalyzed amidation of benzyl alcohol with ammonia. | |
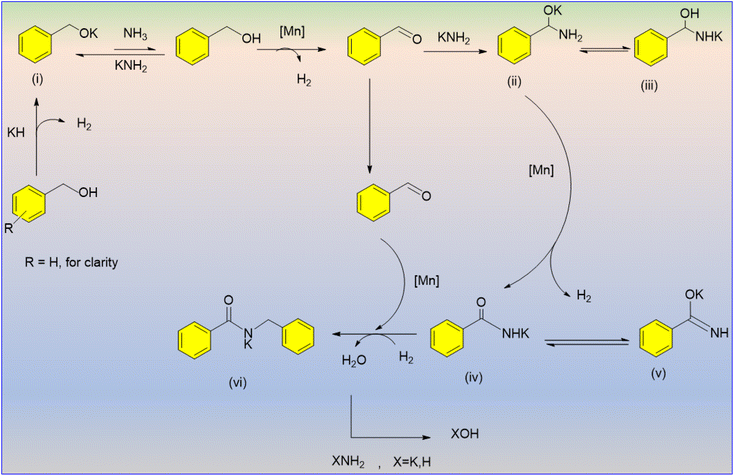 |
| | Scheme 8 Probable mechanistic pathway for the synthesis of secondary amides.33 | |
Sortais et al. described numerous Mn-catalyzed methanol-mediated N-methylations of primary amines using catalytic amounts of base. The researchers developed a new Mn(I) complex with a bis(diaminopyridine)phosphine ligand (PN3P) and studied N-methylation reactions with potassium tert-butoxide (20 mol%) in toluene at 120 °C for 24 h.34 This methodology might be applicable for a variety of functional groups like esters, nitro, ketones, and amides, and afforded moderate to high yields (42–98%) of mono-N-substituted products (Scheme 9). Intriguingly, during the mechanistic investigation, the dearomatized intermediate was isolated from the base-Mn reaction and characterized using X-ray analysis.
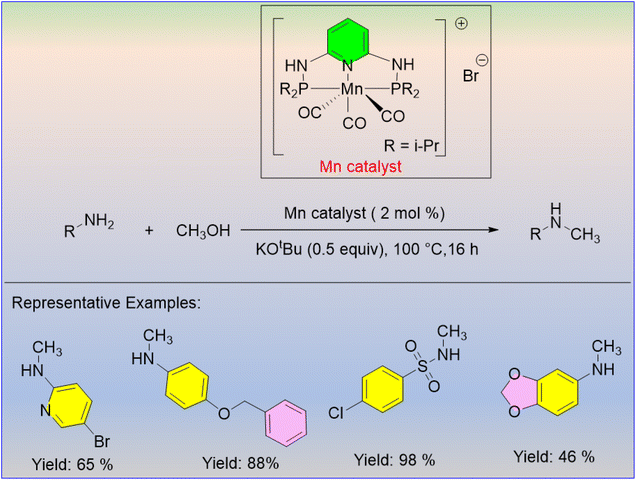 |
| | Scheme 9 Amines and methanol-mediated PN3P–Mn complex-catalysed C–N-bond formation. | |
A unique method for creating C–N bonds using aromatic amines was presented by Elangovan et al. in 2016.35 It was discovered that the catalytic system was very effective for both inter- and intramolecular N-alkylation reactions under favourable conditions and exhibited excellent chemoselectivity. The catalysts become active in the presence of a base, which leads to the deprotonation of the coordinated amines and decoordination of the halide ion. As a result, the formed amido manganese complexes engage in a β-hydrogen elimination reaction with the alcohol to produce the corresponding carbonyl molecule. Manganese hydride complexes eventually lead to imine reduction. Various amines bearing both electron-donating and electron-drawing groups are selectively alkylated with benzyl alcohol to produce N-monoalkylated anilines in high yields (Scheme 10).
 |
| | Scheme 10 Manganese-catalyzed selective N-alkylation reaction of aromatic amines by benzyl alcohol. | |
Madsen's group proposed a manganese(III) porphyrin system as an Mn catalyst for the BH methodology to achieve C–N coupling reactions.36 Several tertiary amines were synthesized by combining secondary amines and benzylic alcohols with Mn catalyst (3 mol%) in the presence of K2CO3 (20 mol%) in mesitylene under reflux conditions (Scheme 11).
 |
| | Scheme 11 Synthesis of tertiary amines using a manganese(III)–porphyrin catalyst. | |
Kempe et al. demonstrated that Mn-pincer catalysts are effective in the BH method for the generation of amines and imines (Scheme 12).37 They obtained different products depending on the base used. Alcohol played the role of an alkylating agent when KOtBu (1 equiv.) was used as a base, but alkylated imine products were obtained when NaOtBu (1.5 equiv.) was used as a base (Scheme 13). This indicates the important contribution of the cation-coordinative interactions. Furthermore, the mechanistic investigation indicates that potassium manganate hydride is more reactive than its sodium counterpart in the transformation of imines into amines via hydrogenation.
 |
| | Scheme 12 Formation of amines using a manganese catalyst. | |
 |
| | Scheme 13 Manganese-catalyzed synthesis of imines. | |
In 2023, Royo et al. developed the alcohol-mediated N-alkylation of amines using bis-triazolylidene manganese complexes.38 Complex Mn shows surprising activity with the cheap catalyst packing (1.5 mol%) and base (50 mol% of KOtBu) at 100 °C for 2 h to produce the N-alkylated products (Scheme 14). Under similar conditions, substrate scopes were performed for the synthesis of N-alkylated amines with several benzyl and aliphatic alcohols and afforded moderate to good yields. Aliphatic amines like isopropylamine and cyclohexylamine were less reactive towards this N-alkylation reaction.
 |
| | Scheme 14 Alcohol-mediated N-alkylation of amines using a bis-triazolylidene–manganese-catalyst. | |
The selective N-alkylation of various aromatic amines by benzyl alcohol (Scheme 15) demonstrated the effectiveness of the catalytic system.35 Benzyl alcohol, bearing both electron-donating and electron-withdrawing substituents, participated nicely in this reaction, and the desired N-alkylated products were obtained with good to high yields. Hetero-aromatic amines were also equally efficient in this reaction.
 |
| | Scheme 15 Manganese-catalyzed N-alkylation of hetero-aromatic amines with hetero-aromatic as well as aliphatic alcohols. | |
In synthetic organic chemistry, C–C bond formation is always challenging. Several cross-coupling named reactions have been reported in the literature for the C–C bond formation. The catalytic cross-coupling reaction between secondary alcohols and different substituted primary alcohols under similar conditions afforded the desired derivatives in excellent yields (Scheme 16).39 The electron-withdrawing substituent-containing substrate showed comparatively poor yields; however, the overall yields of the products for all substituents were comparable.
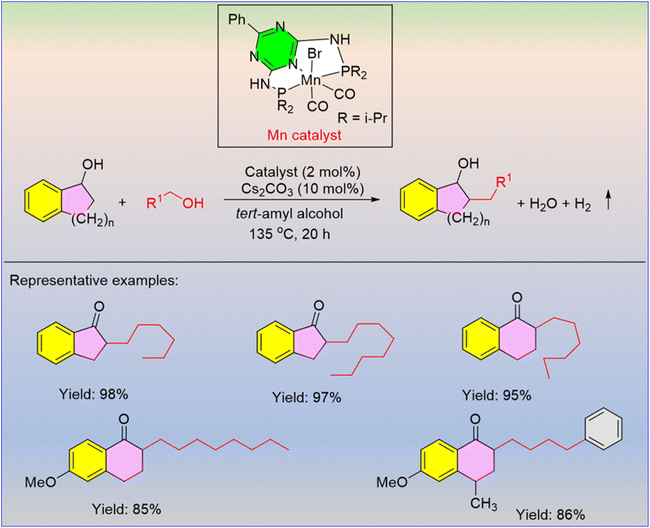 |
| | Scheme 16 Alcohol-mediated manganese-catalyzed N-methylation of primary anilines. | |
The manganese-catalyzed mono-methylation reaction was developed in the presence of KOtBu as the base. The catalyst was highly active for the methylation of both aromatic and hetero-aromatic amines. The reaction was favorable for substrates bearing both electron-donating and electron-withdrawing substituents (Scheme 17).35
 |
| | Scheme 17 Methanol-mediated manganese-catalyzed N-methylation of primary anilines. | |
In 2018, Milstein and coworkers described a partial hydrogen borrowing strategy with a Mn-pincer complex by mixing alcohols and hydrazine to furnish N-substituted hydrazones. Both aliphatic and benzylic alcohols were successfully reacted with hydrazine using Mn(t-Bu-PNN)(CO)2Br (Mn 10, 3 mol%) and a catalytic amount of KOtBu (5 mol%) at 110 °C.40 Benzylic alcohols bearing electron-donating or electron-withdrawing substituents afforded 65–92% yields of the product within 24 h (Scheme 18). Nonetheless, aliphatic alcohols such as 1-octanol and 1-hexanol needed 36 h to give the analogous products with 65% and 77% yields, respectively.
 |
| | Scheme 18 Manganese-catalyzed coupling of primary alcohols and hydrazine. | |
In 2020, Morrill et al. developed a methanol-mediated one-pot synthesis of N-methylarylamines using nitroarenes as the starting material.41 When substituted nitroarenes were methylated with methanol under optimized conditions (5 mol% Mn, 2 equiv. of KOH at 110 °C for 16 h), moderate to good yields of N-methylamines were furnished (Scheme 19).
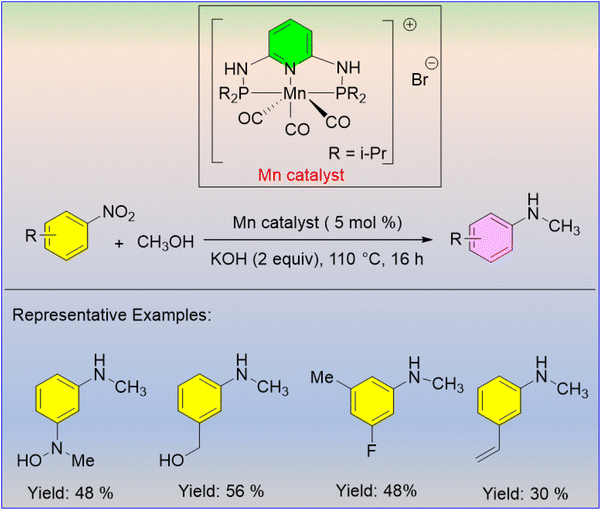 |
| | Scheme 19 Methanol-mediated manganese-catalyzed N-methylation of nitroarenes. | |
Recently, the organosols of transition-metal oxides have attracted considerable interest in synthetic organic chemistry. The authors successfully prepared superparamagnetic β-MnO2 nanoparticles in toluene solvent with a specific size and band gap. The Mn-catalyst-based nanoparticles were used as the photocatalyst for the oxidative phenol coupling reaction between β-naphthol, which afforded BINOL in a reasonable yield at room temperature (Scheme 20).42
 |
| | Scheme 20 MnO2-catalyzed oxidative phenol coupling reaction. | |
Ke et al. demonstrated an interesting example of alcohol-mediated N-alkylation of amines using a phosphine-free Mn(I)–NHC catalyst at room temperature.43 The coupling of different aromatic amines with aliphatic and benzylic alcohols was investigated using a bis-NHC–manganese complex. The combination of 1.5 mol% of catalyst and potassium tert-butoxide (1 equiv.) as base was used as the standardized reaction condition to afford N-alkylated amines in 40–93% yield (Scheme 21). For the selective N-methylated products, a temperature of 100 °C was used.
 |
| | Scheme 21 Alcohol-mediated N-alkylation of amines using a manganese catalyst. | |
The same group identified an attractive opportunity for the N-alkylation of amines using alcohols in the presence of phosphine-free Mn(I)–NHC catalyst at room temperature.43 The bis-NHC–manganese complex was efficiently employed for the coupling of different aromatic amines with aliphatic and benzylic alcohols. It was observed that 1.5 mol% of Mn catalyst loading in the presence of potassium tert-butoxide (1 equiv.) at room temperature furnished the desired N-alkylated amines in good yield (Scheme 22). However, the N-methanol-mediated methylation of anilines required a temperature of 100 °C to yield the selective N-methylated products.
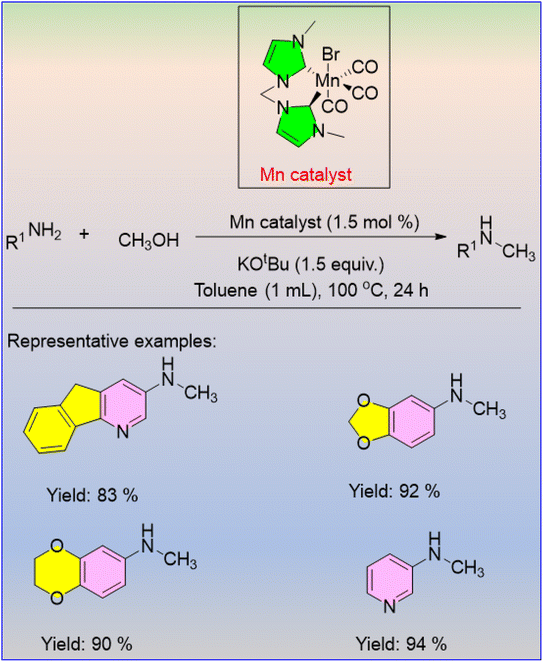 |
| | Scheme 22 N-alkylation reaction of amines with alcohols using the Mn–NHC catalyst. | |
Selective construction of carbon–carbon single and double bonds is of paramount interest in organic synthesis. Werner and their co-workers reported the manganese-catalysed selective formation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–C bonds through the reaction between alcohols with phosphorus ylides via borrowing hydrogen as well as acceptorless dehydrogenative coupling (Scheme 23).44 The excess amount of KOtBu was required for completion of the reaction. Different kinds of alcohols reacted with various phosphorous ylides under the optimized reaction conditions, and the desired products were obtained in good to high yields. The conversion of secondary ylides with alcohols led to good yields of the unsaturated products. These observations show that the borrowing hydrogen technique is disadvantaged owing to the failure of the aforesaid manganese catalyst to hydrogenate the highly substituted α,β-unsaturated carbonyl compound.
C and C–C bonds through the reaction between alcohols with phosphorus ylides via borrowing hydrogen as well as acceptorless dehydrogenative coupling (Scheme 23).44 The excess amount of KOtBu was required for completion of the reaction. Different kinds of alcohols reacted with various phosphorous ylides under the optimized reaction conditions, and the desired products were obtained in good to high yields. The conversion of secondary ylides with alcohols led to good yields of the unsaturated products. These observations show that the borrowing hydrogen technique is disadvantaged owing to the failure of the aforesaid manganese catalyst to hydrogenate the highly substituted α,β-unsaturated carbonyl compound.
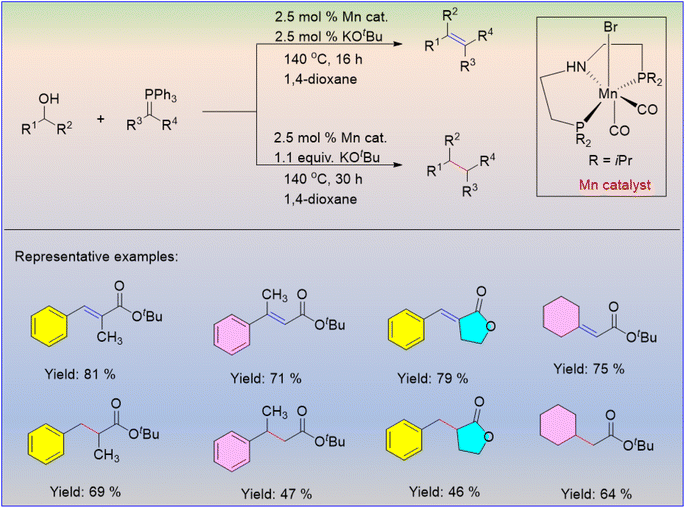 |
| | Scheme 23 Manganese-catalyzed construction of carbon–carbon single and double bonds. | |
In 2020, Maji and his research group developed bidentate amine-based ligands containing manganese(I) catalyst and performed the N-alkylation reaction of aromatic amines with benzylic alcohols (Scheme 24).45 Under the optimized reaction conditions (140 °C, 24 h), the Mn catalyst (2 mol%) was successfully utilized for the coupling of several electron-donating and withdrawing groups containing primary amines and aromatic alcohols, using KOtBu (40 mol%) in toluene to furnish the corresponding secondary amines in up to 98% yield.
 |
| | Scheme 24 The alcohol-mediated N-alkylation reaction of primary anilines using a bidentate Mn-catalyst. | |
Mn-catalyzed C–X bond formation from carbonyl compounds
Escande et al. reported an eco-catalyst obtained using a low-cost photo extraction method in alignment with green chemistry principles.46 Compared to other catalysts such as Cu(OAc)2, ZnCl2, and InCl3, which rely on solvents to facilitate the reaction, this study demonstrated Eco-Mn's high catalytic efficiency in facilitating reductive amination processes. Ketones were reductively aminated in a solvent-free procedure using Eco-Mn derived from a plant growing on mining sites in New Caledonia. The reducing agent in this transformation is the Hantzsch ester (HEH). When cyclic and aliphatic ketones were combined with cyclic and aliphatic amines, the catalyst demonstrated excellent functional group tolerance and was extremely productive. Cyclic ketones produced the highest yields for anilines with electron-withdrawing and electron-donating substituents, whereas aliphatic ketones provided only limited yields (Scheme 25).
 |
| | Scheme 25 Eco-Mn/HEH-mediated reductive amination of ketones with amines. | |
Among the C–C bond formation methods, the alkylation of ketones at the α position is a significant route for the synthesis of several biologically active, heterocyclic compounds and natural product building blocks. Gunanathan and coworkers reported Mn(I)-catalyzed C–C bond formation to obtain selective α-alkylated products (Scheme 26).39 Catalytic cross-coupling of aromatic ketones with the substituted primary alcohols afforded the desired products with good to excellent yield.
 |
| | Scheme 26 Manganese-catalyzed C–C bond formation between carbonyls with alcohols. | |
The plausible reaction mechanism for this manganese-catalysed borrowing hydrogen approach is depicted in Scheme 27.39 The base-mediated deprotonation and debromination of the Mn catalyst resulted in the dearomatized coordinatively unsaturated complex [A]. The activation of alcohol by complex [A] forms the alkoxo complex [B] via proton transfer to complex [A]. The resulting alkoxo complex [B] produces aldehyde and a saturated monohydrido manganese complex [C]. Then, a base-catalysed aldol condensation between aldehyde and acetophenone produces an α,β-unsaturated carbonyl compound, which is then hydrogenated by the manganese hydride complex [C] and affords the alkylated product with the regeneration of the catalytically active complex [A].
 |
| | Scheme 27 Plausible mechanism for the Mn-catalysed cross-coupling of primary alcohols with ketones via the borrowing-hydrogen approach. | |
Wei et al. described a simple alkylation pathway for amines in the presence of a precatalyst that utilized molecular dihydrogen as a reductant.47 To enable alkylation, aldehydes were reductively aminated, with the manganese pyridinyl phosphine complex acting as a pre-catalyst (Scheme 28). Here, the reaction temperature decreased to 80 °C and even 50 °C without having any negative effects on the catalyst's activity, and an adequate yield was still attained. To achieve the best results, the two-step alkylation procedure began with the condensation of an aldehyde and amine, and the resulting imine was treated with a precatalyst, a base, and H2. Fortunately, this reaction delivers high yields for the alkylation of a variety of amines containing both electron-donating and electron-withdrawing groups.
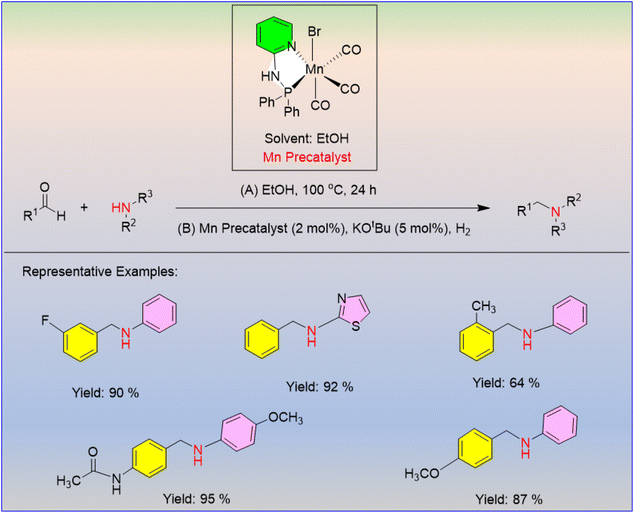 |
| | Scheme 28 Manganese-mediated reductive amination of aldehydes with amines. | |
Mn-catalyzed C–X bond formation from alkenes and alkynes
Alcohols and alkenes are the most frequently used functional groups in synthetic organic transformations. The Zhang group reported Mn-catalyzed coupling between vinylarenes and aliphatic alcohols for the first time to afford oxyalkylated products of the vinylarene in a moderate yield (Scheme 29).48 TBHP played the role of oxidizing agent and radical initiator, as suggested by the proposed mechanism. From the above reaction, it was found that the presence of one extra alkyl group in the alcohol provided a higher yield.
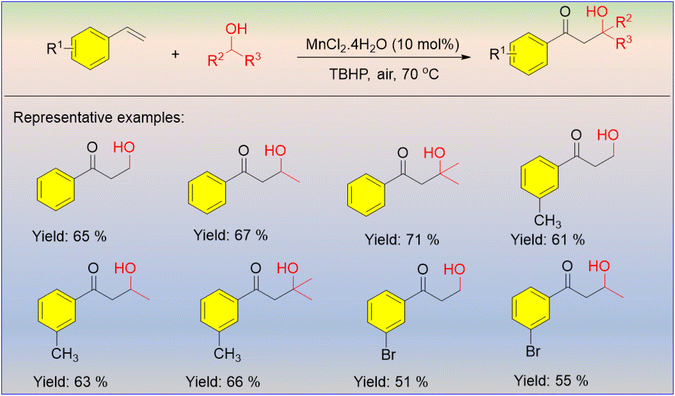 |
| | Scheme 29 MnCl2-catalyzed C–C bond formation through the coupling reaction between vinylarenes and aliphatic alcohols. | |
In recent times, transition metal-based hydroperoxidation under aerobic conditions has been frequently used. Makino et al. developed the Mn-catalyzed hydroperoxidation of carbon–carbon double bonds of olefins using N-hydroxyphthalimide and molecular oxygen to afford the oxidized product (Scheme 30).49 The reaction was performed at room temperature with the direct involvement of molecular oxygen present in the air. The required catalyst amount for this reaction was very low. In general, the substituted benzene afforded the product with excellent yield as compared to the unsubstituted one.
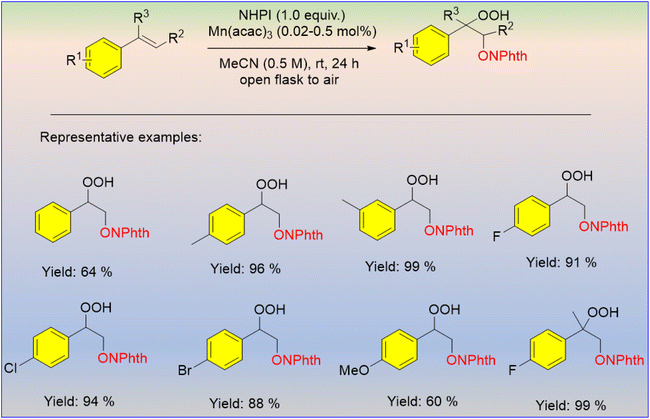 |
| | Scheme 30 Manganese-catalyzed hydroperoxidation of olefins. | |
Alkenyl arylamines are very important precursors for the synthesis of pharmaceutically active ingredients like carbamazepine, opipramol and indopan. Also, the presence of this moiety in various heterocycles, which are a part of various biologically active compounds, has drawn much attention. Here, Sun and coworkers reported the Mn(II)-catalysed ortho-alkenylation of aromatic amines (Scheme 31), which has significant application potential in reproductive diseases.50 Various aromatic amines and aromatic alkynes were reacted using MnCl2 as a catalyst to afford vinyl N-substituted aniline derivatives. These derivatives are useful for the growth of sperm; this was investigated through experiments on male rats as a mammalian model. The presence of the substituents on the aromatic amines or aryl acetylenes mutually affected the yield of the products.
 |
| | Scheme 31 Mn(II)-catalysed ortho-alkenylation of aromatic amines. | |
Mn-catalyzed C–X bond formation through benzylic and allylic C–H functionalization
Huang et al. published a sophisticated procedure for the aliphatic C–H oxidation reaction using an Mn catalyst and sodium azide as the azide source.51 This technique is precise and has a straightforward operational process for converting C–H bonds from a wide range of primary, secondary, and benzylic compounds to their corresponding azides (Scheme 32). This methodology has vast applications in organic synthesis, chemical biology, and drug development. In the azidation of celestolide, the chiral Mn-salen catalyst exhibited an enantioselectivity of about 70%, which motivated them to try to broaden the substrate range of the catalyst to produce more enantioselective products in the future.
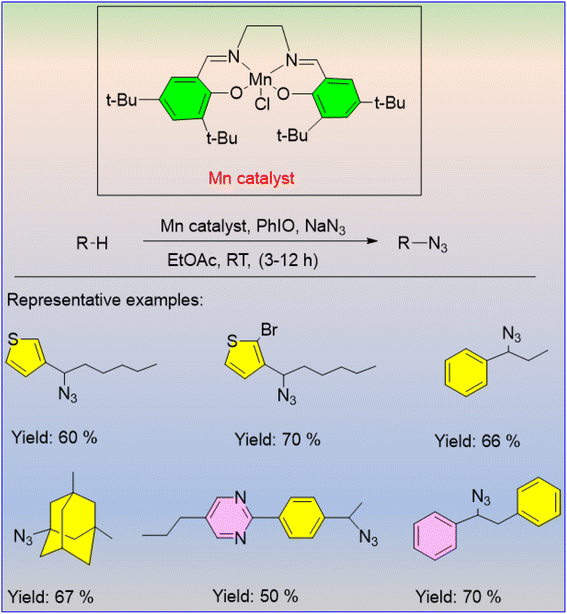 |
| | Scheme 32 Mn(salen)Cl-complex-catalysed C–H azidation using NaN3. | |
Clark et al. designed a [MnIII (ClPc)] catalyst that was used for an intermolecular benzylic C–H amination, having excellent selectivity as well as tolerance for a wide range of functional groups.52 When the substrate contains a variety of benzylic sites, the reaction demonstrates site preference by specifically aminating at the least hindered and most electron-rich site (Scheme 33).
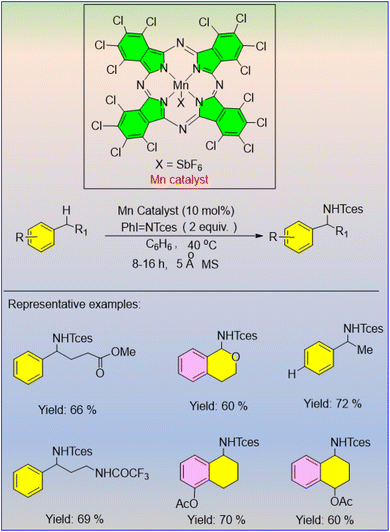 |
| | Scheme 33 Manganese-catalyzed intermolecular benzylic C–H amination reaction. | |
Paradine et al. reported an unusual manganese-based C–H amination catalyst [Mn(tBupc)] in 2015 that can functionalize several types of C(sp3)–H bonds inside the molecule, including primary aliphatic and propargylic C–H bonds, which are often challenging to achieve with metallonitrene-based catalysis (Scheme 34).53 [Mn(tBuPc)] promotes intramolecular C–H amination while maintaining strong chemoselectivity for allylic C–H bonds, which is a selectivity pattern typically associated with iron catalysts; it has a broad range of functional group resistance and displays stereospecificity in a complex molecular environment.
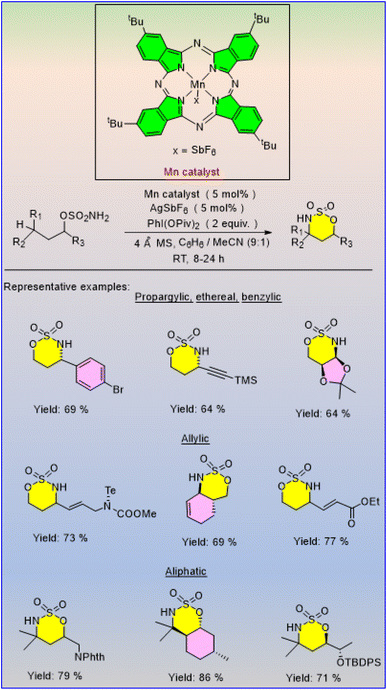 |
| | Scheme 34 [Mn(tBuPc)]Cl-catalyzed C–H amination. | |
A chiral Mn(salen) complex was synthesized by Kohmura and Katsuki and used to catalyze the asymmetric sp3 C–H bond amination reaction.54 The most effective catalyst for the benzylic amination of indan with Mn (salen) was the chiral 3,3′,5,5′-tetrabromosubstituted (salen)manganese(III) complex (Scheme 35).
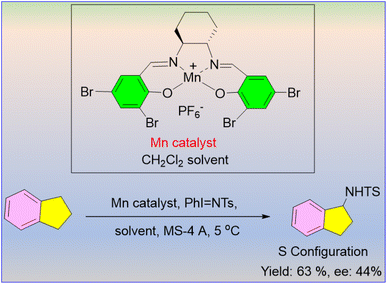 |
| | Scheme 35 Manganese-catalyzed asymmetric amination of indan. | |
The catalyst efficiently achieves high enantioselectivity for structurally different substrates through benzylic or allylic amination instead of aziridination. The yield was determined using the amount of PhINTs in the reaction mixture (Scheme 36).54 This strategy may proceed via nitrene transfer, in which nitrene is produced by coordinating PhINTs with an Mn catalyst.
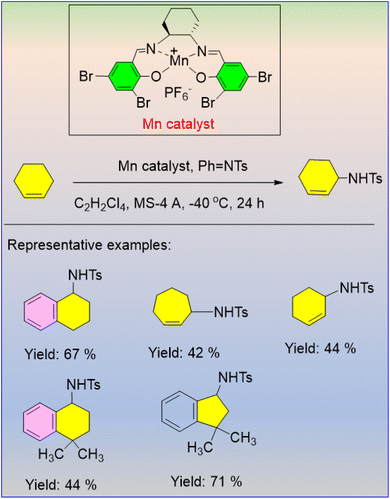 |
| | Scheme 36 Manganese-complex-catalysed asymmetric C–H amination reaction in the presence of 1,1,2,2-tetrachloroethane. | |
The Mn(OAc)3-catalyzed selective amination of benzylic C(sp3)–H bonds was discovered by Zhang et al.55 The authors observed that the selectivity of the reaction increased with the addition of DDQ. The secondary amides with good to excellent yields were produced under ambient conditions (Scheme 37). The reaction performs well in producing a wide range of functional amides in appropriate yields, using a diverse set of primary, secondary, and tertiary alkyl, alkenyl, benzyl, and aryl nitriles. Interestingly, this amidation reaction results in a broad range of secondary amides.
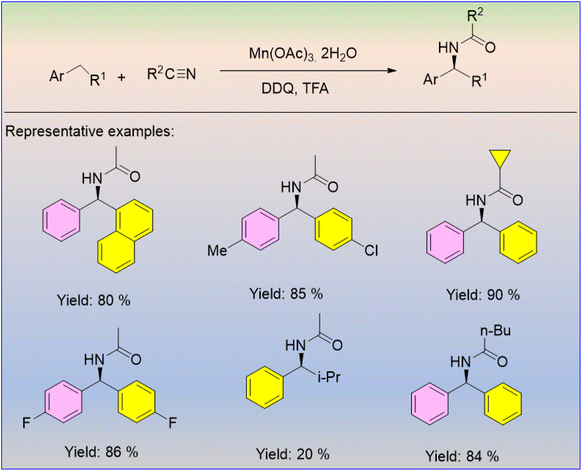 |
| | Scheme 37 Mn(OAc)3-mediated selective amination of benzylic C(sp3)–H bonds. | |
The process occurs through a radical mechanism, with DDQ mainly oxidising the substrate (Scheme 38). This route utilizes TFA to activate the hydrolysis of the nitrilium cation for the synthesis of the amide. The amination of inert C(sp3)–H bonds using this synthetic methodology is superior to the existing methods.55
 |
| | Scheme 38 Proposed mechanistic pathway for the selective amination of benzylic C(sp3)–H bonds.54 | |
The catalyst, in addition to Brønsted and Lewis acids, effectively prevents C–H amination of unavailable tertiary amines and heterocycles (Scheme 39).52 The procedure suggests that the main intermediate is an electrophilic metallonitrene. The reaction proceeds in a sequential C–H amination, with the breaking of the C–H bond being the slowest step, which determines the rate of the reaction.
 |
| | Scheme 39 C–H amination reaction catalysed by a manganese complex. | |
The catalytic application of MnBr(CO)5 as a catalyst for the C–H sulfoxidation of different aromatic ketones was developed by Kong and Xu in 2018.56 Regardless of the type of substituents, aromatic ketones with electron-donating and electron-withdrawing groups at the meta and para positions produced the required sulfonamides in good yields when they were combined with phenyl sulfonyl azide (Scheme 40). Both aliphatic and aromatic sulfonyl azides were employed as amination reagents, and major amounts of sulfonamidated compounds were produced (Scheme 41).56 The produced nitrene intermediate is inserted into the ketone's C–H bond to carry out this reaction. The critical phase in the MnMe(CO)5 reactive intermediate that was created in the reaction mixture is used in the aromatic metalation process, which is driven by the ketone. It is interesting to note that this technique has a high functional group tolerance, and the ortho C–H bond sulfoxidation shows good regioselectivity.
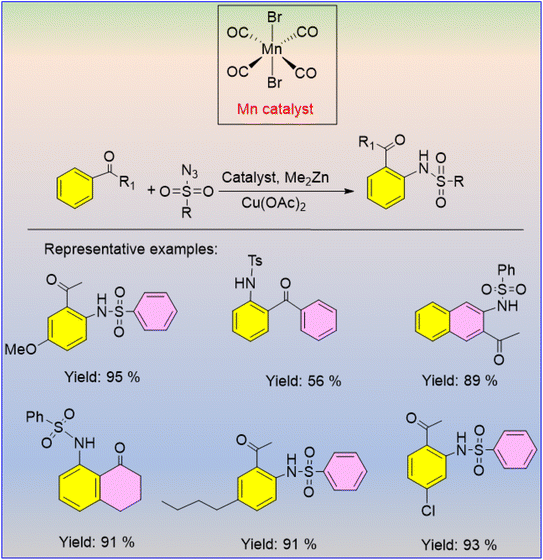 |
| | Scheme 40 Sulfonamidation of aromatic ketones catalysed by a manganese catalyst. | |
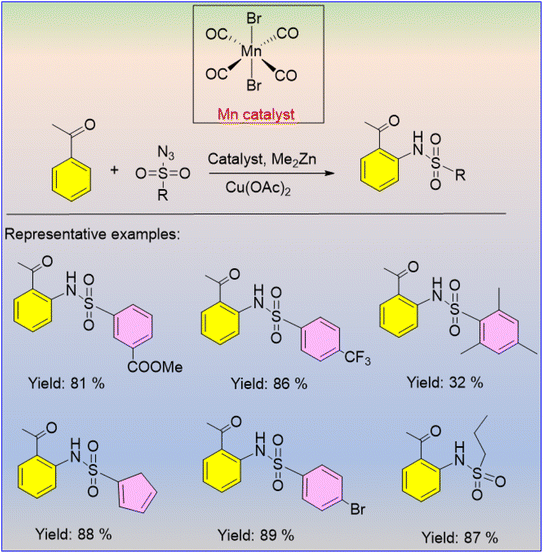 |
| | Scheme 41 Manganese-catalysed scope for the sulfonamidation of different sulfonyl azides. | |
Mn-catalyzed C–X bond formation through heterocyclic C–H functionalization
Though it is difficult to directly functionalize indoles to indolyl benzothiazoles via Mn-catalysed C–H amination, the authors designed appropriate surfactants to make it possible. However, the presence of a Mn(II) catalyst was discovered to be necessary for the additional functionalisation of indolyl benzothiazoles in order to construct biologically relevant tris-heterocyclic frameworks. This synthetic methodology started from 2-(indol-3-yl)benzothiazole and enabled the chemoselective and regioselective construction of the C–N bond (Scheme 42).57 The reaction involves a sp2 C–H amination catalysed by MnBr2 (10 mol%) in the presence of BathoPh (bathophenanthroline) ligand. The reaction involves an sp2 C–H amination catalysed by Mn(II). When Mn catalysts coordinate with nitrogen atoms in substrates, they form a five-membered intermediate. This intermediate then coordinates with pyridone through nitrogen and oxygen atoms to form a Mn(III) complex. This intricate final product is produced by an irreversible proto-demetalation and C–H amination of this complex.
 |
| | Scheme 42 MnBr2-catalyzed construction of tris-heterocyclic frameworks via the indole C2–H amination process. | |
In 2010, Kim et al. discovered a Co and Mn-based catalytic system for the direct formation of the C–N bond in azoles using amines as the nitrogen source.58 For this reaction to proceed smoothly, the authors used peroxide and an acid additive under mild conditions. Tert-butyl hydroperoxide solution (T-HYDRO) was the most productive oxidant in this reaction and the products were obtained in high yields. The Mn catalyst reacts with T-HYDRO to generate radicals, and therefore, T-HYDRO behaves as a powerful oxidant. The reaction occurred via a simple radical mechanism. The resultant radical is highly responsible for the construction of the C–N bond of the desired product. Interestingly, the Mn(OAc)2 catalyst was found to be the most effective catalyst, rather than Co(OAc)2, for productive amination products using primary amines as the nitrogen source (Scheme 43). The products were successfully obtained when 2-amino benzoxazole derivatives were substituted with n-pentyl, n-isobutyl, and cyclohexyl groups. The authors also reported that when optically active primary amines were used as the amine source in the reaction with benzoxazoles, no racemization products were obtained.
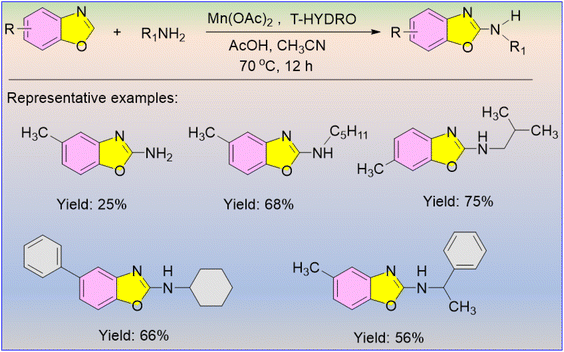 |
| | Scheme 43 Mn(OAc)2-catalyzed C–H amination of benzoxazoles using primary amines. | |
Singh et al. utilised molecular oxygen as the only oxidant for producing biologically active 2-aminoazole derivatives, establishing the process as environmentally friendly.59 The heterogeneous Cu–MnO catalyst was easily separated from the reaction mixture after the completion of the reaction and produced comparatively higher yields than the γ-MnO2 catalyst (Scheme 44). The incorporation of catalytic copper in γ-manganese oxide improves azole oxidative amination activity, highlighting the importance of copper percentage. Amination of benzothiazole and benzoxazole with –Cl and –CH3 substituents produced good yields using both cyclic and acyclic secondary amines.
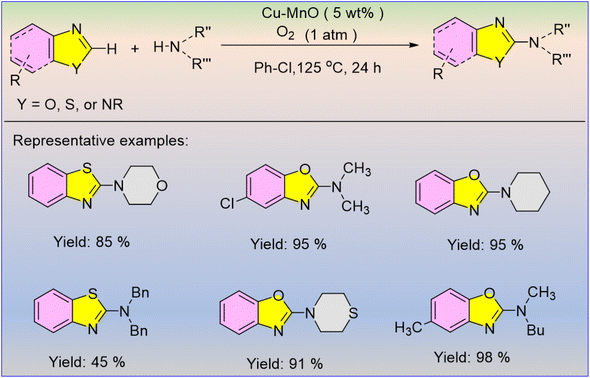 |
| | Scheme 44 Heterogeneous Cu–MnO-catalyzed direct oxidative C–H amination of azoles. | |
The catalyst was most effective with benzoxazole; however, it can also react with primary amines for higher yields. The investigation demonstrated that both O2 and catalyst are required for benzothiazole amination, and a tentative mechanism is proposed (Scheme 45).59 The Cu–MnO catalyst allows for the coordination of azoles with copper, resulting in this reaction. The hydrogen released in the initial stage might be utilised in the third step to produce intermediate (III) by regenerating the catalyst Cu–MnO.
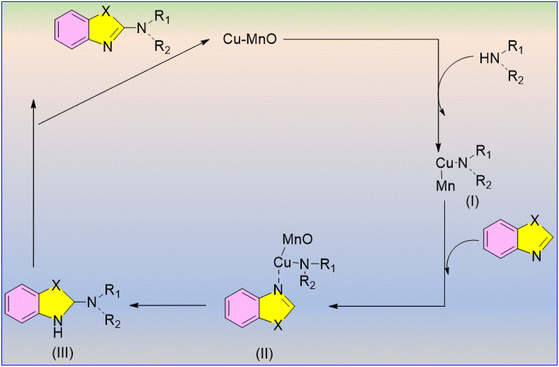 |
| | Scheme 45 Proposed reaction pathway for the oxidative C–H amination of azoles.59 | |
Synthetically significant organic transformations for the formation of novel C–C bonds via Mn-catalysed C–H activation reaction have drawn immense attention from synthetic chemists. McGlacken and co-workers reported an efficient synthetic methodology for the manganese-catalysed C2 allylation of indoles in an eco-friendly aqueous medium (Scheme 46).60 Indoles bearing various electron-donating and electron-withdrawing substituents participated efficiently and gave the desired products in moderate to good yields. Indoles bearing the –NO2 group also participated in the reaction, but the desired product was obtained in only 20% yield. Various sensitive functional groups such as –CHO, –CONH2, and –CO2Et were well tolerated, exhibiting their potential for synthetic utility in pharmaceutical chemistry.
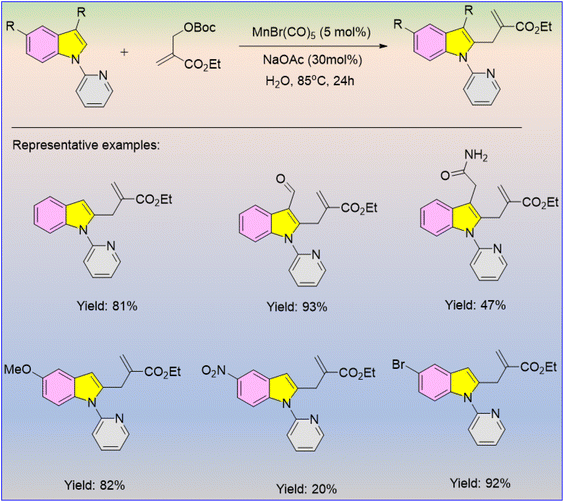 |
| | Scheme 46 MnBr(CO)5-catalyzed C2 allylation of indoles with ester. | |
A straightforward and practically simple method for the amination of 2-amino benzoxazole derivatives, which are the main building blocks of a significant number of physiologically active compounds, was described by Pal et al. in 2014.61 Using molecular oxygen as an eco-friendly oxidant, the authors developed a heterogeneous porous MnO2 catalyst for the formation of C–N bonds via the direct amination of benzoxazole through C–H activation (Scheme 47). The heterogeneous catalyst was easily separated from the reaction mixture and can be used repeatedly to produce C–N bonds. The authors observed that when substituted and unsubstituted benzoxazole were reacted with various primary and secondary amines, the corresponding products were obtained in good to excellent yields, but the reactions with sterically inhibited secondary amines generated poor yields.
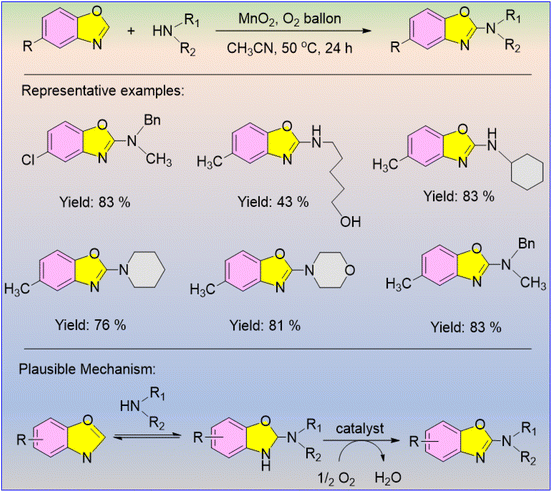 |
| | Scheme 47 MnO2-catalyzed C–H amination of benzoxazole. | |
The reaction is also compatible with alcohol functional groups, demonstrating the broad utility of this amination reaction strategy. The authors also proposed a contingent mechanism in this reaction, where the 2-amino benzoxazolidine intermediate was obtained via amine attack on benzoxazole (Scheme 47).61 The resultant 2-amino benzoxazolidine is then re-aromatized in the presence of aerial oxygen as an eco-friendly oxidant and MnO2 as a heterogeneous catalyst.
The Mn-catalyzed formation of the C–X bond through the cleavage of a strained ring
Various natural products contain cyclopentene precursors that have been frequently used in pharmaceutical and medicinal chemistry. Hence, cyclopentene is of significant interest. The Mn-catalyzed acceptorless-dehydrogenative coupling through a radical-initiated ring expansion rearrangement of cyclopropylmethanol with methyl ketone under basic conditions afforded a plethora of acyl cypentene derivatives in reasonable yields, with water and hydrogen gas as side products (Scheme 48).62 It was observed that the products were obtained in higher yields with electron-donating substituents than those with electron-withdrawing substituents.
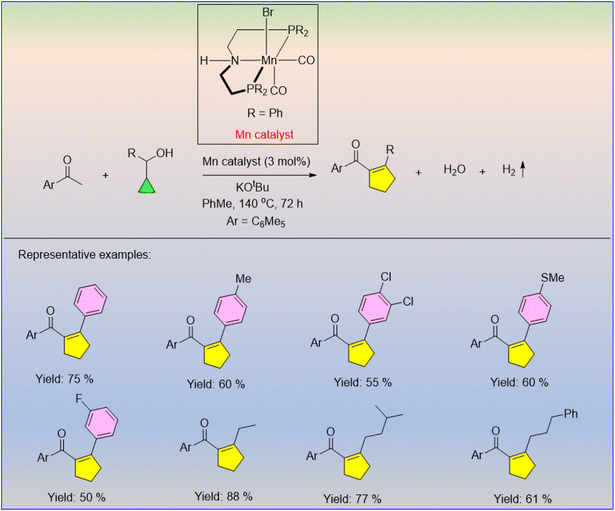 |
| | Scheme 48 Manganese-catalyzed ring expansion of cyclopropylmethanol through the acceptorless-dehydrogenative coupling strategy. | |
Alkylamines are the fundamental structural unit in natural products, biologically active compounds and pharmaceutically active ingredients. An effective Mn(III)-catalyzed hydrazination of cyclobutanols was reported, which involved C–C bond cleavage, where cyclobutanols afforded a series of alkyl hydrazines (Scheme 49).63 The depicted mechanism involves the addition of an alkyl carbon radical to the azodicarboxylate (Scheme 50). The naphthalene moiety containing cyclobutanol afforded the desired product in a smaller yield than the substituted or unsubstituted aryl groups.
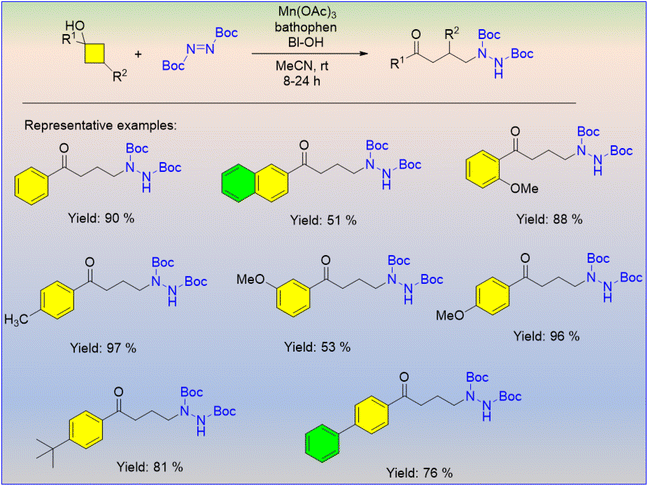 |
| | Scheme 49 Manganese-catalyzed synthesis of alkyl hydrazines through the C–C bond cleavage of cyclobutanols. | |
 |
| | Scheme 50 Proposed reaction pathway for the C–C bond cleavage of cyclobutanols.63 | |
The C–S bond is very common in natural products, pharmaceutically active compounds, and organic materials. Mn(III)-catalyzed regioselective C–S bond formation was developed via C–C bond cleavage of cyclobutanols (Scheme 51).64 Differently substituted cyclobutanols were reacted with bisaryl disulfanes using Mn(III) catalyst to afford a group of thioether derivatives. The plausible mechanism involves radical-mediated tandem C–C bond cleavage and C–S bond formation (Scheme 52). The substrates containing substituents in the ortho or para (except p-tolyl) positions have lower yields.
 |
| | Scheme 51 Mn(III)-catalyzed regioselective formation of C–S bonds via C–C bond cleavage of cyclobutanols. | |
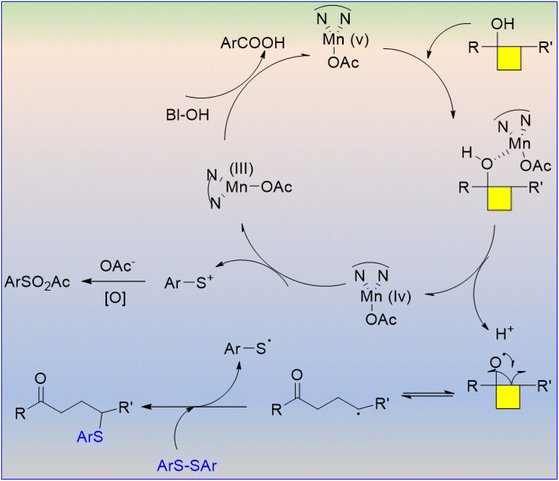 |
| | Scheme 52 Proposed reaction pathway for C–S bond formation via C–C bond cleavage of cyclobutanols.64 | |
Mn-catalyzed synthesis of heterocyclic frameworks
Heterocyclic frameworks are essential to functional materials, pharmaceuticals, and natural products. Manganese-driven organic synthesis has appeared as an economically efficient and environmentally benign approach for synthesizing a wide range of nitrogen-, oxygen-, and sulfur-containing heterocyclic frameworks. Annulation, cyclization, or multicomponent strategies are involved in constructing the complex ring systems under mild reaction conditions. Organic synthesis using manganese catalysis provides cost-effective and eco-friendly substitutes for precious noble metals and also offers exceptional reactivity patterns, including the functionalization of C–H bonds and dehydrogenative coupling strategies.
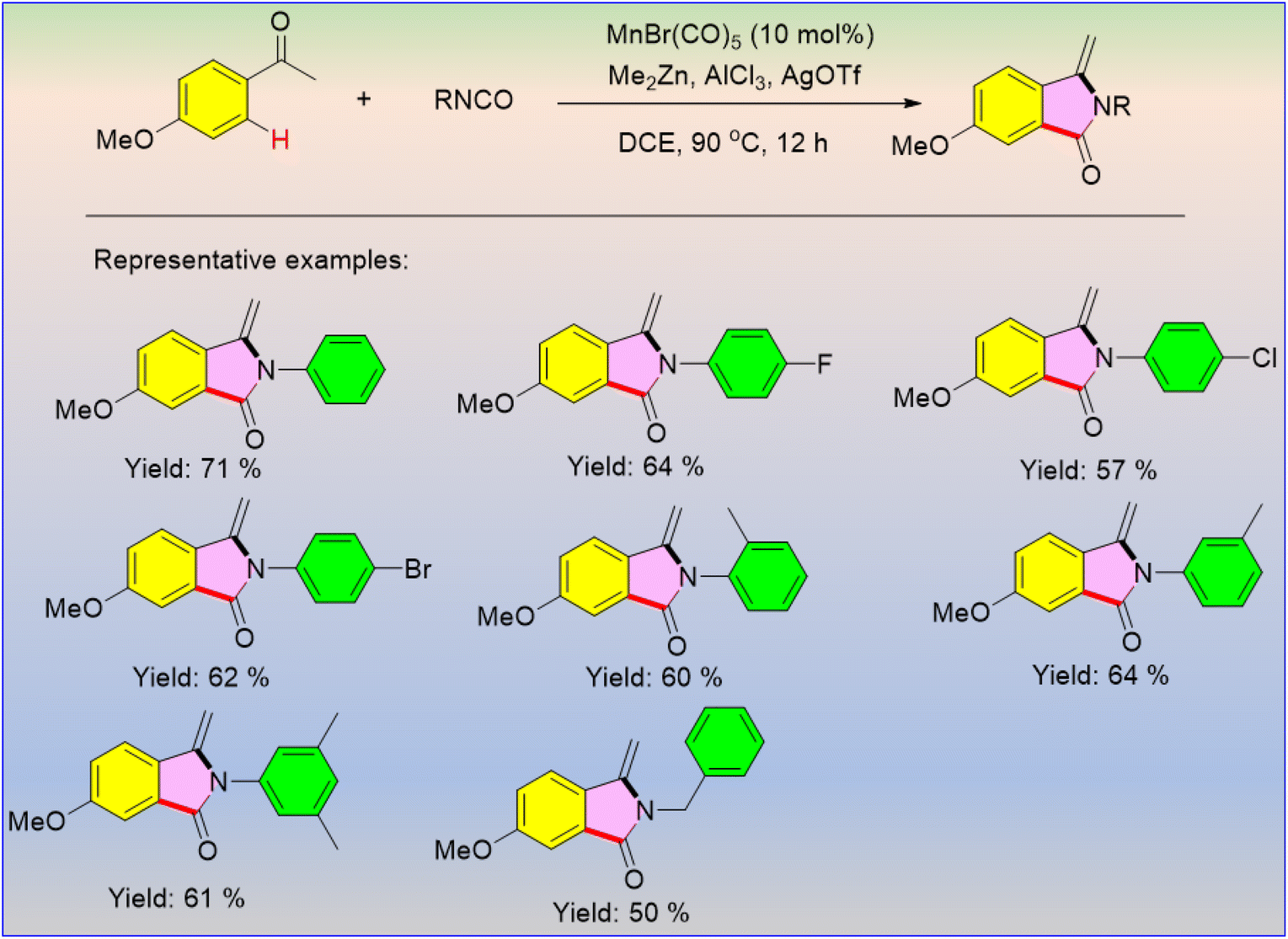
There were no literature reports on the manganese catalytic cycle for the reactions between ketones and isocyanates using transition metal catalysts, but recently, Wang et al. developed a trio of Me2Zn/AlCl3/AgOTf that activates manganese catalysis for the further reaction between ketones and isocyanates under heating conditions to afford functionalized phthalimidines (Scheme 53).65 The reaction involved [3 + 2] cyclization via inert C–H activation. Interestingly, the products with electron-donating or electron-withdrawing substituents have lower yields than the unsubstituted ones.
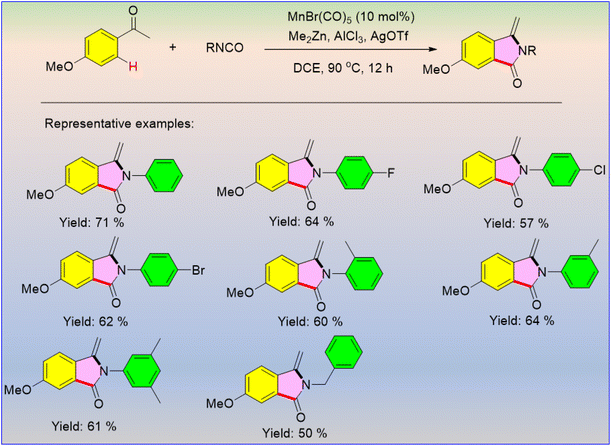 |
| | Scheme 53 MnBr(CO)5-catalyzed [3 + 2] cyclization of ketones with isocyanates. | |
A simple chemical pathway for the synthesis of 1-unsubstituted-2-aryl-1H-indazoles was revealed by Cao and Duan.66 This efficient approach involves microwave irradiation of N-acetyl hydrazones with MnO2 supported on silica as the catalyst, followed by C–H amination (Scheme 54). The generation of cyclized products in this reaction was found to require a high loading percentage of easily accessible MnO2 at higher temperatures. Moreover, unfavorable products are produced when MnO2 is present and acting as an oxidant. However, the use of silica-supported MnO2, which acts as a heterogeneous Mn catalytic system in the reaction, allows for the efficient oxidation of the substrate at lower temperatures under microwave irradiation. The silica-supported MnO2 allowed the reaction to continue with full reactant consumption. The electronic effects of the substituents play an important role in determining the regioselectivity of the reaction. The orientation of cyclization is governed primarily by the electronegativity of the substituents rather than the configuration of the starting materials. Substituents on the phenyl rings influence both the direction of cyclization and the E–Z isomerization.
 |
| | Scheme 54 Heterogeneous SiO2-supported MnO2-catalysed construction of 3,6 di-substituted indazoles. | |
An effective method was developed for generating pyrroles from amino alcohols and secondary alcohols using an Mn catalyst under environmentally friendly conditions.67 A range of amino alcohols were investigated with Mn catalyst (0.5 mol%) and KOtBu (1.5 equiv.) in 2-MeTHF under reflux conditions, and the analogous pyrroles were isolated in up to 93% yield (Scheme 55). It should be noted that the same pincer ligand-containing Co and Fe complexes were inactive in pyrrole synthesis under similar reaction conditions.
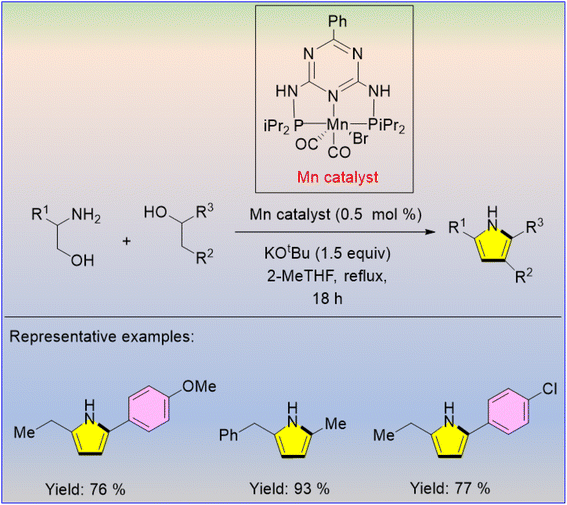 |
| | Scheme 55 Alcohols and aminoalcohol-mediated pyrrole synthesis using a manganese catalyst. | |
The first chiral manganese(III) porphyrin catalyst to use aziridination of styrene-like substrates was described by Lai et al. in 1997.68 The investigation showed that alkenes without an allylic hydrogen atom produce aziridine, while alkenes with an allylic hydrogen yield allylic amination. The reaction provided only mild enantioselectivity (up to 40% to 68%). The position and electronic nature of substituents affected enantioselectivity, and o-bromostyrene (Scheme 56) produced a satisfactory outcome. This method was found to be the most effective for the catalytic allylic amination of alkenes at the time, based on yield and turnover numbers.
 |
| | Scheme 56 Chiral manganese(III) porphyrin-catalyzed asymmetric aziridination reaction using PhINTs. | |
Mixing the catalyst and PhINTs in CH2Cl2 at room temperature generates a novel Mn species, MnIV-PhINTs, that can transfer nitrene to an alkene. The spectroscopic and organic product analyses indicate the presence of a MnIV intermediate in the reaction process that can successfully catalyse nitrene transfer to alkenes (Scheme 57).68
 |
| | Scheme 57 Proposed reaction pathway for the Mn-catalyzed enantioselective aziridination reaction.68 | |
Isonitriles are used as versatile building blocks for the design of nitrogen-containing compounds, especially amides and various heterocycles, which are of great significance in biological and pharmaceutical chemistry. Liu et al. introduced a Mn(III)-catalyzed radical-promoted cyclization reaction to afford a series of 2-functionalized quinolones (Scheme 58).69 The regiospecific 6-endo-trig radical cyclization reaction under heating conditions between o-vinyl arylisonitriles and arylboronic acids afforded 2-substituted quinoline. It was found that phenylboronic acids containing electron-donating and electron-withdrawing groups at the para-position were obtained in moderate to excellent yields.
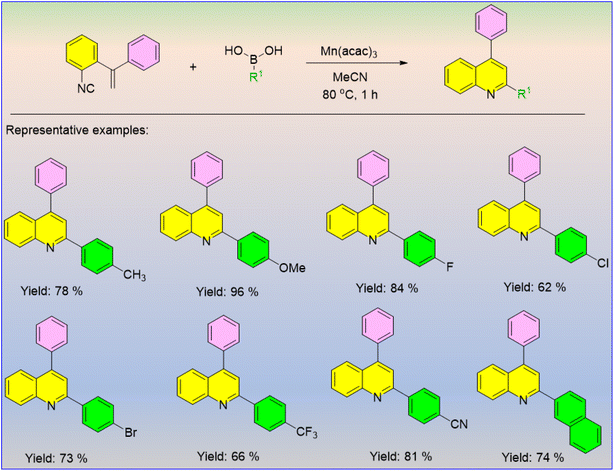 |
| | Scheme 58 Mn(acac)2-catalyzed construction of 2-functionlized quinolones between o-vinyl arylisonitriles and arylboronic acids. | |
Alternate pyrimidines were synthesized by the coupling of different amidines with various alcohols using 2 mol% of Mn catalyst and 1.1–1.5 equiv. of KOtBu in 1,4-dioxane at 120 °C for 20 h, furnishing good to excellent yields (Scheme 59).70
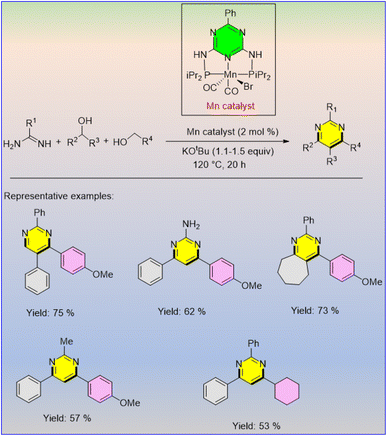 |
| | Scheme 59 Manganese-complex-catalyzed construction of pyrimidines in the presence of KOtBu. | |
Carbonyl compounds like aldehydes and ketones are easily affordable and are potential moieties for different organic transformations. Liu and co-workers reported the Mn-catalyzed [3 + 2] annulation reaction between aldehyde and ketone to produce isobenzofurans, followed by oxidation to obtain the di-keto derivatives (Scheme 60).71 Triphenylborane plays a dual role in this reaction: it acts as an additive that promotes the C–H activation and, as a Lewis acid, it activates the aldehydes. It was found that the aromatic ketones with substituents in the para or meta positions were produced in higher yields as compared to the ketones with alkyl substituents.
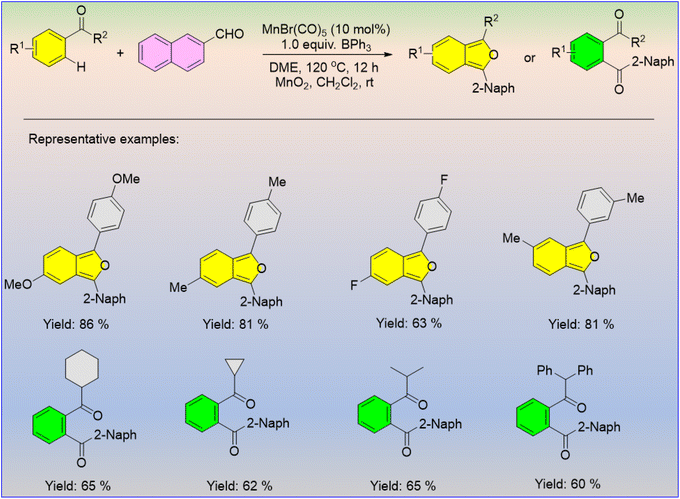 |
| | Scheme 60 MnBr(CO)5-catalyzed [3 + 2] annulation reaction between aldehydes and ketones. | |
Kempe and co-workers reported the synthesis of pyrimidine derivatives via the manganese-catalysed multicomponent strategy of amidines and alcohols.70 This approach followed both C–C and C–N bond formation simultaneously. A series of amidines bearing various functional groups was selectively coupled with various alcohols in the presence of 2 mol% of manganese catalyst and 1.1 to 1.5 equiv. of KOtBu as the effective base in 1,4-dioxane solvent at 120 °C for 20 h. The desired substituted pyrimidine derivatives were afforded in good to excellent yields (Scheme 61).
 |
| | Scheme 61 Manganese-catalyzed direct synthesis of pyrimidines in the presence of KOtBu. | |
Timmons et al. used an inexpensive manganese dioxide catalyst to produce 4-toluene sulfonyl-3-trichloromethyl-4,5 imidazolines.72 In general, α,β-unsaturated ketones are more reactive than esters with similar structures. Compounds with di-substitution at the terminal position yielded the best results (Scheme 62). The combination of three reactants with a catalyst and 4 Å molecular sieves yields valuable products for peptidomimetic studies. The diamination of TsNCl2 with α,β-unsaturated esters and ketones in acetonitrile produced stereoselective and regioselective products. In the presence of a catalyst, TsNCl2 and acetonitrile served as electrophilic and nucleophilic nitrogen sources, respectively. The [2 + 3] cycloaddition mechanism occurs in the reaction shown in Scheme 63. The SN2 reaction between the chlorine anion and 1N-(p-tosyl)imidazolinium (B) produces intermediate C. MnO2 acts as a catalyst, accelerating the formation of intermediate A, as well as the deprotonation of intermediates E and F. The deprotonation of intermediate F allows for the third chlorination on the imidazoline ring of the methyl group.
 |
| | Scheme 62 MnO2-mediated di-amination reaction of α, β-unsaturated esters and ketones. | |
 |
| | Scheme 63 Proposed reaction pathway for the di-amination process.72 | |
In the last few decades, the applications of the 3d transition metals have been less highlighted than those of the 4d or 5d transition metals. As a 3d transition metal, Mn has an important role in C–H activation. Here, Sun and co-workers developed the Mn(II)-catalyzed dehydrogenative annulation of N-aryl anilines with alkenes or alkynes for the regioselective synthesis of quinoline derivatives, which have significant medicinal importance. The Mn(II)-catalyzed dehydrogenative annulation of N-aryl anilines and aryl acetylenes furnished a group of functionalized quinoline derivatives in excellent yields (Scheme 64).73 Here, aryl acetylenes showed better reactivity than aryl olefins. With the standardized conditions, each substituent on the benzene ring provided similar yields.
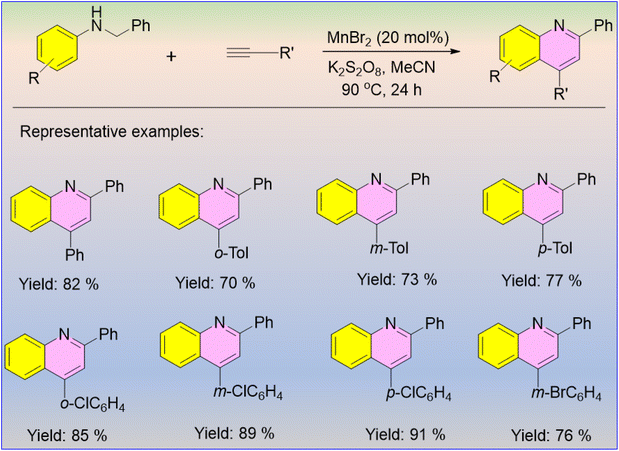 |
| | Scheme 64 MnBr2-catalyzed dehydrogenative annulation of N-aryl anilines with alkynes. | |
Kumar et al. recently produced 2-(indoyl-3yl)benzothiazoles in water by using the TPGS-750 M surfactant.57 The reaction proceeded successfully without using any additives at room temperature. Even after four successive reactions, the yield of the reaction using TGPS-750 M nanomicelles in water did not decrease significantly. Electronically different (–Cl, –Br, –CF3, alkyl) 2-aminothiophenols and 3-carboxaldehyde produced the various indol-3yl-benzothiazoles as the desired products (Scheme 65).
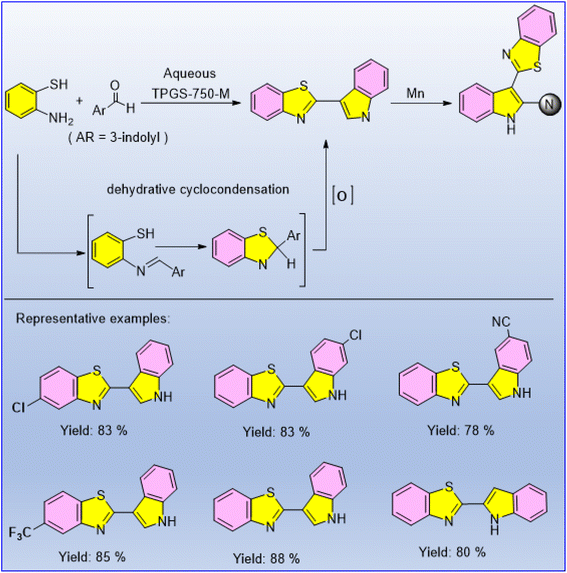 |
| | Scheme 65 Micellar-mediated synthesis of 3-indolylbenzothiazoles using a manganese catalyst. | |
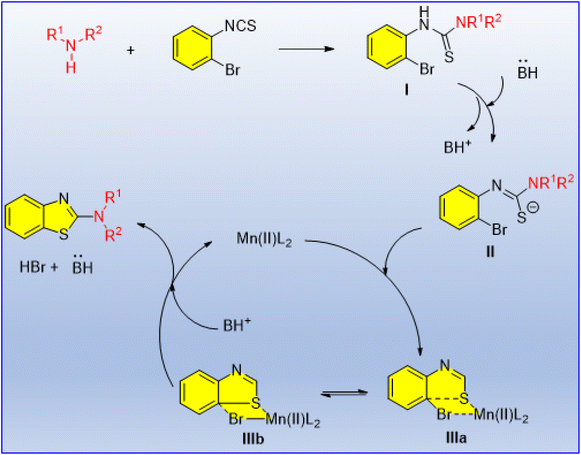
Fused heterocycles have always attracted significant attention in the agro, pharma and dye industries. Among them, 2-aminobenzothiazoles have important agrochemical and pharmaceutical applications. Anilkumar et al. developed a Mn(II)-catalyzed method for the first time in the literature for the synthesis of 2-aminobenzothiazoles. 2-Bromophenyl isothiocyanate and various functionalized amines were reacted using cost-effective and easily available MnCl2·4H2O as a catalyst to furnish a series of 2-aminobenzothiazole derivatives (Scheme 66).74 The amines with straight-chain alkyl groups gave the product in poor yield as compared to the amines with heteroatom-containing cyclic alkyl groups. The possible mechanistic pathway starts with the generation of the intermediate 2-bromophenylthiourea I, which involves the nucleophilic addition of amine to 2-bromophenyl isothiocyanate (Scheme 67). In the next step, I is converted into an anionic sulfide species II in the presence of a base. After that, the manganese(II) complex adds to intermediate II to form a four-membered transition state III. It has been assumed that III exists in equilibrium between IIIa and IIIb. When sulphur is more nucleophilic, as in IIIb, it will attack the electrophilic carbon-leaving bromine by an SNAr mechanism. Finally, the desired product is obtained from IIIb with the release of the catalytically active Mn(II) complex.
 |
| | Scheme 66 Synthesis of 2-aminobenzothiazole derivatives using a manganese catalyst. | |
 |
| | Scheme 67 Plausible reaction mechanism for the synthesis of 2-aminobenzothiazole derivatives.74 | |
Miscellaneous
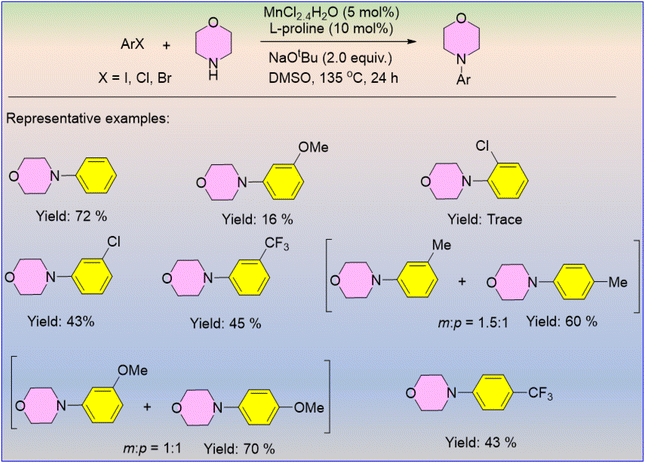
The amination of aryl halides has been frequently applied in the synthesis of organic compounds that contain the N-aryl moiety, which are present in many bioactive pharmaceuticals and conducting materials. The Mn(II)/L-Proline catalyst has been developed by the Teo research group to generate several N-arylated products by the reaction between aliphatic amine and aryl halides using NaOtBu as a base (Scheme 68).75 Substituted aryl halides furnished the products in poor yields with respect to the unsubstituted aryl halide. Only meta-amination products were observed from ortho-substituted aryl halides due to steric hindrance. However, a mixture of meta- and para-aminated products was obtained from para-substituted aryl halides, likely due to the predominance of a reaction pathway involving a benzyne intermediate.
 |
| | Scheme 68 MnCl2-catalyzed N-arylation of morpholine using NaOtBu. | |
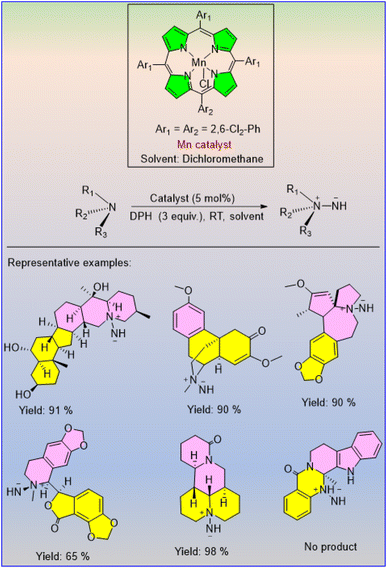
According to Zhang et al., natural aliphatic tertiary alkaloids (R3N) were converted directly into R3N–NH.76 Under mild reaction conditions, this [Mn(TDCPP)Cl]-catalyzed N-amination process offers a simple and practical way to produce unprotected alkaloid aminimides in high yields (Scheme 69).
 |
| | Scheme 69 Manganese-catalyzed formation of unprotected aminimides from alkaloids. | |
There are many literature reports on the formation of C–C bonds, with alkoxycarbonylations being one of the important methods. Here, an organo(alkoxy)borate salt [A] is efficiently formed using a stoichiometric ratio of an organoborane and an alkoxide salt (Scheme 70).77 At first, Mn(CO)5Br reacts with the organo(alkoxy)borate salt [A] to produce a Mn–alkoxy complex [B]. Then, the intramolecular transfer of the alkyl group from BEt3 to the carbonyl center results in complex [C]. Finally, the desired isopropyl propionate ester is obtained from complex [C] via the nucleophilic attack of the alkoxide on the electrophilic acyl carbon.
 |
| | Scheme 70 Mn-catalyzed carbonylation reaction. | |
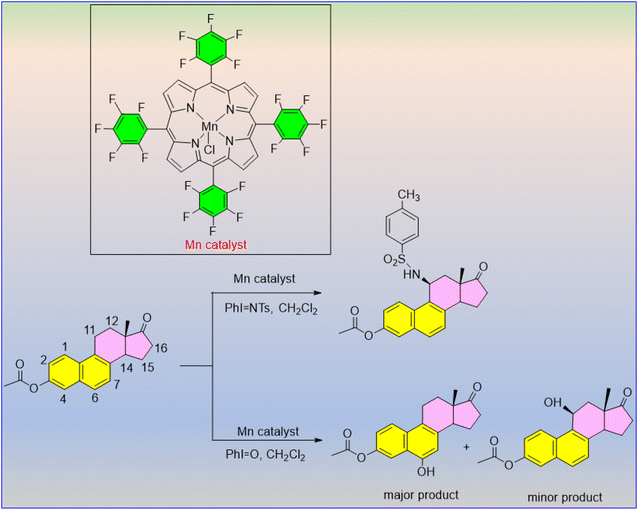
Yang et al. reported a hydroxylation and amidation reaction methodology for an aromatic steroid, equilenin, using N-tosyliminophenyliodinane (PhINTs) and iodosobenzene (PhIO), which provide the nitrogen and oxygen in the transformation.78 A readily available manganese porphyrin catalyst is used to catalyze this extremely regio- and stereoselective reaction. The 6-hydroxylated and 11-β-amidation products are the two main products for the hydroxylation and amidation reaction, respectively (Scheme 71).
 |
| | Scheme 71 Manganese porphyrin complex-catalyzed hydroxylation and amidation reaction. | |
Conclusion and future outlooks
Efficient manganese-driven catalytic methodologies have rapidly advanced the platform for enabling the efficient formation of C–X (X = C, N, O, S) bonds and the construction of heterocyclic frameworks. The unique features of manganese catalysts, such as their earth abundance, low toxicity, and diverse oxidation states, allow their wide applicability in various organic transformations under mild and efficient conditions. Significant recent progress has demonstrated their usefulness across phenols, alcohols, alkenes, carbonyl compounds, allylic, benzylic, and heterocyclic C–H bonds, and strained-ring substrates, highlighting both the scope and effectiveness of manganese-catalyzed methodologies.
In spite of these accomplishments, challenges remain. The main areas for future exploration include increasing the substrate scope, improving stereoselectivity and regioselectivity, and developing additional ligand and catalytic systems. The thoughtful mechanistic exploration of various manganese-catalyzed methods remains restricted; therefore, further investigations are required to exploit the exceptional reactivity of manganese. Furthermore, manganese-driven catalysis, such as photo-redox strategies, flow chemistry, and electrochemical approaches, could also improve the general efficiency and versatile sustainability of these methods.
Looking forward, the development of manganese-catalyzed strategies for the synthesis of libraries of complex molecules, late-stage functionalization, and sustainable heterocycle synthesis is desired for continued progress. The unique combination of cost-effectiveness, environmental compatibility, and mechanistic versatility provides a great platform for manganese catalysis as a promising alternative with respect to traditional noble-metal systems, with substantial activity to make a significant impact on pharmaceutical chemistry, agrochemistry, and materials chemistry. Continuous discovery in this field is likely to expand the toolbox of synthetic chemists and contribute significantly to the advancement of green and sustainable chemistry.
Conflicts of interest
There are no conflicts to declare.
Data availability
No primary research results, software or codes have been included, and no new data were generated or analysed as part of this review.
Acknowledgements
Dr A. K. Das gratefully acknowledges the Department of Science & Technology and Biotechnology, Govt. of West Bengal for financial grant (Grant No. 2147(Sanc.)/STBT-11012(25)/1/2024-ST SEC).
References
-
(a) Y. Obora, Recent Advances in α-Alkylation Reactions using Alcohols with Hydrogen Borrowing Methodologies, ACS Catal., 2014, 4, 3972–3981 CrossRef CAS;
(b) G. E. Dobereiner and R. H. Crabtree, Dehydrogenation as a substrate-activating strategy in homogeneous transition-metal catalysis, Chem. Rev., 2010, 110, 681–703 CrossRef CAS PubMed;
(c) A. K. Das, S. Ali, A. Misra, S. Saikh, S. Dhibar, S. K. Panja, A. Das, G. Ghatak, L. K. Rathore, A. Bera, S. Bhar and S. Biswas, Eco-friendly cyanation strategies of aryl halides using recyclable nickel nanocatalysts with promising antibacterial and antioxidant activities, Mater. Adv., 2026, 7, 2349–2361 RSC;
(d) J. H. Docherty, T. M. Lister, G. Mcarthur, M. T. Findlay, P. Domingo-Legarda, J. Kenyon, S. Choudhary and I. Larrosa, Transition-Metal-Catalyzed C–H Bond Activation for the Formation of C–C Bonds in Complex Molecules, Chem. Rev., 2023, 123, 7692–7760 CrossRef CAS PubMed;
(e) A. Das, D. Chavda, M. Manna and A. K. Das, Dual-active Hf(iv)–organic framework for the detection of FOX-7 and as a heterogeneous catalyst for Knoevenagel condensation, New J. Chem., 2024, 48, 18249–18260 RSC.
-
(a) J. Rayadurgam, S. Sana, M. Sasikumar and Q. Gu, Palladium catalyzed C–C and C–N bond forming reactions: an update on the synthesis of pharmaceuticals from 2015–2020, Org. Chem. Front., 2021, 8, 384–414 RSC;
(b) P. Devendar, R. Y. Qu, W. M. Kang, B. He and G. F. Yang, Palladium-Catalyzed Cross-Coupling Reactions: A Powerful Tool for the Synthesis of Agrochemicals, J. Agric. Food Chem., 2018, 66, 8914–8934 CrossRef CAS PubMed.
- S. Genç, B. Arslan, S. Gulcemal, S. Gunnaz, B. Çetinkaya and D. Gucemal, Iridium(I)-Catalyzed C–C and C–N Bond Formation Reactions via the Borrowing Hydrogen Strategy, J. Org. Chem., 2019, 84, 6286–6297 CrossRef PubMed.
- D. A. Colby, R. G. Bergman and J. A. Ellman, Rhodium-Catalyzed C−C Bond Formation via Heteroatom-Directed C−H Bond Activation, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed.
- Y. Liu, K. P. Shing, V. K. Y. Lo and C. M. Che, Iron- and Ruthenium-Catalyzed C–N Bond Formation Reactions. Reactive Metal Imido/Nitrene Intermediates, ACS Catal., 2023, 13, 1103–1124 CrossRef CAS.
-
(a) A. Das and A. K. Das, A Functionalized Hf(IV)–Organic Framework Introducing Efficient, Recyclable, and Size Selective Heterogeneous Catalyst on MPV Reduction, New J. Chem., 2023, 47, 5347–5355 RSC;
(b) A. K. Das, S. Nandy and S. Bhar, Chemoselective and ligand-free aerobic oxidation of benzylic alcohols to carbonyl compounds using alumina-supported mesoporous nickel nanoparticle as an efficient recyclable heterogeneous catalyst, Appl. Organomet. Chem., 2021, 35, e6282 CrossRef CAS.
-
(a) A. K. Das, S. Ali, A. Misra, S. Islam, B. Kar, S. Biswas, G. Ghatak, D. Mal, M. Shit, M. Dolai and A. Das, Green Synthesized Biogenic Ag Nanoparticles With Enhanced Antibacterial, Antifungal, Antibiofilm, and Antioxidant Activities: Catalytic Applications in the ipso-Hydroxylation of Aryl Boronic Acids, Appl. Organomet. Chem., 2025, 39, e7796 CrossRef CAS;
(b) A. K. Das, N. Sepay, S. Nandy, A. Ghatak and S. Bhar, Catalytic efficiency of β-cyclodextrin hydrate-chemoselective reaction of indoles with aldehydes in aqueous medium, Tetrahedron Lett., 2020, 61, 152231 CrossRef CAS;
(c) A. K. Das and K. Sarkar, Emerging Trends in Two- and Three-Component Efficient Synthetic Strategies for Functionalised and Fused Quinoxalines, New J. Chem., 2025, 49, 12898–12930 RSC.
-
(a) A. K. Das, A. Misra, S. Ali, S. Saikh, S. Dhibar, K. K. Banerjee, G. Ghatak, D. Mal, M. Shit and S. Biswas, Investigating the potent antibacterial, antibiofilm, antidiabetic, and antioxidant activities of biosynthesized iron oxide nanoparticles: recyclable catalyst for ammoxidation of aromatic aldehydes, RSC Adv., 2025, 15, 35844–35858 RSC;
(b) A. K. Das, S. Saikh, A. Misra, S. Ali, P. Pradhan, N. Sepay, S. Dhibar, M. Afzal, J. Abbas and N. Sepay, Heterogeneous Nickel Nanoparticles Catalysed Ligand-Free Hydroxylation of Aryl halides in Aqueous Medium and DFT Investigation, J. Mol. Struct., 2026, 1352, 144012 CrossRef CAS;
(c) A. K. Das, S. Nandy and S. Bhar, Cu(OAc)2 catalysed aerobic oxidation of aldehydes to nitriles under ligand-free conditions, RSC Adv., 2022, 12, 4605–4614 RSC.
-
(a) Z. Wang, H. Xu, X. Han, S. Fan and J. Zhu, Manganese-Catalyzed Cycloalkene Ring Expansion Synthesis of Azaheterocycles, Org. Lett., 2024, 26, 8559–8564 CrossRef CAS PubMed;
(b) H. Liu, T. Tang, B. Li and B. Wang, Manganese(I)-catalyzed nucleophilic addition of C(sp3)–H bonds to aldehydes, Chem. Commun., 2024, 60, 5066–5069 RSC.
-
(a) S. A. Brunclik, A. A. Opalade and T. A. Jackson, Electronic structure contributions to O–O bond cleavage reactions for MnIII-alkylperoxo complexes, Dalton Trans., 2023, 52, 13878–13894 RSC;
(b) D. V. Kumar and B. Sundararaju, Manganese-Catalyzed Z-Selective Allylation of Indoles with Allenyl Derivatives, J. Org. Chem., 2024, 89, 10087–10092 CrossRef PubMed.
- P. S. Kulyabin, O. V. Magdysyuk, A. B. Naden, D. M. Dawson, K. Pancholi, M. Walker, M. Vassalli and A. Kumar, Manganese-Catalyzed Synthesis of Polyketones Using Hydrogen-Borrowing Approach, ACS Catal., 2024, 14, 10624–10634 CrossRef CAS PubMed.
-
(a) Y. Li, H. Wang, Y. Li, Y. Li, Y. Sun, C. Xia and Y. Li, Manganese-Catalyzed [4 + 2] Annulation of N–H Amidines with Vinylene Carbonate via C–H Activation, J. Org. Chem., 2021, 86, 18204–18210 CrossRef CAS PubMed;
(b) H. M. Qian, S. Jiang, Y. Zi, S. J. Ji and X. P. Xu, Mn(III) catalyzed cascade cross-coupling/annulation/C(O)-C bond insertion/rearrangement: Access to multi-substituted indolenines in water, Green Synth. Catal., 2025 DOI:10.1016/j.gresc.2025.03.011.
-
(a) S. Ali, A. Rani and S. Khan, Manganese-catalyzed C-H functionalizations driven via weak coordination: Recent developments and perspectives, Tetrahedron Lett., 2022, 97, 153749 CrossRef CAS;
(b) H. Yue, C. Zhu, L. Huang, A. Dewanji and M. Rueping, Advances in allylic and benzylic C–H bond functionalization enabled by metallaphotoredox catalysis, Chem. Commun., 2022, 58, 171–184 RSC.
- B. Zhao, B. Prabagar and Z. Shi, Modern strategies for C–H functionalization of heteroarenes with alternative coupling partners, Chem, 2021, 7, 2585–2634, DOI:10.1016/j.chempr.2021.08.001.
- L. Huan and C. Zhu, Manganese-catalyzed ring-opening chlorination of cyclobutanols: regiospecific synthesis of γ-chloroketones, Org. Chem. Front., 2016, 3, 1467–1471 RSC.
- M. M. El-bendary, A. Akhdhar, E. M. M. Ali, B. Davaasuren, M. Jaremko and B. A. Babgi, Synthesis, structural characterization and antitumor activities of manganese and cobalt isothiocyanate complexes with 2,2’-bipyridine, J. Coord. Chem., 2024, 77, 563–576 CrossRef CAS.
- S. S. Awaad, M. O. Sarhan, W. R. Mahmoud, T. Nasr, R. F. George and H. H. Georgey, New 2-aminobenzothiazole derivatives: Design, synthesis, anti-inflammatory and ulcerogenicity evaluation, J. Mol. Struct., 2023, 1291, 136042 CrossRef CAS.
- M. S. Gomaa, J. L. Armstrong, B. Bobillon, G. J. Veal, A. Brancale, C. P. F. Redfern and C. Simons, Novel azolyl-(phenylmethyl)]aryl/heteroarylamines:
Potent CYP26 inhibitors and enhancers of all-trans retinoic acid activity in neuroblastoma cells, Bioorg. Med. Chem., 2008, 16, 8301 CrossRef CAS PubMed.
- P. Yogeeswari, D. Srisam, L. R. J. Suniljit, S. Kumar and J. Stables, Anticonvulsant and neurotoxicity evaluation of some 6-chlorobenzothiazolyl-2-thiosemicarbazones, Eur. J. Med. Chem., 2002, 37, 231–236 CrossRef CAS PubMed.
- S. J. Hays, M. J. Rice, D. F. Ortwine, G. Johnson, R. D. Schwartz, D. K. Boyd, L. F. Copeland, M. G. Vartanian and P. A. Boxer, Substituted 2-Benzothiazolamines as Sodium Flux Inhibitors: Quantitative Structure-Activity Relationships and Anticonvulsant Activity, J. Pharm. Sci., 1994, 83, 1425–1432 CrossRef CAS PubMed.
- S. D. Dogan, M. G. Gündüz, S. B. Ugur, H. Dogan, C. Ozkul and Y. Çetinkaya, Copper-Oxone Promoted Oxidative C−H Functionalization: Synthesis of 2-Aminobenzothiazoles and Evaluation of Their Antimicrobial Activities, ChemistrySelect, 2021, 6, 4382 CrossRef CAS.
- V. S. Patil, K. P. Nandre, S. Ghosh, V. J. Rao, B. A. Chopade, B. Sridhar, S. V. Bhosale and S. V. Bhosale, Synthesis, crystal structure and antidiabetic activity of substituted (E)-3-(Benzo [d]thiazol-2-ylamino) phenylprop-2-en-1-one, Eur. J. Med. Chem., 2013, 59, 304–309 CrossRef CAS PubMed.
- E. Hockly, J. Tse, A. Barker, D. Moolman, J. Beunard, A. Revington, K. Holt, S. Sunshine, H. Moffitt, K. Sathasivam, B. Woodman, E. Wanker, P. Lowden and G. Bates, Evaluation of the benzothiazole aggregation inhibitors riluzole and PGL-135 as therapeutics for Huntington's disease, Neurobiol. Dis., 2006, 21, 228 CrossRef CAS PubMed.
- M. Scheetz, D. Carlson and M. Schinitsky, Frentizole, a novel immunosuppressive, and azathioprine: their comparative effects on host resistance to Pseudomonas aeruginosa, Candida albicans, herpes simplex virus, and influenza (Ann Arbor) virus, Infect. Immun., 1977, 15, 145 CrossRef CAS.
- M. Harriet, F. Bret and B. Paul, A Review of its Pharmacodynamic and Pharmacokinetic Properties and Therapeutic Potential in Amyotrophic Lateral Sclerosis, Drugs, 1996, 52, 549–563 CrossRef PubMed.
- J. Van Heusden, R. Van Ginckel, H. Bruwiere, P. Moelans, B. Janssen, W. Floren, B. J. Van der Leede, J. Van Dun, G. Sanz, M. Venet, L. Dillen, C. Van Hove, G. Willemsens, M. Janicot and W. Wouters, Inhibition of all-TRANS-retinoic acid metabolism by R116010 induces antitumour activity, Br. J. Cancer, 2002, 86, 605–611 CrossRef CAS PubMed.
- J. Yu and X. Jiang, Synthesis and perspective of organosulfur chemicals in agrochemicals, Adv. Agrochem, 2023, 2, 3–14 CrossRef.
- B. G. Reed-Berendt and L. C. Morrill, Manganese-Catalyzed N-Alkylation of Sulfonamides Using Alcohols, J. Org. Chem., 2019, 84, 3715–3724 CrossRef CAS PubMed.
- L. M. Azofra, M. A. Tran, V. Zubar, L. Cavallo, M. Rueping and O. El-Sepelgy, Conversion of racemic alcohols to optically pure amine precursors enabled by catalyst dynamic kinetic resolution: experiment and computation, Chem. Commun., 2020, 56, 9094 RSC.
- J. Neumann, S. Elangovan, A. Spannenberg, K. Junge and M. Beller, Improved and General Manganese-Catalyzed N-Methylation of Aromatic Amines Using Methanol, Chem.–Eur. J., 2017, 23, 5410–5413 CrossRef CAS PubMed.
- L. Homberg, A. Roller and K. C. Hultzsch, A Highly Active PN3 Manganese Pincer Complex Performing N-Alkylation of Amines under Mild Conditions, Org. Lett., 2019, 21, 3142–3147 CrossRef CAS PubMed.
- D. Wei, P. Yang, C. Yu, F. Zhao, Y. Wang and Z. Peng, N-Alkylation of Amines with Alcohols Catalyzed by Manganese(II) Chloride or Bromopentacarbonylmanganese(I), J. Org. Chem., 2021, 86, 2254–2263 CrossRef CAS PubMed.
- P. Daw, A. Kumar, N. A. E-Jalapa, Y. B. David and D. Milstein, Direct Synthesis of Amides by Acceptorless Dehydrogenative Coupling of Benzyl Alcohols and Ammonia Catalyzed by a Manganese Pincer Complex: Unexpected Crucial Role of Base, J. Am. Chem. Soc., 2019, 141, 12202–12206 CrossRef CAS PubMed.
- A. Bruneau-Voisine, D. Wang, V. Dorcet, T. Roisnel, C. Darcel and J. B. Sortais, Mono-N-methylation of anilines with methanol catalyzed by a manganese pincer-complex, J. Catal., 2017, 347, 57–62 CrossRef CAS.
- S. Elangovan, J. Neumann, J.-B. Sortais, K. Junge, C. Darcel and M. Beller, Efficient and selective N-alkylation of amines with alcohols catalysed by manganese pincer complexes, Nat. Commun., 2016, 7, 12641 CrossRef PubMed.
- K. Azizi, S. Akrami and R. Madsen, Manganese(III) Porphyrin-Catalyzed Dehydrogenation of Alcohols to form Imines, Tertiary Amines and Quinolines, Chem.–Eur. J., 2019, 25, 6439–6446 CrossRef CAS PubMed.
- R. Fertig, T. Irrgang, F. Freitag, J. Zander and R. Kempe, Manganese-Catalyzed and Base-Switchable Synthesis of Amines or Imines via Borrowing Hydrogen or Dehydrogenative Condensation, ACS Catal., 2018, 8, 8525–8530 CrossRef CAS.
- S. Friaes, C. S. B. Gomes and B. Royo, Bis-Triazolylidenes of Manganese and Rhenium and Their Catalytic Application in N-Alkylation of Amines with Alcohols, Organometallics, 2023, 42, 1803–1809 CrossRef CAS.
- S. S. Gawali, B. K. Pandia, S. Pal and C. Gunanathan, Manganese(I)-Catalyzed Cross-Coupling of Ketones and Secondary Alcohols with Primary Alcohols, ACS Omega, 2019, 4, 10741–10754 CrossRef CAS PubMed.
- U. K. Das, Y. Ben-David, Y. Diskin-Posner and D. Milstein, N-Substituted Hydrazones by Manganese-Catalyzed Coupling of Alcohols with Hydrazine: Borrowing Hydrogen and Acceptorless Dehydrogenation in One System, Angew. Chem., Int. Ed., 2018, 57, 2179–2182 CrossRef CAS PubMed.
- B. G. Reed-Berendt, N. Mast and L. C. Morrill, Manganese-Catalyzed One-Pot Conversion of Nitroarenes into N-Methylarylamines Using Methanol, Eur. J. Org Chem., 2020, 1136–1140 CrossRef CAS.
- S. Jana, S. Pande, A. K. Sinha and T. Pal, Synthesis of Superparamagnetic β-MnO2 Organosol: a Photo-Catalyst for Oxidative Phenol Coupling Reaction, Inorg. Chem., 2008, 47, 5558–5560 CrossRef CAS PubMed.
- M. Huang, Y. Li, Y. Li, J. Liu, S. Shu, Y. Liu and Z. Ke, Room temperature N-heterocyclic carbene manganese catalyzed selective N-alkylation of anilines with alcohols, Chem. Commun., 2019, 55, 6213–6216 RSC.
- X. Liu and T. Werner, Selective Construction of C−C and C=C Bonds by Manganese Catalyzed Coupling of Alcohols with Phosphorus Ylides, Adv. Synth. Catal., 2021, 363, 1096–1104 CrossRef CAS.
- K. Das, A. Kumar, A. Jana and B. Maji, Synthesis and characterization of N,N-chelate manganese complexes and applications in Csingle bondN coupling reactions, Inorg. Chim. Acta, 2020, 502, 119358 CrossRef CAS.
- V. Escande, A. Velati, C. Garel, B.-L. Renard, E. Petit and C. Grison, Phytoextracted mining wastes for ecocatalysis: Eco-Mn®, an efficient and eco-friendly plant-based catalyst for reductive amination of ketones, Green Chem., 2015, 17, 2188–2199 RSC.
- D. Wei, A. B. Voisine, D. A. Valyaev, N. Lugan and J. B. Sortais, Manganese catalyzed reductive amination of aldehydes using hydrogen as a reductant, Chem. Commun., 2018, 54, 4302 RSC.
- W. Zhang, N. X. Wang, C. B. Bai, Y. J. Wang, X. W. Lan, Y. Xing, Y. H. Li and J. L. Wen, Manganese-Mediated Coupling Reaction of Vinylarenes and Aliphatic Alcohols, Sci. Rep., 2015, 5, 15250 CrossRef PubMed.
- D. Yamamoto, M. Soga, H. Ansai and K. Makino, Manganese-catalysed hydroperoxidation of carbon–carbon double bonds using molecular oxygen present in air and hydroxylamine under ambient conditions, Org. Chem. Front., 2016, 3, 1420–1424 RSC.
- J. Yang, X. Wu, B. Yang, Y. Liu, R. Cheng, Z. Gong and F. Sun, Mn(II)-Catalysed ortho-alkenylation of aromatic amines and its application in reproductive diseases, RSC Adv., 2021, 11, 164–167 RSC.
- X. Huang, T. M. Bergsten and J. T. Groves, Manganese-Catalyzed Late-Stage Aliphatic C–H Azidation, J. Am. Chem. Soc., 2015, 137, 5300 CrossRef CAS PubMed.
- J. R. Clark, K. Feng, A. Sookezian and M. C. White, Manganese-catalysed benzylic C(sp3)–H amination for late-stage functionalization, Nat. Chem., 2018, 10, 583 CrossRef CAS PubMed.
- S. M. Paradine, J. R. Griffin, J. Zhao, A. L. Petronico, S. M. Miller and M. C. White, A manganese catalyst for highly reactive yet chemoselective intramolecular C(sp3)–H amination, Nat. Chem., 2015, 7, 987 CrossRef CAS PubMed.
- Y. Kohmura and T. Katsuki, Mn(salen)-catalyzed enantioselective C-H amination, Tetrahedron Lett., 2001, 42, 3339–3342 CrossRef CAS.
- Y. Zhang, J. Dong, L. Liu, L. Liu, Y. Zhou and S. F. Yin, Manganese (III) acetate catalyzed oxidative amination of benzylic C (sp3)–H bonds with nitriles, Org. Biomol. Chem., 2017, 12, 2897–2901 RSC.
- X. Kong and B. Xu, v Manganese-Catalyzed ortho-C-H Amidation of Weakly Coordinating Aromatic Ketones, Org. Lett., 2018, 20, 4495 CrossRef CAS PubMed.
- S. Kumar, D. P. Satpute, G. N. Vaidya, M. Nagpur, S. K. Lokhande, D. Meena and D. Kumar, Micellar catalysis enabled synthesis of indolylbenzothiazoles and their functionalization via Mn(II)-catalyzed C2–H amination using pyridines, Tetrahedron Lett., 2020, 61, 152017 CrossRef CAS.
- J. Y. Kim, S. H. Cho, J. Joseph and S. Chang, Cobalt- and Manganese-Catalyzed Direct Amination of Azoles under Mild Reaction Conditions and the Mechanistic Details, Angew. Chem., 2010, 122, 10095–10099 CrossRef.
- H. Singh, P. Pal, C. Sen, A. B. Panda and S. C. Ghosh, Heterogeneous Cu-MnO-Catalyzed Direct C−H Amination of Azoles Using O2 as the Sole Oxidant, Asian J. Org. Chem., 2017, 6, 702–706 CrossRef CAS.
- S. L. Ko, E. Courtney, M. Light, D. Jones and G. P. McGlacken, Manganese-catalysed C2 allylation and deuteration of indoles in water, Tetrahedron Green Chem, 2023, 2, 100019 CrossRef.
- P. Pal, A. K. Giri, H. Singh, S. C. Ghosh and A. B. Panda, Heterogeneously Porous γ-MnO2-Catalyzed Direct Oxidative Amination of Benzoxazole through C-H Activation in the Presence of O2, Chem.–Asian J., 2014, 9, 2392–2396 CrossRef CAS PubMed.
- K. Sarkar, P. Behera, L. Roy and B. Maji, Manganese catalyzed chemo-selective synthesis of acyl cyclopentenes: a combined experimental and computational investigation, Chem. Sci., 2024, 15, 14287–14294 RSC.
- D. Wang, R. Ren and C. Zhu, Manganese-Promoted Ring-Opening Hydrazination of Cyclobutanols: Synthesis of Alkyl Hydrazines, J. Org. Chem., 2016, 17, 8043–8049 CrossRef PubMed.
- R. Ren, Z. Xu and C. Zhu, Manganese-catalyzed regiospecific sp3 C–S bond formation through C–C bond cleavage of cyclobutanols, Chem. Commun., 2016, 52, 8160–8163 RSC.
- J. Huo, Y. Yang and C. Wang, Manganese-Catalyzed [3 + 2] Cyclization of Ketones and Isocyanates via Inert C–H Activation, Org. Lett., 2021, 23, 3384–3388 CrossRef CAS PubMed.
- S. Cao and W. Duan, Microwave assisted solvent-free C-H amination by silica-supported manganese dioxide, Tetrahedron Lett., 2016, 57, 2390–2394 CrossRef CAS.
- F. Kallmeier, B. Dudziec, T. Irrgang and R. Kempe, Manganese-Catalyzed Sustainable Synthesis of Pyrroles from Alcohols and Amino Alcohols, Angew. Chem., Int. Ed., 2017, 56, 7261–7265 CrossRef CAS PubMed.
- T. S. Lai, H. L. Kwong, C. M. Che and S. M. Peng, Catalytic and asymmetric aziridination of alkenes catalysed by a chiral manganese porphyrin complex, Chem. Commun., 1997, 24, 2373 RSC.
- Y. Liu, S. J. Li, X. L. Chen, L. L. Fan, X. Y. Li, S. S. Zhu, L. B. Qu and B. Yu, Mn(III)-Mediated Regioselective 6-endo-trig Radical Cyclization of o-Vinylaryl Isocyanides to Access 2-Functionalized Quinolines, Adv. Synth. Catal., 2020, 362, 688–694 CrossRef CAS.
- N. Deibl and R. Kempe, Manganese-Catalyzed Multicomponent Synthesis of Pyrimidines from Alcohols and Amidines, Angew. Chem., Int. Ed., 2017, 56, 1663–1666 CrossRef CAS PubMed.
- T. Liu, Y. Hu, Y. Yang and C. Wang, Manganese-Catalyzed Deoxygenative [3+2] Annulations of Ketones and Aldehydes via C–H Activation, CCS Chem., 2020, 2, 749–757 Search PubMed.
- C. Timmons, L. M. Mcpherson, D. Chen, H. X. Wei and G. Li, Manganese (IV) oxide-catalyzed electrophilic diamination of electron-deficient alkenes provides an easy synthesis of α,β-diamino acid and ketone derivatives for peptidomimetic studies, J. Pept. Res., 2005, 66, 249–254 CrossRef CAS PubMed.
- C. Wang, J. Yang, X. Meng, Y. Sun, X. Man, J. Li and F. Sun, Manganese(ii)-catalysed dehydrogenative annulation involving C–C bond formation: highly regioselective synthesis of quinolines, Dalton Trans., 2019, 48, 4474–4478 RSC.
- T. Aneeja, A. Chandravarkar and G. Anilkumar, A tandem strategy for the synthesis of 2-aminobenzothiazoles via manganese catalyzed C-S bond formation, Catal. Commun., 2024, 187, 106875 CrossRef CAS.
- F. F. Yong and Y. C. Teo, Manganese-catalyzed cross-coupling reactions of aliphatic amines with aryl halides, Tetrahedron Lett., 2010, 51, 3910–3912 CrossRef CAS.
- S. Zhang, Y. Liu, F. Xing and C. M. Che, Direct preparation of unprotected aminimides (R3N+–NH−) from natural aliphatic tertiary alkaloids (R3N) by [Mn(TDCPP)Cl]-catalysed N-amination reaction, Chem. Commun., 2020, 56, 9102 RSC.
- R. V. Putten, G. A. Filonenko, A. M. Krieger, M. Lutz and E. A. Pidko, Manganese-Mediated C–C Bond Formation: Alkoxycarbonylation of Organoboranes, Organometallics, 2021, 40, 674–681 CrossRef PubMed.
- J. Yang, R. Weinberg and R. Breslow, The hydroxylation and amidation of equilenin acetate catalyzed by chloro [5,10,15,20-tetrakis(pentafluorophenyl)porphyrinato]manganese(III), Chem. Commun., 2000, 7, 531–532 RSC.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Kumares Sarkar
b,
Suchandra Bhattacharjee
c,
Sushmita Gajurel
c,
Subhendu Dhibar
d,
Sumit Kumar Panja
*a,
Kumares Sarkar
b,
Suchandra Bhattacharjee
c,
Sushmita Gajurel
c,
Subhendu Dhibar
d,
Sumit Kumar Panja