DOI:
10.1039/D6RA01901C
(Paper)
RSC Adv., 2026,
16, 26526-26545
Development of an AQbD-guided sustainable RP-HPLC method for simultaneous quantification of saracatinib and rapamycin in lipid–polymer hybrid nanocarriers
Received
5th March 2026
, Accepted 13th May 2026
First published on 18th May 2026
Abstract
Saracatinib (SRC) and Rapamycin (RAP) are promising targeted anticancer agents that act through dual Src/Abl kinase inhibition and mTOR pathway suppression, respectively, demonstrating synergistic efficacy in head and neck cancer models. To overcome limitations such as poor tumor selectivity, low solubility, and systemic toxicity, both drugs were co-encapsulated into lipid–polymer hybrid nanoparticles (LPHNPs) to enhance bioavailability and therapeutic performance. However, no validated analytical approach has been reported for their simultaneous quantification in such nanocarrier systems. This research presents the development and optimization of a robust, sustainable and green reverse-phase high-performance liquid chromatography (RP-HPLC) method using an Analytical Quality by Design (AQbD) strategy. A Box–Behnken design was utilized to systematically evaluate critical method parameters (CMPs) and establish a design space, ascertaining consistent performance. Chromatographic separation was achieved under optimized isocratic RP-HPLC conditions with baseline resolution of both analytes. SRC and RAP were eluted at 3.787 and 9.462 minutes, respectively. The validated method showed outstanding linearity over the studied concentration, with an r2 of 0.9999 for SRC and 1 for RAP. The % RSD below 2% indicated high precision, robustness and complied with international regulatory standards, specifically the ICH Q2(R2) validation requirements. Furthermore, the method's applicability was confirmed by determining the drug entrapment efficiency (DEE) and monitoring in vitro drug release profiles of LPHNPs. Environmental sustainability was assessed using complementary green analytical metrics, including AGREE (0.68), AGREEprep (0.63), and ComplexMoGAPI (81), indicating a favourable sustainability profile with moderate-to-high environmental compatibility. Overall, the study delivers a validated, regulatory-compliant, and moderately green RP-HPLC method for the simultaneous quantification of dual-drugs, supporting the advancement of nanocarrier-based combination therapies in HNC management.
1. Introduction
Head and neck cancers (HNCs) constitute a diverse group of tumors that arise from the mucosal epithelial layer of the oral cavity, larynx, pharynx, salivary glands, lips, and paranasal sinuses.1–6 Globally, they contribute to over 800![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 new cases and approximately 400000 deaths each year, representing a major public health challenge.6–8 Despite advancements in therapeutic modalities—such as surgery, systemic chemotherapy, and radiotherapy—the prognosis for advanced-stage HNC remains poor due to high recurrence rates, resistance to conventional treatments, and dose-limiting toxicities.9–11 Standard chemotherapeutic agents such as cisplatin and molecular-targeted drugs like cetuximab have shown limited success, often hampered by poor tumor specificity and severe systemic side effects.12–16 These limitations highlight the need for novel therapeutic approaches and more effective drug delivery platforms that can enhance selectivity, reduce off-target toxicity, and improve therapeutic outcomes.17–21
000 new cases and approximately 400000 deaths each year, representing a major public health challenge.6–8 Despite advancements in therapeutic modalities—such as surgery, systemic chemotherapy, and radiotherapy—the prognosis for advanced-stage HNC remains poor due to high recurrence rates, resistance to conventional treatments, and dose-limiting toxicities.9–11 Standard chemotherapeutic agents such as cisplatin and molecular-targeted drugs like cetuximab have shown limited success, often hampered by poor tumor specificity and severe systemic side effects.12–16 These limitations highlight the need for novel therapeutic approaches and more effective drug delivery platforms that can enhance selectivity, reduce off-target toxicity, and improve therapeutic outcomes.17–21
Saracatinib (SRC), a potent dual inhibitor of Src and Abl kinases, and Rapamycin (RAP), an mTOR pathway inhibitor, have gained attention for their ability to disrupt key oncogenic pathways involved in tumor growth, angiogenesis, and metastasis in HNC.22–26 However, their clinical application is restricted by poor aqueous solubility, suboptimal pharmacokinetics, and non-specific biodistribution. To address these shortcomings, nanotechnology-driven drug delivery systems (DDS) have gained considerable attention. Among them, lipid–polymer hybrid nanoparticles (LPHNPs) have emerged as next-generation carriers, combining the advantages of both liposomes and polymeric nanoparticles.27–29 The lipid shell offers improved biocompatibility and stability, while the polymeric core provides structural integrity and sustained drug release. In this study, we developed a co-encapsulated LPHNP formulation of SRC and RAP to enhance their therapeutic efficacy in HNC treatment.
Despite the growing research on dual-drug delivery systems, there remains a significant analytical gap: no validated AQbD driven reverse-phase high-performance liquid chromatography (RP-HPLC) method has yet been established for estimating SRC and RAP simultaneously, particularly within a complex nanocarrier matrix. Such a method is essential for accurately assessing entrapment efficiency, evaluating in vitro drug release, and pharmacokinetic profiling, which are crucial for both formulation development and regulatory approval. While several RP-HPLC methods have been reported for kinase inhibitors or conventional drug combinations, few studies have addressed simultaneous multi-analyte quantification in structurally distinct dual-drug nanocarrier systems using a systematic AQbD-driven framework.30–35 Unlike conventional formulation-oriented assays that focus primarily on quantitation, the present work advances analytical science by integrating design-space-driven method development, multivariate robustness optimization, and green analytical metrics into a single lifecycle-oriented platform. This positions the method not merely as a formulation support tool, but as an innovative analytical strategy for complex combination nanomedicines. In parallel with quality-driven method development, Green Analytical Chemistry (GAC) has emerged as an important paradigm aimed at reducing hazardous solvent use, energy demand, and waste generation while maintaining analytical reliability. This is particularly relevant for chromatographic techniques such as HPLC, which often rely on substantial organic solvent consumption. Accordingly, incorporation of green assessment metrics has become increasingly important for evaluating the sustainability profile of analytical methods using structured criteria related to solvent toxicity, resource consumption, waste generation, and operational safety. In the present study, sustainability considerations were integrated through complementary greenness assessment tools to support the development of an environmentally conscious analytical method. To further position the novelty and analytical advantages of the present method, a comparative benchmarking of previously reported chromatographic methods and the proposed AQbD-based RP-HPLC method is presented in Table 1, which shows previously reported methods largely focus on single-analyte assays, bioanalytical applications, or individually incorporate AQbD, dual-drug analysis, or greenness evaluation. In contrast, the present method uniquely integrates simultaneous dual-drug quantification in LPHNPs with AQbD-driven optimization, comprehensive validation, and complementary greenness assessment, highlighting its broader analytical and sustainability advantages.36–39
Table 1 Comparative benchmarking of previously reported RP-HPLC/UPLC methods and the proposed analytical method
| Author details |
Analyte(s) |
Matrix/application |
AQbD/DoE |
Run time (min) |
Analytical performance |
Validation |
Greenness assessment |
Key distinction vs. present method |
| Kunte et al. (2025)34 |
Capecitabine |
Tablet dosage form |
Yes (BBD-AQbD) |
<10 |
Robust green RP-HPLC |
ICH |
NEMI, GAPI, ComplexGAPI, AGREE, AGREEprep |
Single-analyte dosage form method |
| Kothapalli & Vasanthan (2026)35 |
Trigonelline |
Bulk and nano systems |
Yes (CCD-AQbD) |
5.6 |
Efficient green separation |
ICH |
Multi-tool greenness metrics |
Single-analyte method |
| Sobhani et al. (2013)40 |
Rapamycin |
Bulk/analytical assay |
No |
Short |
Rapid reproducible RP-HPLC; LOQ 25 ng mL−1 |
ICH-type validation |
No |
Single-analyte assay without AQbD optimization or nanocarrier application |
| Svensson et al. (1997)41 |
Rapamycin |
Whole blood |
No |
<10 |
Sensitive UV-HPLC |
Bioanalytical |
No |
Therapeutic monitoring only |
| Attwa & Kadi et al. (2026)42 |
Saracatinib |
Human liver microsomes |
No |
1.0 |
Ultra-fast UPLC-MS/MS |
FDA bioanalytical |
Green extraction/solvent minimization |
Single-analyte metabolic stability method |
| Rathee et al. (2023)43 |
Posaconazole + hemp seed oil |
Nanomicelles |
Yes (BBD) |
8 |
Dual-drug separation |
ICH + forced degradation |
Not reported |
Different therapeutic system; no integrated sustainability assessment |
| Kumar et al. (2023)31 |
Lenvatinib + biochanin A |
PLGA nanoparticles and cubosomes |
Yes (BBD-QbD) |
Rapid |
Dual-drug method; Rs 6.7 |
ICH + USP |
Not reported |
Dual-drug comparator but lacks comprehensive greenness evaluation |
| Present method |
SRC + RAP |
LPHNPs (dual-drug nanocarrier) |
Yes (AQbD-BBD) |
∼18 |
Rs > 18 simultaneous separation |
ICH Q2(R2) |
AGREE + AGREEprep + ComplexGAPI |
Integrates dual-drug nanocarrier quantification, design-space optimization and sustainability assessment |
The present research aimed to develop a robust, eco-friendly, and regulatory-compliant reverse-phase high-performance liquid chromatography method for simultaneously quantifying SRC and RAP in LPHNPs using the AQbD framework. The methodology includes defining an Analytical Target Profile (ATP), assessing the systematic risk assessment, and optimizing critical method variables through a BBD to define the Analytical Design Space (ADS). The method was subsequently validated in compliance with the international regulatory standards as outlined in International Council for Harmonisation Q2(R2) guidelines to comprehensively evaluate its performance characteristics and ensure regulatory compliance. Key parameters of validation—namely linearity, accuracy, precision, and robustness—were systematically assessed using predefined experimental protocols and statistical criteria.44 In addition, the method's environmental sustainability was assessed using a green analytical chemistry tools, supporting its application in sustainable pharmaceutical development. This integrated analytical approach not only facilitates precise quantification of dual drugs in nanocarrier systems but also aligns with the current trends toward green and quality-centric analytical sciences.
2. Experimental
2.1 Materials
Saracatinib (MW: 542.0 g mol−1; purity > 98%; white crystalline powder) was procured from TCI Chemicals, Japan, while rapamycin (off-white powder, purity > 98%; 914.2 g mol−1) was kindly provided by Biocon Biologics, Bengaluru, Karnataka, India. Chromatographic-grade solvents like acetonitrile (ACN) and methanol were purchased from Sigma-Aldrich Chemicals Private Limited, Bengaluru, Karnataka, India. A 12 kilo Dalton molecular weight cut-off of dialysis membrane was procured from Merck Life Science, Mumbai, Maharashtra, India. BASF, Mumbai, India, generously supplied the Soluplus. Furthermore, DSPE-PEG2000 and soy lecithin were provided as kind gift samples from Lipoid, Germany. A 150 × 4.6 mm Agilent XDB C18 column (120 Å, 5 µm) was procured from Agilent Technologies, United States. Meanwhile, the 0.45 µm membrane filter was sourced from Riviera Glass Private Limited, Mumbai, Maharashtra, India. Additional analytical-grade reagents, including orthophosphoric acid, sodium hydroxide, and potassium dihydrogen phosphate, were procured from Sigma-Aldrich Private Limited, Bengaluru, Karnataka, India. Milli-Q water used throughout the experiments was prepared in-house utilizing a Milli-Q filtration unit (Millipore GmbH, Germany).
2.2 Instrumentation
A Shimadzu Corporation (Japan) HPLC system was employed for analysis, configured with an LC20-AD solvent delivery pump, a CTO-10ASvp column oven, an SIL20-AC HT autosampler, and a SPD-M20A detector. The system was operated, and data were processed employing LabSolutions (Shimadzu Corporation, Japan). For precise weighing of samples and standards, an analytical balance that has been well calibrated (Sartorius Mechatronics, India) was utilized. The mobile phase was processed via a 0.45 µm membrane filter (Riviera Glass, India) and sonicated in a bath sonicator (Labtronics, India) for efficient degassing prior to use.
2.3 Preparation of analytical standard and tests samples for HPLC analysis
To prepare the primary stock (1 mg mL−1), accurately 10 mg each of Saracatinib (SRC) and Rapamycin (RAP) were dissolved individually in Milli-Q water (10 mL). After sonication to achieve complete dissolution, the primary stock solutions were diluted with Milli-Q water to obtain 500 µg mL−1 working stocks.
2.4 Determination of isosbestic wavelength
A stock solution containing both saracatinib and rapamycin was prepared in ACN to obtain a concentration of 1 mg mL−1. Aliquots of this solution were subsequently analyzed employing a PerkinElmer LAMBDA 365+ double-beam UV-vis spectrophotometer, with measurements conducted in quartz cuvettes over the wavelength spectrum of 200–800 nm to determine the maximum absorption (λmax), utilizing ACN as the blank. The isosbestic point of the two drugs was identified through overlay analysis of their UV spectra. The isosbestic point provides several advantages for simultaneously estimating two drugs.45–47 It offers certain benefits that include simpler data interpretation and assures that system suitability parameters are met. It also helps to specifically identify two or more active components that are in a single formulation. Hence, the isosbestic point was selected.
2.5 AQbD-based optimization of the chromatographic parameters
The initial phase of the AQbD approach involves defining the ATP to establish clear objectives for method development.38,48–51 This systematic strategy proceeds through sequential steps, including setting method goals, identifying potential risks, and pinpointing critical method parameters (CMPs) as well as critical quality attributes (CQAs) using design of experiments software (DoE).36,37,52 Design Expert® (v23.1, Stat-Ease® 360, USA) was used to construct the response surface methodology (RSM) by using the experimental values. A second-order design, specifically the Box–Behnken design, was applied, which is obtained from incomplete factorial designs and utilizes three levels for each factor.53,54 For optimizing the chromatographic conditions for simultaneously estimating both the drugs, a three factor, three levelled BBD was employed.55–57 The experimental design consisted of three parts, with one factor fixed at the zero level and the other two factors set at three levels. A second-order polynomial equation was analyzed employing the DoE software to evaluate the quadratic response surfaces. The Box–Behnken design ensured a cost-effective optimization process by requiring fewer runs than full factorial designs to accurately characterize the analytical design space. In the current study, 15 runs having 3 centre points were used. The quadratic equation shown below explains the implied model.| | |
Y = b0 + b1A + b2B + b3C + b12AB + b13AC + b23BC + b11A2 + b22B2 + b33C2
| (1) |
where Y is a response variable that corresponds with the combination of factors at each level; b0 represents the intercept; the terms b1 to b33 represent regression coefficients that are calculated on the basis of experimentally observed values, i.e., responses; and factor A, factor B, and factor C denote the coded values of independent factors.58,59 Experiments employing one-factor-at-a-time (OFAT) were carried out initially to understand individual factor influence and to define the appropriate ranges for the independent variables used in the BBD. As a prominent response surface methodology, the BBD optimizes experimental conditions by positioning experimental points at the multidimensional cube's edge midpoints and at the centre. This strategic placement minimizes the total number of required runs without compromising the predictive accuracy of the resulting models. All runs were randomized to eliminate bias due to uncontrolled variability. This research employed a three-level BBD to systemically optimize independent factors; concentration of ACN (% v/v) in mobile phase: A, mobile phase flow rate (mL min−1): B, and injection volume (µL): C. The selected dependent variables were retention time (tR) of SRC and RAP (R1 and R2, respectively), peak area of SRC and RAP (R3 and R4, respectively), and resolution (R5). On the basis of the desirability function derived from the model, optimal method conditions were selected for further validating the method and analytical application. In comparison to other experimental designs, Box Behnken design exhibits significant benefits. It consists of three components, 12 runs, and 3 replicates at the centre point, using less energy and time. Each element is also investigated and categorized at 3 fundamental levels.60 SI Table S1 provides an overview of the independent factors and dependent responses employed in Box Behnken design.
To optimize the chromatographic parameters, a desirability function approach was used to concurrently optimize multiple response variables. Each response were assigned individual desirability functions (di) based on predefined goals, including minimization of retention times (R1 and R2) and maximization of peak areas (R3 and R4) and resolution (R5). Each response was transformed into a desirability value between 0 (completely undesirable) and 1 (fully desirable) using Derringer's desirability function. The lower and upper bounds for each response were defined based on experimentally observed ranges obtained from the Box–Behnken design. Equal importance weights were assigned to all responses to ensure balanced optimization. The overall desirability (D) was determined as the geometric mean of individual desirability functions, expressed as:
| | |
D = (d1 × d2 × d3 × d4 × d5)1/5
| (2) |
where
d1–
d5 represent individual desirability values corresponding to each response. This approach ensures simultaneous optimization of all critical analytical attributes while maintaining method robustness.
2.6 Validation of HPLC method
This is a systematic process that provides a high degree of confidence that the developed method is acceptable for its specified use. The optimized reverse phase-HPLC was validated according to International Council for Harmonisation Q2(R2).44 The developed method was evaluated for linearity, precision, accuracy, limits of detection and quantification (LOD and LOQ), solution stability, robustness, and system suitability, thereby verifying the method to be reliable and reproducible for routine analysis.38,61–64
2.6.1 Linearity. Linearity was established over six different concentration levels from the limit of detection to 140% of 500 µg mL−1 (i.e., target concentration). The drug concentration (x) was plotted against the peak area (y) to generate calibration curves. The linear regression equations and correlation coefficient (r2) values were calculated to evaluate the strength of the linear relationship. The findings verified that the method produces concentration-dependent responses within the established range.
2.6.2 Accuracy. Method accuracy was evaluated via recovery studies at three distinct concentrations—80%, 100%, and 120%—by spiking the prepared formulation with known amounts of the standard drugs. The recovery (%) of SRC and RAP was determined. The acceptable recovery range was set between 98% and 102%, in accordance with the ICH framework, indicating the reliability of the method in estimating the analytes without interference from formulation excipients.
2.6.3 Precision of analytical method. Method repeatability was analyzed by assessing six replicate samples, each having 500 µg mL−1 of SRC and RAP. Intraday precision and interday precision were analyzed at three concentrations (100, 300, and 500 µg mL−1) of both drugs. For intraday precision, triplicate samples were analyzed at multiple time points on the same day to evaluate short-term variability, whereas interday precision was determined by evaluating triplicate samples on different days to confirm the method's consistency and reproducibility over time. For each assessment, the percent relative standard deviation (% RSD) was calculated, with an acceptance criterion of less than 2%, according to ICH guidelines, indicating good precision.
2.6.4 Specificity of analytical method. Specificity was assessed by analyzing blank/placebo formulation (drug-unloaded LPHNPs) and drug-loaded LPHNP samples under the optimized chromatographic conditions. Blank placebo samples containing all formulation excipients but excluding the analytes were prepared and processed similarly to test samples to evaluate potential interference arising from excipients at the retention times corresponding to SRC and RAP. Drug-loaded LPHNP samples containing both analytes were analyzed to verify simultaneous separation and detect any possible co-eluting matrix components. Chromatograms were examined for retention time consistency, peak symmetry, baseline separation, and presence or absence of interfering peaks at the analyte retention times. Resolution between SRC and RAP peaks was determined as part of specificity assessment, with adequate separation considered indicative of selective quantification. In addition, specificity was further supported through photodiode array (PDA) spectral evaluation by examining spectral consistency across the peaks of both analytes to assess peak homogeneity and exclude evidence of co-eluting components.
2.6.5 Determination of limit of detection (LOD) and limit of quantification (LOQ). The limit of detection (LOD) and limit of quantification (LOQ) were determined based on signal-to-noise criteria. The obtained LOD and LOQ values indicate that the method is sufficiently sensitive for the detection and quantification of SRC and RAP at low concentrations. These were calculated using eqn (3) and (4) as per guidelines prescribed by ICH.| |
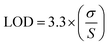 | (3) |
| |
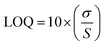 | (4) |
Here, σ represents the standard deviation (SD) of the intercept of the calibration curve, whereas S corresponds to the slope of the regression line.
2.6.6 Evaluation of solution stability. The solution stability of saracatinib and rapamycin was analyzed over a period of 72 hours at 2–8 °C (under refrigerated conditions). Sample solutions were kept in sealed containers and were subjected to analysis at predetermined time points of 0, 12 h, 24 h, 48 h, and 72 h to systematically monitor any changes over time. These controlled storage conditions were maintained to ensure that any measurable change in analyte concentration could be accurately attributed to intrinsic stability rather than external factors. At each specified time point, the samples were assessed employing the validated method, and the results were compared to the initial concentration. Stability was deemed acceptable if the percentage difference in peak area remained within the acceptance criterion of ±2%, signifying no substantial degradation.
2.6.7 Robustness. To evaluate the robustness of the optimized chromatographic method, small deliberate changes were introduced to critical parameters, including the temperature of the column, the pump flow rate, and the concentration of formic acid (FA) in the mobile phase. These controlled adjustments were designed to mimic small operational changes that may occur during daily laboratory practice and to evaluate their influence on analytical performance. The peak area's % RSD was calculated for each condition, with values below 2% considered acceptable, indicating the consistency and reliability of the analytical method under a varied environment.
2.6.8 Assessment of system suitability. The suitability of the system was evaluated by injecting six replicate standard solutions containing SRC and RAP. For each injection, critical parameters were calculated, including retention time to assess reproducibility of analyte elution, resolution to confirm adequate separation between peaks, and theoretical plate count as an indicator of column efficiency. Additionally, the capacity factor was determined to evaluate appropriate analyte retention, and the tailing factor was calculated to assess peak symmetry and overall chromatographic performance. The obtained values were compared with predefined system suitability acceptance criteria. Compliance with these limits confirmed the stability, efficiency, and reliability of the analytical setup, ensuring its suitability for accurate and precise quantitative analysis of SRC and RAP.
2.7 Formulation of SRC + RAP-loaded LPHNPs
Dual drug-loaded lipid polymer hybrid nanoparticles were formulated using a straightforward and reproducible film hydration method, with slight modifications from previously reported protocols.65,66 Briefly, the SRC and RAP were dissolved together in methanol at 1 mg mL−1 each, along with 20 mg mL−1 of Soluplus. The drug–polymer solution (10 mL) was subjected to complete evaporation of solvent under reduced pressure using a rotary vacuum evaporator, resulting in a thin, uniform film along the inner surface of the flask. This film was subsequently hydrated using Milli-Q water (10 mL) containing 0.1 mg mL−1 of DSPE-mPEG-2000 and 3 mg mL−1 of soy lecithin, serving as the lipid phase. Self-assembly of the LPHNPs was achieved by magnetically stirring the resulting mixture at a rate of 1200 rpm for a duration of 30 minutes at ambient temperature.67 The prepared nanosuspension was maintained under optimized storage conditions to preserve its physicochemical integrity until subsequent characterization and analytical testing.
2.8 Analysis of entrapment efficiency (%) and drug content (%)
The drug entrapment efficiency (% EE) for SRC + RAP in nanocarrier was determined by separating the free (unentrapped) drug from the supernatant by employing centrifugation (5810R, Eppendorf, Germany). The sample underwent centrifugation at a speed of 5000 rpm for 1 hour for the separation of the free drug. An aliquot of 10 µL of supernatant was then diluted with 1 mL of ACN to obtain a suitable concentration within the analytical range. The resulting sample was filtered to remove any residual particulates, and the resulting concentrations of both drugs were estimated with the help of the developed method.28,66 The % EE was calculated using eqn (5).| |
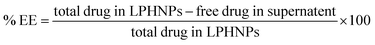 | (5) |
The drug content (% DC) and drug loading (% DL) were estimated by first preparing the nanoparticle sample for analysis. Approximately 0.1 mL of the formulation was accurately measured and mixed with 0.9 mL of ethanol to facilitate complete disruption of the carrier system and ensure thorough dissolution of the encapsulated SRC and RAP, and then vortexed for 3 minutes to obtain a homogeneous solution and promote efficient drug release from the matrix of LPHNPs, followed by filtering through a syringe filter (0.22 µm) to eliminate any residual particulates that might hinder the chromatographic performance. The filtrate was analyzed using validated method.66 The % drug content and % drug loading were estimated using the following eqn (6) and (7).29
| |
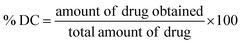 | (6) |
| |
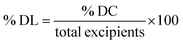 | (7) |
2.9 In vitro drug release
As previously reported,29,68–70 the dialysis membrane bag method was used to determine the in vitro release (%) of SRC and RAP from the nanocarrier. A 12 kilo Dalton dialysis membrane was employed. Phosphate buffer solutions (PBS) at pH 5.4 and 7.4 were employed as the dissolution media to simulate the tumor microenvironment and normal physiological conditions, respectively.28,71 The dialysis setup was maintained under continuous magnetic stirring at 100 rpm while the temperature of the media was maintained at 37 ± 2 °C. Sink conditions were maintained by withdrawing 1 mL aliquots and replacing with fresh media predetermined intervals (0, 0.25, 0.5, 1, 2, 4, 8, 12, 24, 48, and 72 h). The withdrawn samples were subsequently analyzed for cumulative drug release profile using developed RP-HPLC system (LC-2030C, Shimadzu, Japan).72
2.10 Evaluation of method greenness using green analytical chemistry (GAC) principles
The environmental sustainability of the developed analytical method was assessed using three complementary GAC tools, namely the Analytical GREEnness Metric (AGREE), Complex Modified Green Analytical Procedure Index (ComplexMoGAPI), and Analytical GREEnness Metric for Sample Preparation (AGREEprep). The AGREE tool was applied to assess the overall greenness of the analytical method based on the twelve principles of GAC, where each principle was scored and weighted according to its relevance, generating an integrated greenness score along with a visual pictogram representation. In parallel, the AGREEprep tool was employed to specifically evaluate the sustainability of the sample preparation procedure using ten criteria related to sample preparation placement, use of hazardous materials, waste generation, material sustainability, sample economy, throughput, level of automation, energy requirements, post-sample preparation configuration, and operator safety. Additionally, the ComplexMoGAPI tool was used to provide a holistic pictographic assessment of the entire analytical workflow, including sample preparation, instrumentation, reagents, occupational hazards, purification, and waste generation. The greenness profiles generated by these tools were interpreted through their respective numerical scores and color-coded visual outputs (green, yellow, and red zones) to comprehensively assess the environmental impact and overall sustainability of the developed analytical method.73–83
2.11 Statistical analysis
Statistical analyses were performed using GraphPad Prism (10.6.1, GraphPad Software, USA). Values expressed as mean ± SD (n = 3–15). To assess the significant differences among groups, one-way ANOVA was carried out, followed by Dunnett's test to compare each test group with the control. A p-value less than 0.05 was considered to be statistically significant, with each level of significance assigned by asterisks: *p < 0.05, **p < 0.01, ***p < 0.001, **p < 0.0001, while “ns” denoted results that were not significant.
3. Results and discussion
3.1 UV-visible spectral analysis for determination of isosbestic point
The UV absorption spectra of SRC and RAP were recorded over 200–800 nm range to identify a suitable wavelength for simultaneous quantification. Overlay analysis of the spectra revealed an isosbestic point at 267 nm, corresponding to the intersection of the absorbance curves of both analytes, indicating equal molar absorptivity (SI Fig. S1). The selection of the isosbestic wavelength offers a key analytical advantage by enabling simultaneous quantification of both drugs without the need for wavelength switching or multi-wavelength detection, thereby simplifying the analytical method and improving reproducibility. This approach is particularly beneficial for dual-drug systems where spectral overlap may otherwise complicate quantification. However, careful evaluation of the spectral profiles indicates that the absorbance curve of rapamycin exhibits a relatively steep slope in the vicinity of 267 nm. As a result, small deviations in detector wavelength could potentially lead to comparatively higher variability in RAP response. This highlights an inherent trade-off associated with the use of an isosbestic point, where analytical simplicity may come at the expense of increased sensitivity to wavelength variation for one of the analytes. Despite this consideration, the isosbestic wavelength was retained due to its overall advantages in method simplicity, consistency, and suitability for simultaneous estimation. Furthermore, the use of a photodiode array (PDA) detector with high wavelength accuracy and stability minimizes the practical impact of such variations under controlled analytical conditions. Alternatively, selection of a spectrally flatter wavelength region could reduce sensitivity to wavelength fluctuations; however, this may compromise detection sensitivity or require more complex analytical strategies. Therefore, the isosbestic point at 267 nm was considered an optimal compromise between analytical robustness, sensitivity, and operational simplicity. Nonetheless, wavelength robustness remains an important consideration and may be further explored in future method optimization studies.
3.2 Analytical method development using AQbD framework
Based on the AQbD strategy outlined in Section 2.5, risk assessment identified three critical method parameters (A: % ACN, B: flow rate and C: injection volume), which were subsequently optimized through BBD. The resulting analytical design space enabled simultaneous achievement of target retention behaviour, peak response and resolution. Detailed risk prioritization and model optimization outcomes are discussed in later sections.
3.3 Identification of CMPs through risk analysis
During RP-HPLC method development, the critical method parameters that could potentially influence analytical performance were carefully identified following the risk-based principles described in ICH Q9 framework.84 A structured risk analysis was performed for the independent variable (Xn), classifying the associated risk to each method response (Yn) as low, medium, or high. The results of this evaluation are depicted in SI Table S2. The risk assessment identified three high-risk factors: A (% acetonitrile), B (flow rate), and C (injection volume)-due to their pronounced effect on chromatographic responses. These factors significantly affected critical analytical attributes, including tR of SRC (R1) and RAP (R2), peak area of SRC (R3) and RAP (R4), and resolution (R5). Considering their substantial impact on analytical performance, these parameters were chosen as independent variables for subsequent optimization, and their corresponding experimental levels is presented in SI Table S1.
3.4 Statistical optimization
A three-factor, three-level BBD generated an experimental matrix comprising 15 runs. This design incorporated three centre points to specifically assess the reproducibility and inherent variability of the analytical procedure. Each of these 15 predicted run conditions was evaluated using reverse-phase HPLC, and the obtained results were analyzed to identify ideal chromatographic conditions. Table 2 depicts the results of each trial and the effect of individual as well as interactive variables on method performance. Considerable variation in the responses across the experimental domain confirmed that the selected factor ranges were appropriate for modelling and optimization. The centre-point replicates exhibited minimal variability, supporting good experimental reproducibility.
Table 2 Factor levels and their corresponding observed responses in BBD
| Runs |
Mobile phase conc. (A), % |
Flow rate (B), mL min−1 |
Injection volume (C), µL |
Retention time of SRC (R1)a, min |
Retention time of RAP (R2)a, min |
Peak area of SRC (R3)a, AU |
Peak area of RAP (R4)a, AU |
Resolution (R5)a |
| Mean ± SD; n = 3. |
| 1 |
20 |
1 |
10 |
3.76 ± 0.02 |
9.45 ± 0.05 |
5424 ± 37 |
19327 ± 136 |
18.9 ± 0.1 |
| 2 |
30 |
0.8 |
10 |
4.10 ± 0.03 |
10.29 ± 0.06 |
5548 ± 42 |
19756 ± 187 |
20.6 ± 0.1 |
| 3 |
20 |
0.8 |
5 |
4.19 ± 0.03 |
10.54 ± 0.08 |
5248 ± 37 |
18682 ± 167 |
21.0 ± 0.1 |
| 4 |
20 |
1 |
10 |
3.80 ± 0.02 |
9.48 ± 0.05 |
5464 ± 32 |
19461 ± 176 |
19.1 ± 0.1 |
| 5 |
30 |
1 |
5 |
4.31 ± 0.04 |
10.84 ± 0.07 |
5067 ± 27 |
18058 ± 193 |
21.6 ± 0.1 |
| 6 |
10 |
1.2 |
10 |
3.12 ± 0.02 |
7.85 ± 0.04 |
5024 ± 41 |
17879 ± 161 |
15.7 ± 0.1 |
| 7 |
20 |
1.2 |
5 |
3.50 ± 0.03 |
8.79 ± 0.05 |
4519 ± 36 |
16084 ± 136 |
17.6 ± 0.1 |
| 8 |
30 |
1 |
15 |
4.16 ± 0.04 |
10.45 ± 0.07 |
6087 ± 49 |
21686 ± 203 |
20.8 ± 0.1 |
| 9 |
20 |
1.2 |
15 |
3.53 ± 0.03 |
8.88 ± 0.06 |
5693 ± 40 |
20298 ± 219 |
17.7 ± 0.1 |
| 10 |
20 |
1 |
10 |
3.78 ± 0.03 |
9.47 ± 0.06 |
5447 ± 24 |
19383 ± 184 |
19.4 ± 0.1 |
| 11 |
10 |
1 |
15 |
3.57 ± 0.03 |
8.98 ± 0.05 |
5912 ± 37 |
21056 ± 221 |
17.9 ± 0.1 |
| 12 |
30 |
1.2 |
10 |
3.98 ± 0.04 |
10.01 ± 0.08 |
4812 ± 25 |
17155 ± 157 |
19.9 ± 0.1 |
| 13 |
10 |
0.8 |
10 |
3.98 ± 0.04 |
10.01 ± 0.09 |
5492 ± 38 |
19556 ± 189 |
19.9 ± 0.1 |
| 14 |
10 |
1 |
5 |
3.61 ± 0.02 |
9.08 ± 0.07 |
4900 ± 51 |
17469 ± 127 |
18.1 ± 0.1 |
| 15 |
20 |
0.8 |
15 |
4.07 ± 0.03 |
10.24 ± 0.08 |
6193 ± 57 |
22037 ± 237 |
20.4 ± 0.1 |
ANOVA demonstrated that the developed quadratic models were statistically significant for all responses (p < 0.05), confirming that variations in chromatographic performance were meaningfully driven by changes in factor levels. The models exhibited high adjusted and predicted R2 values with close agreement between them, indicating strong explanatory and predictive capability (SI Table S3). Furthermore, adequate precision values for R1 to R5 were 21.94, 22.24, 24.88, 25.17, and 20.09, respectively, exceeded the threshold value of 4 for all responses, confirming adequate signal-to-noise ratio and model discrimination. The results affirm the validity of the quadratic polynomial model's suitability for optimizing the method. To represent the impact of experimental factors on the critical analytical attributes, a series of reduced quadratic models was established (eqn (8)–(12)), which were derived based on statistically significant regression coefficients.
| | |
R1 = +3.78 + 0.2825A − 0.2760B − 0.0340C + 0.1865AB − 0.0290AC + 0.0385BC + 0.0522A2 − 0.0373B2 + 0.0807C2
| (8) |
| | |
R2 = +9.47 + 0.7102A − 0.6938B − 0.0855C + 0.4693AB − 0.0728AC + 0.0967BC + 0.1476A2 − 0.0774B2 + 0.2191C2
| (9) |
| | |
R3 = +5444.89 + 23.42A − 303.82B + 518.93C − 66.97AB + 1.81AC + 57.00BC − 73.92A2 − 152.02B2 + 120.68C2
| (10) |
| | |
R4 = +19390.50 + 86.77A − 1076.91B + 1847.89C − 231.22AB + 10.32AC + 214.55BC − 256.02A2 − 547.90B2 + 432.65C2
| (11) |
| | |
R5 = +19.12 + 1.41A − 1.38B − 0.1716C + 0.9155AB − 0.1273AC + 0.1920BC + 0.1662A2 − 0.2550B2 + 0.3147C2
| (12) |
In addition to conventional statistical parameters, the predictive performance of the developed models was rigorously evaluated using model validation metrics, including Root Mean Square Error (RMSE) and Predicted Residual Error Sum of Squares (PRESS). The RMSE values were found to be low across all responses, with values of 0.0675 and 0.1673 for the retention times of SRC and RAP, respectively, while peak area responses exhibited RMSE values of 80.98 (SRC) and 284.54 (RAP), and the resolution model showed an RMSE of 0.3647. These low RMSE values indicate high predictive accuracy and minimal deviation between experimental and predicted responses. Consistently low PRESS values further confirmed the strong predictive capability of the developed quadratic models. Model adequacy was further validated through residual diagnostics and graphical analysis (SI Fig. S3). The predicted versus actual plots demonstrated a strong correlation between experimental and predicted values (Fig. 1), while residuals versus predicted plots showed random dispersion around zero without any discernible pattern (SI Fig. S4), indicating the absence of systematic bias. Additionally, normal probability plots of residuals exhibited near-linear behaviour, confirming the normal distribution of errors. Collectively, these statistical and graphical evaluations substantiate the adequacy, robustness, and predictive reliability of the developed models for analytical method optimization.
 |
| | Fig. 1 Predicted vs. actual graphs for the responses (a) SRC retention time; (b) RAP retention time; (c) SRC peak area; (d) RAP peak area; and (e) resolution. | |
Statistical modelling via BBD yielded polynomial regression equations that successfully mapped the influence of three independent variables on the dependent responses. These response variables were tR of SRC and RAP (R1 and R2, respectively), peak areas (R3 and R4), and resolution (R5). The model for R1 (SRC tR) revealed that ACN percentage (A) had a positive effect (with increased organic content prolonging retention time), while flow rate (B) had a negative influence (higher flow rates shortened tR due to reduced interaction time). A significant positive AB interaction indicated a synergistic influence on retention. A comparable pattern was evident for R2 (RAP tR), with increasing ACN concentration, prolonged retention, while higher flow rates shortened it. Additionally, the quadratic equations for A2 and C2 revealed a curve in the response surface, highlighting the existence of an optimal zone for each variable. With regard to peak area (R3 for SRC and R4 for RAP), they were primarily influenced by flow rate and injection volume. RAP demonstrated a particularly pronounced positive dependency on injection volume, which suggests that detector response is highly sensitive to volume variations. Both the model of peak area also included significant quadratic terms, especially B2 and C2, indicating non-linear effects that further reinforced the need for careful control of these parameters to maximize both signal intensity as well as analytical sensitivity.
For resolution (R5) between SRC and RAP, the model captured equal contributions from all dependent variables. The significant AB interaction emphasized the critical role of ACN concentration and flow rate interplay in achieving optimal separation. The presence of strong quadratic effects (A2, B2, C2) pointed to a well-defined optimum zone within the design space, affirming the utility of BBD in identifying and fine-tuning method conditions. By utilizing the developed polynomial models, three-dimensional (3D) response surface plots (Fig. 2) and perturbation plots (Fig. 3) were developed, which visually represented the relationships between variables and responses. These graphs were instrumental in defining the MODR, thus establishing the robustness and reproducibility of the developed method.
 |
| | Fig. 2 Response surface graphs (3D) illustrating the influence of independent variables on responses (labelled R1 through R5). These responses include: (a) SRC retention time; (b) RAP retention time; (c) SRC peak area; (d) RAP peak area; and (e) resolution. | |
 |
| | Fig. 3 Schematic illustration of perturbation graphs demonstrating the effect of independent factors on selected responses (labelled R1 through R5). These responses include: (a) SRC retention time; (b) RAP retention time; (c) SRC peak area; (d) RAP peak area; and (e) resolution. Curves correspond to variation in A: ACN concentration, B: flow rate, and C: injection volume. The x-axis denotes deviation from the reference point in coded units, where −1, 0, and +1 correspond to the actual values of 10%, 20%, and 30% ACN; flow rates of 0.8, 1.0, and 1.2 mL min−1; and 5, 10, and 15 µL injection volume, respectively. | |
Statistical analysis of the models demonstrated their high predictive power, as evidenced by low p-values, significant F-values, and high coefficients of determination (R2), all of which validate the derived regression equations. These combined outcomes showcase the effective use of the AQbD approach for developing an analytical method for co-quantification of SRC and RAP, particularly within the context of complex nanocarrier-based formulations.
3.4.1 Desirability and identification of optimum method condition. Using the AQbD framework, the chromatographic conditions were optimized employing desirability. The optimization criteria were defined such that retention times of SRC and RAP were minimized within the experimental range to ensure rapid analysis, while peak areas and resolution were maximized to achieve high sensitivity and efficient separation. The bounds for each response were selected based on the observed minimum and maximum values obtained from experimental runs. Equal weights and importance were assigned to all responses, ensuring that no single parameter disproportionately influenced the optimization outcome. The desirability ramp functions were applied accordingly to convert each response into an individual desirability scale prior to calculating the composite desirability (SI Table S4).The desirability score, calculated as a single metric integrating all responses, reached a peak score of 0.977 (Fig. 4). This near-unity result indicates an optimal balance among the CQAs, confirming the suitability and robustness of the final conditions. It is important to note that while baseline chromatographic separation typically requires a resolution (Rs) value of approximately 2, the optimized method yielded a significantly higher resolution (∼19). In the present study, resolution was treated as a maximization objective to ensure complete and reliable separation of the two structurally distinct and hydrophobic analytes within a complex nanocarrier matrix. Considering the potential presence of formulation excipients, degradation products, or matrix-related interferences, a higher resolution was deemed advantageous to enhance method selectivity, peak purity, and analytical robustness. Although a target-based optimization strategy (e.g., Rs = 8–10) could potentially reduce analysis time or solvent consumption, the selected chromatographic conditions resulted in a total run time of 18 minutes, which was considered acceptable in view of the improved separation efficiency and reliability of the method. The increased resolution provides an additional safety margin against co-elution, particularly important for routine analysis of complex formulations. Therefore, open-ended maximization of resolution was preferred to prioritize robustness and method reliability over marginal gains in analysis time. Nevertheless, adopting a target-based resolution criterion may be explored in future studies to further optimize run time and enhance the green analytical profile of the method. Based on the optimized desirability function, the optimum chromatographic conditions were determined as follows. A 150 mm × 4.6 mm, 5 µm, 120 Å Agilent XDB-C18 column was pre-selected based on its established suitability for reversed-phase separation of hydrophobic compounds and was used as the stationary phase throughout the study. The choice of column was not included as a variable within the AQbD optimization, which focused on CMPs such as concentration of mobile phase, flow rate, and injection volume. Simultaneous quantification of SRC and RAP without co-elution was achieved using an isocratic mobile phase comprising ACN and 0.1% FA in water (20:80, v/v), which provided optimal peak symmetry, resolution, and reproducibility, with a 1 mL min−1 of flow rate (for balanced throughput and resolution), and a 10 µL injection volume (for adequate sensitivity). The temperature of the column oven was kept at 25 °C to ensure temperature consistency, minimizing variability in retention times.
 |
| | Fig. 4 (a) Desirability plot showing the combined optimization score for the analytical method; and (b) overlay plot representing the optimal conditions for the developed method. | |
The reliability of the optimized method was validated by carrying out six replicate analyses (n = 6) under these conditions. Experimental response values were compared to the predictions from the regression models. In all cases, the relative error between predicted and actual responses was found to be <10%, confirming the accuracy, precision, and predictive strength of the model. This minimal deviation indicates that the developed model accurately depicts the analytical system and ensures consistent and reproducible performance across repeated runs (Table 3). The chromatogram, presented in Fig. 5, features symmetric, well-separated peaks for both saracatinib and rapamycin. This high-quality separation confirms that the optimized method is fit-for-purpose for routine analysis. The chromatographic separation of SRC and RAP on a C18 stationary phase can be rationalized based on their physicochemical properties, including polarity, lipophilicity, and ionization behaviour. SRC exhibits moderate lipophilicity (logP ∼ 3–4) and contains ionizable functional groups with reported pKa values that allow partial protonation under acidic conditions. In the present study, the mobile phase consisted of 0.1% FA, resulting in a low pH environment where SRC exists predominantly in its protonated and relatively more polar form. This reduces its interaction with the non-polar C18 stationary phase, leading to earlier elution (tR ≈ 3.8 min). In contrast, RAP is a highly lipophilic macrolide compound with a significantly higher logP (∼4.5–5.5) and lacks strongly ionizable groups within the studied pH range. As a result, RAP remains largely non-ionized and exhibits stronger hydrophobic interactions with the C18 stationary phase, resulting in prolonged retention (tR ≈ 9.4 min). Thus, the observed separation is primarily governed by differences in hydrophobicity and ionization behaviour, with SRC eluting earlier due to increased polarity under acidic conditions, while RAP shows delayed elution due to its strong lipophilic character. This physicochemical distinction enables effective resolution between the two analytes under the optimized chromatographic conditions.
Table 3 Checkpoint analysis of critical chromatographic responses under optimized conditions
| Chromatographic responses |
Predicted value |
Observed value |
Relative error |
Desirability |
| Values are presented as mean ± SD; n = 3. |
| Retention time of SRC (min)a |
3.78 ± 0.07 |
3.79 ± 0.02 |
0.1 |
0.977 |
| Retention time of RAP (min)a |
9.47 ± 0.17 |
9.46 ± 0.03 |
0.1 |
| Peak area of SRC (AU)a |
5450 ± 81 |
5441 ± 94 |
9 |
| Peak area of RAP (AU)a |
19409 ± 285 |
19303 ± 258 |
106 |
| Resolutiona |
19.1 ± 0.4 |
18.9 ± 0.1 |
0.2 |
 |
| | Fig. 5 RP-HPLC chromatogram demonstrating simultaneous separation of SRC and RAP in a standard solution (500 µg mL−1 each), having retention times of 3.787 minutes for SRC and 9.462 minutes for RAP. | |
3.5 Validation of RP-HPLC method
The developed method's validation was performed in accordance to the International Council for Harmonisation Q2(R2) framework,44 ensuring that all critical performance characteristics met established regulatory standards. A detailed overview of validation results is depicted in Table 4.
Table 4 A detailed overview of validation parameters
| Sr. no. |
Parameters |
Saracatinib (SRC)a |
Rapamycin (RAP)a |
| Mean ± SD; n = 3. |
| 1 |
Retention time (tR) |
3.79 ± 0.02 |
9.46 ± 0.03 |
| 2 |
Peak area |
3.84 × 106 ± 1.0 × 104 |
5.94 × 106 ± 2.3 × 104 |
| 3 |
Theoretical plates (N) |
28460 ± 130 |
77630 ± 360 |
| 4 |
Tailing factor (T) |
1.36 ± 0.02 |
1.80 ± 0.03 |
| 5 |
Capacity factor (k′) |
1.43 |
| 6 |
Resolution |
18.9 ± 0.1 |
| 7 |
Linearity equation |
y = 7709.9x − 11831 |
y = 11617x + 37014 |
| 8 |
R2 |
0.9999 |
1 |
| 9 |
Accuracy (% recovery) |
| 80% |
98.9 ± 0.6 |
100.2 ± 0.5 |
| 100% |
100.3 ± 0.3 |
99.9 ± 0.1 |
| 120% |
99.9 ± 0.2 |
100.4 ± 0.2 |
| 10 |
Precision (% relative standard deviation) |
| Intra-day precision (µg mL−1) |
| 100 |
0.4 |
0.7 |
| 300 |
0.4 |
0.4 |
| 500 |
0.3 |
0.4 |
| Inter-day precision (µg mL−1) |
| 100 |
0.7 |
0.8 |
| 300 |
0.4 |
0.5 |
| 500 |
0.3 |
0.4 |
| 11 |
LOD (µg mL−1) |
1.33 |
0.68 |
| 12 |
LOQ (µg mL−1) |
4.0 |
2.04 |
3.5.1 Linearity. Linearity of the RP-HPLC method for SRC and RAP was assessed using six standard concentration levels, each injected in triplicate, ranging from the limit of detection to 140% of 500 µg mL−1 (i.e., target concentration). Curves were generated by plotting drug concentration against the corresponding peak area, thereby establishing the proportional correlation between concentration of analyte and response of the detector across the specified range. The method demonstrated excellent linearity, with r2 of 0.9999 for SRC and 1.000 for RAP, reflecting a strong concentration–response relationship. The regression equations obtained from the calibration curves were found to be y = 7709.9x − 11831 for SRC and y = 11617x + 37014 for RAP (SI Fig. S2). These results confirm the linear nature of the method and its suitability for simultaneously estimating both drugs within the desired range.
3.5.2 Accuracy. Method accuracy was assessed by performing recovery studies conducted at 80%, 100%, and 120% of the target concentration. Known quantities of standard drugs were spiked into the pre-analyzed sample matrix and measured in triplicate. The recorded percentage recoveries-99.86 to 100.26% for SRC, and 99.87 to 100.36% for RAP (Table 4)-demonstrate the accuracy of the developed method. The mean recoveries ranged between 98% and 102% were within the acceptable range. The strong agreement between experimental results and theoretical values confirms the accuracy of the method and its reliability for quantifying both drugs simultaneously.
3.5.3 Precision of the analytical method. The precision of the optimized method was systematically evaluated by assessing repeatability, intraday precision, and interday precision to ensure the consistency and reproducible nature of the obtained results. Repeatability of the analytical method was evaluated by analyzing six replicate analyses of the same samples at a concentration of 500 µg mL−1, and the results are presented in Table 5. The % RSD values for peak area were below 2% for both analytes, indicating acceptable repeatability of the method. Interday and intraday precision studies were assessed at three distinct concentrations, and the resulting data are depicted in Table 4. Across all evaluated conditions, the % RSD remained below 2% confirm that the developed method produces reproducible results for simultaneously estimating SRC and RAP.
Table 5 Repeatability of the developed RP-HPLC method (n = 6)
| Parameter |
SRC |
RAP |
| Mean peak area |
3.84 × 106 |
5.94 × 106 |
| SD |
1.0 × 104 |
2.3 × 104 |
| % RSD |
0.3% |
0.4% |
3.5.4 Specificity of the analytical method. The specificity of the developed RP-HPLC method was evaluated based on the ability to distinctly resolve SRC and RAP without interference under the optimized chromatographic conditions. The chromatograms obtained for blank and formulation demonstrated well-resolved, symmetric peaks with no observable co-elution at the respective retention times (Fig. 6). The high resolution achieved between the analytes (Rs > 18) further confirms effective separation and absence of peak overlap. In addition, peak characteristics and spectral consistency (as evidenced by the chromatographic separation and peak characteristics observed under optimized conditions) support that the detected peaks correspond exclusively to the analytes of interest without interference from potential co-eluting components. Although placebo or blank formulation interference studies were not explicitly performed, the chromatographic behaviour and peak characteristics indicate that the method possesses adequate specificity for simultaneous estimation of SRC and RAP.
 |
| | Fig. 6 Representative specificity chromatograms of (top) blank/placebo LPHNP matrix showing absence of interfering or co-eluting excipient peaks, and (bottom) drug-loaded LPHNPs demonstrating well-resolved, selective peaks of SRC (3.96 min) and RAP (9.46 min). | |
3.5.5 LOD and LOQ. The LOD and LOQ for both SRC and RAP were determined to evaluate the sensitivity of the method. The values for LOD and LOQ for SRC and RAP were computed using eqn (3) and (4). The values for LOD were 1.33 µg mL−1 for SRC and 0.68 µg mL−1 for RAP, and the LOQ values were 4.00 µg mL−1 for SRC and 2.04 µg mL−1 for RAP, indicating that the developed HPLC method is sufficiently sensitive for detecting and quantifying both drugs at low concentrations.
3.5.6 Solution stability. The solution stability study was performed over 72 hours under refrigerated storage conditions, and the results are depicted in Table 6. Solutions were maintained in tightly closed jars and examined at 0, 12, 24, 48, and 72 hours. No notable changes in peak area or tR were observed during the study period, and the % RSD for SRC and RAP was found to be less than 2%, indicating stability of the solutions over 72 hours under the specified conditions.
Table 6 Solution stability of SRC and RAP (n = 3)
| Time (h) |
SRC (peak area; AU) |
RAP (peak area; AU) |
| 0 |
3.97 × 106 |
5.99 × 106 |
| 12 |
3.92 × 106 |
5.94 × 106 |
| 24 |
3.88 × 106 |
5.90 × 106 |
| 48 |
3.83 × 106 |
5.87 × 106 |
| 72 |
3.83 × 106 |
5.85 × 106 |
| Mean peak area |
3.89 × 106 |
5.91 × 106 |
| SD |
6.2 × 104 |
5.8 × 104 |
| % RSD |
1.6 |
0.9 |
3.5.7 Robustness. The robustness of the developed method was assessed by individually varying key chromatographic conditions, such as flow rate (±0.1 mL min−1), column oven temperature (±2 °C), and FA concentration in the mobile phase (±0.1%), while maintaining all other conditions unchanged. The effects of these variations on tR and peak area of both SRC and RAP were systematically assessed. The % RSD values for peak areas under all modified conditions were found to be below 2%, indicating that the developed method is robust and efficiently produces consistent and reliable results despite minor operational fluctuations (SI Table S5). Although wavelength variation was not experimentally included as a robustness parameter, its potential impact was critically considered based on the spectral characteristics of the analytes. In particular, the relatively steep absorbance profile of RAP near the selected isosbestic wavelength (267 nm) may increase sensitivity to minor wavelength deviations. However, the use of a photodiode array detector with high wavelength accuracy and stability minimizes such variations under normal operating conditions. This aspect has been discussed to provide a comprehensive understanding of method robustness.
3.5.8 System suitability. System suitability parameters were evaluated under optimized chromatographic parameters, and the results are summarized in Table 4. The method demonstrated acceptable theoretical plate count, tailing factor, and resolution, indicating that the system performance was adequate for reliable analysis.
3.6 Drug entrapment efficiency and loading analysis of optimized LPHNPs
The encapsulation efficiency values of 79.65 ± 1.12% (SRC) and 85.28 ± 1.26% (RAP), the results strongly suggest the effective incorporation of these two hydrophobic agents into the matrix of LPHNP. High % EE also suggests minimal loss of drug during the process of formulation and indicates a strong interaction between the drug and the hybrid matrix, likely due to the amphiphilic nature that favours encapsulation. The drug content (% DC) values were found to be consistent with the entrapment efficiency i.e. 78.96 ± 1.05% (SRC) and 84.84 ± 1.35% (RAP), indicating minimal drug loss during formulation and processing. The % DL of the optimized SRC and RAP-LPHNPs was determined to be 8.04 ± 0.78% and 9.36 ± 0.98%, respectively. These values reflect the effective drug entrapment per unit weight of the LPHNPs and suggest efficient utilization of the carrier material. The high drug loading increases the therapeutic payload, offering a promising strategy for reducing dosage volume and improving patient compliance in cancer therapy.
3.7 In vitro release of LPHNPs
As depicted in Fig. 7, the optimized LPHNP formulation provided a biphasic release profile of SRC and RAP over 72 hours. The system demonstrated a rapid initial release, with over 50% of the total drug load released within the first 8 hours. This burst effect can be due to the immediate diffusion or desorption of drug molecules that were weakly associated with or adsorbed onto the nanoparticle surface rather than being fully entrapped within the matrix. Following this, a sustained and controlled release phase was observed, characterized by a gradual diffusion of the remaining drug. Importantly, the drug release study was conducted at two different pH levels—pH 5.4 (tumor microenvironment) and pH 7.4 (physiological pH)—to evaluate the responsiveness of the formulation. Over the 72 hour study, the release of SRC and RAP was significantly higher at pH 5.4 (approximately 90%) in comparison to pH 7.4 (only about 45%). This enhanced release in acidic conditions is linked to the destabilization of the polymer–lipid matrix in the slightly acidic tumor environment, which consequently accelerates drug diffusion. The resultant pH-responsive biphasic release is a highly desired characteristic for cancer treatment, as it ensures targeted delivery at the tumor site while preventing off-target leakage during systemic transport.
 |
| | Fig. 7 % cumulative drug release of SRC (a), and RAP (b) at two different pH levels (5.4 and 7.4). Values are depicted as mean ± SD, n = 3. | |
3.8 Greenness assessment
The greenness and sustainability of the developed analytical method were comprehensively assessed using three complementary GAC assessment tools, namely the AGREE, AGREEprep, and the ComplexMoGAPI.75,77,79–83 As illustrated in the AGREE pictogram (Fig. 8a), the developed method achieved an overall greenness score of 0.68, indicating moderate-to-good environmental performance and substantial alignment with GAC principles. The central score is supported by the peripheral segment analysis, where most criteria exhibited favourable green ratings, particularly those related to direct analysis, minimal sample handling, automation, derivatization avoidance, and operator safety. However, some limitations were observed in principles related to waste generation, use of renewable reagents, and multi-analyte capability, represented by yellow-to-red segments, which moderately lowered the overall score. These findings suggest that while the method demonstrates good sustainability characteristics, certain aspects still offer room for further greening.
 |
| | Fig. 8 Greenness evaluation of the developed analytical method using complementary GAC assessment tools: (a) AGREE based on the twelve GAC principles; (b) AGREEprep based on ten sustainability criteria; and (c) ComplexMoGAPI. | |
To specifically assess the environmental sustainability of the sample preparation procedure, AGREEprep was employed (Fig. 8b), yielding an overall score of 0.63, further indicating moderate green performance for the extraction and preparation workflow. In the AGREEprep assessment, the weights assigned to each criterion were determined based on their relative importance in promoting the use of safer reagents and materials, minimizing hazardous inputs, and encouraging the generation of benign wastes, in accordance with green analytical chemistry priorities. The AGREEprep radial diagram showed particularly strong performance in sample consumption, energy consumption, and operator safety (green zones), whereas hazardous material usage and waste generation were identified as comparatively weaker criteria (yellow/orange zones). Despite these moderate penalties, the sample preparation protocol maintained efficient resource utilization, low energy demand, and minimal operational risk, supporting the sustainability of this stage of the analytical process.
Further holistic assessment using ComplexMoGAPI (Fig. 8c) provided a broader pictographic evaluation of the complete analytical workflow, including sample preparation, instrumentation, reagents, and waste management. The method attained a ComplexMoGAPI score of 81, with a predominance of green fields and only limited yellow/red zones in the pictogram. This high ComplexMoGAPI score confirms a favourable ecological profile, particularly with respect to high analytical yield, simplified work-up, low energy consumption, and minimal occupational hazards, while indicating only minor opportunities for improvement.
Taken together, the combined results from AGREE (0.68), AGREEprep (0.63), and ComplexMoGAPI (81) demonstrate that the developed method possesses moderate-to-high environmental compatibility, with especially strong performance in operational safety, energy efficiency, and procedural simplification, though some improvement in waste minimization and reagent sustainability remains possible. The use of these three complementary tools strengthens the reliability of the greenness assessment and confirms the developed method as a sustainable and environmentally conscious approach for pharmaceutical analysis. Detailed greenness assessment parameters are presented in Tables 7 and 8.
Table 7 Assessment of green analytical chemistry of RP-HPLC method using the AGREE approach, presenting individual principle scores, with equal weights to all components
| Sl. no. |
Principle/criterion |
Score |
| 1 |
Use of direct analytical techniques to minimize or eliminate additional sample preparation steps |
0.85 |
| 2 |
Emphasis on reducing both sample volume and overall sample quantity |
1.0 |
| 3 |
Preference for in situ measurements wherever feasible |
0.66 |
| 4 |
Integration of analytical operations to enhance energy efficiency and reduce reagent consumption |
1.0 |
| 5 |
Recommendation of automated and miniaturized analytical techniques |
1.0 |
| 6 |
Recommendation of automated and miniaturized analytical techniques |
1.0 |
| 7 |
Avoidance of excessive waste generation and implementation of appropriate waste disposal practices |
0 |
| 8 |
Preference for simultaneous multi-parameter or multi-analyte analysis over sequent single-analyte methods |
0.38 |
| 9 |
Recommendation to minimize overall energy consumption |
0.5 |
| 10 |
Preference for reagents derived from renewable resources |
0 |
| 11 |
Replacement of hazardous reagents with eco-friendly alternatives |
1.0 |
| 12 |
Adoption of protective measures to enhance safety of operator |
0.8 |
| |
Average greenness score |
0.68 |
Table 8 Assessment of green analytical chemistry of RP-HPLC method using the AGREEprep approach, presenting individual principle scores
| Sl. no. |
AGREEprep criterion |
Score |
Weight |
| 1 |
Positioning of sample preparation step |
0.66 |
1 |
| 2 |
Use of hazardous chemicals/materials |
0.33 |
5 |
| 3 |
Sustainability, renewability, and reuse potential of materials |
0.50 |
4 |
| 4 |
Waste production generated during sample preparation |
0.15 |
5 |
| 5 |
Sample size economy |
1.00 |
4 |
| 6 |
Sample processing throughput |
0.64 |
2 |
| 7 |
Degree of integration and automation |
0.50 |
1 |
| 8 |
Energy requirements during preparation |
1.00 |
4 |
| 9 |
Analytical configuration after sample preparation |
0.50 |
2 |
| 10 |
Safety considerations for the operator |
1.00 |
5 |
| |
Average score |
0.63 |
|
4. Conclusions
A novel, robust, and environmentally conscious RP-HPLC method was developed successfully for the simultaneous quantification of saracatinib and rapamycin using an AQbD-guided framework, wherein BBD enabled systematic optimization of CMPs and establishment of a reliable analytical design space. The optimized chromatographic parameters provided efficient separation of both analytes with excellent resolution, while maintaining a strong balance between analytical performance, robustness, and practical applicability for complex nanocarrier systems. Validation in accordance with International Council for Harmonization Q2(R2) guidelines confirmed the method to be precise, accurate, sensitive, specific, and reproducible, with all performance characteristics meeting regulatory acceptance criteria and supporting its suitability for routine qualitative and quantitative analysis. Statistical modelling further reinforced the predictive capability and reliability of the method, while greenness assessments using AGREE, AGREEprep, and ComplexMoGAPI demonstrated favourable environmental compatibility, highlighting the improved sustainability of the developed analytical procedure.
Beyond its application in formulation characterization, the present work contributes to analytical chemistry innovation by advancing beyond conventional empirical multi-analyte methods through integration of AQbD-driven design-space development, multivariate robustness optimization, and sustainability-oriented method evaluation within a unified lifecycle-based analytical platform. The establishment of a MODR, combined with high-resolution simultaneous analysis of two structurally distinct hydrophobic agents in a complex nanocarrier matrix, underscores the broader analytical relevance of the method. These features position the proposed approach not merely as a routine formulation assay, but as an innovative analytical strategy for complex combination nanomedicines.
The developed method was further demonstrated to be suitable for critical formulation applications, including drug entrapment and in vitro release studies, supporting its utility in advanced nanocarrier-based drug delivery research and pharmaceutical quality control. Nevertheless, although the method showed excellent performance for standard solutions and laboratory-prepared LPHNP formulations, its applicability to complex biological matrices such as plasma or tissues was beyond the scope of the present study. Future translation toward pharmacokinetic or bioanalytical applications will require additional validation addressing matrix effects, extraction recovery, endogenous interference, and analyte stability in accordance with regulatory bioanalytical guidelines. Optimization of appropriate sample preparation strategies, including protein precipitation or solid-phase extraction, would also be necessary for such applications. Overall, the proposed method represents a sustainable, regulatory-compliant, and analytically innovative platform with relevance extending beyond formulation analytics to broader pharmaceutical analysis, offering a valuable contribution toward modern, green, and quality-centric analytical science.
Author contributions
Mohit Angolkar: conceptualization, methodology, software, data curation, writing—original draft preparation; Sharanya Paramshetti: software, data curation, writing—original draft preparation; Darshan Patil: software, data curation, writing—review and editing; Asha Spandana K. M.: validation, formal analysis, investigation, visualization, supervision, funding acquisition; Riyaz Ali M. Osmani: project administration, investigation, resources, visualization, supervision, funding acquisition; H. V. Gangadharappa: validation, visualization, formal analysis, investigation; Adel Al Fatease: writing—review and editing, resources, investigation; Ali H. Alamri: methodology, resources, formal analysis, Umme Hani: validation, visualization, funding acquisition. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
Data are contained within the article and supplementary information (SI). Supplementary information: Fig. S1. Illustration of the isosbestic point in the UV absorption spectra of SRC (black line) and RAP (red line); Fig. S2. Linearity of SRC and RAP; (a) chromatograms of various concentrations ranging from the LOD to 140% of the desired concentration (i.e. 500 µg mL−1) for both drugs, and (b) standard calibration curve for both SRC and RAP with their respective regression equations. The findings are depicted as mean ± SD, n = 3; Fig. S3. Normal plots of residuals for the responses (a) SRC retention time; (b) RAP retention time; (c) SRC peak area; (d) RAP peak area; and (e) resolution; Fig. S4. Residual vs. Predicted plots for the responses (a) SRC retention time; (b) RAP retention time; (c) SRC peak area; (d) RAP peak area; and (e) resolution; Table S1. BBD variables for chromatographic factors and their level; Table S2. Risk assessment for critical method parameters vs. HPLC method responses; Table S3. Regression terms for responses of BBD; Table S4. Desirability function parameters for optimization; Table S5. Robustness data of various factors. See DOI: https://doi.org/10.1039/d6ra01901c.
Acknowledgements
The authors express heartfelt gratitude toward the Leadership and Management of JSS College of Pharmacy, Mysuru and JSS Academy of Higher Education and Research (JSS AHER), Mysuru, Karnataka, India, for providing all the necessary facilities and support for completion of the research work. The authors extend their appreciation to the Deanship of Research and Graduate Studies at King Khalid University, Abha, for funding this work through Large Research Project under grant no. RGP2/22/47. Authors would also like to acknowledge the generous help and support extended by the staff and technical experts of University Sophisticated Instrumentation Center (USIC), JSS AHER, Mysuru, Karnataka, India.
References
- L. Q. M. Chow, N. Engl. J. Med., 2020, 382, 60–72 Search PubMed.
- M. D. Mody, J. W. Rocco, S. S. Yom, R. I. Haddad and N. F. Saba, Lancet, 2021, 398, 2289–2299 CrossRef PubMed.
- A. Argiris, M. V Karamouzis, D. Raben and R. L. Ferris, Lancet, 2008, 371, 1695–1709 CrossRef CAS PubMed.
- E. Crozier and B. D. Sumer, Med. Clin. North Am., 2010, 94, 1031–1046 CrossRef PubMed.
- N. W. Johnson and H. K. Amarasinghe, Head and Neck Cancer, 2011, pp. 1–40 Search PubMed.
- D. E. Johnson, B. Burtness, C. R. Leemans, V. W. Y. Lui, J. E. Bauman and J. R. Grandis, Nat. Rev. Dis. Primers, 2020, 6, 92 Search PubMed.
- D. Stepnick and D. Gilpin, Semin. Plast. Surg., 2010, 24, 107–116 CrossRef PubMed.
- T. M. B. Rezende, M. d. S. Freire and O. L. Franco, Cancer, 2010, 116, 4914–4925 CrossRef PubMed.
- A. Guidi, C. Codecà and D. Ferrari, Med. Oncol., 2018, 35, 37 CrossRef.
- E. Alsahafi, K. Begg, I. Amelio, N. Raulf, P. Lucarelli, T. Sauter and M. Tavassoli, Cell Death Dis., 2019, 10, 540 Search PubMed.
- P. Zolkind, J. J. Lee, R. S. Jackson, P. Pipkorn and S. T. Massa, Head Neck, 2021, 43, 89–97 CrossRef PubMed.
- E. S. Davidi, T. Dreifuss, M. Motiei, E. Shai, D. Bragilovski, L. Lubimov, M. J. J. Kindler, A. Popovtzer, J. Don and R. Popovtzer, Head Neck, 2018, 40, 70–78 Search PubMed.
- V. Subbiah, J. E. Grilley-Olson, A. J. Combest, N. Sharma, R. H. Tran, I. Bobe, A. Osada, K. Takahashi, J. Balkissoon, A. Camp, A. Masada, D. J. Reitsma and L. A. Bazhenova, Clin. Cancer Res., 2018, 24, 43–51 CrossRef CAS PubMed.
- J. Yang, Z. Ju and S. Dong, Drug Deliv., 2017, 24, 792–799 CrossRef CAS.
- J. Weiss, J. Gilbert, A. M. Deal, M. Weissler, C. Hilliard, B. Chera, B. Murphy, T. Hackman, J. J. Liao, J. Grilley Olson and D. N. Hayes, Oral Oncol., 2018, 84, 46–51 CrossRef CAS PubMed.
- M. J. Awan, L. Nedzi, D. Wang, V. Tumati, B. Sumer, X. J. Xie, I. Smith, J. Truelson, R. Hughes, L. L. Myers, P. Lavertu, S. Wong and M. Yao, Ann. Oncol., 2018, 29, 998–1003 CrossRef CAS PubMed.
- T. T. Wu and S. H. Zhou, Int. J. Med. Sci., 2015, 12, 187–200 CrossRef CAS PubMed.
- Y. Zhao, H. Chen, X. Chen, G. Hollett, Z. Gu, J. Wu and X. Liu, Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol., 2017, 9, e1469 CrossRef PubMed.
- C. Hoffmann, C. Shen and C. Le Tourneau, Curr. Opin. Oncol., 2022, 34, 177–184 CrossRef PubMed.
- S. R. Panikkanvalappil, M. A. El-Sayed and I. H. El-Sayed, Head and Neck Cancer: Multimodality Management,
2nd edn, 2016, pp. 827–844 Search PubMed.
- M. Angolkar, S. Paramshetti, D. Patil, K. M. Asha Spandana, R. A. M. Osmani, H. V. Gangadharappa, A. Al Fatease, A. H. Alamri, U. Hani and A. Rajput, Pathol. Res. Pract., 2026, 282, 156453 CrossRef CAS PubMed.
- Saracatinib|C27H32ClN5O5|CID 10302451 – PubChem, https://pubchem.ncbi.nlm.nih.gov/compound/Saracatinib#section=MeSH-Entry-Terms, accessed 1 September 2025 Search PubMed.
- Rapamycin (TN)|C51H79NO13|CID 5497196 – PubChem, https://pubchem.ncbi.nlm.nih.gov/compound/Rapamycin-_TN, accessed 1 September 2025 Search PubMed.
- L. Lang, C. Shay, Y. Xiong, P. Thakkar, R. Chemmalakuzhy, X. Wang and Y. Teng, J. Hematol. Oncol., 2018, 11, 85 CrossRef PubMed.
- L. Lang, C. Shay, X. Zhao, Y. Xiong, X. Wang and Y. Teng, J. Hematol. Oncol., 2019, 12, 132 CrossRef CAS PubMed.
- M. M. Abu-Khalaf, M. A. Baumgart, S. N. Gettinger, I. Doddamane, D. P. Tuck, S. Hou, N. Chen, C. Sullivan, K. Lezon-Geyda, D. Zelterman, C. Hatzis, H. Deshpande, M. P. Digiovanna, M. Azodi, P. E. Schwartz and L. N. Harris, Cancer, 2015, 121, 1817–1826 CrossRef CAS.
- C. O. Silva, J. O. Pinho, J. M. Lopes, A. J. Almeida, M. M. Gaspar and C. Reis, Pharmaceutics, 2019, 11, 22 CrossRef CAS PubMed.
- H. Sarma, A. Dutta, A. Bharali, S. S. Rahman, S. Baruah, N. Biswas and B. P. Sahu, Drug Dev. Ind. Pharm., 2024, 50, 856–864 CrossRef CAS PubMed.
- R. Dave, U. Shah, A. Patel, R. Patel and M. Patel, J. Drug Deliv. Sci. Technol., 2025, 105, 106644 CrossRef CAS.
- A. M. Kashid and D. P. Kaldate, Discover Chemistry, 2025, 2, 218 CrossRef.
- A. Kumar, S. Jat, P. Kumar and A. Gulbake, Future J. Pharmaceut. Sci., 2023, 9, 110 CrossRef.
- R. S. Tambare, S. R. Shahi, V. C. Gurumukhi, S. M. Kakade and G. G. Tapadiya, Heliyon, 2024, 10, e39172 CrossRef CAS.
- A. Shamim, M. J. Ansari, A. Aodah, M. Iqbal, M. Aqil, M. A. Mirza, Z. Iqbal and A. Ali, ACS Omega, 2023, 8, 21618–21627 CrossRef CAS.
- R. Kunte, P. Sabale, K. Somkuwar, V. Sawale and V. Sabale, Chromatographia, 2025, 88, 877–889 CrossRef CAS.
- P. Kothapalli and M. Vasanthan, BMC Chem., 2026, 20, 46 CrossRef CAS PubMed.
- S. N. Adin, I. Gupta, M. Aqil, M. Mujeeb and A. K. Najmi, Biomed. Chromatogr., 2023, 37(8), e5648 CrossRef CAS PubMed.
- K. Y. Patel, Z. R. Dedania, R. R. Dedania and U. Patel, Future Journal of Pharmaceutical Sciences, 2021, 7, 141 CrossRef.
- A. H. Akabari, S. K. Patel, K. V. Shah, A. Patel, D. Modi, A. K. Sen, V. D. Ramani, D. P. Shah and S. P. Patel, Discover Chemistry, 2025, 2, 1 CrossRef.
- S. Paramshetti, M. Angolkar, D. Patil, A. S. K. M., R. A. M. Osmani, H. V. Gangadharappa, A. Al Fatease, A. H. Alamri and U. Hani, Microchem. J., 2025, 219, 116242 CrossRef CAS.
- H. Sobhani, A. Shafaati, N. Nafissi-Varcheh and R. Aboofazeli, Iran. J. Pharm. Res., 2013, 12, 77 CAS.
- J. O. Svensson, C. Brattström and J. Säwe, Ther. Drug Monit., 1997, 19, 112–116 CrossRef CAS.
- M. W. Attwa and A. A. Kadi, Anal. Methods, 2026, 18, 1695–1708 RSC.
- A. Rathee, P. Solanki, S. Verma, D. Vohora, M. J. Ansari, A. Aodah, K. Kohli and Y. Sultana, ACS Omega, 2023, 8, 30057–30067 CrossRef CAS PubMed.
- ICH Q2(R2) Validation of analytical procedures – Scientific guideline | European Medicines Agency (EMA), https://www.ema.europa.eu/en/ich-q2r2-validation-analytical-procedures-scientific-guideline, accessed 30 June 2025 Search PubMed.
- R. Kamal, P. Paul, D. Mukherjee, T. G. Singh, R. Bhatia and A. Awasthi, Sep. Sci. Plus, 2025, 8(4), e70042 CrossRef CAS.
- M. G. Fawzy, H. Saleh, A. Reda and E. A. Bahgat, Spectrochim. Acta, Part A, 2022, 283, 121585 CrossRef CAS.
- E. H. Mohamed, A. Hamza, A. Reda, O. Adel and S. Atef, Curr. Pharm. Anal., 2020, 16, 254–261 CrossRef CAS.
- A. Shamim, M. J. Ansari, A. Aodah, M. Iqbal, M. Aqil, M. A. Mirza, Z. Iqbal and A. Ali, ACS Omega, 2023, 8, 21618–21627 CrossRef CAS.
- M. Sha’at, A. F. Spac, I. Stoleriu, A. Bujor, M. S. Cretan, M. Hartan and L. Ochiuz, Pharmaceutics, 2022, 14, 1187 CrossRef.
- S. Jani, U. Shah, J. Patel and M. Dua, Green Analytical Chemistry, 2025, 13, 100271 CrossRef.
- P. Usgaonkar, A. Adhyapak and R. Koli, Sep. Sci. Plus, 2024, 7, e202400155 CrossRef CAS.
- P. Paul, R. Kamal, T. G. Singh, A. Awasthi and R. Bhatia, Sep. Sci. Plus, 2025, 8(7), e70097 CrossRef CAS.
- G. E. P. Box and D. W. Behnken, Technometrics, 1960, 2, 455–475 CrossRef.
- M. Saha, A. Gupta, S. Shetty, S. Mutalik, K. Nandakumar, H. Raghu Chandrashekar, N. Dhas and S. Moorkoth, Chromatographia, 2024, 87, 533–548 CrossRef CAS.
- K. Patel, U. A. Shah and C. N. Patel, Future Journal of Pharmaceutical Sciences, 2023, 9, 57 CrossRef.
- G. Kumar, P. Mullick, K. Nandakumar, S. Mutalik and C. M. Rao, Chromatographia, 2022, 85, 629–642 CrossRef CAS.
- P. Alam, F. Shakeel, M. Taleuzzaman, A. I. Foudah, M. H. Alqarni, T. M. Aljarba, S. Alshehri and M. M. Ghoneim, Processes, 2022, 10, 1082 CrossRef CAS.
- A. Kramar, S. Turk and F. Vrečer, Int. J. Pharm., 2003, 256, 43–52 CrossRef CAS PubMed.
- A. P. Munasur, V. Pillay, D. J. Chetty and T. Govender, Int. J. Pharm., 2006, 323, 43–51 CrossRef CAS PubMed.
- D. Bei, J. Marszalek and B. B. C. Youan, AAPS PharmSciTech, 2009, 10, 1032–1039 CrossRef CAS PubMed.
- V. Parab Gaonkar and K. Hullatti, J. Liq. Chromatogr. Relat. Technol., 2021, 44, 95–102 CrossRef CAS.
- Y. Zhu, J. Qin, W. Wu and L. Cai, Front. Chem., 2024, 12, 1450692 CrossRef CAS PubMed.
- U. Chaudhari, J. K. Sahu and P. R. Dande, Drug Metab. Bioanal. Lett., 2023, 16, 140–152 CrossRef CAS PubMed.
- H. Nsairat, W. Alshaer, Z. Lafi, S. Ahmad, A. Al-Sanabrah and M. El-Tanani, Bioanalysis, 2023, 15, 1393–1405 CrossRef CAS PubMed.
- M. Ersoz, A. Erdemir, S. Derman, T. Arasoglu and B. Mansuroglu, Pharm. Dev. Technol., 2020, 25, 757–766 CrossRef CAS PubMed.
- P. Sathishbabu, U. Hani, C. Shakeela, P. R. Hemanth Vikram, M. Ghazwani, R. A. M. Osmani, B. M. Gurupadayya and D. V. Gowda, J. Chromatogr. B, 2022, 1212, 123483 CrossRef CAS PubMed.
- R. Takayama, Y. Inoue, I. Murata and I. Kanamoto, Colloids Interfaces, 2020, 4, 28 CrossRef CAS.
- K. Avgoustakis, J. Controlled Release, 2002, 79, 123–135 CrossRef CAS PubMed.
- M. Zahiri, S. M. Taghdisi, K. Abnous, M. Ramezani and M. Alibolandi, Expert Opin. Drug Delivery, 2021, 18, 1309–1322 CrossRef CAS PubMed.
- J. P. Carmona-Almazán, A. B. Castro-Ceseña and S. A. Aguila, Nanoscale, 2024, 16, 15801–15814 RSC.
- R. S. Jadon, G. Sharma, N. K. Garg, N. Tandel, K. R. Gajbhiye, R. Salve, V. Gajbhiye, U. Sharma, O. P. Katare, M. Sharma and R. K. Tyagi, Colloids Surf., B, 2021, 203, 111760 CrossRef CAS PubMed.
- M. G. Shivananjegowda, U. Hani, R. A. M. Osmani, A. H. Alamri, M. Ghazwani, Y. Alhamhoom, M. Rahamathulla, S. Paranthaman, D. V. Gowda and A. Siddiqua, Pharmaceutics, 2023, 15, 221 CrossRef CAS PubMed.
- A. M. Michael, H. M. Lotfy and C. K. Nessim, Microchem. J., 2023, 190, 108669 CrossRef CAS.
- L. Yin, L. Yu, Y. Guo, C. Wang, Y. Ge, X. Zheng, N. Zhang, J. You, Y. Zhang and M. Shi, J. Pharm. Anal., 2024, 14, 101013 CrossRef PubMed.
- N. Sammut Bartolo, L. L. Gallo, K. Szyrner, P. I. Buhagiar and J. Vella Szijj, Anal. Methods, 2024, 16, 5931–5942 RSC.
- E. Y. Santali, I. A. Naguib, A. M. Alshehri, Y. A. Alzahrani, A. E. Alharthi, T. S. Alosaimi, B. D. Alsayali, I. Alsalahat, A. Almahri, M. A. S. Abourehab and F. F. Abdallah, Separations, 2022, 9, 275 CrossRef CAS.
- F. Pena-Pereira, W. Wojnowski and M. Tobiszewski, Anal. Chem., 2020, 92, 10076–10082 CrossRef CAS PubMed.
- K. M. Kelani, E. S. Elzanfaly, A. S. Saad, M. K. Halim and M. B. El-Zeiny, Microchem. J., 2021, 171, 106826 CrossRef CAS.
- Y. Syrgabek, M. Alimzhanova, P. A. García-Encina, J. J. Jiménez and R. López-Serna, Trends Environ. Anal. Chem., 2023, 39, e00206 CrossRef CAS.
- S. I. Kaya, G. Ozcelikay-Akyildiz and S. A. Ozkan, Green Analytical Chemistry, 2024, 11, 100159 CrossRef.
- F. R. Mansour, K. M. Omer and J. Płotka-Wasylka, Green Analytical Chemistry, 2024, 10, 100126 CrossRef.
- F. Pena-Pereira, M. Tobiszewski, W. Wojnowski and E. Psillakis, Adv. Sample Prep., 2022, 3, 100025 CrossRef.
- W. Wojnowski, M. Tobiszewski, F. Pena-Pereira and E. Psillakis, TrAC, Trends Anal. Chem., 2022, 149, 116553 CrossRef CAS.
- Step, ICH Guideline Q9(R1) on Quality Risk Management, 2025 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Sharanya Paramshetti
a,
Sharanya Paramshetti











