DOI:
10.1039/D6RA01760F
(Paper)
RSC Adv., 2026,
16, 20087-20102
Engineering the A-site cations in (Na/K/Rb)3InBr6 halides for enhanced optoelectronic and thermoelectric performance in renewable energy devices
Received
28th February 2026
, Accepted 1st April 2026
First published on
17th April 2026
Abstract
Perovskite halides are a subclass of perovskite materials that are widely known for their exceptional compositional tunability and structural flexibility. This manuscript reports first principles analysis of the (Na/K/Rb)3InBr6 perovskite halides. The structural analysis for all three halides is validated based on the optimization curves and formation energies. The obtained tolerance factors and octahedral titling indicate minimum distortions in the formation of the cubic structure of (Na/K/Rb)3InBr6. The stress-energy tensor matrix is employed to assess the elastic constants of A3InBr6 (A = Na, K, and Rb). The transverse rigidity, longitudinal stiffness and ductility decline when the A-site cation is exchanged from Na to Rb. The elastic anisotropy computed using the ELATE software indicates that among the investigated compounds, K3InBr6 displays comparatively reduced anisotropy and a more uniform elastic response, but Na3InBr6 and Rb3InBr6 show pronounced directional changes. This highlights the function of the alkali-ion size in modifying the mechanical properties of these halide systems. Direct bandgaps of 3.37 eV, 3.84 eV and 3.86 eV were determined for Na3InBr6, K3InBr6 and Rb3InBr6, respectively. The partial density of states plots indicate that for all studied halides, the Br-p states have a high contribution to the valence band. Optical analysis of Na3InBr6, K3InBr6 and Rb3InBr6 reveals a prominent peak and electronic excitations in the UV region. The Seebeck coefficient of the studied halides is high at low temperatures, while the electrical and thermal conductivities increase with temperature. The ZT values suggest that Na3InBr6, K3InBr6 and Rb3InBr6 are strong candidates for thermoelectric devices.
Introduction
Renewable energy sources are being developed as feasible and ecologically benign options for meeting the energy needs of the world. Global population growth and industrialization have resulted in a huge increase in energy demand.1 In this technological age, the world faces numerous problems, one of which is the attainment of clean and affordable energy, which has to be addressed as soon as possible.2 The intensive use of fossil fuels contributes significantly to carbon emissions, which are harmful for humans and all living beings.3,4 The negative impact of the intensive use of fossil fuels as an energy source has disrupted the climate equilibrium.5 The world is experiencing extreme weather occurrences as a result of global warming and human activity. The only way to attain zero carbon and fulfill the uninterrupted energy supply is to search for materials that are cost-effective and environmentally benign.6–8 It is now a top priority of researchers to develop renewable energy solutions as the consumption of large amounts of energy generated from fossil fuels is creating environmental problems.9–11 The search for unique materials with specified qualities is critical for advancing research and technology within materials research.12–14 Solar energy, in particular, is an important and long-term option that has lately been identified as the most cost-effective method of electricity generation.15,16 Understanding the technical applications, such as deep-UV nonlinear optical responses, is vital for evaluating material performance and inherent qualities.17 An extraordinary class of materials, known as perovskite materials, has been in demand due their wide range of applications and structural flexibility.18,19 In recent years, there has been an explosion of research on perovskite materials in photovoltaics and photoluminescence.20,21 Furthermore, halide perovskites with alkali metal ions possess the tendency to enhance the material brightness, bandgap and moisture resistance.22–24 Multiple perovskite halides have been investigated in the literature, such as lead-free perovskites Cs3Ag2X5 (X = I, Br, and Cl), by analyzing the GGA and HSE06 potentials, demonstrating that these halides exhibit strong prominent peaks in the UV region.25 RbZnX3 (X = Br, Cl, and F) halides possess indirect bandgaps of 0.103 eV, 1.387 eV and 3.63 eV, respectively, making them promising candidates for photovoltaic applications.26 The halides AlSnX3 (X = F, Cl, Br and I) were analyzed using different potentials. Analysis of all optical properties reveal that the studied halides exhibit excellent optical responses in the visible region.27 Double perovskites Z2NaCrCl6 (Z = K and Rb) were investigated using first principles analysis. Both studied halides are thermodynamically and mechanically stable.28 The electronic analysis of K2TlZI6 (Z = Al and In) suggests that the materials are viable for photovoltaic applications.29 A computational method was employed to investigate the physical attributes of Cs2KAsA6 (A = Cl, Br, and I). The mechanical attributes suggest that both halides are ductile and are viable for renewable energy devices.30 Using SCAPS-1D simulations, the properties of X2SnBr6 revealed direct bandgaps and a power energy conversion efficiency up to 30.62%.31 Thermoelectric analysis of Na2Sn(Cl/Br)6 reveals that both halides possess favorable ZT values.32 The BoltzTraP code was used to assess the transport performance of K2ReX6. The studied halides exhibit high ZT values which make them effective thermoelectric materials.33 The FP-LAPW approach is used for studying the physical attributes of LiMgI3 and NaMgI3 halides. First principles investigation reveals that both halides are ductile and exhibit indirect bandgaps. Furthermore, the thermoelectric properties reveal favorable transport parameters at room temperature, making them potential materials for green energy.34 DFT analysis of A2AlInI6 halides indicates strong absorption in the visible region and promising photovoltaic efficiencies determined via SLME. For all halides, the ZT value is approximately 0.7, revealing their potential for solar–thermoelectric renewable energy applications.35 The physical properties of Cs2GaBiX6 halides were predicted using different functionals. Comprehensive analysis of these attributes suggests that these halides are optically and thermally stable.36 DFT simulations of A2AgRhF6 (A = Na and Rb) show that they are indirect semiconductors with strong absorption in the UV-vis region. Thermoelectric analysis of the reported halides exhibits high ZT values, which indicate strong potential thermoelectric applications.37 As a thermoelectric attribute, Li2ATlCl6 (A = Na and K) demonstrates high thermoelectric performance at 1200 K.38 The physical attributes of Z2BIrCl6 (Z = Cs and Rb and B = Na and K) were determined using the mBJ approximation. The obtained bandgaps suggest that the material possesses strong absorption in the visible range.39 The CASTEP code was used to evaluate the physical attributes of AgSrM3. Evaluation of the mechanical properties indicates that the studied halides are ductile.40 The phonon curves of Cs2SiCl6, Cs2GeCl6, and Cs2SnCl6 indicate the structural stability of these halides.41 The present study introduces (Na/K/Rb)3InBr6 halides as a previously unexplored class of indium-based bromide perovskite-related materials, which are investigated via first principles analysis. This work provides the first systematic comparison of alkali-metal substitution at the A-site and its impact on structural stability and lattice distortions. The study reveals composition-dependent electronic bandgap tunability, which allows for controlled regulation of the optoelectronic properties. Comprehensive mechanical investigation verifies the elastic stability and ductile behavior, which have not been described before for this family. The optical response exhibits high visible-light absorption and low reflectance, indicating their potential for optoelectronic devices. These findings identify (Na/K/Rb)3InBr6 halides as innovative, stable, and adjustable materials for optoelectronic and energy-related applications.
Computational approach
Unlike traditional wave-function-based quantum mechanical approaches, which are computationally prohibitively expensive for complicated materials, DFT reduces the many-body problem of interacting electrons and nuclei to an effective single-particle formulation.42 This simplification is accomplished by the Kohn–Sham formalism, which allows for efficient and reliable evaluation of electronic structures. In this study, the Wien2K code is utilized to analyze the physical properties of (Na/K/Rb)3InBr6 halides. The Kohn–Sham equations are solved using the FP-LAPW method, and electron–ion interactions are better described using the mBJ potential.43 The halides are structurally optimized using the PBE-GGA potential. The elastic characteristics are determined by evaluating the three independent elastic constants using the second-order stress–strain relation. To achieve accurate self-consistent field convergence, we set the computational parameters to Gmax = 14 (a.u.)−1, Lmax = 12, and Kmax × RMT = 7. To prevent charge leakage and muffin-tin sphere overlap, the cut-off energy of −8 Ry was adopted. The muffin-tin radii for Na, K, Rb, In and Br are set to 2.47, 2.41, 1.53, 1.51, and 1.78 (a.u.)−1, respectively. To generate well-converged electronic structures, a dense k-point mesh with 1300 points in the first Brillouin zone is used. The SCF convergence criterion was set at 0.1 mRy for the total energy and 0.0001e for the electronic charge. The VESTA software was used to visualize the crystal formations. Furthermore, the optical response of (Na/K/Rb)3InBr6, including the dispersion and polarizability, were investigated using the Kramers–Kronig relationships.
Structural analysis and volume optimization
Modifying the structure of a material can alter its physical attributes drastically. In halide perovskites, cation and anion replacement can alter the material's structural parameters which can affect its physical attributes. In this section, we discuss the structural parameters of (Na/K/Rb)3InBr6 halides. The studied halides possess the general formula of double perovskite, A3BX6, in which the A-site is occupied by sodium, potassium and rubidium, the B-site is occupied by indium and the X-site contains bromine. The structural properties of the studied halides (Na/K/Rb)3InBr6 are investigated using the VESTA software.44 The atomic positions are obtained as Na/K/Rb (1/4, ¼, ¼), K (0, 0, ½), In (0, 0, 0) and Br (0, 0, 0.254). The bond lengths for Na–Br, K–Br, Rb–Br and In–Br are given as 3.57715 Å, 3.8513 Å, 4.05498 Å and 3.05329 Å, respectively. The computed atomic bond lengths between the A-site cations and X-site halide increase when the A-site cation is changed from Na to K and from K to Rb. This increment in the bond length indicates lattice expansion and reduced coulombic interaction strength between the A-site cation (Na, K, Rb) and X-site anion (Br). Fig. 1(a–c) presents visual representations of the crystal structures of Na3InBr6, K3InBr6 and Rb3InBr6. The crystal structures were adjusted to achieve a stable geometry by reducing the total energy and atomic forces while maintaining structural stability.45 This optimization produces precise equilibrium lattice parameters and bond angles, all of which are required to properly predict the material's electrical, mechanical, and optical properties. The obtained optimization curves for Na3InBr6, K3InBr6 and Rb3InBr6 are presented in Fig. 1. The most stable configuration for the studied halides is obtained by fitting the optimization data into the following Birch–Murnaghan equation as follows:| | 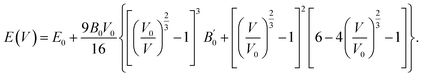 | (1) |
 |
| | Fig. 1 Unit cells and optimization curves of (a) Na3InBr6 (b) K3InBr6 and (c) Rb3InBr6. | |
The obtained values from curve fitting for the studied halides are presented in Table 1. The obtained value of the lattice constant increases when the A-site cation is substituted as Na → K → Rb, related to expansion of the crystal structure. This suggests that further replacement of the heavier cation at the A-site in the studied halide can further increase the lattice constant of the studied halides. The reported bulk modulus suggests a decreasing trend in the materials resilience towards external strain when the A-site cation is replaced as Na → K → Rb. The negative energies at equilibrium volumes determined for Na3InBr6, K3InBr6 and Rb3InBr6 suggest that the materials are structurally stable. The thermodynamic stability of the studied halides is assessed based on the formation energy, computed as follows:
| | | Ef = E(Na/K/Rb)3InBr6 − [3ENa/K/Rb − EIn − 6EBr]. | (2) |
Table 1 Optimized structural attributes of (Na/K/Rb)3InBr6
| Perovskites |
Na3InBr6 |
K3InBr6 |
Rb3InBr6 |
|
a = b = c (Å) |
11.11 |
11.645 |
11.917 |
| Angle |
90° |
| Symmetry/space group |
Cubic/Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m-225 m-225 |
|
B (GPa) |
22.49 |
18.96 |
18.079 |
|
B
|
5 |
5 |
5 |
|
V
0 (a.u3) |
2314.96 |
2664.43 |
2855.55 |
|
E
0 (Ry) |
−44021.773 |
−46660.083 |
−60936.23 |
|
E
F (Ry per atom) |
−1.705 |
−1.815 |
−1.787 |
|
τ
|
0.88 |
0.86 |
0.88 |
|
µ
|
0.412 |
0.416 |
0.428 |
The negative formation energies of the material suggest that the material is thermodynamically stable. This suggests that the material is energetically favorable under equilibrium conditions.46 Here, E(Na/K/Rb)3InBr6 is the ground-state energy, computed from the optimization curves; the individual energies for Na, K, Rb, In and Br are given as ENa/K/Rb, EIn and EBr. The computed formation energies for Na3InBr6, K3InBr6 and Rb3InBr6 are negative, which suggests that these halides are energetically favorable under equilibrium conditions. The tolerance factor for the studied halides was also computed to assess the structural stability. For cubic materials, the tolerance factor is suggested to be in the range of 0.8–1.11.47 The octahedral tilting is a measure of the stability of BX6 octahedra, which lies in the range of 0.40–0.5 (ref. 48) for cubic materials with minimum distortions. The computed tolerance factor and octahedral tilting for Na3InBr6, K3InBr6 and Rb3InBr6 suggest minimum distortions and complete structural stability.
Phonon dispersion curves
Phonon dispersion curves are essential for assessing the dynamic stability of a material by identifying the presence or absence of imaginary frequencies. The phonon dispersion curves of Na3InBr6, K3InBr6, and Rb3InBr6 are plotted along the high-symmetry path (Γ–X–M–Γ–R–X) in Fig. 2. The curves demonstrate the dynamic stability of all three compounds, as no imaginary (negative) frequencies are observed across the Brillouin zone. Each system exhibits three acoustic branches originating from the Γ-point in the low-frequency region, corresponding to collective lattice vibrations, followed by multiple optical branches at higher frequencies due to the presence of multiple atoms in the unit cell. Additionally, the presence of moderately dispersive and partially flat optical modes suggests localized vibrations and enhanced phonon scattering, which can contribute to reduced lattice thermal conductivity. These features highlight the stability and potential suitability of these materials for thermoelectric and energy-related applications.
 |
| | Fig. 2 Phonon curves for determining the stability criteria of (a) Na3InBr6 (b) K3InBr6 and (c) Rb3InBr6. | |
Mechanical properties
The mechanical properties of a material describe the material's resistance and resilience to external deformation.49 A material's ability to expand and contract in the provided elastic limit can be determined by assessing its mechanical properties. The mechanical properties are analyzed using the elastic constant as a metric. These elastic constants are independent and are obtained according to the material's symmetry. The stress and strain of the material are proportional to each other within the provided elastic limit, which is defined by Hooke's law. The second-order strain-energy equation is written as a quadratic expansion of the strain tensor, which relates the elastic constants to the stored energy due to deformation.50 The elastic strain energy is described by the second-order strain-energy equation as a quadratic expansion of strain tensor components. In this approach, the elastic constants act as coefficients that link the applied strain to the energy stored during deformation. As a result, the equation establishes a basic relationship between the microscopic elastic constants and macroscopic mechanical response of a material to tiny, reversible deformations. The equation is given as follows:| | 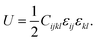 | (3) |
For the studied halides, three constants C11, C12 and C44 are obtained via the Thomas Charpin's technique.45,47 The calculated elastic constants for Na3InBr6, K3InBr6 and Rb3InBr6 fit Born's stability criteria. The elastic constant C11 indicates the material's ability to resist being stretched and compressed in one direction. C12 expresses how much the material's shape is altered in one direction when its pushed and pulled in another direction, whereas C44 defines how hard it is to slide one layer of the material over another.47 A higher C11 implies great resistance to uniaxial deformation, whereas lower C44 values imply moderate shear stiffness, which is good for structural flexibility and potential mechanical robustness under external stress. Among the studied halides, C11 for Na3InBr6 indicates the highest resistance to being stretched and compressed in one direction, as compared to K3InBr6 and Rb3InBr6. The value of C12 also indicates that Na3InBr6 possesses higher capacity for being pulled or pushed in another direction as compared to K3InBr6 and Rb3InBr6. Based on the computed value of C44, Rb3InBr6 demonstrates the highest level of shear deformation, as compared to K3InBr6 and Na3InBr6. The bulk modulus represents a material's capacity to be compressed when external pressure is applied.51 The bulk modulus for Na3InBr6 is moderately larger than that of K3InBr6 and Rb3InBr6, which suggests that more external pressure is required to compress Na3InBr6 than K3InBr6 and Rb3InBr6. The Na cation at the A-site of the material corresponds to stronger interatomic bonding and more compact crystal formation while remaining stable under high pressures. The toughness and stiffness of a material is indicated by the Young's modulus.52 The computed values of the Young's modulus suggest that Rb3InBr6 is stiffer and more rigid than Na3InBr6 and K3InBr6. This implies that an increase in the A-site cation ionic size can increase the material's stiffness. The shear modulus expresses the material's ability to resist shear deformation.53 When the A-site cation size is increased from Na to K and Rb, the computed shear modulus increases moderately, suggesting that Rb3InBr6 is more resistant to shear deformation, as compared with Na3InBr6 and K3InBr6. The anisotropy factor describes whether a material exhibits anisotropic or isotropic mechanical behavior. For a cubic system, an anisotropy factor of A = 1 indicates isotropic character, meaning the mechanical properties are independent of direction. Any deviation of A from 1 reflects anisotropic behavior.54 The anisotropy factor for the three halides indicates their anisotropic nature, indicating that the mechanical attributes are independent of the crystallographic directions. The Pugh's ratio is a common parameter indicating a material's ductile and brittle nature. Ductile materials are relatively tough and possess higher plastic deformation as compared to brittle materials, which are fragile and can easily break.55,56 This distinction results from the materials' differing atomic bonding and deformation mechanisms. Ductile materials can undergo extensive plastic deformation via dislocation motion prior to fracture, allowing them to absorb more energy under stress. Brittle materials, on the other hand, have restricted dislocation activity and fracture abruptly when the elastic limit is reached. As a result, brittle materials are often less appropriate for applications that require impact loading or mechanical stress fluctuations. The critical Pugh's ratio (B/G) for ductile materials should be greater than 1.75.57 The obtained values for the studied halides (Na3InBr6, K3InBr6 and Rb3InBr6) indicates their ductile nature as the Pugh's ratio is higher than the critical value of 1.75. When the A-site is substituted from Na to K and Rb, the material's ductility declines as the ionic size increases. The Poisson's ratio is another parameter that further confirms the material's ductility and brittleness suggested by the computed Pugh's ratio.56 The computed Poisson's ratio suggests that Na3InBr6, K3InBr6 and Rb3InBr6 are all ductile, as their Poisson's ratio is higher than 0.26. The Cauchy's pressure is another term that also aids in assessing a material's ductility and brittleness. A positive Cauchy's pressure suggests ductile nature, whereas brittleness is indicated by negative values of Cauchy's pressure.58 The computed values of the Cauchy's pressure for Na3InBr6, K3InBr6 and Rb3InBr6 suggest that the studied halides are ductile, consistent with the Pugh's and Poisson's ratios. The hardness factor reflects the materials ability to resist localized plastic deformations such as scratching and abrasion.59,60 The obtained values of the hardness factor imply that Rb3InBr6 is more resistant to localized deformation as compared to K3InBr6 and Na3InBr6. This indicates that increasing the cation size at the A-site of the studied material can lead to higher resistance to localized plastic deformations such as scratching and abrasion. The machinability index indicates the level of ease through which the material can be manufactured or processed.61 The machinability index shows a declining trend, which suggests that as the A-site cation is replaced by larger ionic-size cations, its level of ease in manufacturing declines. Moreover, it is more convenient to manufacture Na3InBr6 as compared to K3InBr6 and Rb3InBr6. Elastic wave propagation provides a direct measure of the material's wave velocity. Furthermore, it indicates the material's stiffness and rigidity.62 The longitudinal velocity represents the speed of compressional elastic waves passing through the material. Larger longitudinal velocities suggest stronger interatomic bonding and greater stiffness. The computed longitudinal velocities reveal that K3InBr6 possesses stronger bonding and is stiffer than Na3InBr6 and Rb3InBr6. The transverse velocity reflects the speed of shear elastic waves and is directly related to the shear rigidity. Higher transverse velocities suggest that the material exhibits better resistance to shear deformation. The obtained transverse velocities suggest that Rb3InBr6 possesses higher resistance to shear deformation while Na3InBr6 possesses the lowest shear stability. The mean sound velocity combines both longitudinal and transverse wave contributions. It is an important parameter, which is crucial for assessing the thermal and vibrational properties of a material.63 The obtained values of the mean sound velocity indicate that Rb3InBr6 possesses stronger phonon propagation, whereas Na3InBr6 shows comparatively compliant lattice dynamics. The Debye temperature of a material describes the temperature required for activating the vibrational modes in the lattice. Moreover, stronger bonding and better thermal conductivity are indicated by a higher Debye temperature.64 From the obtained values for the studied halides, it is noticeable that K3InBr6 exhibits stronger atomic interactions and stiffer lattice vibrations. At the melting temperature, the crystal breaks down into the liquid phase, reflecting its thermal stability. High melting temperatures suggest that a material possesses better thermal robustness.65,66 The obtained melting temperature revealed that Na3InBr6 possesses better thermal stability than K3InBr6 and Rb3InBr6 (Table 2).
Table 2 Mechanical properties of Na3InBr6, K3InBr6 and Rb3InBr6
| Parameters |
Na3InBr6 |
K3InBr6 |
Rb3InBr6 |
|
C
11 (GPa) |
41.803 |
35.802 |
39.43 |
|
C
12 (GPa) |
12.81 |
10.52 |
7.365 |
|
C
44 (GPa) |
2.88 |
5.569 |
5.747 |
|
B (GPa) |
22.49 |
18.96 |
18.079 |
|
E (GPa) |
16.2 |
20.5 |
22.68 |
|
S (GPa) |
5.88 |
7.77 |
8.79 |
|
A
|
0.19 |
0.43 |
0.35 |
|
B/G |
3.82 |
2.43 |
2.05 |
|
v
|
0.37 |
0.31 |
0.29 |
|
C
p (GPa) |
9.93 |
4.96 |
1.62 |
|
H
v
|
0.512 |
0.99 |
1.23 |
|
µ
m
|
7.80 |
3.41 |
3.14 |
|
v
l (m s−1) |
4140.3 |
4197.28 |
4045.54 |
|
v
t (m s−1) |
1823.75 |
2161.99 |
2198.33 |
|
v
m (m s−1) |
2058.76 |
2420.92 |
2452.55 |
|
θ
D (K) |
161.48 |
181.25 |
179.45 |
|
T
m (K) |
800.056 |
764.58 |
786.03 |
Elastic anisotropy
Elastic anisotropy quantifies the difference in the mechanical characteristics across crystallographic orientations. An anisotropy of 1 suggests that a material is totally isotropic, whereas deviations imply directional dependence of the elasticity. The elastic anisotropy of the studied halides was further assessed by employing the ELATE software.67 This software generates 2D and 3D images of variation in the material's mechanical properties in the crystallographic direction, which helps in describing the bond strength and mechanical stability. Fig. 2 demonstrates the 2D and 3D visualization of the Young's modulus, linear compressibility, Shear modulus and Poisson's ratio for Na3InBr6, K3InBr6 and Rb3InBr6. The Young's modulus quantifies the material's directional-dependent stiffness. A perfectly spherical shape indicates that a material is elastically isotropic, whereas lobed or non-spherical shapes indicate that a material is elastically anisotropic. Among the studied halides, K3InBr6 exhibits a smooth and more symmetric surface, suggesting lower anisotropy and uniform stiffness, whereas Na3InBr6 and Rb3InBr6 exhibit stronger anisotropy. The linear compressibility indicates the amount of compression a material can endure in one direction when external pressure is applied. The linear compressibility plots suggest that the obtained linear compressibility of all studied halides is less directionally dependent, as compared to the Young's modulus. Among the studied halides, Rb3InBr6 possesses enhanced linear compressibility compared to Na3InBr6 and K3InBr6. The shear modulus reflects the resistance to shape changes, which is critical for mechanical rigidity. The cross-like patterns in the 2D projections and the multi-lobed 3D surfaces show a considerable directional dependence. This anisotropy results from non-uniform resistance to shear deformation across distinct planes. Among the studied halides, K3InBr6 exhibits balanced shear behavior and Na3InBr6 and Rb3InBr6 display stronger anisotropy. Lastly, we assessed the transverse strain response to applied longitudinal stress, which is known as the Poisson's ratio. The very non-spherical, petal-like forms imply strong anisotropy in v. Directional variations indicate that bonding and angular forces vary greatly with orientation. For the studied halides, a variation in the ductility was noted in all crystallographic directions. In conclusion, all studied halides are elastically anisotropic despite being cubic. K3InBr6 exhibits the strongest isotropic mechanical response, suggesting better mechanical uniformity compared with Na3InBr6 and Rb3InBr6 (Fig. 3).
 |
| | Fig. 3 Elastic anisotropy plots of (a) Na3InBr6, (b) K3InBr6 and (c) Rb3InBr6. | |
Electronic properties
The nature of a material is determined by its electronic properties, and collectively, these properties govern the material's electronic behavior. Moreover, the electronic properties of the halides can be manipulated by altering the A-site cation or X-site anion, as these modifications alter the crystal structure and orbital hybridization in the material. The electronic properties of (Na/K/Rb)3InBr6 were obtained via the mBJ approximation and are presented in Fig. 4. All three studied halides exhibit no band-crossing at the Fermi level, indicating their semiconducting nature. A visible gap in the valence and conduction bands is visible for the studied halides, indicating no metallic behavior. The comparatively flat bands in both the valence and conduction regions indicate localized electronic states, which are typical of ionic halide perovskites. The obtained electronic bandgaps for Na3InBr6, K3InBr6 and Rb3InBr6 are 3.37 eV, 3.84 eV and 3.86 eV, as depicted in Fig. 4(a–c). All studied halides have direct bandgaps. This systematic increase in the bandgap from Na3InBr6 to Rb3InBr6 is due to lattice expansion and reduced In–Br orbital hybridization. Furthermore, this change in the bandgap is also evident through the TDOS plots for all studied halides. The TDOS plots for Na3InBr6, K3InBr6 and Rb3InBr6 also show a rising trend, which suggests that A-site substitution significantly modifies the electronic states by reducing orbital overlap, improving the ionic nature of the material, which leads to wide bandgaps. The band structure and TDOS investigations show that Na3InBr6, K3InBr6 and Rb3InBr6 are wide-gap semiconductors with tunable electronic characteristics caused by A-site substitution. The ability to modify the bandgap by alkali-metal cation engineering makes these materials potential candidates for optoelectronic, insulating, and UV-related device applications that require chemical stability and band-gap control.
 |
| | Fig. 4 Electronic properties of (a) Na3InBr6 (b) K3InBr6 and (c) Rb3InBr6. | |
The partial density of states plots for Na3InBr6, K3InBr6 and Rb3InBr6 are presented in Fig. 4(a–c). For Na3InBr6, the Na-s, Na-p, In-s and In-d orbitals have a minor contribution to the valence band, whereas a high contribution from the Br-s and Br-p states is observed. In the conduction band, the Br-s states are more prominent, along with a minor contribution from the In-s states, as depicted in Fig. 5(a). Fig. 5(b) demonstrates the partial density of states plot for K3InBr6, which indicates a minor contribution of the K-s, K-p, In-s and In-p orbitals to the valence band, whereas the majority contribution is from the Br-s and Br-p states. The conduction band has a high contribution from the Br-s states with a minor contribution from the K-p states. The valence band of Rb3InBr6 has a minor contribution from the Rb-s, Rb-p, In-s, In-p and In-d states, whereas the major contribution is from the Br-s and Br-p states. Furthermore, the conduction band has a large contribution from Br-s states, with minor contribution of In-s and In-d states, as depicted in Fig. 5(c).
 |
| | Fig. 5 PDOS plots of (a) Na3InBr6, (b) K3InBr6 and (c) Rb3InBr6. | |
Optical properties
The material's interaction with light gives rise to multiple optical phenomena, which aids in predicting the material's suitability in various light-based devices.68 Optical analysis of the studied halides is performed using the complex Kramer–Kronig dielectric equations. The dielectric function exhibits real and the imaginary parts; it describes the material's dispersive behavior and optical absorption. The real and imaginary parts are expressed as follows:| | 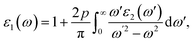 | (4) |
| | 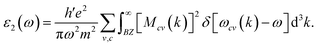 | (5) |
Fig. 6 presents an optical analysis of Na3InBr6, K3InBr6 and Rb3InBr6. ε1(ω) for the studied halides is presented in Fig. 6(a), from which the static values are obtained as 2.39, 2.18 and 2.14 for Na3InBr6, K3InBr6 and Rb3InBr6, respectively. The decline in the static value is connected to the Penn's model in which the static value of the studied material is inversely related to the material's bandgap.69 With increasing energy, the ε1(ω) plot shows minor peaks in the UV region at 3.90 eV, 4.44 eV and 4.42 eV, whereas maximum peaks are observed at 6.02 eV, 6.10 eV and 6.02 eV for Na3InBr6, K3InBr6 and Rb3InBr6, respectively. This means that the drop in the static dielectric constant is due to bandgap widening, which is compatible with Penn's concept, in which a bigger bandgap reduces the low-energy electronic polarizability. Minor peaks in the lower-UV region correlate to mild interband electronic transitions, but prominent maxima at higher UV energies imply substantial interband transitions from the valence to conduction states. These properties demonstrate that the optical response of Na3InBr6, K3InBr6 and Rb3InBr6 is dominated by high-energy electronic excitations. The halides are optically active largely in the UV region and transparent in the visible range. ε2(ω) for the studied halides is presented in Fig. 6(b). The ε2(ω) plot shows minor peaks at 4.53 eV, 4.72 eV and 4.66 eV for Na3InBr6, K3InBr6 and Rb3InBr6, respectively. As the energy increases, major peaks of the imaginary part appear in the UV region at 6.27 eV, 6.24 eV and 6.16 eV, whereas the maximum peaks are observed at 9.78 eV, 8.50 eV and 8.34 eV for Na3InBr6, K3InBr6 and Rb3InBr6, respectively. This trend suggests that the weak interband electronic transitions near the band edge cause the low-intensity peaks, but the intense UV peaks are caused by high-energy valence-to-conduction-band transitions. The shift and lowering of the maximum peak energies from Na to Rb imply increased polarizability and systematic alteration of the electronic structure via A-site substitution. Overall, the optical absorption of these halides is dominated by UV excitations, confirming their visual transparency. The refractive index defines the material's ability to slow down the speed of light as it moves through it.70 The refractive index of the studied halides is plotted in Fig. 6(c). The n(ω) at zero frequency is determined as 1.54, 1.47 and 1.46 for Na3InBr6, K3InBr6 and Rb3InBr6, whereas significant peaks are observed in the UV region at 4.06 eV, 4.55 eV and 4.50 eV, respectively. The maximum peaks of n(ω) for Na3InBr6, K3InBr6 and Rb3InBr6 are observed at 6.08 eV, 6.13 eV and 6.02 eV, respectively. The decrease in the static refractive index from Na3InBr6 to Rb3InBr6 indicates reduced electronic polarizability, which aligns with the growing bandgap trend. The pronounced UV peaks are caused by strong interband electronic transitions that promote light–matter interaction at higher photon energy. As a result, these halides show minimal dispersion in the visible and strong optical activity in the UV region. The extinction coefficient of optical materials is directly correlated with the imaginary part of the complex refractive index and indicates the loss of light intensity as a result of absorption within the medium. The extinction coefficient for the studied halides is presented in Fig. 6(d), where the minor extinction peaks for Na3InBr6, K3InBr6 and Rb3InBr6 are observed at 4.58 eV, 4.74 eV and 4.66 eV, respectively. As the energy increases, significant peaks appeared in the UV region at 6.35 eV, 6.29 eV and 6.21 eV, respectively. The maximum peaks of the extinction coefficient for Na3InBr6, K3InBr6 and Rb3InBr6 are observed at 12.23 eV, 9.75 eV and 12.80 eV, respectively. This suggests that mild optical losses occur near the band-edge as a result of tiny interband transitions, whereas substantial absorption and attenuation dominate in the UV region due to high electrical excitation. Thus, Na3InBr6, K3InBr6 and Rb3InBr6 have little extinction in the visible region, indicating their applicability for UV optoelectronic applications. The optical reflectivity of a material is often defined as its potential to reflect incident light. The reflectivity plot for the studied halides is shown in Fig. 6(e). The static value of the reflectivity for Na3InBr6, K3InBr6 and Rb3InBr6 is observed at 0.046, 0.037 and 0.035, respectively, whereas as the energy of the major peaks increased for Na3InBr6, occurring at 4.20 eV, 6.29 eV and 12.50 eV. For K3InBr6, prominent peaks are observed at 4.58 eV and 6.27 eV, whereas for Rb3InBr6, prominent peaks are observed at 4.50 eV and 6.16 eV. For all studied halides, the maximum reflectivity peaks are observed at 13.56 eV. This suggests that low static reflectivity confirms little light reflection in the low-energy region, implying high optical transparency. The optical conductivity measures a material's capacity to conduct electricity in response to an incident electromagnetic field at optical frequencies. It is derived from electronic interband and intraband transitions and represents frequency-dependent charge-carrier dynamics. The optical conductivity plots for Na3InBr6, K3InBr6 and Rb3InBr6 are demonstrated in Fig. 6(f). The optical conductivity plots show minor peaks for Na3InBr6, K3InBr6 and Rb3InBr6 at 4.58 eV, 4.72 eV and 4.66 eV, whereas strong peaks are observed at 6.27 eV, 6.24 eV and 6.16 eV, respectively. The maximum peaks of the optical conductivity for Na3InBr6, K3InBr6 and Rb3InBr6 are observed at 9.83 eV, 9.70 eV and 8.39 eV, respectively. The absorption coefficient demonstrates a material's ability to absorb electromagnetic radiation per unit path length in the material. The absorption coefficient for Na3InBr6, K3InBr6 and Rb3InBr6 is plotted in Fig. 6(g). Minor peaks of the absorption coefficient for Na3InBr6, K3InBr6 and Rb3InBr6 are observed at 4.55 eV, 4.69 eV and 4.72 eV, whereas prominent peaks are observed at 12.20 eV, 9.78 eV and 9.37 eV, respectively, in the UV region. The maximum absorption coefficient peaks for Na3InBr6, K3InBr6 and Rb3InBr6 are observed at 13.56 eV, 12.99 eV and 12.93 eV, respectively. The energy loss refers to the amount of energy dissipated by fast-moving electrons as they go through a material due to inelastic scattering processes. The energy loss plot for Na3InBr6, K3InBr6 and Rb3InBr6 is presented in Fig. 6(h). Minor energy loss peaks are observed at 4.72 eV, 4.80 eV and 4.72 eV, whereas prominent peaks appear at 6.21 eV, 6.38 eV and 6.40 eV, respectively. The maximum L(ω) for Na3InBr6, K3InBr6 and Rb3InBr6 is observed at 13.04 eV, 12.20 eV and 10.70 eV, respectively. This indicates that the low-energy loss features are caused by mild interband electronic excitations, but the pronounced peaks are caused by valence electron collective plasma oscillations. The shift in the highest loss peak from Na3InBr6 to Rb3InBr6 suggests a change in the plasma frequency and electron density due to A-site cation replacement. It is noted that the optical spectra indicate that A-site cation substitution (Na → K → Rb) primarily tunes the orbital hybridization indirectly through lattice expansion and structural distortion. Because the A-site cations (Na, K, Rb) have negligible direct contribution near the band-edges, their increasing ionic size weakens the In–Br orbital overlap by enlarging the bond lengths. This reduced hybridization leads to slight bandgap modification and a shift in the optical transitions, which is reflected in the graphs as peak shifts toward lower energies (red-shift) in the dielectric function, absorption coefficient, and energy loss spectra. Consequently, the refractive index and reflectivity also decrease slightly with heavier cations, confirming that A-site tuning controls the optical response via structural modulation rather than direct electronic contribution.
 |
| | Fig. 6 Optical properties of (Na/K/Rb)3InBr6. | |
Thermoelectric properties
Thermoelectric materials have been increasingly explored for the past few decades for their energy conversion applications. Thermoelectric materials possess the ability to directly transform the thermal power obtained from sun to electrical power.71,72 Utilizing the most efficient techniques to harvest electricity from the least expensive sources has been discovered to be very important by material scientists. Therefore, it becomes vital to clarify the operation of devices in order to explain their fundamental qualities and practical position. Waste heat and solar energy collection have enormous potential.73 Researchers are now considering materials that can effectively convert waste energy into green energy. Perovskite materials with their versatile crystal uniqueness and tunable properties have emerged as promising candidates for thermoelectric devices. Their flexibility allows for compositional engineering that can aid in optimizing the thermoelectric parameters, which are key parameters in assessing the thermoelectric efficiency. In this context, halide-based perovskite possess higher thermal stability, which makes them suitable high-performance thermoelectric devices.73 The sun is one of the main renewable energy sources. However, recent research has demonstrated that combining organic and inorganic components can result in novel halide perovskites.74 The effectiveness of thermoelectric materials is assessed based on several thermoelectric properties, which are computed with the help of semiclassical Boltzmann theory implemented in the BoltzTraP code.75 The thermoelectric properties of Na3InBr6, K3InBr6 and Rb3InBr6 are presented in Fig. 7. The electrical conductivity describes a material's ability to transfer charge carriers under an applied electric field. Materials with narrow bandgaps possess higher electrical conductivity and insulators or wide-bandgap semiconductors possess lower electrical conductivity. For the studied halides, the electrical conductivity shows an increasing trend as the temperature is increased, and the maximum values for Na3InBr6, K3InBr6 and Rb3InBr6 are obtained as 1.44 × 1019 1 Ω−1 m−1, 1.02 × 1019 1 Ω−1 m−1 and 8.21 × 1018 1 Ω−1 m−1 at 800 K, respectively, as depicted in Fig. 7(a). These results demonstrate that Na3InBr6 has a larger effective charge-carrier concentration, as compared to K3InBr6 and Rb3InBr6, suggesting more efficient charge transport at elevated temperatures. The thermal conductivity is also an important factor in analyzing the materials' thermoelectric performance. The thermal conductivity of the semiconducting materials comprises the lattice and electronic thermal conductivity, whereas the code we have employed only determines the electronic thermal conductivity.76 The thermal conductivity of the studied halides follows an increasing trend as the temperature is increased, and the maximum values obtained at 800 K are 55.21 × 1014 W m−1 s−1 K−1, 2.64 × 1014 W m−1 s−1 K−1 and 1.60 × 1014 W m−1 s−1 K−1 for Na3InBr6, K3InBr6 and Rb3InBr6, respectively, as shown in Fig. 7(b). This implies that the studied halides become more efficient in conducting heat at higher temperatures, with Na3InBr6 exhibiting the highest thermal conductivity among the series. The trend also suggests that the A-site cation influences heat transport, decreasing from Na to Rb due to the increasing lattice size and associated phonon scattering. The Seebeck coefficient for Na3InBr6, K3InBr6 and Rb3InBr6 is plotted in Fig. 7(c). The Seebeck coefficient demonstrates a material's ability to generate voltage in the presence of a temperature gradient.77 A higher Seebeck coefficient suggests that the material possesses higher capability for converting waste heat into electricity. The Seebeck coefficient has a maximum value of 215.67 µ(V K−1) at 50 K for Na3InBr6, which declines to 181.98 µ(V K−1) at 800 K. For K3InBr6, the maximum value is observed at 50 K as 215.67 µ(V K−1), which declines to 154.61 µ(V K−1) at 800 K. For Rb3InBr6, the maximum Seebeck coefficient is observed at 193.17 µ(V K−1) at 100 K, which declines to 131.96 µ(V K−1) at 800 K, as demonstrated in Fig. 7(c). All studied halides exhibit high thermoelectric efficiency at 50 K, whereas with a significant increase in temperature, the Seebeck coefficient declines. The Seebeck coefficient further demonstrates that Na3InBr6 is an effective material for converting waste heat to energy as compared to K3InBr6 and Rb3InBr6. The power factor indicates a material's efficiency to convert waste heat into electricity.78 The power factor's maximum value for Na3InBr6 is obtained as 4.78 × 1011 W m−1 s−1 K−1 at 800 K, whereas for K3InBr6, the maximum value is observed at 600 K as 2.55 × 1011 W m−1 s−1 K−1, which slightly declines to 2.43 × 1011 W m−1 s−1 K−1 when the temperature is raised to 800 K. For Rb3InBr6, the maximum value of the power factor is observed at 400 K as 1.87 × 1011 W m−1 s−1 K−1, which declines to 1.43 × 1011 W m−1 s−1 K−1 when the temperature is raised to 800 K, as shown in Fig. 7(d). The figure of merit is the most important parameter and plays a crucial role in assessment of the thermoelectric performance.79 High values of ZT indicate whether the material has high thermoelectric efficiency. Among the studied halides, the maximum value of ZT for Na3InBr6 is observed as 0.738 at 100 K, which declines to 0.72 at 250 K, and then increases to 0.732 when the temperature is raised to 800 K. The maximum value of ZT for K3InBr6 is obtained as 0.78 at 50 K, which drops to 0.74 at 200 K; when the temperature is increased, the ZT value remains steady till 600 K and a slight decline is observed in the value to 0.73 at 800 K. The ZT of Rb3InBr6 follows a parabolic path as the temperature is raised, and the maximum ZT is reported as 0.74 K at 550 K, which declines to 0.71 when the temperature is raised to 800 K, as illustrated in Fig. 7(e). The obtained values of ZT suggest that K3InBr6 and Na3InBr6 are viable for converting waste energy at elevated temperatures, whereas Rb3InBr6 is beneficial at moderate temperatures.
 |
| | Fig. 7 Thermoelectric properties of (Na/K/Rb)3InBr6. | |
Using Slack's equation, the lattice thermal conductivity (κl) was calculated, as illustrated in Fig. 8(a), and is given by the following expression:
| | 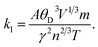 | (6) |
 |
| | Fig. 8 (a) Lattice thermal conductivity and (b) ZT plots of Na3InBr6, K3InBr6 and Rb3InBr6. | |
A represents a collection of physical constants (A ≈ 3.1 × 10−8), γ is the Grüneisen parameter, θD denotes the Debye temperature, and V corresponds to the atomic volume. The phonon vibrations exhibit a rapid increase, followed by a sharp decline over the temperature range of 100–800 K, suggesting an inverse correlation with the electronic contribution to the thermal conductivity for all materials. We investigated the influence of lattice vibrations on the ZT values to enhance their accuracy (Fig. 8(b)). The ZT values show a steady increase as the temperature rises. At 800 K, a ZT of 0.65, 0.7 and 0.73 is obtained for Na3InBr6, RB3InBr6 and K3InBr6, respectively. It can be observed that the inclusion of lattice vibrations has only a minimal effect on the ZT values across all the studied materials.
Conclusion
This paper presents a first principles analysis of perovskite halides (Na/K/Rb)3InBr6 by employing the mBJ approximation and FP-LAPW approach. The negative ground state and formation energies indicate complete structural and thermodynamic stability. The studied halides exhibit cubic formation with minimum distortions, as predicted via the tolerance factor and octahedral tilting. The elastic constants computed using the stress-energy tensor matrix exhibits a decreasing trend with the replacement the A-site cation from Na to Rb. The material exhibits ductile behavior, which declines gradually when the A-site cation is replaced in order of Na → K → Rb. The elastic anisotropy estimated using the ELATE software shows that among the examined compounds, K3InBr6 has a lower anisotropy and a more uniform elastic response, whereas Na3InBr6 and Rb3InBr6 have significant directional shifts. The electronic properties indicate direct bandgaps of 3.37 eV, 3.84 eV, and 3.86 eV for Na3InBr6, K3InBr6 and Rb3InBr6, respectively. The optical properties of Na3InBr6, K3InBr6 and Rb3InBr6 showed strong dispersion and polarization peaks in the UV region, making these materials potential candidates for ultraviolet optoelectronic applications, such as UV photonic devices. Furthermore, the thermoelectric properties of Na3InBr6, K3InBr6 and Rb3InBr6 indicate that these halides are excellent candidates for the conversion of waste energy into renewable energy.
Conflicts of interest
The authors have no conflict of interest.
Data availability
Data will be available from the authors upon reasonable request.
Acknowledgements
This work was supported and funded by the Deanship of Scientific Research at the Imam Mohammad Ibn Saud Islamic University (IMSIU) (grant number IMSIU-DDRSP2603).
References
- Z. Zang,
et al., Study of structural, electronic, phonon, thermodynamic, and hydrogen storage properties of hydride Rb2AsSnH6 perovskites: DFT insights, Int. J. Hydrogen Energy, 2025, 133, 225–234 CrossRef CAS.
- H. Rached,
et al., Future insights into double perovskites Ba2AlTMO6 (TM= W, Re, and Os) for sustainable and clean energy production: a DFT investigation using GGA, TB-mBJ, and HSE06 methods, J. Inorg. Organomet. Polym. Mater., 2025, 1–22 Search PubMed.
- M. Manzoor,
et al., Probing the optoelectronic and thermoelectric performance of inorganic halide perovskite Rb2KInI6 for renewable energy applications via DFT computations, J. Inorg. Organomet. Polym. Mater., 2025, 35(5), 3296–3312 CrossRef CAS.
- M. S. Uddin,
et al., Achieving over 28% efficiency in inorganic halide perovskite Ca3AsI3: optimization of electron transport layers via DFT, SCAPS-1D, and machine learning, J. Phys. Chem. Solid., 2025, 200, 112622 CrossRef CAS.
- A. Et-Taleb,
et al., Novel KSiCl3 and LiSiBr3 Lead-free Halide Perovskite for Photovoltaic and Photocatalytic Applications: A DFT-based Study, Silicon, 2025, 1–16 Search PubMed.
- R. Fatima,
et al., First-principles insights to probe structural and opto-electronic properties of AgYF3 (Y= Mg, Sr) halide perovskites with variety of DFT methods, Chem. Phys., 2025, 588, 112443 CrossRef CAS.
- M. Zafar,
et al., Comparative analysis of band gap using different approximations, structural, mechanical and optical behaviour analysis of lead free double halide perovskites Cs2AgBiBr6 using DFT approach, Solid State Commun., 2025, 397, 115825 CrossRef CAS.
- C. Tu,
et al., Engineered thermal conduction pathways in high-performance flexible PEG phase-change composites enabling efficient thermal storage and thermoelectric conversion, Chem. Eng. J., 2026, 172574 CrossRef CAS.
- B. L. Farah,
et al., RbNbX3 (X= cl, Br, I) lead-free halide perovskites: a DFT study of structural, elastic, electronic, and thermoelectric properties for energy applications, Chem. Phys., 2025, 598, 112843 CrossRef CAS.
- W. Zhao,
et al., TiN supported 3D directional tubular skeleton encapsulating phase change materials for efficient solar-thermal energy conversion and storage, Sol. Energy, 2025, 302, 114071 CrossRef CAS.
- L. Su,
et al., An Advanced Energy-Coordinated Control for Half-Bridge Submodule-based Centralized DC Chopper in Offshore Wind MMC-HVDC Systems, IEEE Trans. Power Electron., 2026, 1–15 CAS.
- M. Tarekuzzaman,
et al., DFT analysis of structural, electronic, optical, and thermodynamic properties of LiXI3 (where X= Ca, Sr, Ba) halide perovskites for optoelectronics, Sci. Rep., 2025, 15(1), 25304 CrossRef PubMed.
- H. Murtaza,
et al., Exploring hydrogen storage attributes of alkali metal XNH6 (X= Li, Na, K) perovskite hydrides using DFT calculations, J. Power Sources, 2025, 641, 236788 CrossRef CAS.
- Y. Hao,
et al., Characteristics of carrier localization and their effects on minority carrier lifetime in InAs/In0. 5Ga0. 5As0. 5Sb0. 5 type II superlattices, Appl. Phys. Lett., 2025, 127(8), 082105 CrossRef CAS.
- H. Fatihi,
et al., Enhancing Solar cell efficiency: a comparative study of lead-free double halide perovskites R b 2 C u A s B r 6 and R b 2 T l A s B r 6 using DFT and SLME methods, J. Inorg. Organomet. Polym. Mater., 2025, 35(2), 964–977 CrossRef CAS.
- A. Shakoor,
et al., DFT Investigation of Structural, Electronic, Optical, Mechanical, and Thermoelectric Properties of Halide Perovskites XZnCl3 (X= Ag, Au, Cu), Phys. B Condens. Matter, 2025, 417309 CrossRef CAS.
- F. Li,
et al., Covalently bonded fluorine optimizing deep-ultraviolet nonlinear optical performance of fluorooxoborates, Sci. Bull., 2024, 69(9), 1192–1196 CrossRef CAS PubMed.
- J. Khan,
et al., Mechanical Stability and Optoelectronic Potential of Halide Perovskites AXCl3 (A= Ca, Sr; X= K, Rb): A DFT Study, Computational Condensed Matter, 2025, e01095 CrossRef.
- X. Wang,
et al., Recent advances and optoelectronic applications of Cu–Ag–Bi–I quaternary lead-free perovskites, Semicond. Sci. Technol., 2025, 40(4), 043002 CrossRef CAS.
- D. Abdullah and D. C. Gupta, Engineering, investigation of structural, electronic, optical, and elastic properties of Ru-based inorganic halide perovskites ARuBr3 (A= K, rb, Cs) via DFT computations, Arabian J. Sci. Eng., 2025, 50(9), 6823–6833 CrossRef CAS.
- X. Han,
et al., Preparation of highly conductive PEDOT: PSS hole transport layer by simple treatment with ethanol for Sn–Pb perovskite solar cells, Semicond. Sci. Technol., 2025, 40(9), 095015 CrossRef CAS.
- A. Ullah,
et al., Unlocking the physical properties of RbXBr3 (X= Ba, Be) halide perovskites for potential applications: DFT study, Inorg. Chem. Commun., 2025, 173, 113879 CrossRef CAS.
- A. Shakoor,
et al., DFT investigation of structural, electronic, optical, mechanical, and thermoelectric properties of halide perovskites XZnCl3 (X= Ag, Au, Cu), Phys. B Condens. Matter, 2025, 712, 417309 CrossRef CAS.
- P. Sharma and D. Vashisht, Ag-based halide perovskites AgEX3 (E= Ge, Sn; X= Cl, Br, I): DFT-driven exploration of structural, optoelectronic, and thermal properties for photovoltaic applications, Next Mater., 2025, 9, 101058 CrossRef CAS.
- U. Chakma,
et al., Eco-Friendly Halide Perovskites Cs3Ag2X5 (X= Cl, Br, I): DFT Study on Electronic Structure, Optical and Thermodynamic Properties, Elastic Constants, and Toxicity, ChemistrySelect, 2025, 10(27), e02706 CrossRef CAS.
- M. B. H. Parosh,
et al., DFT Investigations of Non-Toxic Perovskites RbZnX3 (X= F, Cl, and Br): Analyzing the Structural, Electrical, Optical, Mechanical, and Thermodynamic Properties for Suitable Optoelectronic Applications, Int. J. Quant. Chem., 2025, 125(4), e70014 CrossRef CAS.
- M. A. Takin,
et al., Effect of halogen variation on the optoelectronic properties of AlSnX3 (X= F, Cl, Br and I) perovskites: a first-principles DFT investigation, Bull. Mater. Sci., 2025, 48(4), 147 CrossRef CAS.
- J. Abbas,
et al., A comprehensive DFT analysis of structural, thermoelectric, optoelectronic, and magnetic properties of X2NaCrCl6 (X= K, Rb) halide perovskites, Multiscale and Multidiscip. Model. Exp. and Des., 2025, 8(10), 1–19 Search PubMed.
- S. Al-Qaisi,
et al., Comprehensive DFT study of K2TlZI6 (Z= Al, In) double perovskites: atructural stability and potential for optoelectronic and thermoelectric energy harvesting, Phys. B Condens. Matter, 2025, 710, 417239 CrossRef CAS.
- S. Ahmad,
et al., DFT study on elastic, structural, and optoelectronic properties of Cs2KAsA6 (A= Cl, Br, I) halide double perovskites: potential for green energy applications, J. Inorg. Organomet. Polym. Mater., 2025, 1–12 Search PubMed.
- M. F. Rahman,
et al., Achieving efficiency above 30% with new inorganic cubic perovskites X 2 SnBr 6 (X= Cs, Rb, K, Na) via DFT and SCAPS-1D, Phys. Chem. Chem. Phys., 2025, 27(2), 1155–1170 RSC.
- Z. Khan,
et al., Exploring Sn-based vacancy-ordered halide double perovskites Na2Sn (Cl/Br) 6 for optoelectronic, thermoelectric, and solar-driven hydrogen reduction applications, Chem. Pap., 2025, 1–21 Search PubMed.
- M. E. A. El Goutni,
et al., Designing K2ReX6 (X= Cl, Br) Perovskites for Energy Conversion: A DFT-Based Study on Structural Stability and Functional Properties, ChemistrySelect, 2025, 10(28), e01505 CrossRef CAS.
- M. Manzoor,
et al., A DFT calculations of mechanical, optoelectronic and transport properties of cubic AMgI3 (A= Li/Na) halides for photovoltaic and energy conversion applications, J. Inorg. Organomet. Polym. Mater., 2025, 35(2), 795–812 CrossRef CAS.
- N. Israr,
et al., A2AlInI6 (A= K, Rb, Cs) Double Perovskite Halides for Renewable Energy Applications: A DFT Study on Stability, Light Absorption, and Thermoelectric Performance, J. Inorg. Organomet. Polym. Mater., 2025, 1–16 Search PubMed.
- M. Shakil,
et al., Computational investigation of newly proposed double halide perovskites Cs2GaBiX6 (X= Cl, Br and I) with enhanced optoelectronic properties for green energy harvesting and photocatalytic applications, J. Phys. Chem. Solid., 2025, 201, 112638 CrossRef CAS.
- M. Jamil,
et al., Screening the structural, tuning band gaps, optical and thermal properties of inorganic A2AgRhF6 (A= Na, Rb) double perovskites halide using DFT calculations, J. Inorg. Organomet. Polym. Mater., 2025, 35(4), 2734–2745 CrossRef CAS.
- Q. Xiao,
et al., Probing the opto-electronic, thermoelectric, thermodynamic and elastic responses of lead-free double perovskite Li2ATlCl6 (A= Na and K) for potential photovoltaic and high-energy applications: A DFT study, Mater. Sci. Eng., B, 2025, 312, 117860 CrossRef CAS.
- M. Tarekuzzaman,
et al., DFT analysis of the physical properties of direct band gap semiconducting double perovskites A 2 BIrCl 6 (A= Cs, Rb; B= Na, K) for solar cells and optoelectronic applications, RSC Adv., 2025, 15(17), 13643–13661 RSC.
- M. Riaz,
et al., Screening the Geometrical, Thermodynamic, Mechanical, and Bond Population Characteristics of Inorganic Halide Perovskites AgSrX3 (X= Cl, Br, I) for Photovoltaic Applications: A DFT Insight, Chem. Eng. Res. Des., 2025, 222, 227–234 CrossRef CAS.
- M. S. Hayat and R. A. Khalil, A DFT engineering of double halide type perovskites Cs2SiCl6, Cs2GeCl6, Cs2SnCl6 for optoelectronic applications, Solid State Commun., 2023, 361, 115064 CrossRef CAS.
- Z. Zhang,
et al., Real-space Kohn–Sham density functional theory for complex energy applications, Chem. Commun., 2025, 61, 10273–10286 RSC.
- H. A. Abdulhussein,
et al., A comprehensive analysis of the structural, phonon, electronic, mechanical, optical, and thermophysical properties of cubic Ca3SbX3 (X= Cl, Br): DFT-GGA and mBJ studies, Mater. Sci. Semicond. Process., 2025, 187, 109133 CrossRef CAS.
- K. Momma and F. Izumi, VESTA: a three-dimensional visualization system for electronic and structural analysis, Appl. Crystallogr., 2008, 41(3), 653–658 CrossRef CAS.
- H. Murtaza,
et al., The effect of pressure in tuning the optoelectronic, thermoelectric and hydrogen storage properties of RbCaH3 for renewable energy applications, J. Energy Storage, 2025, 134, 118183 CrossRef CAS.
- N. Rahman,
et al., DFT insights on the future prospects of Ba2PrXO6 (X= Ir, Pt) double perovskites for high-energy applications, J. Inorg. Organomet. Polym. Mater., 2025, 35(2), 1439–1452 CrossRef CAS.
- H. Murtaza,
et al., First-principles prediction of the optoelectronic, mechanical, thermodynamic and hydrogen storage attributes of double perovskite Rb2NaXH6 (X= Al, In) hydrides, J. Inorg. Organomet. Polym. Mater., 2025, 35(4), 2877–2888 CrossRef CAS.
- F. Han,
et al., Enhancing Spin–Orbit Torque Through Octahedral Tilt/Rotation Relaxation in CaRuO3 Films for Efficient Magnetization Switching, Adv. Funct. Mater., 2025, 2504842 CrossRef CAS.
- M. Cawkwell and M. Zecevic, Second-and Third-Order Elastic Constants of Inert and Energetic Molecular Crystals From Density Functional Theory, Propellants, Explos., Pyrotech., 2025, e12084 Search PubMed.
- L. K. Bhaskar,
et al., Probing elastic isotropy in entropy stabilized transition metal oxides: experimental estimation of single crystal elastic constants from polycrystalline materials, Acta Mater., 2025, 288, 120871 CrossRef CAS.
- N. L. Muttalib and B. Abdullah, A novel model for bulk modulus and bond energy calculation of Si and its binary compounds, tetrahedral ternary semiconductors in bulk and nanocrystals, Phys. Chem. Chem. Phys., 2025, 27(15), 7805–7815 RSC.
- T. Zhang,
et al., Mechanical properties of minerals in high-sulfur bauxite from DFT calculation, Miner. Eng., 2025, 232, 109572 CrossRef CAS.
- D. Tufail,
et al., DFT study of alkaline earth metals NaXH 3 (X= Be, Mg, Ca, Sr) for hydrogen storage capacity, RSC Adv., 2025, 15(1), 337–347 RSC.
- A. Awais,
et al., First-principles study of lead-free Na2TmAgCl6 and Na2TmCuCl6 double halide perovskites for photovoltaic and thermoelectric applications, J. Mol. Graph. Model., 2025, 109157 CrossRef CAS.
- A. Es-Smairi,
et al., DFT insights into the structural, stability, elastic, and optoelectronic characteristics of Na2LiZF6 (Z= Ir and Rh) double perovskites for sustainable energy, J. Comput. Chem., 2025, 46(8), e70097 CrossRef CAS PubMed.
- H. Murtaza,
et al., The excellent hydrogen storage performance of structural, mechanical and dynamical stable perovskite XCH3 (X= Li, Na, K) hydrides: First-principles investigations, Int. J. Hydrogen Energy, 2025, 179, 151447 CrossRef CAS.
- M. S. U. Sahar,
et al., A DFT study to evaluate the modulation in the band gap, elastic, and optical performances of RbCdF 3 under the influence of stress, Mater. Adv., 2025, 6(17), 5959–5970 RSC.
- H. Murtaza,
et al., Excellent hydrogen storage capabilities and optoelectronic attributes of XClH 6 (Li, Na, and K) perovskite hydrides for green energy technologies, RSC Adv., 2025, 15(38), 31609–31619 RSC.
- M. Akhtar,
et al., The determination of the mechanical, optoelectronic, structural and transport attributes of double perovskite A2InGaBr6 (A= K, Rb, Cs) halides for renewable energies: a DFT study, ChemPhysChem, 2025, 26(5), e202400891 CrossRef CAS PubMed.
- H. Murtaza,
et al., Harnessing the physical attributes of mechanical, structural and dynamical stable alkali-metal perovskite (Rb/Cs) NH6 for sustainable hydrogen storage, Chem. Phys., 2025, 877, 142282 CAS.
- M. A. Ullah,
et al., An approach towards next-generation hydrogen storage: a DFT study on A 2 LiTiH 6 (A= K, Ca) perovskite hydrides, RSC Adv., 2025, 15(46), 38714–38728 RSC.
- M. Mizanuzzaman,
et al., DFT based investigation of Se doped MgMo6S8-ySey as promising cathode materials for Mg-ion battery application, Sci. Rep., 2025, 15(1), 30817 CrossRef CAS PubMed.
- N. Roktim,
et al., A DFT Study to Investigate the Stability and Physical Properties of Y2AB (A= Te, Tl) Phases, Papers Search PubMed , https://www.ssrn.com.
- S. Hussain,
et al., Study of structural, electronic, optical, mechanical, and thermodynamic properties of perovskite RbX2Ta3O10 (X= Ca and Mg) materials for photocatalytic applications: a DFT insights, J. Inorg. Organomet. Polym. Mater., 2025, 35(1), 384–395 CrossRef CAS.
- B. Ahmed,
et al., Unveiling the potential of XInH3 (X= Rb and Cs): A DFT study for solid state hydrogen storage applications, Chem. Phys., 2025, 588, 112441 CrossRef CAS.
- H. Murtaza,
et al., Tunable physical properties of A-site double perovskite KLaYBiO6 under hydrostatic pressure: implications for renewable energy technologies, Int. J. Mod. Phys. B, 2025, 2550289 CrossRef CAS.
- N. J. Nisha,
et al., Novel Lead-Free Perovskites FrZnX3 (X= F, Cl, and Br) for Optoelectronics Applications: A DFT Study, Int. J. Energy Res., 2025, 2025(1), 5549809 CrossRef CAS.
- A. Kumar, M. Kumar and R. Singh, Magnetic, opto-electronic, and thermodynamic properties of half-metallic double perovskite oxide, Ba2YbTaO6: a density functional theory study, J. Mater. Sci. Mater. Electron., 2021, 32(10), 12951–12965 CrossRef CAS.
- M. Hassan, A. Shahid and Q. Mahmood, Structural, electronic, optical and thermoelectric investigations of antiperovskites A3SnO (A= Ca, Sr, Ba) using density functional theory, Solid State Commun., 2018, 270, 92–98 CrossRef CAS.
- M. Rabbi,
et al., DFT prediction of double perovskites A2B′RhCl6 (A= Cs/Rb; B′= Na/K) for green energy technology, Comput. Condens. Matter, 2025, e01093 CrossRef.
- M. Tauqeer, T. Anwar and S. U. Asif, Designing Stable Intermetallic Zr6MTe2 (M= Co, Ni) Materials: DFT Insights into Electronic Structure, Plasmonic Response, and Thermoelectric Transport, J. Electron. Mater., 2025, 1–14 Search PubMed.
- H. Murtaza,
et al., Scrutinize the physical attributes of thermodynamically and elastically stable double perovskite oxides Ba2CdXO6 (X= Mo, U) for optoelectronics, photocatalytic and green technology, Comput. Mater. Sci., 2024, 232, 112674 CrossRef CAS.
- R. Sharma,
et al., A DFT investigation of CsMgX3 (X= Cl, Br) halide perovskites: electronic, thermoelectric and optical properties, Comput. Theor. Chem., 2021, 1204, 113415 CrossRef CAS.
- A. Nazir,
et al., A DFT exploration of the optoelectronic and thermoelectric features of a novel halide double perovskite A 2 YAuI 6 (A= Rb, Cs) for solar cell and renewable energy applications, Mater. Adv., 2024, 5(10), 4262–4275 RSC.
- S. Azam, S. A. Khan and S. J. O. Goumri-Said, DFT combined to Boltzmann transport theory for optoelectronic and thermoelectric properties investigations for monoclinic metallic selenide: Cu5Sn2Se7, Optik, 2016, 127(13), 5472–5478 CrossRef CAS.
- S. M. Qaid,
et al., Correction: predictive analysis of the physical characteristics of mechanically stable Cs2Rb B′ I6 (B′= Al, Ge, Tl) for photovoltaics and optoelectronic applications: a DFT study, J. Mater. Res., 2025, 40(2), 310 CrossRef CAS.
- M. Manzoor,
et al., A comprehensive DFT insight on thermoelectric response of halide perovskites TlGeZ3 (Z= Cl, Br and I) for renewable energy devices, J. Inorg. Organomet. Polym. Mater., 2025, 1–14 Search PubMed.
- A. M. Alsuhaibani,
et al., High thermoelectric and opto-electronic properties of Ba3NX3 (X= F, Cl) perovskite: insights from DFT computation, Radiat. Phys. Chem., 2024, 225, 112129 CrossRef CAS.
- D. Abdullah and D. C. Gupta, Analyzing the structural, optoelectronic, and thermoelectric properties of InGeX3 (X= Br) perovskites via DFT computations, Sci. Rep., 2024, 14(1), 23575 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Mohamed A.
Habib
b,
Quratul
Ain
a,
Mohamed A.
Habib
b,
Quratul
Ain












