DOI:
10.1039/D6RA01650B
(Paper)
RSC Adv., 2026,
16, 18709-18723
DNA/BSA binding, antidiabetic and antioxidant studies of copper(II) complexes derived from ONO-tridentate Schiff bases and diimines as auxiliary ligands
Received
25th February 2026
, Accepted 1st April 2026
First published on 10th April 2026
Abstract
Six new copper(II) complexes with general molecular formula [CuL1(Phen)] (1), [CuL2(Phen)] (2), [CuL3(Phen)] (3), [CuL1(DM-Phen)] (4), [CuL2(DM-Phen)] (5) and [CuL3(DM-Phen)] (6) (L = (E)-1-(((4-chloro-2-hydroxyphenyl)imino)methyl)naphthalen-2-ol (L1), (E)-1-(((2-hydroxy-5-methylphenyl)imino)methyl)naphthalen-2-ol (L2), (E)-1-(((2-hydroxy-4-nitrophenyl)imino)methyl)naphthalen-2-ol (L3), Phen = 1,10-phenanthroline and DM-Phen = 2,9-dimethyl-1,10-phenanthroline) have been synthesized and characterized by spectroscopic as well as crystallographic techniques. Molecular structures of 2, 4, 5 and 6 revealed that they are mononuclear species where the copper(II) center is five-coordinated to a pair of oxygen atoms as well as one nitrogen atom from the ligands (L1–L3) and to a pair of nitrogen atoms from the auxiliary ligand (Phen or DM-Phen) conforming to a distorted square pyramid. The binding affinities of ligands L1–L3 and copper(II) complexes 1–6 with calf thymus DNA (ctDNA) and bovine serum albumin (BSA) were evaluated using UV-visible absorption spectroscopy. For ctDNA, all compounds showed hypochromism and bathochromic shifts suggesting intercalative binding, with Kb values ranging from 1.49 × 104 to 2.74 × 105 M−1. The complexes exhibited 3–18-fold higher affinity than ligands, especially those with 2,9-dimethyl-1,10-phenanthroline coligands. BSA titrations of complexes 1–6 revealed hyperchromic shifts at ∼280 nm, with Kb values of 9.59 × 104 to 2.41 × 105 M−1. These moderate-to-strong bindings were enhanced by lipophilic substituents that promote hydrophobic and π-stacking interactions in protein pockets. All of the compounds inhibit α-amylase better than acarbose with complex 5 (IC50 = 0.274 mM) showing the highest activity. For the α-glucosidase assay, only complexes 2 (IC50 = 0.055 mM) and 4 (IC50 = 0.054 mM) outshined acarbose (IC50 = 0.059 mM) while other compounds showed moderate to good α-glucosidase inhibition activity. Antioxidant activities of the free ligands showed better activity than that of the metal complexes with none of the compounds performing better than quercetin in all of the assays explored for this study.
1 Introduction
The research on the design and development of metallodrugs with multifunctional therapeutic activities continues to gain significant interest in the field of bioinorganic and medicinal chemistry.1,2 Among metal ions explored in metal-based drugs, copper has been unique due to its important role in biological systems. Copper exists in crucial enzymes (i.e., ceruloplasmin, laccase, superoxide dismutase, ascorbate oxidase, among others).3,4 These enzymes are essential in energy metabolism and nervous system development.5 It is found in humans either as Cu2+ (oxidized form) or Cu+ (reduced form). While high doses of copper are toxic, copper(II) ions are less toxic than copper(I) ions at appropriate concentrations. This is because, in aqueous solutions, copper(II) is the most stable oxidation state, while copper(I) is highly prone to oxidation (less stable) and not easy to develop into reliable oral medication. In pharmaceuticals, copper-based drugs or supplements are mainly copper(II) salts or complexes, i.e., copper(II) glycinate and copper(II) citrate, both common in supplements.6,7 Copper(II) participates in diverse physiological processes, including enzymatic catalysis, electron transfer, and oxidative stress regulation, making copper-based complexes potential candidates for therapeutic application.8
Schiff base ligands have been widely explored as chelate ligands in coordination chemistry.9,10 They are well known for their structural versatility, robust metal-binding ability, and tunable electronic properties. The bio-active azomethine functional group, which allows nucleophilic and electrophilic reactions at the electron-rich nitrogen and electron-deficient carbon atoms, respectively, has been linked to the inherent therapeutic qualities of Schiff bases.11,12 Copper(II) complexes derived from Schiff bases have been reported to display a myriad of medicinal properties, which include anticancer, antioxidant, antimicrobial, and antifungal with enhanced medicinal activity compared to the free ligands.13,14 However, the incorporation of planar nitrogen heterocyclic (N-heterocyclic) i.e., 2,2-bipyridine, 1,10-phenanthroline, and 2,9-dimethyl-1,10-phenanthroline, is known to further enhance the biological activities of copper(II) complexes. These N-heterocyclic ligands are well known for their strong π–π stacking interaction with nucleic acid–base pairs of DNA, hence facilitating DNA intercalation.15
DNA binding studies play an important role in developing DNA molecule probes and chemotherapeutics.16,17 It also helps in understanding the mechanisms of action of metal-based drugs, as interaction with DNA can influence replication and transcription processes.18 Strong binding affinities for DNA via intercalative, groove-binding, or electrostatic modes have been shown by copper(II) complexes, especially those with Schiff bases and N-heterocyclic ligands and these affinities do corroborate with biological activities.19–23 Hence, the need to evaluate the DNA binding potential of newly synthesized compounds is crucial to elucidate possible mechanisms of biological activity. Similarly, BSA binding studies are equally vital, as bovine serum albumin (BSA), a model plasma protein, transports metal complexes in blood, influencing their bioavailability, distribution, and toxicity. Copper(II) Schiff base complexes often exhibit strong BSA affinity through hydrophobic interactions or hydrogen bonding with its Sudlow sites, which correlates with enhanced therapeutic efficacy.24 These studies provide insights into drug–protein interactions, complementing DNA binding data for multifunctional agents.
In addition to the DNA and BSA binding potential of copper(II) complexes, they have also attracted interest as antidiabetic agents, through the inhibition of carbohydrate-hydrolyzing enzymes such as α-glucosidase and α-amylase enzymes.25–27 Inhibiting these enzymes can reduce postprandial hyperglycemia by retarding the rate at which dietary starch breaks down to glucose in the body and this has been validated as an effective strategy to manage diabetes.28 It is important to note that, drugs like acarbose and miglitol have been successfully used to treat diabetes by acting as α-glucosidase and α-amylase inhibitors. However, adverse effects associated with their usage i.e., stomach discomfort, meteorism, and diarrhea due to undigested carbohydrate fermenting in the bowel, have significantly decreased their use as antidiabetic medication.29,30 Hence, the need for new α-glucosidase and α-amylase inhibitors with little or no side effects.
Interestingly, oxidative stress has been associated with the progression and complications of diabetes, highlighting the relevance of antioxidant activity in potential antidiabetic drugs.31–33 Aside from that free radicals are closely linked to the cause of diabetes, they are also associated to deadly diseases such as liver cirrhosis, cancer, atherosclerosis among others.34 Hence, an antioxidant drug could play a crucial chemoprotective role in the treatment of diabetes and other diseases. Copper(II) complexes do display radical scavenging activity due to facile redox cyclic between Cu(II) and Cu(I) states, contributing to their therapeutic relevance.14 Copper(II) complexes derived from Schiff bases have demonstrated significant antioxidant activities in multiple radical scavenging assays.9,14,35
In this study, the ligand design was based on introducing controlled variations in electronic, steric and physicochemical properties. Chloro and nitro substituents were selected as electron-withdrawing groups, while the methyl group was included as an electron-donating substituent to facilitate comparison of electronic effects on the resulting copper(II) complexes. Furthermore, 1,10-phenanthroline and 2,9-dimethyl-1,10-phenanthroline were used as co-ligands to probe steric and lipophilic influences, as methyl substitution at the 2 and 9 positions may increase steric hindrances and hydrophobicity. These variations were considered in relation to their potential impact on biological interactions, including DNA binding affinity and inhibitory activity against α-amylase and α-glucosidase enzymes. Herein, we report the synthesis and characterization of copper(II) complexes derived from ONO-donor Schiff base ligands and N-heterocyclic (1,10-phenanthroline or 2,9-dimethyl-1,10-phenanthroline) as coligands. Their DNA/BSA binding, antidiabetic, and antioxidant potential are investigated to evaluate their suitability as multifunctional bioactive agents.
2 Experimental
2.1 Reagents
The reagents used for the preparation of the metal complexes are 1,10-phenanthroline (≥99%), 2,9-dimethyl-1,10-phenanthroline (≥98%), 2-hydroxy-4-nitroaniline (98%), 2-amino-5-chlorophenol (97%), 2-hydroxy-5-methylaniline (97%), acetic acid, and copper(II) acetate monohydrate (≥98%). Solvents such as dimethyl sulfoxide (DMSO), methanol and diethyl ether are A.C.S. grade and used as obtained. All reagents are purchased from Merck.
2.2 Instrumentation
The FT-IR spectra for complexes 1–6 were obtained using a PerkinElmer Universal A.T.R. spectrum 100 FT-IR spectrometer, and the elemental/combustion analysis for the complexes was carried out using a Vario elemental E.L. cube CHNS analyser. Shimadzu UV-Vis-NIR spectrophotometer was used to obtain the UV-Visible spectra of all the complexes and Waters Synapt G2 coupled to a Waters UPLC., ESI probe, ESI Positive, Cone Voltage 15 V was used to analyze the mass spectra for the complexes. The JEOL FA200 spectrometer was used to record the electron paramagnetic resonance (EPR) spectra for compounds 1–6.
2.3 Preparation of complexes 1–6
We have previously reported the synthetic procedures as well as fully characterized the Schiff base ligands L1–L3 used in the preparation of complexes 1–6.36 For the preparation of complexes 1–6, we followed the procedure reported by Kiran et al.37 with slight modification. 0.751 mmol (0.150 g) of copper(II) acetate monohydrate was dissolved in 20 mL of methanol in 100 mL round bottom flask and stirred until complete dissolution. Thereafter, 0.751 mmol of methanolic solution of 1,10-phenanthroline or 2,9-dimethyl-1,10-phenanthroline was added to the copper(II) salt solution and heated to around 40–50 °C while stirring vigorously for 1 h. For the 1,10-phenanthroline, the solution turned deep blue, while the one for 2,9-dimethyl-1,10-phenanthroline, it turned light green. 0.751 mmol of L1–L3 dissolved in 20 mL of dimethyl sulfoxide is added to the reaction mixture in a round bottom flask and further stirred for another 1 h. After 10 min of stirring, a reddish-brown precipitate crashed out of the reaction mixture, which was later washed with methanol and rinsed four times with diethyl ether. The obtained products were dried in an oven at 60–70 °C.
2.3.1 Synthesis of Cu(L1)(Phen) (1). FT-IR ν (cm−1): (νC–H = 3054), (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N = 1600), (νCC = 1576), (νC–N = 1395), (νC–O = 1291), and (νCu–N = 557). ES-TOF MS: m/z(%): [M + H]+, 539.0454, [M + Na-naph]+, 436.9912 MS calc. analysis: 538.04 (100.0%), 540.04 (44.6%). Anal. calc. for C29H18ClCuN3O20.5C2H6OS: C, 62.28, H, 3.66, N, 7.26. Found: C, 62.30, H, 3.89, N, 7.46.
N = 1600), (νCC = 1576), (νC–N = 1395), (νC–O = 1291), and (νCu–N = 557). ES-TOF MS: m/z(%): [M + H]+, 539.0454, [M + Na-naph]+, 436.9912 MS calc. analysis: 538.04 (100.0%), 540.04 (44.6%). Anal. calc. for C29H18ClCuN3O20.5C2H6OS: C, 62.28, H, 3.66, N, 7.26. Found: C, 62.30, H, 3.89, N, 7.46.
2.3.2 Synthesis of Cu(L2)(Phen) (2). FT-IR ν (cm−1): (νC–H = 3051), (νCN = 1600), (νCC = 1583), (νC–N = 1388), (νC–O = 1299), and (νCu–N = 557). ES-TOF MS: m/z(%): [M + H]+, 519.100, [M + Na-naph]+, 417.0460 MS calc. analysis: 518.09 (100.0%), 520.09 (44.6%), 519.10 (32.4%). Anal. calc. for C30H21CuN3O2: C, 69.42, H, 4.08, N, 8.10. Found: C, 69.30, H, 3.42, N, 8.04.
2.3.3 Synthesis of Cu(L3)(Phen) (3). FT-IR ν (cm−1): (νC–H = 3054), (νCN = 1598), (νCC = 1584), (νC–N = 1365), (νC–O = 1284), and (νCu–N = 572). ES-TOF MS: m/z(%): [M + H]+, 550.0701, [M + Na-naph]+, 448.0151 MS calc. analysis: 549.06 (100.0%), 551.06 (44.6%), 550.07 (31.4%). Anal. calc. for C29H18CuN4O40.5C2H6OS: C, 61.17, H, 3.59, N, 9.51. Found: C, 61.29, H, 3.87, N, 9.82.
2.3.4 Synthesis of Cu(L1)(DMPhen) (4). FT-IR ν (cm−1): (νC–H = 3100–3000), (νCN = 1601), (νCC = 1587), (νC–N = 1394), (νC–O = 1290), and (νCu–N = 569). ES-TOF MS: m/z(%): [M + H]+, 567.0773, [M + Na-ArCl]+, 479.1292 MS calc. analysis: 566.07 (100.0%), 568.07 (44.6%), 567.07 (33.5%). Anal. calc. for C29H18CuN4O40.5C2H6OS: C, 64.22, H, 4.06, N, 7.11. Found: C, 64.44, H, 4.26, N, 7.24.
2.3.5 Synthesis of Cu(L2)(DMPhen) (5). FT-IR ν (cm−1): (νC–H = 3048), (νCN = 1601), (νCC = 1585), (νC–N = 1389), (νC–O = 1275), and (νCu–N = 555). ES-TOF MS: m/z(%): [M + Na-ArCH3]+, 479.1297 [M + H]+, 547.12 MS calc. analysis: 546.1 (100.0%), 548.12 (44.6%), 547.13 (34.6%). Anal. calc. for C32H25CuN3O20.3C2H6OS: C, 68.63, H, 4.73, N, 7.38. Found: C, 68.30, H, 4.78, N, 7.24.
2.3.6 Synthesis of Cu(L3)(DMPhen) (6). FT-IR ν (cm−1): (νC–H = 3054), (νCN = 1600), (νCC = 1587), (νC–N = 1367), (νC–O = 1277), and (νCu–N = 571). ES-TOF MS: m/z(%): [M + Na-ArNO2]+, 479.1288 MS calc. analysis: 577.09 (100.0%), 579.09 (44.6%), 578.10 (33.5%). Anal. calc. for C31H22CuN4O40.5C2H6OS: C, 62.88, H, 4.08, N, 9.08. Found: C, 62.74, H, 3.92, N, 9.21.
2.4 Single crystal X-ray crystallography
Crystals with dimensions of 0.258 × 0.075 × 0.032 mm3 (for 2), 0.231 × 0.117 × 0.107 mm3 (for 4), 0.483 × 0.221 × 0.074 mm3 (for 5), and 0.153 × 0.140 × 0.039 mm3 (for 6) were mounted on a Bruker DUO Apex II X-ray diffractometer equipped with an INCOATEC IµS HB microfocus tube (MoKα radiation λ = 0.71073 Å) fitted with a multilayer monochromator. Data were captured with a CCD (charge-coupled device) area detector. Data collection was carried out at 100 K using an Oxford Cryosystems cryostat (700 series Cryostream Plus) attached to the diffractometer. Data collection and reduction were carried out using the Bruker software package APEX4,38 using standard procedures. All structures were solved and refined using SHELX-201639 employed within the X-Seed40 environment. Hydrogen atoms were placed in calculated positions using riding models. Diagrams for the crystal structures were obtained by Mercury software.41 Table 1 details the structural data together with the refinement parameters.
Table 1 X-ray crystal structural data together with refinement parameters collections for complexes 2, 4, 5 and 6
| |
2 |
4 |
5 |
6 |
| Empirical formula |
C31H23Cl2CuN3O2 |
C32H23Cl4CuN3O2 |
C33H29CuN3O3 |
C32H24Cl2CuN4O4 |
| Formula weight |
603.96 |
686.87 |
579.13 |
662.99 |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Triclinic |
| Space group |
P21/c |
P21/c |
P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a/Å |
10.1131(6) |
10.3155(3) |
9.6459(6) |
7.418(4) |
| b/Å |
18.2779(10) |
22.5459(6) |
20.2675(13) |
13.351(7) |
| c/Å |
14.6964(8) |
13.4635(4) |
14.4479(9) |
15.437(8) |
| α/° |
90 |
90 |
90 |
93.220(16) |
| β/° |
105.1330(10) |
107.6710(10) |
108.352(2) |
103.676(15) |
| γ/° |
90 |
90 |
90 |
103.705(16) |
| Volume/Å3 |
2622.4(3) |
2983.49(15) |
2680.9(3) |
1433.1(14) |
| Z |
4 |
4 |
4 |
2 |
| ρcalc/g cm−3 |
1.530 |
1.529 |
1.435 |
1.536 |
| µ/mm−1 |
1.072 |
1.126 |
0.856 |
0.995 |
| F(000) |
1236 |
1396 |
1204 |
678 |
| Crystal size/mm3 |
0.258 × 0.075 × 0.032 |
0.231 × 0.117 × 0.107 |
0.483 × 0.221 × 0.074 |
0.153 × 0.140 × 0.039 |
| 2Θ range for data collection/° |
1.817 to 26.410 |
1.806 to 26.386 |
1.793 to 26.420 |
1.581 to 26.582 |
| Index ranges |
−12 ≤ h ≤ 12 |
−12 ≤ h ≤ 12 |
−12 ≤ h ≤ 11 |
−9 ≤ h ≤ 9 |
| −22 ≤ k ≤ 22 |
−28 ≤ k ≤ 28 |
−25 ≤ k ≤ 25 |
−16 ≤ k ≤ 16 |
| −18 ≤ l ≤ 18 |
−16 ≤ l ≤ 16 |
−18 ≤ l ≤ 17 |
−19 ≤ l ≤ 19 |
| Reflections collected |
53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 446 446 |
67118 |
64228 |
29441 |
| Independent reflections |
5383 [Rint = 0.0653] |
6105 [Rint = 0.0410] |
5477 [Rint = 0.0477] |
5874 [Rint = 0.0949] |
| Data/restraints/parameters |
5383/0/353 |
6105/0/381 |
5477/0/366 |
5874/718/418 |
| Goodness-of-fit on F2 |
1.056 |
1.031 |
1.040 |
0.992 |
| Final R indexes [I ≥ 2σ(I)] |
R1 = 0.0391, wR2 = 0.0910 |
R1 = 0.0424, wR2 = 0.1077 |
R1 = 0.0479, wR2 = 0.1139 |
R1 = 0.0590, wR2 = 0.1492 |
| Final R indexes [all data] |
R1 = 0.0602, wR2 = 0.1008 |
R1 = 0.0610, wR2 = 0.1216 |
R1 = 0.0614, wR2 = 0.1210 |
R1 = 0.0951, wR2 = 0.1714 |
| Largest diff. peak and hole (e Å−3) |
0.453 and −0.399 |
0.516 and −0.370 |
1.063 and −0.442 |
0.399 and −0.949 |
2.5 DNA binding study
The DNA binding study was done using modified literature procedures.42,43 UV-visible absorption spectroscopy was employed to investigate the binding affinity of ligands L1–L3 and complexes 1–6 with calf thymus DNA (ctDNA). The ctDNA stock solution was prepared by dissolving an appropriate amount in phosphate buffer saline (pH 7.2), with its concentration quantified spectrophotometrically using the molar absorptivity coefficient ε260 = 6600 M−1 cm−1. The purity of the ctDNA solution was confirmed by a UV absorbance ratio A260/A280 > 1.8, verifying the absence of protein contamination. Binding interactions were evaluated by maintaining a fixed compound concentration of 40 µM while incrementally adding ctDNA (0–25 µM). The compound-DNA mixtures were incubated for 5 min at 25 °C prior to recording absorption spectra. Intrinsic binding constants Kb were determined by nonlinear fitting of the data to the Wolfe–Shimmer equation (eqn (1)). The experiments were performed in triplicate to confirm the precision and accuracy of the procedure and in all the experiments on the interaction of the compounds with ctDNA, the final concentration of DMSO in the incubation mixture was ∼2% (v/v).| |
 | (1) |
where A is the absorbance recorded in the presence of an added guest, A0 is the absorbance of the receptor in the absence of a guest, and Amax is the absorbance in the presence of added [complex]n.
2.6 Protein binding studies
The interactions of complexes 1–6 were performed using the UV-vis absorption titration method.44 BSA stock solutions were prepared by dissolving the protein in phosphate-buffered saline (pH 7.2) at 25 °C under constant stirring, with concentrations determined spectrophotometrically using the molar absorptivity coefficient ε280 = 44300 M−1 cm−1. The complexes' stock solutions (1 × 103 mM) were dissolved in DMSO. Titrations were done by adding varying complex concentrations (0–10 µM) to a fixed BSA concentration (6 µM), followed by 5 min incubation at 25 °C. The experiments were performed in triplicate to confirm the precision and accuracy of the procedure, and in all the experiments on the interaction of the compounds with ctDNA, the final concentration of DMSO in the incubation mixture was ∼1% (v/v). Absorption at λmax = 280 nm was measured, and the binding constant Kb was calculated using the Benesi–Hildebrand equation (eqn (2)) from the intercept to the slope ratio of 1/[A − A0] vs. 1/[complex].| |
 | (2) |
where A and A0 represent the absorbance of BSA in the presence and absence of complex, respectively.
2.7 Antidiabetes studies
2.7.1 Inhibition of α-amylase activity. α-Amylase is a calcium metalloenzyme and a key digestive enzyme that breaks down carbohydrates into glucose causing postprandial blood sugar spikes in diabetes patients. Inhibiting this enzyme is a crucial therapeutic strategy for type 2 diabetes. The ability of acarbose and the compounds to inhibit the α-amylase was determined using the method according to Ibitoye et al.45 with slight modification. 25 µL of varied concentrations of the compounds were added to 175 µL of distilled water in a test tube. After adding 200 µL of the 2.5 mg mL−1 pancreatic α-amylase solution in 0.1 M sodium phosphate buffer (pH 6.8), the resulting solution was incubated for 20 minutes at 37 °C. Then, 0.15 mL of a 1% starch solution was added to initiate the enzyme's catalytic activity. After allowing the reaction to proceed for 15 minutes under the same previous conditions, it was terminated by adding 0.4 mL of dinitrosalicylic acid reagent. The whole solution was boiled in a water bath for 10 minutes before 1 mL of distilled water was added. Finally, the absorbance of the coloured solution was read in a microplate reader at 540 nm against a blank solution lacking the test samples.
2.7.2 Inhibition of α-glucosidase activity. α-Glucosidase is an enzyme in the small intestine that breaks down complex carbohydrates into glucose, causing post-meal blood sugar spikes. This enzyme is responsible for the final step of carbohydrate digestion into glucose, allowing absorption into the bloodstream. Inhibiting α-glucosidase in the human body results in lower and slower rise in blood glucose level after meals.30 The ability of acarbose and the compounds to inhibit α-glucosidase activity was determined using the protocol according to Olofinsan et al.46 where p-nitrophenyl-α-D-glucopyranoside (pNPG) was employed as enzyme substrate. 25 µL of each compound or acarbose was diluted with 100 µL of distilled water in a 96-well plate. The solution was then incubated with the enzyme solution, which contained 0.1 M sodium phosphate buffer (pH 6.8) with yeast α-glucosidase (1.5 U mL−1). The mixture was incubated for 15 minutes at 37 °C. The enzyme reaction was initiated by adding pNPG (2 mg mL−1), and the reaction was monitored for 1 minute. The optical density was then measured in a microplate reader at 405 nm.
2.8 Antioxidant studies
2.8.1 Nitric oxide production inhibitory capacity. While sodium nitroprusside (Na2[Fe(CN)5NO]) at physiological pH releases nitric oxide, the ability of the compounds to inhibit this process was determined using the method described by Beseni et al.47 with minor modifications. 100 µL of the compounds or quercetin solutions at 250–2000 µM were incubated with a Na2[Fe(CN)5NO] solution (5 mM) in PBS for 1 hour and 30 minutes at 37 °C in Eppendorf tubes. 50 µL of each resulting solution was pipetted to the 96-well plate in triplicate before the addition of 50 µL of Griess reagent. The optical density of the pink chromogen developed after this step was measured in a microplate reader at 540 nm against a blank sample containing distilled water instead of the test samples.
2.8.2 Free radical neutralising ability. The interaction of the synthesized compound with 2,2-diphenyl-1-picrylhydrazyl (DPPH) was conducted according to the protocol of Changlian et al.48 with little modification. DPPH powder was initially dissolved in methanol to achieve a final concentration of 0.3 mM in an amber bottle. Then, 100 µL of the compound's solution and the free radical solution were incubated in a 96-well plate in the dark for 20 minutes. The degree of bleaching of the initial colour of the DPPH solution was measured in microplate reader at 517 nm against a blank solution lacking the test compound or quercetin.
2.8.3 Ferric ion reductive capacity. The ability of the compounds to reduce ferric ion in comparison with quercetin at 250–2000 µM concentration was assessed using the Beseni et al.47 method with slight modifications. 0.1 mL of each compound solution was added to an equal volume of potassium ferricyanide, followed by an hour of equilibration at 50 °C in an incubator. After adding 50 µL of distilled water and the same volume of 10% trichloroacetic acid, the mixture was subjected to centrifugation at 4000g for 15 minutes. Then, 150 µL of each sample was added to 50 µL of FeCl3 (0.1%) in a 96-well plate. The absorbance of the resulting mixture was read at 700 nm in a microplate reader.
3 Results and discussion
3.1 Preparation of complexes 1–6
The free ligands L1–L3 explored in synthesizing complexes 1–6 were prepared by the condensation reaction between 2-hydroxy-1-napthaldehyde and respective primary amines viz; 2-amino-5-chlorophenol (L1), 2-hydroxy-5-methylaniline (L2) and 2-hydroxy-4-nitroaniline (L3) in ratio 1:1 at room temperature. The reaction between ligand (L1/L2/L3), copper(II) salt (Cu(CH3COO)2.H2O) and N,N′-adducts ligands (1,10-phenanthroline or 2,9-dimethyl-1,10-phenanthroline) at 40–50 °C in 1:1:1 respectively affords complexes 1–6 (Scheme 1) in good yield. The resulting copper(II) complexes are reddish brown in colour and very stable at room temperature. Generally, the complexes dissolve readily in DMSO, dichloromethane as well as chloromethane, while they do not dissolve readily in tetrahydrofuran and protic solvents i.e., ethanol and methanol. They are completely insoluble in hexane.
 |
| | Scheme 1 Synthetic route for the preparation of complexes 1–6. | |
3.2 Spectroscopic elucidation of complexes 1–6
3.2.2 Electron spin resonance (ESR) spectroscopy. The paramagnetic nature of complexes 1–6 were confirmed by ESR techniques, which were carried out by recording the ESR spectra at X-band frequency for the complexes in their powder state and are shown in Fig. 2 and S8. It could be observed that all the spectra are of strong intensity, affirming the ESR sensitivity of copper(II) complexes, and they are also known to possess characteristic g-values. A g-factor of approximately 2.0 (1.9728 for 1, 1.9817 for 2, 1.9901 for 3, 1.9788 for 4, 1.9684 for 5 and 1.9844 for 6) which is equivalent to that of a free electron (g = 2.0023). The absence of forbidden ΔMs = ±2 line (half field signal) in all the spectra rules out the possibility of a dimer resulting from a pronounced Cu⋯Cu interaction.49,50
 |
| | Fig. 2 ESR spectra of complexes 1–4. | |
3.3 Single crystal X-ray structures
We used the single crystal X-ray crystallography technique to determine the structures of complexes 2, 4, 5 and 6. Single crystals suitable for this experiment were obtained by the slow evaporation of 4:1 dichloromethane: methanol or chloromethane: methanol solution. Fig. 3 shows the crystal structures of the complexes using an ORTEP diagram with 50% probability thermal ellipsoids, while Table 2 details selected bond angles as well as bond lengths. Complexes 2, 4 and 5 crystallize as monoclinic with a space group of P21/c for complexes 2 and 4 and P21/n for 5 while complex 6 crystallizes as triclinic with a space group of P. Generally, the asymmetric unit of the complexes contain a whole molecule with one molecule of solvent i.e. one molecule of dichloromethane for 2 and 6, one molecule of chloromethane for 4, and one molecule of methanol for 6. All the crystal structures are mononuclear species in which the copper(II) ion is five-coordinated to a pair of oxygen atoms as well as one nitrogen atom from the Schiff base ligand (L1–L3) and to a pair of nitrogen atoms from the auxiliary ligand (1,10-phenanthroline or 2,9-dimethyl-1,10-phenanthroline) conforming to a distorted square pyramid with O1N7O2N1 atoms forming the basal plane for Cu1. The apical Cu1–N2 bond length for 2, 5 and 6 and Cu1–N1 for 3 are longer than other two Cu–N bond lengths. For example, the Cu1–N2 bond length is 2.272(2)° in complex 2, while the ones for Cu1–N1 as well as Cu1–N7 are 2.049(2)° and 1.944(2)° respectively. We used the structural parameter index (eqn (3)) for five coordinated complexes51 to affirm the geometry of 2, 4, 5 and 6.| |
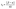 | (3) |
β and α are the largest coordination angles at the metal center. For τ5 = 1 and τ5 = 0, the geometries of the complex are trigonal bipyramidal and perfect square pyramidal, respectively. The calculated τ5 values for complexes 2, 4, 5 and 6 are 0.01, 0.05, 0.04, and 0.05, respectively, and these fit into distorted square pyramidal attributed to the geometry of the complexes. The observed bond lengths and bond angles for the complexes are comparable to the ones of related copper(II) complexes with the same geometry.52
 |
| | Fig. 3 Crystal structure of copper(II) complexes 2, 4, 5, and 6 with thermal ellipsoids drawn at 50% probability. Hydrogen atoms were removed for clarity. | |
Table 2 Selected bond lengths (Å) and bond angles (°) for complexes 2, 4, 5 and 6
| Parameters |
2 |
4 |
5 |
6 |
| C8–N7 |
1.296(4) |
1.294(4) |
1.301(4) |
1.311(4) |
| C10–O2 |
1.295(4) |
1.302(4) |
1.302(3) |
1.298(5) |
| C1–O1 |
1.328(3) |
1.321(4) |
1.330(3) |
1.316(4) |
| Cu1–O1 |
1.933(2) |
1.977(2) |
1.954(2) |
1.942(3) |
| Cu1–O2 |
1.918(2) |
1.930(2) |
1.918(2) |
1.911(3) |
| Cu1–N7 |
1.944(2) |
1.945(3) |
1.955(2) |
1.955(3) |
| Cu1–N1 |
2.049(2) |
2.303(2) |
2.026(2) |
2.052(3) |
| Cu1–N2 |
2.272(2) |
2.045(3) |
2.288(3) |
2.323(4) |
|
| Bond angles |
| O2–Cu1–O1 |
171.20(8) |
168.09(9) |
166.63(9) |
171.4(1) |
| N2–Cu1–N1 |
77.75(8) |
76.9(1) |
78.0(1) |
76.4(1) |
| N7–Cu1–O1 |
85.32(8) |
83.68(9) |
84.4(1) |
84.6(1) |
| N7–Cu1–O2 |
92.23(8) |
90.92(9) |
92.4(1) |
91.8(1) |
| O2–Cu1–N2 |
91.64(8) |
91.2(1) |
98.18(9) |
92.0(1) |
| O1–Cu1–N1 |
90.18(8) |
94.24(9) |
91.6(1) |
89.0(1) |
3.4 DNA binding study of ligands L1–L3 and complexes 1–6
The UV-visible absorption spectroscopy technique is commonly employed to study the noncovalent DNA binding modes for potential metallodrugs, particularly those targeting DNA replication in anticancer therapies. In this study, we evaluate the binding affinities of ligands L1–L3 and complexes 1–6 to calf thymus DNA (ctDNA) by monitoring their spectral changes upon incremental ctDNA addition. All the compounds displayed absorption bands between 447 and 508 nm, showing hypochromism of 9.44–40.69% and bathochromic shifts of 2–9 nm with increasing ctDNA concentration (Fig. 4 and S15–S22). These observations suggest a potential intercalative binding, wherein the aromatic π-systems of the ligands stack with DNA base pairs, reducing transition probabilities. The pronounced hypochromism and red shifts signify strong π–π interactions, common in phenanthroline-containing copper complexes. Using the Wolfe–Shimmer equation (eqn (1)), the intrinsic binding constant (Kb) was calculated to quantify the binding interactions of the compounds.
 |
| | Fig. 4 Electronic absorption spectrum of complex 5 in the absence (dashed line) and the presence of different concentrations of ctDNA. | |
The intrinsic binding constants (Kb) of L1–L3 and complexes 1–6 (Table 3) revealed moderate to strong DNA affinities. The Kb values for L1, L2 and L3 were 1.49 × 104 M−1, 2.46 × 104 M−1, and 1.56 × 104 M−1, respectively (L2 > L3 > L1; Table 3), reflecting the ligands' moderate DNA affinities.
Table 3 The binding parameters of L1–L3 and complexes 1–6 interactions with CT-DNA and bovine serum albumina
| Cpd. |
DNA |
BSA |
| Kb (×104 M−1) |
λmax (nm) |
Spec. Shift (nm) |
%Hypo |
Kb (×105 M−1) |
λmax (nm) |
Spec. Shift (nm) |
%Hyper |
| Kb = intrinsic binding constant; the Kb values shown are mean ± standard deviation. |
| L1 |
1.49 ± 0.01 |
471 |
3(red) |
40.69 |
— |
— |
— |
— |
| L2 |
2.46 ± 0.06 |
446 |
3(red) |
36.96 |
— |
— |
— |
— |
| L3 |
1.56 ± 0.01 |
508 |
9(red) |
9.44 |
— |
— |
— |
— |
| 1 |
4.73 ± 0.08 |
504 |
3(red) |
30.63 |
1.81 ± 0.03 |
280 |
7(blue) |
64.42 |
| 2 |
7.15 ± 0.01 |
483 |
2(red) |
32.14 |
2.41 ± 0.06 |
280 |
4(blue) |
51.52 |
| 3 |
9.31 ± 0.11 |
507 |
3(red) |
22.83 |
1.53 ± 0.01 |
280 |
8(blue) |
56.01 |
| 4 |
17.5 ± 0.20 |
462 |
2(red) |
29.73 |
1.57 ± 0.07 |
280 |
3(blue) |
61.55 |
| 5 |
27.4 ± 0.20 |
459 |
5(red) |
36.65 |
1.46 ± 0.02 |
280 |
8(blue) |
60.53 |
| 6 |
24.8 ± 0.05 |
490 |
3(red) |
26.03 |
0.96 ± 0.03 |
280 |
4(blue) |
54.28 |
Ligand L2, bearing a methyl substituent, exhibited the highest affinity due to enhanced π-electron density and hydrophobic interactions with DNA base pairs. In contrast, L1 (chloro-substituted) and L3 (nitro-substituted) showed lower affinities, likely due to the electron-withdrawing groups reducing π-stacking efficiency. Complexes 1–6 showed enhanced binding affinity, with Kb values ranging between 4.73 × 104 and 2.74 × 105 M−1 in the order 5 > 6 > 4 > 3 > 2 > 1. Complex 5 exhibited the highest affinity (Fig. 4), attributable to the synergistic effect of the methyl group on its Schiff base ligand and the 2,9-dimethyl-1,10-phenanthroline coligand, which enhances hydrophobicity and π-stacking relative to unsubstituted phenanthroline.
The obtained Kb values of 1–6 show that the coordination of L1–L3 to the copper(II) centre increased their binding affinities by 3 to 18-fold compared to the free ligands, suggesting metal-assisted stabilization of the potential intercalative mode. Notably, complexes 4–6 (with 2,9-dimethyl-1,10-phenanthroline as co-ligand) show 2–3 times higher Kb values than 1–3, due to methyl-induced hydrophobicity, as such encouraging π-stacking with DNA bases. Overall, the Kb values within 104–105 M−1 recorded for the compounds suggest their non-covalent intercalative binding modes, aligning with literature reports for mixed-ligand copper-phen Schiff base complexes (Kb = ∼104–106 M−1),53,54 positioning these compounds as classical intercalators suitable for biomedical applications.
3.5 Bovine serum albumin interaction study
Drug molecule binding to plasma proteins offers useful information about drug distribution, transportation, and excretion, which is crucial in drug discovery and design. Bovine serum albumin (BSA), the most abundant plasma protein, serves as a cost-effective and structurally similar model to human serum albumin for drug–protein interaction studies. In this study, UV-visible absorption spectroscopy was employed to evaluate the binding affinity of copper(II) complexes 1–6 to BSA. The binding study was monitored by the conformational changes in BSA characterized by its intense absorbance at 280 nm, attributed to the polarity of tyrosine and tryptophan residues. Ligands L1–L3 were excluded, as their ctDNA binding affinities were lower than those of the complexes.
Upon titration of increasing complexes 1–6 concentrations into fixed BSA solutions, continuous hyperchromic shifts were observed between 260 and 300 nm, with hyperchromism of 51.52–64.42% and hypsochromic shifts of 3–8 nm, indicating alterations in the protein microenvironment (Fig. 5 and S23–S27).
 |
| | Fig. 5 Electronic absorption spectra of BSA in the absence (dashed line) and the presence of different concentrations of complex 2. | |
Binding constants (Kb) were calculated from the intercept-to-slope ratio of linear plots of 1/(A − A0) vs. 1/[complex], where A and A0 are the absorbances with and without complex, respectively. The Kb values for complexes 1–6 ranged from 9.59 × 104 M−1 to 2.41 × 105 M−1 (Table 3), falling within the typical 104 to 106 M−1 range for drug–protein interactions. This suggests their moderate to strong binding to protein, as such, suitable for pharmacological applications.
Complex 2 exhibited the highest binding affinity (Kb = 2.41 × 105 M−1, Fig. 5), followed by complex 1 (1.81 × 105 M−1, Fig. S23) and complex 4 (1.57 × 105 M−1, Fig. S24), highlighting the influence of ligand substituents and co-ligands. In complex 2 (bearing L2 with an electron-donating methyl group on the aniline), the methyl group enhances lipophilicity, thus promoting stronger hydrophobic interactions and π–π stacking with BSA's aromatic residues in hydrophobic pockets like subdomain IIA. For complex 1, the planarity of the unsubstituted 1,10-phenanthroline co-ligand could have influenced the insertion into the binding pocket. Similarly, complex 4's affinity can be related to the steric bulk and hydrophobic effects of the 2,9-dimethyl substituents on 1,10-phenanthroline, which promote stable interactions within the BSA subdomain. These trends correspond with literature reports on copper(II) Schiff base complexes, where substituents modulate binding through steric and electronic effects.55,56 Overall, the Kb values position complexes 1–6, especially 1, 2, and 4, as viable drug carriers with protein, strong enough for transport and allowing free complex release for bioactivity.
3.6 In vitro antidiabetes activity
3.6.1 α-Amylase inhibition activity. In diabetes patients, α-amylase inhibitors are used to retard, or slow down dietary starch digestion, and this technique has been primarily used to manage type 2 diabetes by reducing post-meal blood sugar spikes.57 Both the ligands (L1–L3) and the copper(II) complexes (1–6) displayed α-amylase inhibitory potential, and the results are quantified using IC50 values as shown in Table 4. The IC50 is the concentration of drug required for 50% inhibition. The lower the IC50 value, the better the α-amylase inhibitory efficiency. We observed that all the compounds displayed better α-amylase inhibition activity than the standard drug, acarbose, and the activity of the free ligands were greatly improved upon coordination with the copper(II) ion. For example, the IC50 for ligand L1 is 0.78 mM while the one for complex 1 is 0.33 mM. It was suggested that this might be due to an increase in lipophilicity in 1–6 resulting from the complexation of the ligands with Cu2+, which allowed them to penetrate through cell membranes and reach targets (α-amylase) more easily compared to the unbound ligands.58 However, further direct experimental evidence such as cellular uptake/penetration measurements must be carried out to ascertain this inference. It could also be observed that complexes with 2,9-dimethyl-1,10-phenanthroline showed good α-amylase inhibition activity than the ones having 1,10-phenanthroline in their structural backbone. This indicates that the electronic properties of the complexes play an important role in the inhibition of α-amylase. It was suggested that the presence of extra methyl moieties at position 2 and 9 of the 1,10-phenanthroline in complexes 4–6 might contribute to increased lipophilicity, which could, in turn, enhance their penetration and interaction with α-amylase enzymes compared to complexes 1–3 containing unsubstituted 1,10-phenanthroline. Statistically, the inhibitory activity of all the compounds at a 0.5 mM test concentration ranges from 51% to 58%; however, at a 2 mM concentration, complexes 5 and 6 exhibited inhibitory activities of 70% and 71%, respectively (Fig. 6). This trend also reflected in their IC50 values. It could be generalized that, complexes 5 and 6 with IC50 values of 0.27 mM and 0.28 mM respectively, exhibited the best α-amylase enzyme inhibitory activities among the test samples.
Table 4 Antidiabetic potential of free ligands L1–L3 and copper(II) complexes 1–6
| Compounds |
IC50 (mM) |
| α-Amylase |
α-Glucosidase |
| L1 |
0.780 |
0.062 |
| L2 |
0.415 |
0.071 |
| L3 |
0.604 |
0.081 |
| 1 |
0.329 |
0.093 |
| 2 |
0.361 |
0.055 |
| 3 |
0.335 |
0.121 |
| 4 |
0.290 |
0.054 |
| 5 |
0.274 |
0.076 |
| 6 |
0.275 |
0.065 |
| Acarbose |
0.912 |
0.059 |
 |
| | Fig. 6 Comparative α-amylase enzyme inhibitory effect of acarbose in comparison with the synthesised compounds at 0.25–2 mM test concentrations. | |
3.6.2 α-Glucosidase inhibition activity. Another major strategy used in managing diabetes mellitus is to inhibit α-glucosidase, an enzyme also known as maltase.59 α-Glucosidase catalyzes the breaking down of α(1 → 4) glycosidic bonds in starch, resulting in absorbable glucose in the body. Inhibiting the activities of α-glucosidase can directly reduce postprandial blood sugar in the body.57 Herein, we studied the α-glucosidase inhibition capability of ligands L1–L3 and complexes 1–6 and the results are given in Table 4. All the compounds displayed very good α-glucosidase inhibition potential. However, only complexes 2 and 4 with IC50 values of 0.055 mM and 0.054 mM outshined acarbose with IC50 value of 0.058 mM and this suggests that complexes 2 and 4 may have potential as antidiabetic therapeutics. Generally, as the concentration of the compounds increases, the % α-glucosidase inhibition increases, indicating that they are dose-dependent (Fig. 7). Contrary to what was observed for α-amylase inhibition assay, it cannot be outrightly stated that the complexes inhibited α-glucosidase better than the ligands, as there are few cases where the ligands performed better than the metal complexes. This might probably be due to ligands having superior access and binding affinities (via hydrogen bonding) to the enzyme active site when compared to ones of the respective copper(II) complex.
 |
| | Fig. 7 Comparative α-glucosidase enzyme inhibitory effect of acarbose in comparison with the synthesized compounds at 0.25–2 mM test concentrations. | |
3.7 In vitro antioxidant activity
3.7.1 Nitric oxide inhibitory activity. The NO˙ assay is one of the important techniques used for evaluating antioxidant activity of drug molecules due to the fact that it specifically assesses a compound's ability to scavenge reactive nitrogen species (RNS) and modulate inflammation, which are different from reactive oxygen species (ROS) handled by other assays.60 Compounds capable of neutralizing nitric oxide radicals might be considered potential candidates for further investigation as anti-inflammatory agents. This is because chronic inflammatory conditions like arthritis are closely related to excessive nitric oxide radical. In this study, we evaluate the potential of free ligands L1–L3 and copper(II) complexes 1–6 to scavenge NO˙ and the results are given in Table 5 and Fig. 8. All the compounds showed good NO˙ scavenging potential, and it was observed that all the ligands (L1–L3) as well as complex 2 outshined quercetin (the standard). While quercetin has an IC50 value of 0.060 mM, compounds L1, L2, L3 and 2 have IC50 values of 0.050 mM, 0.053 mM, 0.050 mM and 0.049 mM respectively. Generally, the ligands scavenge NO˙ better than the complexes. This might be due to the masking of the hydroxyl group (electron-donating group) in the complexes, which ought to have facilitated electron transfer and in turn stabilizes NO˙.
Table 5 Antioxidant potential of free ligands L1–L3 and copper(II) complexes 1–6
| Compounds |
IC50 (mM) |
| NO˙ |
DPPH |
FRAP |
| L1 |
0.050 |
0.087 |
>5.00 |
| L2 |
0.053 |
0.261 |
4.375 |
| L3 |
0.050 |
0.531 |
3.414 |
| 1 |
0.066 |
0.627 |
>5.00 |
| 2 |
0.049 |
1.222 |
>5.00 |
| 3 |
0.079 |
1.323 |
>5.00 |
| 4 |
0.074 |
2.788 |
>5.00 |
| 5 |
0.065 |
1.001 |
>5.00 |
| 6 |
0.064 |
3.951 |
>5.00 |
| Quercetin |
0.060 |
0.126 |
0.034 |
 |
| | Fig. 8 Effect of the compounds in comparison with quercetin to inhibit nitric oxide formation at 0.25–2 mM test concentrations. | |
3.7.2 DPPH free radical neutralising ability. The antioxidant potential of the ligands (L1–L3) as well as their corresponding copper(II) complexes (1–6) were also evaluated using the DPPH free radical scavenging assay, and the results were expressed as IC50 values as given in Table 5. The antioxidant activities of the compounds were compared to quercetin. All the ligands showed good results. Among all the compounds, L1 exhibited the strongest activity, with an IC50 value of 0.087 mM, which is lower than that of other compounds and quercetin (0.126 mM) inclusive. Similarly to what was observed in the nitric oxide assay, the ligands scavenge the free radical of DPPH better than that of the copper(II) complexes. The reduced antioxidant potential observed for the complexes 1–6 might be attributed to the coordination of the ligand donor atoms to the metal center, which can limit the availability of the hydroxyl group or heteroatom sites responsible for effective hydrogen atom or electron transfer to the DPPH free radicals.61 More so, steric hindrance and changes in electronic distribution following the coordination of copper(II) with the ligands may further impede interaction with the free radical species. Generally, the % free radical scavenging of the compounds increases as the concentration increases from 0.25 mM to 2.00 mM (Fig. 9).
 |
| | Fig. 9 Free radical scavenging effect of quercetin in comparison with the compounds at 0.25–2 mM test concentrations. | |
3.7.3 Ferric ion reducing capacity assay. We used the ferric ion reductive capacity assay to further investigate the compounds' potential as antioxidants. Antioxidants can generally convert ferric ions (Fe3+) to ferrous ions (Fe2+), and the reduction transformation level can be used to assess the antioxidant capability.62 The data in Fig. 10 revealed that at test concentrations ranging from 0.25 to 2 mM, complexes 1–6 exhibited lower Fe3+ reducing properties than compounds L1–L3. While compounds L1, L2, and L3 at 1 mM had estimated activities of approximately 28%, 37%, and 40%, respectively, these properties increased by 52%, 36%, and 37% at the highest test concentration. Despite the antioxidant potency of these synthesized compounds, they could not outperform quercetin, which had higher activities at 2 mM. Nonetheless, the results in Table 5 showed that compounds L2, L3 and quercetin had IC50 values of 4.375 mM, 3.414 mM, and 0.034 mM, respectively, while others have IC50 values greater than 5.00 mM. The IC50 values obtained for complexes 1–6 indicate that, they exhibited very weak or negligible ferric reducing ability under the experimental conditions used for this study.
 |
| | Fig. 10 Fe3+ reducing effect of quercetin in comparison with the compounds at 0.25–2 mM test concentrations. | |
4 Conclusion
In conclusion, six new copper(II) complexes derived from mixed ligands (Schiff bases and diimines) with general molecular formula [CuL1(Phen/DM-Phen)] have been synthesized and characterized by core spectroscopic techniques. The crystallography analysis of complexes 2, 4, 5 and 6 unraveled the geometry around the CuO2N3 of the complexes to be distorted square pyramidal. UV-visible absorption spectroscopy revealed the interactions of ligands L1–L3 and complexes 1–6 with ctDNA and BSA. Spectral analysis showed hypochromism with slight bathochromic shifts, suggesting an intercalation binding mode with ctDNA via classical non-covalent π-stacking. Complexes 1–6 displayed moderate-to-strong binding affinities to ctDNA (Kb = 4.73 × 104 to 2.74 × 105 M−1), better than those of the free ligands L1–L3 (Kb = 1.49 × 104 to 2.46 × 104 M−1). This positions the complexes as promising DNA-targeting agents for anticancer applications. For BSA, complexes 1–6 exhibited strong binding (Kb = 9.59 × 104 to 2.41 × 105 M−1), accompanied by hyperchromic shifts that signal microenvironment alterations and hydrophobic/π–π interactions, particularly driven by lipophilic methyl substituents (highest in complex 2). These affinities lie within optimal pharmacological ranges, facilitating effective protein-potential drug transport and release for bioactivity, especially for complexes 1, 2, and 4. All the complexes displayed good α-amylase and α-glucosidase inhibition potential with complexes 2 and 4 having lower IC50 values than the ones for acarbose in both assays, suggesting them to have potential as antidiabetic therapeutics. Compounds with methyl group substituents displayed better DNA and BSA binding affinities when compared to those with chloro and nitro groups. They also inhibit α-amylase and α-glucosidase more than their counterparts. The ligands exhibited good antioxidant activities compared to copper(II) complexes and also scavenge nitric oxide and DPPH (only L1) radical better than quercetin, the standard used for this study.
Author contributions
Conceptualization, S. D. O. and R. C. L.; methodology; S. D. O., A. A. A., B. O. R. C. L. and K. A. O.; software, S. D. O., K. A. O., and A. A. A., validation, S. D. O., A. A. A., K. A. O., B. O., and R. C. L.; formal analysis; S. D. O., K. A. O., and A. A. A.; resources' R. C. L., B. O., data curation, S. D. O, A. A. A. and K. A. O.; writing original draft preparation; S. D. O., A. A. and K. A. O.; writing review and editing, S. D. O., A. A. A., K. A. O., B. O. and R. C. L., visualization, S. D. O., A. A. A., K. A. O., B. O., and R. C. L., supervision, R. C. L., project administration, R. C. L., funding acquisition, S. D. O and R. C. L.
Conflicts of interest
The authors declare no conflicts of interest.
Data availability
CCDC 2530540 (2), 2530541 (4), 2530542 (5) and 2530543 (6) contain the supplementary crystallographic data for this paper.63a–d
All data generated or analyzed during this study are included in this published article and its supplementary information (SI). Supplementary information (with charaterization data) is available. See DOI: https://doi.org/10.1039/d6ra01650b.
Acknowledgements
The authors acknowledge the funding from the National Research Foundation (grant number: CPRR23041794158), Stellenbosch University, Matieland, 7602, South Africa. The first author is a recipient of a postdoctoral fellowship award of the NRF at Stellenbosch University, Matieland, South Africa.
References
- A. A. Ali, M. Shebl, O. M. Adly, M. S. Mahfouz and R. Fouad, J. Mol. Struct., 1359, 206, 145313 Search PubMed.
- S. D. Oladipo, B. Omondi and C. Mocktar, Polyhedron, 2019, 170, 712–722 CrossRef CAS.
- L. Esmaeili, M. G. Perez, M. Jafari, J. Paquin, P. Ispas-Szabo, V. Pop, M. Andruh, J. Byers and M. A. Mateescu, J. Inorg. Biochem., 2019, 192, 87–97 CrossRef CAS PubMed.
- I. Bento, C. Peixoto, V. N. Zaitsev and P. F. Lindley, Acta Crystallogr., Sect. D:Biol. Crystallogr., 2007, 63, 240–248 CrossRef CAS PubMed.
- F. Tisato, C. Marzano, M. Porchia, M. Pellei and C. Santini, Med. Res. Rev., 2010, 30, 708–749 CrossRef CAS PubMed.
- X. Wu, Y. Zhou, Z. Lu, Y. Zhang, T. Zhang and Q. Jiang, Anim. Biosci., 2024, 38, 530 CrossRef PubMed.
- M. Azeem, K. Li, Y. Qin, L. Dong and W. Li, CrystEngComm, 2021, 23, 1815–1820 RSC.
- I. Iakovidis, I. Delimaris and S. M. Piperakis, Mol. Biol. Int., 2011, 2011, 594529 CrossRef PubMed.
- S. D. Oladipo, R. C. Luckay, K. A. Olofinsan, A. A. Badeji and S. Mokoena, Inorg. Chim. Acta, 2025, 575, 122447 CrossRef CAS.
- T. L. Yusuf, I. Waziri, S. D. Oladipo, M. S. Abd El-Maksoud, A. J. Muller and B. Vatsha, ACS Omega, 2025, 10, 50795–50805 CAS.
- S. D. Oladipo, R. C. Luckay and K. A. Olofinsan, Results Chem., 2025, 16, 102471 CrossRef CAS.
- S. Thakur, A. Jaryal and A. Bhalla, Results Chem., 2024, 7, 101350 CrossRef CAS.
- M. E. Moreno-Narváez, L. González-Sebastián, R. Colorado-Peralta, V. Reyes-Márquez, L. O. Franco-Sandoval, A. Romo-Pérez, J. A. Cruz-Navarro, I. V. Mañozca-Dosman, A. Aragón-Muriel and D. Morales-Morales, Inorganics, 2025, 13, 38 CrossRef.
- R. D. Mohan, S. Sarukrishna, A. S. Babu, D. Santhosh, S. A. Fathima and B. Parvathy, Inorg. Chim. Acta, 2026, 550, 122980 CrossRef.
- A. Gil, M. Melle-Franco, V. Branchadell and M. J. Calhorda, J. Chem. Theor. Comput., 2015, 11, 2714–2728 CrossRef CAS PubMed.
- M. A. Alossaimi, T. Aldakhil, H. Elmansi, F. Belal and G. Magdy, RSC Adv., 2026, 16, 5437–5454 RSC.
- X.-B. Fu, D.-D. Liu, Y. Lin, W. Hu, Z.-W. Mao and X.-Y. Le, Dalton Trans., 2014, 43, 8721–8737 RSC.
- J. C. García-Ramos, R. Galindo-Murillo, F. Cortés-Guzmán and L. Ruiz-Azuara, J. Mex. Chem. Soc., 2013, 57, 245–259 Search PubMed.
- D. Ray, A. Manna, M. F. Ansari, F. Arjmand, Z. Jaglicic and R. Herchel, Dalton Trans., 2026, 55, 3750 RSC.
- R. Ghanta, S. Jhulki, T. Chowdhury, P. Giri, R. Mondal, S. Kundu, S. Dasgupta, U. K. Das, M. Jana and A. Pal, Appl. Organomet. Chem., 2026, 40, e70511 CrossRef CAS.
- S. V. Saravanaselvam, X. Arulanantham, M. Tamilelakkiya, R. Sakthivadivel, S. Ponnusamy, P. Pandi, P. Velusamy, S. E. Muthu and K. A. Kumar, J. Mol. Struct., 2025, 1325, 141000 CrossRef CAS.
- M. Lištiak, J. Valentová, E. Zahradníková, R. Varga, E. Samoľová, M. Litecká, K. Lušpai, M. Puchoňová, M. Malček Šimunková and J. Moncoľ, Eur. J. Inorg. Chem., 2025, 28, e202500143 CrossRef.
- V. Vinayagam, D. Chandren, P. Arthur, S. Theerthagiri and W. Yong, Inorg. Chem. Commun., 2024, 161, 112116 CrossRef CAS.
- M. Maity, U. Pramanik, V. R. Hathwar, P. Brandao, S. Mukherjee, S. Maity, R. Maity, T. Maity and B. C. Samanta, Heliyon, 2022, 8, e11345 CrossRef CAS PubMed.
- N. Yasumatsu, Y. Yoshikawa, Y. Adachi and H. Sakurai, Bioorg. Med. Chem., 2007, 15, 4917–4922 CrossRef CAS PubMed.
- S. D. Oladipo and R. C. Luckay, New J. Chem., 2024, 48, 13276–13288 RSC.
- S. S. Lakshmi, K. Geetha, M. Gayathri and G. Shanmugam, J. Chem. Sci., 2016, 128, 1095–1102 CrossRef CAS.
- T. S. Chaithanya and V. Sabareesh, Pept. Sci., 2026, 118, e70017 CrossRef CAS.
- A. Reuser and H. Wisselaar, Eur. J. Clin. Invest., 1994, 24, 19–24 CrossRef CAS PubMed.
- S. R. Pathak, K. R. Senwar and K. N. Sharma, in Diabetes Mellitus, Elsevier, 2025, pp. 63–78 Search PubMed.
- M. Sabu and R. Kuttan, J. Ethnopharmacol., 2002, 81, 155–160 CrossRef CAS PubMed.
- P. Thakur, A. Kumar and A. Kumar, J. Drug Targeting, 2018, 26, 766–776 CrossRef CAS PubMed.
- A. Ceriello, Diabetes Care, 2003, 26, 1589–1596 CrossRef CAS PubMed.
- M. Valko, C. Rhodes, J. Moncol, M. Izakovic and M. Mazur, Chem.-Biol. Interact., 2006, 160, 1–40 CrossRef CAS PubMed.
- T. Dhanya, M. P. Kurup, K. Rajimon, G. A. Krishna, J. K. Varughese, K. Raghu, S. Philip, K. Divya, M. Augustine and P. Mohanan, Dalton Trans., 2025, 54, 3216–3234 RSC.
- S. D. Oladipo, R. C. Luckay, A. A. Badeji, V. A. Obakachi, K. K. Govender and A. D. Osinubi, Chem. Biodiversity, 2026, 23, e02739 CrossRef CAS PubMed.
- T. Kiran, V. G. Prasanth, M. Balamurali, C. Vasavi, P. Munusami, K. I. Sathiyanarayanan and M. Pathak, Inorg. Chim. Acta, 2015, 433, 26–34 CrossRef CAS.
- APEX4, SAINT, and SADABS, Bruker AXS Inc., Madison, WI, 2022 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- L. J. Barbour, J. Appl. Crystallogr., 2020, 53, 1141–1146 CrossRef CAS.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens and M. Towler, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- A. A. Adeleke, Md. S. Islam, K. Olofinsan, V. F. Salau, C. Mocktar and B. Omondi, New J. Chem., 2021, 45, 17827–17846 RSC.
- T. L. Yusuf, D. C. Akintayo, S. D. Oladipo, A. A. Adeleke, K. Olofinsan, B. Vatsha and N. Mabuba, New J. Chem., 2022, 46, 12968–12980 RSC.
- S. J. Zamisa, A. A. Adeleke, N. Devnarain, M. A. Rhman, P. M. O. Owira and B. Omondi, RSC Adv., 2023, 13, 21820–21837 RSC.
- O. Ibitoye, K. Olofinsan, K. Teralı, U. Ghali and T. Ajiboye, J. Food Biochem., 2018, 42, e12627 CrossRef.
- K. A. Olofinsan, O. L. Erukainure, N. Z. Msomi and M. S. Islam, Asian Pac. J. Trop. Biomed., 2022, 12, 300–311 CrossRef CAS.
- B. K. Beseni, K. A. Olofinsan, V. F. Salau, O. L. Erukainure and M. S. Islam, Asian Pac. J. Trop. Biomed., 2022, 12, 453–465 CrossRef CAS.
- P. Changlian, C. Shaowei, L. Zhifang and L. Guizhu, Europe PMC, 2000, 27, 658–661 Search PubMed.
- M. K. Koley, S. U. Parsekar, N. Duraipandy, M. S. Kiran, B. Varghese, P. T. Manoharan and A. P. Koley, Inorg. Chim. Acta, 2018, 478, 211–221 CrossRef CAS.
- W. A. Munzeiwa, S. D. Oladipo, C. U. Ibeji, C. Mocktar and B. Omondi, J. Inorg. Biochem., 2021, 225, 111600 CrossRef CAS PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- B. Mohan and M. Choudhary, J. Mol. Struct., 2021, 1246, 131246 CrossRef CAS PubMed.
- K. Paliwal, P. Haldar, P. K. S. Antharjanam and M. Kumar, ACS Omega, 2022, 7, 21961–21977 CrossRef CAS PubMed.
- L. Li, Q. Guo, J. Dong, T. Xu and J. Li, J. Photochem. Photobiol., B, 2013, 125, 56–62 CrossRef CAS PubMed.
- S. Mali, S. D. Vatagude, M. Horitani and P. M. Gurubasavaraj, J. Mol. Struct., 2026, 1360, 145599 CrossRef CAS.
- S. K. Tarai, S. Mandal, A. Tarai, I. Som, A. Pan, A. Bagchi, A. Biswas and S. C. Moi, Appl. Organomet. Chem., 2023, 37, e7164 CrossRef CAS.
- H. O. Omoregie, T. L. Yusuf, S. D. Oladipo, K. A. Olofinsan, M. B. Kassim and S. Yousuf, Polyhedron, 2022, 217, 115738 CrossRef CAS.
- A. d. S. Portas, D. C. Miguel, J. K. Yokoyama-Yasunaka, S. R. B. Uliana and B. P. Espósito, J. Biol. Inorg. Chem., 2012, 17, 107–112 CrossRef CAS PubMed.
- S. D. Oladipo, R. C. Luckay, K. A. Olofinsan, V. A. Obakachi, S. J. Zamisa, A. A. Adeleke, A. A. Badeji, S. A. Ogundare and B. P. George, Heliyon, 2024, 10, e23174 CrossRef CAS PubMed.
- U. Rao, B. A. Ahmad and K. S. Mohd, Malays. J. Anal. Sci., 2016, 20, 1191–1202 CrossRef.
- E. Rodríguez-Arce and M. Saldías, Biomed. Pharmacother., 2021, 143, 112236 CrossRef PubMed.
- İ. Gulcin and S. H. Alwasel, Processes, 2025, 13, 1296 CrossRef.
-
(a) CCDC 2530540: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qy7b8;
(b) CCDC 2530541: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qy7c9;
(c) CCDC 2530542: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qy7db;
(d) CCDC 2530543: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qy7fc.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a













