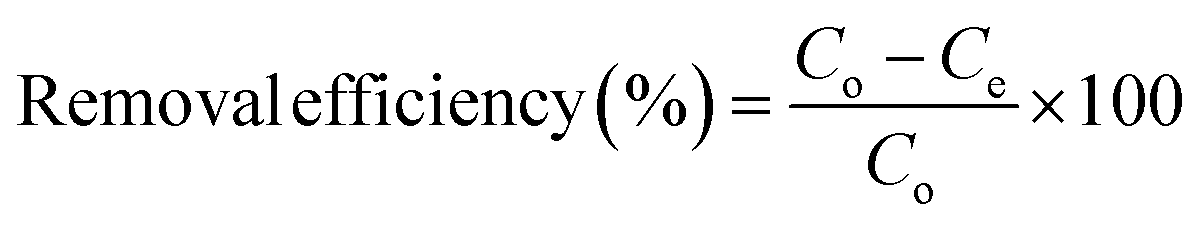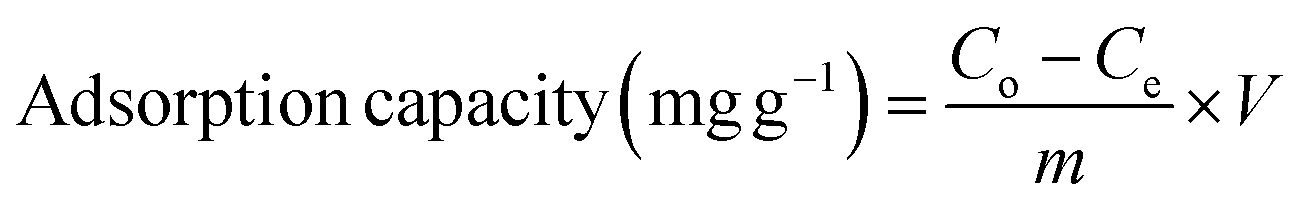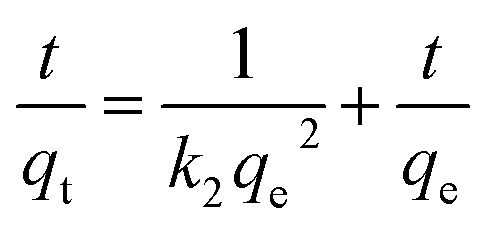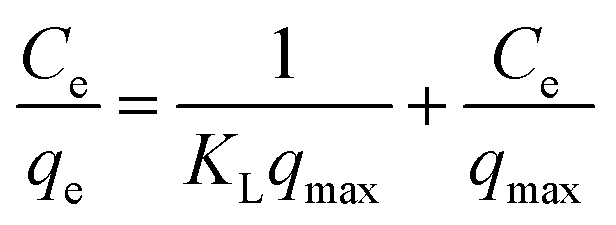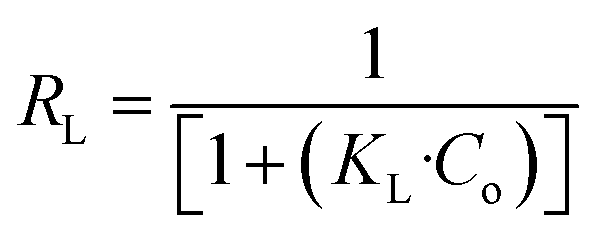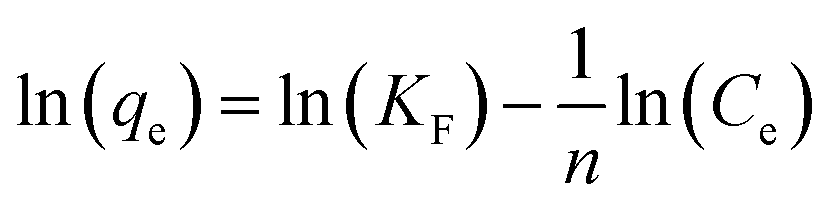DOI:
10.1039/D6RA01669C
(Paper)
RSC Adv., 2026,
16, 27611-27620
Removal of iodine from organic media using diethylene triamine-grafted vinylbenzyl chloride-divinylbenzene resin
Received
25th February 2026
, Accepted 8th May 2026
First published on 22nd May 2026
Abstract
Iodine released from different industrial activities and nuclear fuel reprocessing poses significant health and environmental risks due to its long half-life and high volatility, demanding efficient materials for its removal. In the current study, vinylbenzyl chloride-divinylbenzene-based resin is synthesized and functionalized with diethylene triamine for the removal of iodine from organic media. The synthesis success of the resin beads is confirmed using FTIR, CHNS, and TGA analyses, which revealed the presence of a characteristic N–H band, an increase in nitrogen content, and enhanced thermal stability of the aminated resin. In batch experiments, the aminated resin demonstrated an improved iodine removal efficiency (almost complete removal) compared to the vinylbenzyl chloride-divinyl-based pristine resin (10%), for an initial iodine concentration of 200 mg L−1 in n-hexane at an equilibrium time of 7 h. The adsorption efficiency of the aminated resin is found to be dependent on both the contact time and the initial iodine concentration. The adsorption process follows the pseudo-second-order kinetic model. The Langmuir isotherm model provided the best fit for explaining the adsorption mechanism of iodine on aminated resin. The maximum adsorption capacity of the aminated resin is found to be 54.64 mg g−1. The nearly complete removal of 129I is highly promising for reducing nuclear-industry waste challenges and significantly reducing waste volume.
1. Introduction
Electricity is important for modern society, supporting healthcare, transportation, communication, domestic needs, and industrial activities. The global demand is continuously increasing as a result of technological advancement, urbanization, and rapid population growth. To meet this increasing requirement, various sources have been used for the generation of electricity, including nuclear power, renewable energy, and fossil fuels.1 However, concerns related to environmental sustainability and carbon emissions have catalyzed the search for low-carbon energy alternatives. Amid rising demands, nuclear energy, as a sustainable and clean energy source, has experienced widespread global expansion.2–5 Currently, 11% of global electricity is being generated from around 450 nuclear power reactors.6,7 The rapid growth of the nuclear industry has led to the substantial accumulation of nuclear waste, accounting for tens of thousands of tons each year worldwide.8 Nuclear fission reactions in nuclear power plants produce large amounts of radioisotopes, which can easily become a part of the food chain and end up in the human body. A key safety concern is managing radioactive waste, particularly volatile fission products like 3H, Cs137, 85Kr, 131I, and 129I.9–11
Radioisotopes of iodine with mass numbers 125, 129, and 131–135 are of great significance due to their toxicology, as 131I emits strong radiation (606 keV β-particles and 364 keV γ-rays) with a short half-life of 8 days.7 Among all the isotopes of iodine in radioactive waste management, 129I is regarded as a major safety concern with the exceptionally long half-life of 15.7 million years.12–14 Due to its slow decay rate, mobility, and migration characteristics, 129I persists over an extended timescale and poses long-term radiological and environmental risk.12,15–17 The other non-nuclear sources of iodine, primarily in the form of organic iodides and molecular iodine, include food processing, pharmaceuticals, industry, hospitals, and chemical plants that eventually become part of the environment.14 Wastewater with iodine may become a part of groundwater and enter the food cycle, causing thyroid cancer in humans.18 Hazards of exposure to low concentrations of iodine can lead to ocular damage, ocular irritation, and cutaneous burns. Furthermore, exposure to iodine vapors can result in irritation of the eyes and throat. Longer exposure to iodine also causes reproductive hazards.19
Radioactive iodine species commonly found in nuclear power plants' off-gas streams or solid waste include diatomic iodine (I2), organic iodides, such as methyl iodide (CH3I) or ethyl iodide (CH3CH2I). In addition, inorganic iodine compounds, including hydrogen iodide (HI), hypoiodous acid (HOI), and iodine cyanide (ICN), may also be present, though typically in trace concentrations due to their lower formation rates under reactor conditions.14 In water, iodine can also be found in the form of iodide, hypoiodite, and iodate ion (i.e. I−, IO−, and IO3−).20
Currently, several techniques are being employed to mitigate iodine from the environment. These methods include membrane filtration (ion exchange, nanofiltration, reverse osmosis, and electrodialysis), adsorption, and electrochemical methods.21 All these methods have their pros and cons. Adsorption is widely preferred due to its high potency in decontamination processes, lower energy demand, cost-effectiveness, and scalability.22,23 Different types of adsorbents have been synthesized and explored, particularly for iodine mitigation.
The conventional approach for capturing radioactive iodine relies on Ag-based adsorbents.24,25 These adsorbents are expensive and have low efficiency for iodine.26 Over the past decade, different porous adsorbents such as metal–organic frameworks (MOFs),27–29 and covalent organic frameworks (COFs)30,31 have been considered an efficient adsorbent material for iodine. However, in MOFs, the pore channels bind more stably with iodine molecules, which decreases their desorption ability.32 In addition to this, porous polymers33,34 emerged as some of the most effective materials showing remarkable iodine capture efficiency.35–38 In porous adsorbents, integration of electron-rich heteroatoms (N, O, S, B, and P) plays a decisive role in iodine adsorption. They capture iodine through charge-complex formation within the polymer porous matrix, thereby enhancing both retention stability and adsorption capacity.32,39–41
Several studies have shown that nitrogen-functionalization of porous hosts markedly enhances adsorption performance. For instance, Harijan et al.42 prepared Fe3O4@PPy powder and demonstrated its high absorption capacity (1627 mg g−1) of I2 in the form of polyiodide complex (i.e. I3− and I5−) with polypyrrole. Alsalbokh et al.43 prepared different alumina-supported aminosilanes (i.e. 3-aminopropyl-triethoxysilane, N-methyl aminopropyl-trimethoxy silane, and 3-N,N-dimethylaminopropyl-trimethoxysilane) for I2 removal, showing adsorption capacities of about 124, 223, and 241 mg g−1. Lou et al.44 synthesized a bipyridine-based cage that exhibits good I2 absorption capacity of up to 3230 mg g−1. Li et al.45 synthesized nitrogen-rich covalent organic frameworks (COFs) (i.e. made from tri-formaldehyde-based organic ligands and triple symmetric amines) and found adsorption capacity between ∼2500 and ∼3200 mg g−1. Li et al.46 developed a copper-based metal–organic framework (MOF) with hybrid pyrazole ligands (MOF-303) and exhibited I2 adsorption capacities of 747 and 837 mg g−1 for Cu2+-MOF-303 and Cu0-MOF-303, respectively. These studies highlight that nitrogen atom-rich adsorbents have considerably higher I2 adsorption characteristics.
Chelating agents typically play a crucial role in nuclear decontamination operations due to their high selectivity and ability to form highly stable complexes with various radionuclides.47 Chelating resins exhibit some advantages compared to other adsorbents, offering greater reproducibility, simple handling, and easy transfer procedures.48 Despite their potential, research on the chelating polymer for iodine adsorption has remained only to a limited extent. Ye et al. synthesized a silica-based quaternized poly(4-vinyl pyridine) resin, demonstrating 96% iodine removal within 30 minutes.49 Robshaw et al. synthesized a Cu-based bispicolylamine chelating resin, with high uptake capacity of 2940 mg g−1 and 305 mg g−1 for iodine and iodide.7 For I2 removal, resins with amine functionalities are comparatively less studied. However, Abdelmoaty et al. synthesized two nitrogen-rich porous polymers generated from the building block 4-bis-(2,4-diamino-1,3,5-triazine)-benzene for I2 capture. The material exhibits high capacity and quick reversible uptake for I2, suggesting that porous materials with N species are suitable for I2 adsorption.38 The literature further shows high adsorption efficiency for iodine when using nitrogen-based materials. This paper mainly focuses on the formation of nitrogen-based chelating resin, aimed to balance economic feasibility, adsorption efficiency, and practical applicability, which is further used to remove molecular iodine through an adsorption mechanism.
In this study, a novel chelating polymer is synthesized by the functionalization of vinylbenzyl chloride-divinylbenzene-based resin (PVBCl) with diethylene triamine (DTA) through an easy and efficient synthetic route. The base polymer PVBCl is inexpensive, and subsequent nitrogen functionalization makes the proposed adsorbent economically viable. Pristine PVBCl and aminated resin are tested for I2 removal via a batch adsorption experiment from organic media. Various parameters, such as the effect of contact time, adsorbent dose, and adsorbate concentration, are studied for optimization. In comparison to PVBCl, the aminated resin exhibits considerably higher removal efficiency, nearly complete removal of molecular iodine due to the presence of amine groups on the polymer backbone, which enhances its surface affinity for iodine molecules. The aminated resin is therefore found to be a promising material for iodine uptake with high removal efficiency and adsorption capacity.
2. Materials and methods
2.1 Materials
Chemicals used in the current study include iodine (I2) (Anala R, 99.9%), n-hexane (Merck, 98%), gum arabic (Merck), gelatin (Merck, 99%), and benzoyl peroxide (Merck, 98%). Vinylbenzyl chloride (97%), diethylene triamine (DTA) (99%), methanol (99.8%), cyclohexanone (99%), and divinyl benzene (80%) used in this study are purchased from Sigma-Aldrich. These chemicals are used without further purification.
2.2 Preparation of PVBCl pristine and aminated resin
Poly-vinylbenzyl chloride (PVBCl) resin is synthesized in a single batch via suspension polymerization by reacting vinylbenzyl chloride (VBCl) and divinylbenzene (DVB) monomers.50 Fig. S1 illustrates the synthesis schematic of pristine PVBCl and aminated resins. Before the reaction starts, first the aqueous and organic phases are prepared separately. In the aqueous phase, gum arabic and gelatin, 3 g each, are introduced into a 250 mL beaker having 150 mL of deionized water. This mixture is placed on a hot plate at 70 °C with continuous stirring for 30 min. For the organic phase, 10 mL of VBCl, 3 mL of DVB (i.e., cross-linker), 7 mL of cyclohexanone (i.e. porogen), and 0.1 g of benzoyl peroxide (i.e., initiator) are added to a 50 mL beaker. The organic phase is stirred for 10 min to dissolve benzoyl peroxide. Afterward, both the aqueous and organic phases are mixed in a Pyrex cylindrical reactor. The reactor is placed in a water bath equipped with a mechanical stirrer, and the temperature is set at 80 °C to initiate the reaction. The reaction is continued for 6 h with stirring at 300 rpm. After 6 h, when the reaction is complete, the beads are filtered using cellulose fabric and washed thoroughly using hot water to remove unused ingredients. The beads are finally dried in an oven at 80 °C overnight. The as-prepared resin is named PVBCl pristine resin.
Functionalization of the PVBCl pristine resin is carried out in a single batch to eliminate batch-to-batch variations using diethylenetriamine (DTA). The PVBCl pristine resin possesses a chloro group that can be replaced with an amine group of DTA at room temperature. In brief, 3 g of PVBCl pristine resin is added to 25 mL of methanol, which is left for 1 h for its swelling. Later, 25 mL of DTA is added to the swelled beads in a flask fitted with a condenser to prevent the loss of volatile amine from the reaction mixture. The reaction is carried out for 48 h at room temperature with stirring at 300 rpm. Following this, the beads are filtered and dried overnight at 60 ℃. Both PVBCl pristine and aminated resins are stored in a desiccator for further studies. The schematic illustration of the reaction leading to the amination of PVBCl is shown in Fig. S2.
2.3 Characterization
A morphological analysis of the prepared resin is conducted using the TESCAN MIRA2 FE-SEM with an acceleration voltage of 10 keV. The optical microscope is used to study the size distribution of resin beads. For structural analysis of resin beads, a Thermo Scientific Nicolet iS50 FTIR Spectrophotometer is used in the 4000–1000 cm−1 spectral range. The compositional analysis of resin is carried out using a Thermo Scientific Flash 2000 CHNS-O Analyzer. Thermo-gravimetric analysis (TGA) is performed on METTLER TOLEDO TGA/SDTA851e in an air atmosphere in a 25–600 °C temperature range at a heating rate of 10 °C min−1. The U-2900 Hitachi Spectrophotometer is used for the analysis of iodine throughout the study.
2.4 Spectrophotometric measurement of iodine
A standard calibration curve in a concentration range of 50–600 mg L−1 is drawn (Fig. S3), using absorbance values acquired at 525 nm, to determine the unknown concentration of I2. Standard solution and the adsorption experiments are performed in n-hexane solvent. The obtained calibration equation is y = 0.0036x − 0.0775 (R2 = 0.999). To monitor the overall change in concentration in adsorption experiments, absorbance spectra are also taken in the wavelength range of 300–800 nm.
2.5 Batch adsorption experiment
The I2 adsorption performance in n-hexane using aminated resin is assessed using a batch experiment. For the batch studies, 0.09 g of adsorbent is introduced into 10 mL of a 200 mg L−1 I2 solution. The mixture is shaken at 250 rpm for 7 h to ensure equilibrium is reached. Thereafter, the mixture is centrifuged for 2 min using a laboratory centrifuge (DH-16), and the remaining iodine (I2) concentration in the solution is determined using a UV-visible spectrophotometer.
The percentage removal efficiency (%) and adsorption capacity (mg g−1) are calculated using eqn (1) and (2).
| |
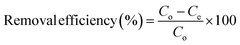 | (1) |
| |
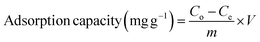 | (2) |
where
Co (mg L
−1) is the initial iodine concentration,
Ce (mg L
−1) is the concentration at equilibrium,
m is the mass of adsorbent, and
V is the volume in liters.
51
To comprehensively examine the adsorption behavior, different factors, including the effect of interaction time, solid-to-liquid ratio (S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :L), and adsorbate concentration, are studied in triplicate to ensure the reliability and reproducibility of results.
:L), and adsorbate concentration, are studied in triplicate to ensure the reliability and reproducibility of results.
2.5.1 Adsorption kinetic experiment (effect of contact time). To analyze the kinetic behavior of I2 adsorption, various kinetic models such as pseudo-first order (PFO), pseudo-second order (PSO), and intra-particle diffusion (IPD) models are applied. These can be represented as:| | |
ln(qe − qt) = ln(qe) − k1t
| (3) |
| |
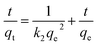 | (4) |
where qe (mg g−1) and qt (mg g−1) are the adsorption capacities at equilibrium, k1 (min−1), k2 (mg g−1 min−1), and kid (mg g−1 min−0.5) are the adsorption constants, and C denotes the intercept.51In the kinetic study, 0.09 g of aminated resin is added to 50 mL of a 200 mg L−1 I2 solution, and the mixture is agitated for 7 h. Samples are collected at time intervals of 0, 60, 120, 180, 240, 300, 360, and 420 min. These samples are centrifuged and analyzed for I2 concentration. The change in solid-to-liquid ratio during the time-optimization parameter is considered negligible because of the relatively small (2 mL) withdrawn volume compared to the total volume of solution.
2.5.2 Optimization of solid-to-liquid ratio. The batch adsorption experiment is further performed to study the effect of the S:L ratio by changing the amount of aminated resin (adsorbent dose). Different adsorbent concentrations (1.5, 3, 5, 7, 9, and 11 g L−1) are added to 10 mL of 200 mg L−1 I2 solution. The mixture is stirred for approximately 7 h, and after centrifugation, the residual concentration is analyzed to determine the optimized adsorbent dosage.
2.5.3 Adsorption isotherm experiment (effect of adsorbate concentration). To examine the maximum adsorption capacity and adsorption mechanism of the prepared adsorbent material, Langmuir, Freundlich, and Temkin isotherm models are applied. The linearized forms of these models are mentioned in eqn (6), (8), and (9), respectively.| |
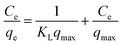 | (6) |
| |
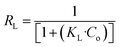 | (7) |
| |
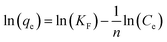 | (8) |
In the Langmuir adsorption model, eqn (6), Ce (mg L−1) is the equilibrium concentration, qe (mg g−1) and qmax (mg g−1) represent equilibrium adsorption capacity and maximum adsorption capacity, KL (L mg−1) denotes the Langmuir constant, and RL in eqn (7) is a dimensionless factor which shows the favorability of the adsorption process. If the value of RL is greater than 1, it indicates unfavorable adsorption; if it is less than 1, it indicates favorable adsorption.51 In the Freundlich adsorption model eqn (8), KF (mg g−1) is indicative of adsorption capacity, and n denotes intensity. In the Temkin adsorption model eqn (9), B = RT/b, where R is the general gas constant, T is temperature (in kelvin), b is the Temkin constant, KT (L g−1), and B (J mol−1) represent the binding constants and heat of adsorption, respectively.In the isotherm study, 0.09 g of adsorbent is added to 10 mL of a different concentration, 50, 100, 200, 400, and 600 mg L−1 I2 solution, and the mixture is agitated for 7 h. The samples are centrifuged and analyzed for the determination of I2 concentration.
3. Results and discussion
3.1 SEM and optical microscopic analysis
Morphological and size distribution analyses of resin beads are conducted before and after functionalization with DTA, as depicted in Fig. 1a–d. The SEM images of PVBCl (Fig. 1a) show a typical spherical morphology.52 While the DTA functionalized an aminated resin (Fig. 1b) shows no significant changes in surface texture or bead size, even the bead shape remains intact with a uniform spherical morphology after functionalization.53,54 This observation demonstrates that the introduction of DTA occurs primarily at the molecular level, altering only the surface chemistry instead of morphological transformation. Similar findings are also reported by Burevska-Atkovska et al.55 Such attainments are consistent with the perception that chemical functionalization predominantly exhibits the changes in surface chemistry and chemical composition of the material, whereas the macroscopic morphology remains unchanged. Additionally, the successful functionalization of PVBCl is validated visually, as indicated by a pronounced color transformation (Fig. S4). PVBCl appeared yellow, while after functionalization with DTA, the material showed a brownish coloration. This distinct contrast in appearance provides initial validation of the successful attachment of the functional group. This implies that the color shift observed in aminated resin is due to chemical modification instead of morphological transformation.
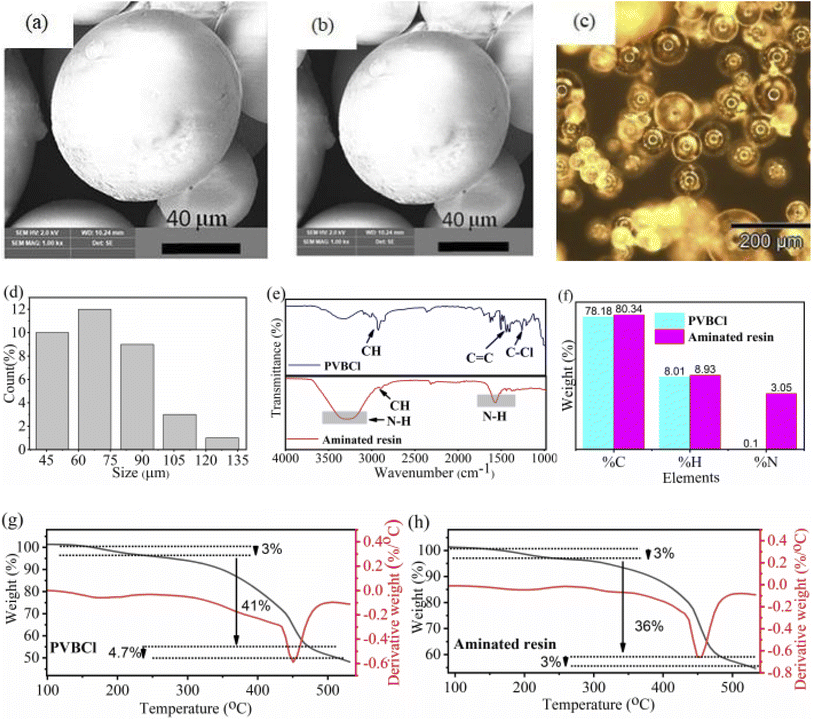 |
| | Fig. 1 SEM images of (a) PVBCl pristine resin (before functionalization), (b) aminated resin (after functionalization), (c) optical image of PVBCl resin beads, (d) histogram of bead's size distribution (PVBCl), (e) FTIR spectra of the PVBCl pristine and aminated resins, (f) organic elemental analysis of PVBCl pristine and aminated resins, (g) TGA-DTG profiles of PVBCl pristine, and (h) TGA-DTG profiles of aminated resin. | |
Optical microscopy is employed to analyze the size distribution of the prepared PVBCl resin. Fig. 1c and d show the optical microscopy image of prepared resin beads and the histogram of the bead's size distribution, respectively. The histogram (Fig. 1d) substantiates that beads of different sizes ranging from 45 to 135 µm exist, with the maximum beads lying between 60 and 75 µm. No noticeable changes are observed after amination, as a comparative image is integrated in Fig. S5 to illustrate this.
3.2 Structural analysis
To examine the structural characteristics of the resins and confirm the successful incorporation of amine group, FTIR analysis of the pristine PVBCl and the aminated resin is conducted in the 4000–1000 cm−1 spectral range, as shown in Fig. 1e. FTIR spectrum of the aminated resin shows the characteristic peak of the PVBCl resin matrix, consisting of aromatic rings with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (aromatic) and C–H stretches at around 1570 cm−1 and 3000 cm−1, respectively.56 The characteristic stretching of C–Cl appears at 1275 cm−1 in PVBCl pristine resin.57 Conversely, the band at 1275 cm−1 almost disappears in aminated resin because of the replacement of the chloro group with DTA functionality after the amination.58,59 Furthermore, the appearance of a broad and high-intensity N–H band centered around 3000–3500 cm−1 confirms the successful amination and synthesis of aminated resin.58,60,61 Additionally, another N–H band appears at around 1600 cm−1, which further validates the confirmation of amination.62 Burevska-Atkovska et al.63 also reported similar spectral features, with a characteristic band at around 3000–3670 cm−1, along with an assignment of 1600 cm−1 to N–H bending vibrations, explicitly confirming the successful introduction of amine functionalities.
C (aromatic) and C–H stretches at around 1570 cm−1 and 3000 cm−1, respectively.56 The characteristic stretching of C–Cl appears at 1275 cm−1 in PVBCl pristine resin.57 Conversely, the band at 1275 cm−1 almost disappears in aminated resin because of the replacement of the chloro group with DTA functionality after the amination.58,59 Furthermore, the appearance of a broad and high-intensity N–H band centered around 3000–3500 cm−1 confirms the successful amination and synthesis of aminated resin.58,60,61 Additionally, another N–H band appears at around 1600 cm−1, which further validates the confirmation of amination.62 Burevska-Atkovska et al.63 also reported similar spectral features, with a characteristic band at around 3000–3670 cm−1, along with an assignment of 1600 cm−1 to N–H bending vibrations, explicitly confirming the successful introduction of amine functionalities.
3.3 Elemental analysis
The organic elemental analysis (OEA) is carried out to further support the presence of DTA functionality in the aminated resin, shown in Fig. 1f. The aminated resin contained an elemental composition of 3.05 wt% N, 80.34 wt% C, and 8.93 wt% H. In contrast, the PVBCl pristine resin contained 0.10 wt% N, 78.18 wt% C, and 8.01 wt% H. The pronounced increase in N content is a distinct marker of the amine functionality in the polymer matrix. As the pristine PVBCl is devoid of nitrogen, the transition to a significant increase in percentage corroborates the successful attachment of DTA on PVBCl.58,59 In addition to N, both H and C contents also show a slight increase after amination, which aligns with the structural participation of the DTA chain, due to the introduction of multiple NH2 units along with additional CH2 groups into the resin backbone. This highlights that this functionalization route possibly anchors DTA moieties onto the PVBCl framework. These findings reveal that successful amination have occurred, and the final material contains amine donor sites, essential for strengthening the polymer's binding affinity for adsorption applications.
3.4 Thermogravimetric analysis (TGA)
To provide a deeper understanding of the thermal stability, degradation pathways, and structural integrity of PVBCl and aminated resin, thermogravimetric analysis (TGA) is performed at a heating rate of 10 °C min−1 from 35 to 530 °C under ambient air conditions (Fig. 1g and h). Both materials show a multistep degradation profile. At onset, weight loss of approximately 3% is observed at temperatures ranging from 130 to 230 °C, which can be attributed to the residual moisture or the desorption of physically absorbed solvent molecules within the polymer matrix. The more pronounced weight loss phase of 41% in PVBCl and 36% in aminated resin, approximately at 250 °C and extending to 450 °C, corresponds to the main chain polymer degradation, including the incorporated DTA groups and the uncross-linked portion of the PVBCl resin in the aminated resin.64 The final stage, initiated at 450 °C and extending beyond 500 °C, represents the thermal degradation of the cross-linked portion of the resins. Aminated resin was thermally more stable in comparison to PVBCl pristine resin, as evidenced by the thermal profiles of resins, which might be due to the incorporation of DTA in the aminated resin. The major decomposition peak of both resins appeared at 450 °C.
3.5 Adsorption study
UV-visible spectra of the pristine and aminated resins are shown in Fig. S6, whereas Fig. 2a shows the comparative I2 removal efficiency between PVBCl and the aminated resin over time. The aminated resin exhibits a substantially higher removal efficiency in comparison to the PVBCl. A substantial divergence in the removal performance of two types of adsorbent samples emerges with increasing time. The PVBCl exhibits poor adsorption behavior due to the unavailability of attached amine groups. It shows the removal efficiency of <10% even after 7 h. This performance can be ascribed to the chemically inert and hydrophobic nature of PVBCl, which is devoid of any functional groups. Conversely, the aminated resin promptly increases removal efficiency, achieving around 56% within the first 60 min. This presents a consistent removal trend, reaching nearly complete removal in 420 min. This demonstrates that the amine functional groups incorporation into the resin through reaction of PVBCl with diethylenetriamine enhances the material's affinity for the adsorption of I2 molecules. These findings corroborate that chemical functionalization of PVBCl through diethylenetriamine converts it from a low-affinity adsorbent into a significantly effective adsorbent material for I2 removal.
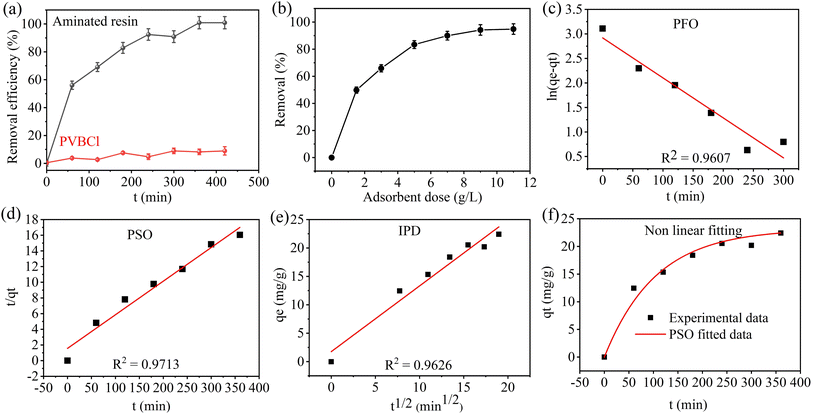 |
| | Fig. 2 (a) Comparative removal efficiency of I2 by PVBCl and aminated resin adsorbent versus time (Co = 200 mg L−1, contact time = 420 min, S:L ratio = 9 g L−1, solution volume = 10 mL, and shaking speed = 250 rpm), and (b) effect of S:L in the range of 1.5 to 11 g L−1 (Co = 200 mg L−1, contact time = 420 min, shaking speed = 250 rpm, solution volume = 10 mL), (c) adsorption kinetics linear model fitting, PFO model, (d) PSO model, (e) IPD model, and (f) non-linear curve fitting of PSO model (Co = 200 mg L−1, and contact time = 420 min, S:L ratio = 9 g L−1, solution volume = 10 mL, shaking speed = 250 rpm). | |
3.5.1 Effect of adsorbent dose. The evaluation of the adsorbent dose is a crucial aspect of adsorption studies, as it clarifies the effect of S:L on both removal percentage and adsorption capacity. The effect of aminated resin dosage on I2 removal is comprehensively examined by varying the adsorbent dose within the range of 1.5 to 11 g L−1 for 420 min, as shown in Fig. 2b. The removal efficiency shows a substantial enhancement with increasing the amount of adsorbent dose. At a dosage of 1.5 g L−1, the removal efficiency is 49.8%; however, it increases to >90% with an increase in the amount of 9 g L−1, and remains consistent, demonstrating no significant enhancement with further increasing the adsorbent amount.
3.5.2 Adsorption kinetics. The iodine adsorption onto the aminated resin is studied through various kinetic models to obtain a detailed understanding of the adsorption mechanism and nature. The linear fitting of experimental data with PFO, PSO, and IPD is shown in Fig. 2c–e, whereas Fig. 2f shows the non-linear fitting of the best fitted model along with the experimental data. The parameters for adsorption kinetics are calculated using eqn (3)–(5), and are presented in Table 1. Among the kinetic models employed, the PSO model demonstrates the highest correlation coefficient of 0.971 and exhibits the strongest agreement between the experimental and calculated adsorption capacities compared to the PFO model. This provides compelling evidence that the dominant mechanism for I2 removal is chemisorption involving a strong chemical interaction, such as a donor–acceptor complex mechanism between the amine functional groups present on the PVBCl matrix and iodine molecules. In sorption studies, chemisorption is generally preferred over physisorption because of the stronger and more specific chemical interactions between the adsorbate and the adsorbent.65
Table 1 Kinetic adsorption parameters PFO, PSO, and IPD for the adsorption of I2 by aminated resin
| Type of kinetic model |
Parameter |
Value |
| PFO |
Experimental qe (mg g−1) |
22.42 |
| Calculated qe (mg g−1) |
18.49 |
| K1 (min−1) |
8.1 × 10−6 |
| R2 |
0.960 |
| PSO |
Experimental qe (mg g−1) |
22.42 |
| Calculated qe (mg g−1) |
23.31 |
| K2 (g mg−1 min−1) |
0.001 |
| R2 |
0.971 |
| IPD |
Kid (mg g−1 min−0.5) |
1.15 |
| C |
1.75 |
| R2 |
0.962 |
3.5.3 Adsorption isotherm. The effect of initial I2 concentration on the adsorption process plays a crucial role in determining the removal efficiency. Efficient I2 removal is generally observed at lower concentrations due to the comparatively large number of available active sites. However, the binding sites become limited at higher concentrations, leading to a decrease in adsorption efficiency, which is shown in Fig. S7, where the color changes from transparent to purple with increasing adsorbate concentration. Moreover, elevated I2 concentrations increase competition among iodine molecules for the same binding sites, thereby hampering the overall uptake. Therefore, adsorption experiments are performed to determine the maximum adsorption capacity of the adsorbent and to analyze the effect of I2 concentration on the overall system's performance. Fig. 3a–c present the linear fitting of Langmuir, Freundlich, and Temkin models, respectively, whereas Fig. 3d illustrates the Langmuir non-linear curve fitted data with experimental data. Among the applied adsorption isotherm models, the Langmuir adsorption model provides the best fit, with the highest correlation coefficient value of 0.937 and a maximum adsorption capacity of 54.64 mg g−1. This model suggests monolayer adsorption of I2 on a homogeneous surface, and its high degree of correlation with experimental data demonstrates that the aminated polystyrene offers uniform and well-defined adsorption sites for iodine. Langmuir, Freundlich, and Temkin isotherm parameters are calculated using eqn (6), (8), and (9) and shown in Table 2. The dimensionless separation factor (RL = 0.60) calculated using eqn (7) and the Freundlich adsorption constant (N = 2.35) further suggests the favorable nature of the adsorption process.
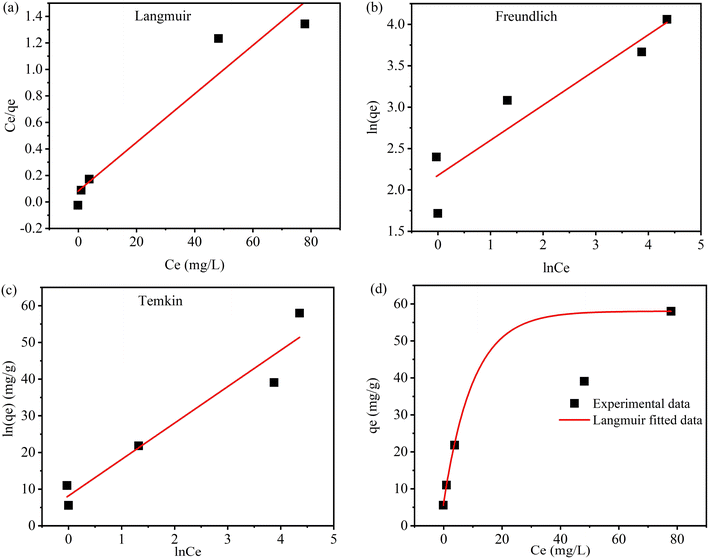 |
| | Fig. 3 Adsorption isotherm linear model fitting of (a) Langmuir adsorption isotherm, (b) Freundlich adsorption isotherm, (c) Temkin adsorption isotherm, (d) and non-linear curve fitting of Langmuir adsorption isotherm model (Co = 200 mg L−1, contact time = 420 min, S:L ratio = 9 g L−1, solution volume = 10 mL, shaking speed = 250 rpm). | |
Table 2 Adsorption isotherm parameters for iodine adsorption onto aminated resin
| Type of isotherm |
Parameter |
Values |
| Langmuir |
Qmax (mg g−1) |
54.64 |
| KL (L mg−1) |
0.21 |
| RL |
0.60 |
| R2 |
0.937 |
| Freundlich |
KF (mg g−1) |
8.81 |
| N |
2.35 |
| R2 |
0.885 |
| Temkin |
KT (L g−1) |
3.35 |
| B (J mol−1) |
9.92 |
| R2 |
0.936 |
3.5.4 Reusability. The reusability of aminated resin for iodine adsorption was sequentially evaluated over four adsorption–desorption cycles, as presented in Fig. 4. For the regeneration of aminated resin, thermal desorption was used due to the sublimable and volatile nature of molecular iodine, particularly the regeneration of spent adsorbent is achieved by vacuum drying at 60 °C for 24 hours. Based on preliminary considerations, the desorption temperature is selected to ensure efficient evaporation of the solvent while concurrently promoting the release of physically adsorbed I2. Increasing the temperature beyond 60 °C facilitates iodine volatilization; nevertheless, it may also lead to deformation or softening of the polymer matrix, ultimately affecting its adsorption performance and structural stability. Therefore, this temperature is considered an optimal compromise between preservation of the material integrity and effective desorption. In the first cycle, aminated resin exhibits nearly complete removal, confirming strong affinity and high accessibility of amine functionalities towards iodine molecules. After regeneration, the second cycle sustained a significant removal efficiency of 90%, substantiating that vacuum drying is a mild and effective regeneration strategy. During the third and fourth cycles, there is a marked decline in removal efficiency, stabilizing at approximately 58%, indicating partial deactivation or progressive saturation of amine active sites. Regardless of the decrease in removal efficiency, the aminated resin after multiple cycles still exhibits reasonable removal, validating its chemical stability, structural stability, and partial regenerability.
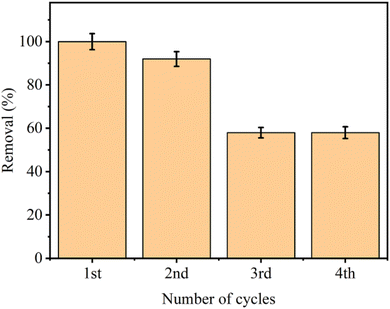 |
| | Fig. 4 Reusability of aminated resin for I2 removal. | |
3.5.5 Proposed adsorption mechanism. Fig. 5 presents the schematic illustration of the adsorption mechanism. The adsorption of I2 onto aminated resin proceeds primarily through chemisorption. This demonstrates that the rate-limiting step involves a chemical interaction, as diethylenetriamine introduces many electron-rich nitrogen atoms onto the polystyrene backbone, thereby increasing the density of potential active sites. Each nitrogen atom, provided with a lone pair of electrons, can serve as an electron donor and participate in charge transfer interactions with I2. Iodine, in non-polar solvents primarily as molecular iodine with accessible antibonding orbitals, is capable of accepting electron density from nitrogen donors, resulting in the formation of a donor–acceptor complex.66 This interaction, without reducing iodine to iodide or fully breaking it, polarizes the I–I bond. When exposed to the surface functionalization, nitrogen atoms donate electron density to the molecules of iodine, leading to comparatively stable N⋯I2 interactions. Avais et al.67 reported similar results in which antibonding orbitals of iodine interact with the lone pair of nitrogen to form stable complexes, although polyiodide formation depends on ionic and solvent conditions. These interactions are stronger than physisorption, consistent with chemisorption via electron donation. The multiple nitrogen sites may promote multidentate binding, increasing both the surface coverage and binding strength. In this way, the aminated resin is effective for iodine removal from non-polar solvents.
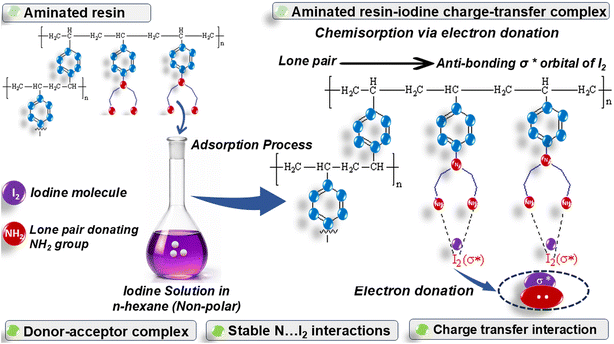 |
| | Fig. 5 Schematic illustration of I2 adsorption on the aminated resin. | |
4. Conclusion
The aminated resin is successfully synthesized from poly-vinylbenzyl chloride (PVBCl) resin using diethylene triamine (DTA) via an efficient and facile synthetic approach. The successful functionalization and synthesis are confirmed through SEM, FTIR, CHNS, and TGA analyses. SEM analysis highlights the typical spherical morphology of the aminated resin; FTIR analysis demonstrates the existence of the N–H band and the disappearance of the C–Cl stretch after functionalization; CHNS analysis shows N content after functionalization of the pristine resin; and TGA analysis further validates the high thermal stability of the aminated resin. The aminated resin shows excellent performance for iodine removal in comparison to the PVBCl pristine resin, and its adsorption efficiency depended on its contact time and the iodine concentration. The enhanced performance of the aminated resin in comparison to the PVBCl pristine resin was attributed to the chelating effect of the introduced DTA, which provided the binding sites to the resin. Adsorption studies revealed that the interaction of iodine and aminated resin is best explained through the Langmuir isothermal model, as they fit the best to experimental data. The adsorption process follows the pseudo-second-order model. The chemical stability of covalently bonded amine functionalities and PVBCl backbone indicates that in non-reactive organic media, aminated resin maintains its structural integrity. This substantiates its potential applicability in iodine decontamination processes under engineered or controlled conditions. However, further studies in aqueous or multi-component systems would have to thoroughly evaluate its performance under nuclear waste and realistic environmental conditions.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
Data will be made available on reasonable request.
Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d6ra01669c.
Acknowledgements
The authors highly acknowledge the Central Analytical Facility Division and Physics Division of the Pakistan Institute of Nuclear Science and Technology (PINSTECH) for helping in the characterization of resins prepared in this study.
References
- N. Armaroli and V. Balzani, Energy Environ. Sci., 2011, 4, 3193–3222 RSC.
- S.-T. Lee and S.-M. Jung, SAGE Open, 2024, 14, 21582440241301468 CrossRef.
- S. Sadekin, S. Zaman, M. Mahfuz and R. P. Sarkar, Energy Proc., 2019, 160, 513–518 CrossRef.
- C. Karakosta, C. Pappas, V. Marinakis and J. Psarras, Renew. Sustain. Energy Rev., 2013, 22, 187–197 CrossRef.
- J. Bruggink and B. D. van Zwaan, Int. J. Glob. Energy Issues, 2002, 18, 151–180 CrossRef.
- M. Mathew, Prog. Nucl. Energy, 2022, 143, 104080 CrossRef CAS.
- T. J. Robshaw, S. M. Griffiths, A. Canner, J. P. Bezzina, A. G. Waller, D. B. Hammond, S. van Meurs and M. D. Ogden, Chem. Eng. J., 2020, 390, 124647 CrossRef CAS.
- D. Mallants and N. Chapman, Nat. Mater., 2020, 19, 959–961 CrossRef CAS PubMed.
- L. M. R. P. W. Sylvester, P. B. J. P. L. Richard and J. Bull, J. Toxicol. Environ. Health, Part A, 1998, 55, 93–106 CrossRef PubMed.
- J. E. Ten Hoeve and M. Z. Jacobson, Energy Environ. Sci., 2012, 5, 8743–8757 RSC.
- P. A. Kharecha and J. E. Hansen, Environ. Sci. Technol., 2013, 47, 4889–4895 CrossRef CAS PubMed.
- F. C. Küpper, M. C. Feiters, B. Olofsson, T. Kaiho, S. Yanagida, M. B. Zimmermann, L. J. Carpenter, G. W. Luther III, Z. Lu and M. Jonsson, Angew. Chem., Int. Ed., 2011, 50, 11598–11620 CrossRef PubMed.
- A. Saiz-Lopez, J. M. Plane, A. R. Baker, L. J. Carpenter, R. Von Glasow, J. C. Gómez Martín, G. McFiggans and R. W. Saunders, Chem. Rev., 2012, 112, 1773–1804 CrossRef CAS PubMed.
- T. Pan, K. Yang, X. Dong and Y. Han, J. Mater. Chem. A, 2023, 11, 5460–5475 RSC.
- W. Xie, D. Cui, S.-R. Zhang, Y.-H. Xu and D.-L. Jiang, Mater. Horiz., 2019, 6, 1571–1595 RSC.
- K. Subrahmanyam, D. Sarma, C. D. Malliakas, K. Polychronopoulou, B. J. Riley, D. A. Pierce, J. Chun and M. G. Kanatzidis, Chem. Mater., 2015, 27, 2619–2626 CrossRef CAS.
- Y. Benamrane, J.-L. Wybo and P. Armand, J. Environ. Radioact., 2013, 126, 239–252 CrossRef CAS PubMed.
- W. Han, W. Clarke and S. Pratt, Ecol. Eng., 2016, 94, 286–294 CrossRef.
- M. Balter, Science, 1995, 270, 1758–1759 CrossRef CAS PubMed.
- M. Zhao, S. Wang, H. Wang, P. Qin, D. Yang, Y. Sun and F. Kong, Environ. Pollut., 2019, 248, 938–946 CrossRef CAS PubMed.
- S. A. Patil, R. R. Rodríguez-Berríos, D. Chavez-Flores, D. V. Wagle and A. Bugarin, ACS ES&T Water, 2023, 3, 2009–2023 Search PubMed.
- S. M. Siddeeg, Mater. Res. Express, 2020, 7, 025038 CrossRef CAS.
- M. A. Tahoon, S. M. Siddeeg, N. Salem Alsaiari, W. Mnif and F. Ben Rebah, Processes, 2020, 8, 645 CrossRef CAS.
- K. W. Chapman, P. J. Chupas and T. M. Nenoff, J. Am. Chem. Soc., 2010, 132, 8897–8899 CrossRef CAS PubMed.
- R. M. Asmussen, J. Matyáš, N. P. Qafoku and A. A. Kruger, Hazard Mater., 2019, 379, 119364 CrossRef CAS PubMed.
- M. Tan, L. Horvàth, P. S. Brunetto and K. M. Fromm, Polymers, 2018, 10, 665 CrossRef PubMed.
- G. M. Espallargas and E. Coronado, Chem. Soc. Rev., 2018, 47, 533–557 RSC.
- K. Shen, L. Zhang, X. Chen, L. Liu, D. Zhang, Y. Han, J. Chen, J. Long, R. Luque and Y. Li, Science, 2018, 359, 206–210 CrossRef CAS PubMed.
- Z. Ji, H. Wang, S. Canossa, S. Wuttke and O. M. Yaghi, Adv. Funct. Mater., 2020, 30, 2000238 CrossRef CAS.
- Y. Lin, X. Jiang, S. T. Kim, S. B. Alahakoon, X. Hou, Z. Zhang, C. M. Thompson, R. A. Smaldone and C. Ke, J. Am. Chem. Soc., 2017, 139, 7172–7175 CrossRef CAS PubMed.
- K. Jie, Y. Zhou, E. Li, Z. Li, R. Zhao and F. Huang, J. Am. Chem. Soc., 2017, 139, 15320–15323 CrossRef CAS PubMed.
- Q. Tao, L. Sun and L. Jing, Sep. Purif. Technol., 2025, 353, 128608 CrossRef CAS.
- X.-H. Xu, Y.-X. Li, L. Zhou, N. Liu and Z.-Q. Wu, Chem. Sci., 2022, 13, 1111–1118 RSC.
- S. Xiong, X. Tang, C. Pan, L. Li, J. Tang and G. Yu, ACS Appl. Mater. Interfaces, 2019, 11, 27335–27342 CrossRef CAS PubMed.
- T. Geng, C. Zhang, M. Liu, C. Hu and G. Chen, J. Mater. Chem. A, 2020, 8, 2820–2826 RSC.
- X. Qian, Z.-Q. Zhu, H.-X. Sun, F. Ren, P. Mu, W. Liang, L. Chen and A. Li, ACS Appl. Mater. Interfaces, 2016, 8, 21063–21069 CrossRef CAS PubMed.
- Y. Zhu, Y.-J. Ji, D.-G. Wang, Y. Zhang, H. Tang, X.-R. Jia, M. Song, G. Yu and G.-C. Kuang, J. Mater. Chem. A, 2017, 5, 6622–6629 RSC.
- Y. H. Abdelmoaty, T.-D. Tessema, F. A. Choudhury, O. M. El-Kadri and H. M. El-Kaderi, ACS Appl. Mater. Interfaces, 2018, 10, 16049–16058 CrossRef CAS PubMed.
- W. Du, Y. Qin, C. Ni, W. Dai and J. Zou, ACS Appl. Polym. Mater., 2020, 2, 5121–5128 CrossRef CAS.
- Y. Li, X. Li, J. Li, G. Cheng and H. Ke, Microporous Mesoporous Mater., 2021, 325, 111351 CrossRef CAS.
- Q. Tao, X. Zhang, L. Jing, L. Sun and P. Dang, Molecules, 2023, 28, 8151 CrossRef CAS PubMed.
- D. K. Harijan, V. Chandra, T. Yoon and K. S. Kim, J. Hazard. Mater., 2018, 344, 576–584 CrossRef CAS PubMed.
- M. Alsalbokh, N. Fakeri, S. Lawson, A. A. Rownaghi and F. Rezaei, Chem. Eng. J., 2021, 415, 128968 CrossRef CAS.
- D. Luo, Y. He, J. Tian, J. L. Sessler and X. Chi, J. Am. Chem. Soc., 2021, 144, 113–117 CrossRef PubMed.
- H. Li, D. Zhang, K. Cheng, Z. Li and P.-Z. Li, ACS Sustainable Chem. Eng., 2023, 6, 1295–1302 CAS.
- M. Li, X. Wang, J. Zhang, Y. Gao and W. Zhang, Appl. Surf. Sci., 2023, 619, 156819 CrossRef CAS.
- J. L. Means and C. A. Alexander, Nucl. Chem. Waste Manage., 1981, 2, 183–196 CrossRef CAS.
- B. C. Monda and A. K. Das, J. Indian Chem. Soc., 2004, 81, 95–110 Search PubMed.
- Z. Ye, L. Chen, C. Liu, S. Ning, X. Wang and Y. Wei, React. Funct. Polym., 2019, 135, 52–57 CrossRef CAS.
- D. Jermakowicz-Bartkowiak, B. Kolarz and A. Serwin, React. Funct. Polym., 2005, 65, 135–142 CrossRef CAS.
- K. Khaliq, A. Khan, S. Shahida, R. Akhtar, M. A. R. Anjum, I. Rafiq, M. Rehan, R. N. Qureshi, S. Iqbal and M. Saifullah, RSC Adv., 2026, 16, 1051–1067 RSC.
- P. Cyganowski, D. Jermakowicz-Bartkowiak and J. Chgcmanowski, Acta Chim. Slov., 2015, 62, 672–678 CrossRef CAS PubMed.
- Y. Zhang, X. Pan and J. Zhu, Polymers, 2021, 13, 1632 CrossRef CAS PubMed.
- R. Rodrigo, C. A. Toro and J. Cuellar, J. Appl. Polym. Sci., 2013, 130, 4054–4065 CrossRef CAS.
- K. Burevska-Atkovska, F. Olivieri, R. Avolio, R. Castaldo, M. Cocca, M. E. Errico, G. Gentile, A. J. C. Grozdanov, S. A. Physicochemical and E. Aspects, Colloids Surf., A, 2024, 700, 134720 CrossRef CAS.
- R. Akhtar, S. Latif, S. A. A. Shah, S. Saeed and A. Aziz, Nucl. Eng. Technol., 2023, 55, 2547–2555 CrossRef CAS.
- M. Wilson, R. Kore, A. Ritchie, R. Fraser, S. Beaumont, R. Srivastava and J. Badyal, Colloids Surf., A, 2018, 545, 78–85 CrossRef CAS.
- M. Rajiv Gandhi, N. Viswanathan and S. Meenakshi, Ind. Eng. Chem. Res., 2012, 51, 5677–5684 CrossRef CAS.
- N. Viswanathan, S. M. Prabhu and S. Meenakshi, J. Fluorine Chem., 2013, 153, 143–150 CrossRef CAS.
- S. Li, C. Cui and H. Hou, Colloid Polym. Sci., 2015, 293, 2681–2688 CrossRef CAS.
- D. Jermakowicz-Bartkowiak, React. Funct. Polym., 2005, 62, 115–128 CrossRef CAS.
- C. Xu, C. Wang, W. Sun, W. Yu, C. Yin, F. Liu, M. Xian and S. Yu, Environ. Sci. Pollut. Res., 2019, 26, 10767–10775 CrossRef CAS PubMed.
- K. Burevska-Atkovska, F. Olivieri, R. Avolio, R. Castaldo, M. Cocca, M. E. Errico, G. Gentile and A. Grozdanov, Colloids Surf., A, 2024, 700, 134720 CrossRef CAS.
- M. E. Mathew, I. Ahmad, S. Thomas, M. B. KASSIM and R. Daik, Sains Malays., 2021, 50, 1767–1773 CrossRef CAS.
- K. Khaliq, M. A. R. Anjum, S. Shahida, R. Akhtar, A. Khan, M. A. Shafiq, I. Rafiq, M. Rehan, R. N. Qureshi and S. Iqbal, RSC Adv., 2025, 15, 14158–14169 RSC.
- H. A. Bent, J. Chem. Rev., 1968, 68, 587–648 CrossRef CAS.
- M. Avais and S. Chattopadhyay, J. Mater. Chem. A, 2022, 10, 20090–20 RSC.
Footnote |
| † The first three authors have contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence d,
Samreen Shehzadid,
Iqra Rafique
d,
Samreen Shehzadid,
Iqra Rafique