DOI:
10.1039/D6RA01242F
(Paper)
RSC Adv., 2026,
16, 27738-27755
Symmetry-dependent electronic reconstruction and intrinsic ultraviolet response in Sr2B′B″O6 (B′ = Ti, Zr; B″ = Sn, Ge) double perovskite oxides: a first-principles study
Received
12th February 2026
, Accepted 18th May 2026
First published on 22nd May 2026
Abstract
Understanding the respective roles of crystal symmetry and B-site chemistry in determining functional properties is essential for the rational design of double perovskite oxides. In this study, we systematically investigated a family of Sr2B′B″O6 (B′ = Ti, Zr; B″ = Sn, Ge) compounds in both the cubic Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m and distorted P21/c phases using density functional theory. All compositions exhibited negative formation energies, confirming thermodynamic stability, with the P21/c phase being energetically favored. The elastic constants and derived mechanical parameters demonstrate mechanical stability in both structures, while symmetry lowering generally promotes ductility and modifies the bonding characteristics. Electronic structure calculations revealed that structural distortion significantly reconstructs the band edges, with most compositions undergoing a direct-to-indirect band-gap transition accompanied by band-gap widening. The valence band maximum is dominated by O-2p states and the conduction band minimum by B′-site d orbitals, indicating a potential p-type semiconducting tendency. In contrast, optical responses are only weakly affected by symmetry change: all compounds exhibit intrinsic strong ultraviolet absorption and visible transparency, while B-site substitution induces systematic blue shifts of the absorption edge. Boltzmann transport calculations further show that thermoelectric properties are largely insensitive to symmetry transition and elemental substitution within the constant relaxation time approximation. These results clarify the distinct influences of structural symmetry and chemical composition on multifunctional behavior in Sr2B′B″O6 double perovskites and provide theoretical guidance for symmetry-informed materials design.
m and distorted P21/c phases using density functional theory. All compositions exhibited negative formation energies, confirming thermodynamic stability, with the P21/c phase being energetically favored. The elastic constants and derived mechanical parameters demonstrate mechanical stability in both structures, while symmetry lowering generally promotes ductility and modifies the bonding characteristics. Electronic structure calculations revealed that structural distortion significantly reconstructs the band edges, with most compositions undergoing a direct-to-indirect band-gap transition accompanied by band-gap widening. The valence band maximum is dominated by O-2p states and the conduction band minimum by B′-site d orbitals, indicating a potential p-type semiconducting tendency. In contrast, optical responses are only weakly affected by symmetry change: all compounds exhibit intrinsic strong ultraviolet absorption and visible transparency, while B-site substitution induces systematic blue shifts of the absorption edge. Boltzmann transport calculations further show that thermoelectric properties are largely insensitive to symmetry transition and elemental substitution within the constant relaxation time approximation. These results clarify the distinct influences of structural symmetry and chemical composition on multifunctional behavior in Sr2B′B″O6 double perovskites and provide theoretical guidance for symmetry-informed materials design.
1. Introduction
With the continuous growth of global energy demand and the increasing prominence of environmental impact issues, the development of clean, efficient, and scalable photoelectric and energy conversion materials has become a key research direction in the field of materials science. Solar energy, due to its abundant resources and the steadily decreasing cost of technology, holds a central position in the renewable energy system. Since the initial development of organic–metal halide perovskite solar cells, multiple rounds of optimization and iteration have significantly improved their photoelectric conversion efficiency in a short period, sparking extensive interest in both fundamental and applied research.1
Double perovskite materials (general formula A2B′B″X6 or oxide A2B′B″O6) exhibit richer compositional and symmetry tuning capabilities compared to single perovskite AX3, due to structural degrees of freedom such as B-site double occupancy, A-site or anion substitution, and octahedral distortion. As a result, these materials have attracted widespread attention in fields such as lead-free optoelectronic candidates, catalysis, and spintronics. Systematic reviews of oxide double perovskites and related studies on halide double perovskites (e.g..Cs2AgBiBr6) indicate that these materials offer significant advantages in chemical stability as well as tunability of electronic and optical properties. However, to realize their application in high-efficiency devices, more precise control and deeper understanding of material composition, defect regulation, and structural symmetry are still required.2
Although a large number of studies have focused on the compositional screening of double perovskite materials and their individual properties—such as band structure, light absorption characteristics, magnetism, or thermoelectric performance—there remains a lack of systematic and in-depth investigation into the coupling mechanisms between space group (crystal symmetry) changes and site substitution, as well as their comprehensive effects on mechanical stability, electronic structure, optical response, and thermoelectric transport properties. Structural details of materials, such as the octahedral configuration, B-site element ordering, and A-site ion size, typically affect the band gap, carrier effective mass, and phonon scattering by modulating soft mode vibrations and phonon spectra. These microscopic variations then collaboratively influence the material's optoelectronic and thermal transport properties. This phenomenon indicates that during material screening, it is insufficient to focus solely on individual properties; instead, a multi-property coupled comprehensive evaluation should be conducted from the perspective of crystal symmetry and microscopic structural distortions.3 In addition, recent high-throughput calculations and thermodynamic stability analyses of A2B′B″O6-type systems have also indicated that thermodynamic feasibility, phase stability range, and synthetic feasibility are key prerequisites for the transition of theoretical candidate materials to experimental validation.4
In this work, we perform a systematic first-principles investigation of Sr2B′B″O6 (B′ = Ti, Zr; B″ = Sn, Ge) double perovskite oxides to elucidate how crystal symmetry and B-site substitution govern structural stability and multifunctional properties. By directly comparing the cubic Fmm and distorted P21/c phases, we evaluate thermodynamic stability, mechanical behavior, electronic structure, optical response, and thermoelectric transport. Particular attention is given to symmetry-induced band-edge reconstruction and its implications for optical and transport properties. This comprehensive analysis allows us to distinguish between properties that are intrinsically determined by chemical composition and those strongly modulated by structural symmetry.5–7
2. Computational details
This study employs the Vienna Ab initio Simulation Package (VASP) to perform density functional theory (DFT) calculations aimed at simulating and analyzing the structure and properties of the double perovskite oxide Sr2B′B″O6 (B′ = Ti, Zr; B″ = Sn, Ge). In the calculations, the exchange-correlation energy is treated using the projector augmented-wave (PAW) pseudopotentials combined with the Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA-PBE). The plane-wave cutoff energy (ENCUT) is set to 500 eV, a value confirmed through convergence testing. The k-point meshes used are 8 × 8 × 8 and 6 × 4 × 3, chosen based on energy and force convergence tests. The energy convergence criterion (EDIFF) is set to 1.0 × 10−5, and the force convergence criterion (EDIFFG) is set to −0.01. To verify the thermodynamic stability of the material at room temperature, ab initio molecular dynamics (AIMD) simulations were conducted based on the relaxed structure. The simulations were performed in the NVT ensemble, with temperature controlled by a Nosé–Hoover thermostat set at 300 K. The time step (POTIM) was 1 fs, and the total number of simulation steps (NSW) was 4300, corresponding to an overall simulation time of approximately 4.3 picoseconds. The simulations provide a finite-time check of stability under ambient conditions and may not capture possible temperature-driven phase transitions at higher temperatures. While phonon dispersion calculations would ideally provide a complete assessment of lattice dynamical stability, the AIMD simulations here offer a qualitative indication of the thermodynamic stability of the materials. Thus, the combination of geometry optimization and AIMD results supports the overall stability trends reported in this work, without implying quantitative dynamical confirmation. The mechanical properties of the material were obtained via the stress–strain method. Specifically, small strains were applied to the PBE-optimized crystal structure, and the resulting stress tensors were calculated to determine the elastic constants. Subsequently, the elastic constants were processed using the Vaspkit software to compute mechanical parameters such as bulk modulus, shear modulus, Young's modulus, and Poisson's ratio, which were used to assess the material's mechanical stability and ductility. In terms of electronic structure, the calculations include total density of states (TDOS), partial density of states (PDOS), band structure (TBAND), and projected band structure (PBAND). Band structure calculations were performed using the PBE, PBE+U, Tran–Blaha modified Becke–Johnson (TB-mBJ), and TB-mBJ+U methods to compare the effects of different functionals on the band structure and band gap. For transition metal elements with localized or semi-localized electronic states, the Hubbard U correction was introduced to improve calculation accuracy. The U values were set to 3.5 eV for Ti and Sn, 3.0 eV for the 4d orbitals of Zr, while no U correction was applied to Ge. The choice of U values follows commonly adopted values in previous studies of transition-metal oxides and was used to improve the description of localized and semi-localized electronic states. For Ti and Zr, the U values account for the localized d orbitals. For Sn, although it is not typically considered a strongly correlated element, a moderate U value was introduced to adjust the relative position of its electronic states and improve the overall band structure description, particularly in systems involving hybridization with transition-metal d orbitals. While the choice of U may affect quantitative results, the comparative trends discussed in this work remain robust.8 The discussion of band-edge reconstruction and band-gap trends in this work is therefore based on scalar-relativistic DFT calculations. Finally, the density of states calculated using the TB-mBJ method was employed to investigate the dominant roles of various elements in the band structure. The calculation of optical properties was based on linear response theory, combining the PBE+U and TB-mBJ+U methods, and the band gap was corrected by introducing a scissors operator to obtain more accurate optical response parameters such as the dielectric function, absorption coefficient, and photoconductivity. The same Hubbard U values as applied in the electronic structure were used for the optical calculations, and while quantitative details may vary with different U values, the main qualitative trends are preserved. For the anisotropic crystal structure P21/c, the optical tensor was calculated and plotted along the xx, yy, and zz directions respectively to reveal its anisotropic characteristics. In this study, Sr2TiGeO6 was chosen as a representative example for detailed P21/c optical analysis due to its typical structural distortion and intermediate band gap among the four compounds. Although the full P21/c optical analysis was not repeated for Sr2TiSnO6, Sr2ZrSnO6, and Sr2ZrGeO6, the similarity in crystal framework and bonding environment suggests that the observed anisotropic trends along the principal axes are qualitatively applicable to these other compositions. Therefore, the conclusions drawn from Sr2TiGeO6 can be reasonably extended to the entire set of double perovskite oxides studied. Thermoelectric properties were calculated using the BoltzTraP program based on the band structure results, combined with semiclassical Boltzmann transport theory under the constant relaxation time approximation. Parameters such as the Seebeck coefficient, electrical conductivity (σ), power factor (PF), and thermal conductivity were computed, and their variations with temperature and carrier concentration were systematically analyzed. Due to the assumed relaxation times and model limitations, these results are presented as approximate estimates, providing qualitative insight rather than quantitatively predictive values.
Scalar relativistic effects were included in all DFT calculations through the use of relativistic pseudopotentials. However, explicit spin–orbit coupling (SOC) was not considered in this work. It is noted that scalar relativistic effects and SOC represent two distinct contributions to relativistic corrections: the former accounts for mass–velocity and Darwin terms, while the latter describes the interaction between electron spin and orbital angular momentum. All discussions of electronic structure in this work are therefore based on scalar relativistic DFT results.
3. Results and discussion
3.1 Structural properties
In order to thoroughly investigate the properties of double perovskite oxides Sr2B′B″O6 (B′ = Ti, Zr; B″ = Sn, Ge), this study employs the Fmm structural model proposed by Hansraj Karwasara for material construction,7 as shown in Fig. 1 and 2. Referring to the P21/c structural configuration in the Materials Project database, material models under the P21/c space group were constructed, as shown in Fig. 3 and 4. All structures were optimized using the PBE functional to obtain the corresponding lattice constants and atomic position parameters of Sr2B′B″O6 (B′ = Ti, Zr; B″ = Sn, Ge). For the Fmm space group, the optimized lattice constants of Sr2TiSnO6, Sr2TiGeO6, Sr2ZrSnO6, Sr2ZrGeO6 are 8.052 Å, 7.788 Å, 8.279 Å, and 8.001 Å, respectively. In the studied cubic double perovskite compounds, the Sr, Ti/Zr, Sn/Ge, and O atoms occupy the 8c site (0.25, 0.25, 0.25), 4b site (0.5, 0, 0), 4a site (0, 0, 0), and 24e site (x, 0, 0), respectively. The variable x takes values of 0.2560, 0.2468, 0.2490, and 0.2399 for Sr2TiSnO6, Sr2TiGeO6, Sr2ZrSnO6, Sr2ZrGeO6, respectively. The results show that when the B′ site is the same and the B″ site is Sn, the lattice constant is larger than when it is Ge; similarly, when the B″ site is the same and the B′ site is Zr, the lattice constant is larger than when it is Ti. This phenomenon is mainly attributed to the effect of atomic size. For the P21/c space group, the optimized lattice constants of Sr2TiSnO6, Sr2TiGeO6, Sr2ZrSnO6, Sr2ZrGeO6 are listed in Table 1, and the atomic occupancy coordinates are detailed in Table 2.
 |
| | Fig. 1 Crystal structures of Sr2B′B″O6 under the Fmm space group: (a)–(d) are Sr2TiSnO6, Sr2TiGeO6, Sr2ZrSnO6, Sr2ZrGeO6. The B′ and B″ cations occupy the octahedral sites, while Sr atoms are located at the A-sites. | |
 |
| | Fig. 2 Projection view of Sr2B′B″O6 structures under the Fmm space group along a specific crystallographic direction: (a)–(d) are Sr2TiSnO6, Sr2TiGeO6, Sr2ZrSnO6, Sr2ZrGeO6. This projection highlights the connectivity of the octahedral B–O framework and the spatial arrangement of the A-site Sr atoms. | |
 |
| | Fig. 3 Crystal structures of Sr2B′B″O6 under the P21/c space group: (a)–(d) are Sr2TiSnO6, Sr2TiGeO6, Sr2ZrSnO6, Sr2ZrGeO6. The distorted octahedral framework reflects the symmetry-lowering effect compared to the cubic phase. | |
 |
| | Fig. 4 Projection view of Sr2B′B″O6 structures under the P21/c space group along a specific crystallographic direction: (a)–(d) are Sr2TiSnO6, Sr2TiGeO6, Sr2ZrSnO6, Sr2ZrGeO6. The projection highlights octahedral tilting and structural distortion in the low-symmetry phase. | |
Table 1 Lattice constants of P21/c space group
| Material |
a |
b |
c |
| Sr2TiSnO6 |
5.68 |
5.68 |
8.04 |
| Sr2TiGeO6 |
5.51 |
5.51 |
7.82 |
| Sr2ZrSnO6 |
5.79 |
5.84 |
8.23 |
| Sr2ZrGeO6 |
5.66 |
5.65 |
8.01 |
Table 2 Atomic positions occupied in the P21/c space group
| Element |
Sr2TiSnO6 |
Sr2TiGeO6 |
Sr2ZrSnO6 |
Sr2ZrGeO6 |
| Sr(4e) |
(0.75,0.02,0.25) |
(0.75,0,0.25) |
(0.74,0.03,0.25) |
(0.75,0,0.25) |
| B’(Ti,Zr)(2a) |
(0.50,0.50,0) |
(0.50,0.50,0) |
(0.50,0.50,0) |
(0.50,0.50,0) |
| B″(Sn,Ge)(2b) |
(0,0.50,0.50) |
(0,0.50,0.50) |
(0,0.50,0.50) |
(0,0.50,0.50) |
| O(4e) |
(0.74,0.78,0.48) |
(0.75,0.75,0.50) |
(0.75,0.78,0.46) |
(0.75,0.75,0.48) |
| O(4e) |
(0.69,0.27,0.47) |
(0.75,0.25,0.50) |
(0.68,0.28,0.46) |
(0.73,0.27,0.48) |
| O(4e) |
(0.80,0.49,0.24) |
(0.76,0.50,0.25) |
(0.82,0.48,0.25) |
(0.80,0.50,0.26) |
Analysis shows that for materials with the P21/c space group, similar to the Fmm cubic structure, when the B′ element is the same and B″ is Sn, the lattice constant is larger than when B″ is Ge. Similarly, when the B″ element is the same and B′ is Zr, the lattice constant is also larger than when B′ is Ti. This phenomenon may be attributed to the influence of atomic size effects.
The geometric stability of the material mainly depends on the degree of matching of the ionic radius Sr2+, Ti4+, Zr4+, Sn4+, Ge4+, O2−. In this paper, the tolerance factor (τ) and the octahedral factor (µ) are used as evaluation indicators.
| |
 | (1) |
| |
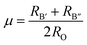 | (2) |
Here,
RA and
RO represent the ionic radii of Sr and O, respectively, while
RB′ and
RB″ represent the ionic radii of elements B′ (Ti, Zr) and B″ (Sn, Ge), respectively. The calculated tolerance factors for Sr
2TiSnO
6, Sr
2TiGeO
6, Sr
2ZrSnO
6 and Sr
2ZrGeO
6 are 0.89, 0.93, 0.87, and 0.90, respectively; the calculated octahedral factors are 0.46, 0.41, 0.50, and 0.47, respectively. It can be seen that the tolerance factors of these materials are all close to 0.9 and meet the range criterion of 0.71 <
τ < 1; as for the octahedral factors, except for a few slightly below the standard of 0.42 <
µ < 0.75, they generally conform to this standard.
4,5 From this, it can be inferred that the octahedral factor of Sr
2TiGeO
6 is slightly smaller, which may lead to minor structural distortions; however, its tolerance factor is closest to 1, indicating that its structure is closer to an ideal cubic lattice. In contrast, Sr
2ZrSnO
6 has a larger octahedral factor but a relatively smaller tolerance factor, exhibiting greater distortion. A comprehensive analysis shows that although these materials can form stable B–O octahedral structures under the
Fmm space group, their tolerance factors all deviate from the ideal value of 1, and their octahedral factors are closer to or even slightly below the lower limit of 0.42. This suggests that they tend to relieve internal stress through rotation and tilting of the octahedra, thereby forming low-symmetry crystal structures such as
P2
1/
c.
Furthermore, to systematically investigate the thermal stability of the materials, this study conducted an in-depth analysis of the binding energy and molecular dynamics behavior of the materials at room temperature (300 K). Specifically, supercells of sizes 2 × 1 × 1 and 2 × 2 × 1 were constructed based on materials with the Fmm space group and P21/c space group, respectively, ensuring that each model contained approximately 80 atoms. Molecular dynamics simulations were performed under the NVT ensemble, using the Nosé–Hoover thermostat to maintain the system temperature steadily at 300 K. The simulation time step was set to 1 femtosecond, with an initial equilibration period of 3 femtoseconds to eliminate initial stress, followed by the collection of trajectory data over 4 picoseconds. To ensure computational accuracy, the AIMD simulations employed the same pseudopotentials as used in structural optimization, and the plane-wave cutoff energy (ENCUT) was appropriately increased to 520 eV to enhance the numerical stability of energy and forces, while also ensuring consistency of the basis set during atomic position changes. Brillouin zone sampling of the supercells was limited to the Γ point only (KPOINTS: 1 × 1 × 1) to reduce computational resource consumption and minimize the risk of structural collapse or severe temperature fluctuations.
Fig. 5 shows the trend of the total energy of the system over time. After the equilibration phase, the total energy of each material exhibits small, bounded thermal oscillations around a certain average value, without any unidirectional drift or divergence over time. Data analysis indicates that the amplitude of energy fluctuations for each curve is controlled within ±1 eV. Given that the supercell operation includes 80 atoms for each material, the energy fluctuation amplitude can be converted to ±0.0125 eV per atom. This value falls within the range of 0.01 to 0.05 eV per atom. Therefore, it can be preliminarily inferred that these eight designed materials possess thermodynamic stability at room temperature (300 K). While explicit phonon dispersion calculations are not performed, the AIMD results provide a qualitative assessment of lattice stability, supporting the overall trends reported in this work without implying full dynamical confirmation.
 |
| | Fig. 5 Total energy evolution of Sr2B′B″O6 supercells during ab initio molecular dynamics (AIMD) simulations at 300 K: (a) Fmm, (b) P21/c. The bounded energy fluctuations indicate thermodynamic stability at room temperature. | |
From the perspective of binding energy, based on the formula:
| |
 | (3) |
where
Ebinding represents the binding energy,
Etot denotes the total energy of the crystal system,
ni indicates the atomic number of a specific element, and
µi signifies the energy of an isolated atom of that element. Isolated atom energy data are shown in
Table 3, while the total crystal system energy and binding energy are presented in
Table 4.
Table 3 Isolated atomic energy table for required elements
| Element |
Sr |
Ti |
Zr |
Sn |
Ge |
O |
| µ(eV) |
−3.27 |
−23.33 |
−17.04 |
−30.65 |
−35.90 |
−39.52 |
Table 4 Total energy and binding energy of material crystal systems
| Materials |
Sr2TiSnO6 |
Sr2TiGeO6 |
Sr2ZrSnO6 |
Sr2ZrGeO6 |
| Fmm, Etot(eV) |
−285.25 |
−289.33 |
−291.24 |
−295.38 |
| Fmm, Eform (eV) |
−12.40 |
−13.58 |
−0.12 |
−1.23 |
| P21/c, Etot (eV) |
−142.80 |
−144.27 |
−146.13 |
−147.69 |
| P21/c ,Eform (eV) |
−154.85 |
−158.27 |
−145.23 |
−148.92 |
We note that using isolated atomic energies as reference states may lead to formation energies that differ quantitatively from those calculated relative to the elements in their stable bulk phases. Therefore, the present definition corresponds to a binding-energy-like quantity rather than a conventional thermodynamic formation energy. Nevertheless, the calculated values provide a qualitative measure of relative stability among different configurations.
Analysis reveals that the calculated binding energies of all configuration materials are negative, indicating that the structures are energetically favorable relative to isolated atoms. However, this does not directly imply thermodynamic stability with respect to decomposition into competing bulk phases. Interestingly, under identical B′ element conditions, selecting Ge as the B″ element yields greater stability than choosing Sn; conversely, under identical B″ element conditions, selecting Ti as the B′ element is more stable than choosing Zr. Furthermore, the binding energy Eform for each material in the P21/c space group is significantly lower than that of corresponding materials in the Fmm space group. This indicates that, within the same computational framework, the P21/c configurations are relatively more energetically favorable.
These results suggest a tendency for the studied double perovskite oxides to favor the lower-symmetry P21/c structure, although this conclusion should be interpreted in a comparative rather than strict thermodynamic sense.
3.2 Mechanical properties
To evaluate the mechanical stability, applicability, and structural integrity of the material, this study analyzes the mechanical behavior of Sr2B′B″O6 by calculating its elastic properties, including parameters such as Young's modulus, bulk modulus, shear modulus, and Poisson's ratio. For the Fmm space group, determining elastic parameters C11, C12, C44 is crucial for analyzing mechanical properties. Table 5 presents the elastic parameters under the Fmm space group. Based on the results in Table 5, it is confirmed that the elastic parameters measured for Sr2B′B″O6 satisfy the mechanical stability conditions for Fmm space group materials,5 specifically:
| C11 − C12 > 0, C11 > 0, C44 > 0, C11 + 2C12 > 0 |
Table 5 Elastic parameters of Fmm space group
| Material |
C11 |
C12 |
C44 |
| Sr2TiSnO6 |
291.85 |
91.25 |
91.04 |
| Sr2TiGeO6 |
180.19 |
89.72 |
106.33 |
| Sr2ZrSnO6 |
292.88 |
80.17 |
77.75 |
| Sr2ZrGeO6 |
295.23 |
99.17 |
97.25 |
The data in Table 6 demonstrate that the Sr2B′B″O6 under the P21/c space group also satisfies the mechanical stability requirements9 specifically:
| Cii > 0 (i = 1, 2, 3, 4, 5, 6) |
| [C11 + C22 + C33 + 2(C12 + C13 + C23)] > 0 |
| C 33C55 − C352 > 0; C44C66 − C462 > 0; C22 + C33 − 2C23 > 0 |
| C22(C33C55 − C352) + 2C23C25C35 − C232C55 − C252C33 > 0 |
| 2[C15C25(C33C12 − C13C23) + C15C35(C22C13 − C12C23) + C25C35(C11C23 − C12C13)] − [C152(C22C33 − C232) + C252(C11C33 − C132) + C352(C11C22 − C122)] + C55, g > 0 |
| g = C11C22C33 − C11C232 − C22C132 − C33C122 + 2C12C13C23 |
Table 6 Elasticity parameters of space groups for P21/c space group
| Material |
C11 |
C22 |
C33 |
C44 |
C55 |
C66 |
C46 |
| Sr2TiSnO6 |
248.69 |
249.28 |
333.74 |
108.18 |
90.09 |
64.36 |
10.94 |
| Sr2TiGeO6 |
260.15 |
244.97 |
279.14 |
132.22 |
107.69 |
60.36 |
3.77 |
| Sr2ZrSnO6 |
279.83 |
373.62 |
407.39 |
108.38 |
90.63 |
103.41 |
9.61 |
| Sr2ZrGeO6 |
300.71 |
325.43 |
323.24 |
128.93 |
104.01 |
84.26 |
−4.05 |
| Material |
C12 |
C13 |
C23 |
C35 |
C25 |
C15 |
g |
| Sr2TiSnO6 |
155.68 |
153.95 |
134.27 |
−13.98 |
5.40 |
2.72 |
16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 735091.28 735091.28 |
| Sr2TiGeO6 |
91.26 |
122.60 |
108.45 |
−0.45 |
1.76 |
2.97 |
11149504.72 |
| Sr2ZrSnO6 |
229.61 |
199.09 |
162.97 |
−1.77 |
6.27 |
2.78 |
13774182.74 |
| Sr2ZrGeO6 |
162.49 |
156.99 |
140.68 |
9.63 |
−4.50 |
−2.85 |
16303978.59 |
Sr2B′B″O6 These elastic parameters are further utilized to derive several additional elastic indices, including Young's modulus (Y), bulk modulus (B), shear modulus (G), Poisson's ratio (υ), Klein's coefficient (K), and Cauchy pressure (CP). Based on the data shown in Table 7, the high bulk modulus (B) indicates that the material exhibits significant compressive resistance under external pressure, demonstrating good stability in both the Fmm space group and the P21/c space group, with the latter exhibiting superior stability. Data on shear modulus (G) reveal a minimum value of 70.77 (P21/c space group Sr2TiSnO6) and a maximum of 97.56 (Fmm space group Sr2ZrGeO6), indicating good rigidity for this material class. Furthermore, except for Sr2TiGeO6, the other three materials exhibit reduced rigidity during the space group transition from Fmm to P21/c, while Sr2TiGeO6 shows a trend toward increased rigidity. In the P21/c space group, results clearly indicate that Zr and Ge elements enhance material rigidity. In contrast, within the cubic Fmm structure, when B′ is Zr and B″ is Ge, material rigidity outperforms that with B″ as Sn; conversely, when B′ is Ti, the opposite trend emerges. This phenomenon is speculated to stem from differences in chemical bonds arising from atomic orbital properties or atomic radius variations.10,11 Regarding the Pugh ratio (B/G), it is used to assess Sr2B′B″O6 brittleness or ductility. If B/G < 1.75 Sr2B′B″O6, the material is deemed brittle; conversely, B/G > 1.75 Sr2B′B″O6 indicates ductile behavior. Concurrently, Poisson's ratio (υ) is introduced as an auxiliary criterion: υ < 0.26 corresponds to brittleness, while υ > 0.26 corresponds to ductility. As the space group transitions from Fmm to P21/c, Sr2B′B″O6 the material properties shift from brittle to ductile. Furthermore, when B′ is Ti, the material exhibits a tendency toward brittleness, and a similar trend is observed when B″ is Ge5,9. Consequently, the most brittle material belongs to the Fmm space group Sr2TiGeO6, while the most ductile material belongs to the P21/c space group Sr2ZrSnO6. Finally, we analyzed bonding characteristics using the Klein parameter (K) and Cauchy pressure (CP). A Klein parameter K close to 1 indicates that structural deformation primarily occurs through bond length changes (i.e., bond angles are relatively flexible), while K close to 0 indicates deformation primarily occurs through bond angle changes (i.e., bond lengths are relatively rigid). Results show that most Sr2B′B″O6 materials exhibit a Klein factor K approaching 1, indicating deformation primarily relies on bond length changes. In contrast,Sr2ZrSnO6 belonging to the Fmm space group has a K value of 0.49, slightly below 0.5, suggesting its deformation involves both bond angle and bond length changes, but with a greater tendency toward bond angle changes.12,13 A negative Cauchy pressure (CP) value indicates predominantly covalent bonding, while a positive value indicates predominantly ionic bonding. Only Sr2TiGeO6 exhibits covalent bonding characteristics; the others are predominantly ionic. Poisson's ratio (υ) between 0.25 and 0.50 indicates the material is stabilized by central forces (termed a central force crystal), while values outside this range indicate stabilization by non-central forces (termed a non-central force crystal). Notably, only Sr2TiGeO6, belonging to the Fmm space group, exhibits non-centripetal force crystal behavior. This explains the previously observed opposite trend in the Fmm space group when B′ is Ti.
Table 7 Sr2B′B″O6 Additional elastic parameters. Y: Young's modulus, υ: Poisson's ratio, K: Klein's coefficient, CP: Cauchy pressure
| |
B |
G |
B/G |
Y |
υ |
K |
CP |
Fm![[3 with combining macron]](https://www.rsc.org/images/entities/b_char_0033_0304.gif) m m |
| Sr2TiSnO6 |
158.11 |
94.64 |
1.67 |
236.69 |
0.25 |
0.55 |
0.20 |
| Sr2TiGeO6 |
119.88 |
75.47 |
1.59 |
187.13 |
0.24 |
0.83 |
−16.60 |
| Sr2ZrSnO6 |
151.07 |
88.16 |
1.71 |
221.40 |
0.26 |
0.49 |
2.40 |
| Sr2ZrGeO6 |
164.52 |
97.56 |
1.69 |
244.38 |
0.25 |
0.58 |
1.90 |
|
| P21/c |
| Sr2TiSnO6 |
189.80 |
70.77 |
2.55 |
197.82 |
0.33 |
1.05 |
47.50 |
| Sr2TiGeO6 |
157.97 |
83.82 |
1.81 |
221.23 |
0.27 |
0.60 |
−41.00 |
| Sr2ZrSnO6 |
247.75 |
78.15 |
2.92 |
228.72 |
0.35 |
1.41 |
121.20 |
| Sr2ZrGeO6 |
207.73 |
92.02 |
2.55 |
245.12 |
0.30 |
0.90 |
33.60 |
3.3 Electronic properties
For the eight Sr2B′B″O6 (B′ = Ti, Zr; B″ = Sn, Ge) material systems designed in this study, four first-principles calculation methods were employed: PBE, PBE+U, Tran–Blaha modified Becke–Johnson (TB-mBJ), and TB-mBJ+U, to systematically compare their electronic structure characteristics. Overall, the widely used generalized gradient approximation (PBE) method, serving as the reference, generally underestimated the band gap. In contrast, the TB-mBJ method significantly enhanced the band gap values and exhibited more reliable band level ordering across all systems. For samples containing localized d orbitals, Hubbard U corrections (PBE+U or TB-mBJ+U) substantially adjust the relative energy positions of d states within the valence or conduction bands, thereby influencing band gap size and the contribution of edge band orbitals.14 Notably, compared to other relevant studies, the band gap values calculated using the TB-mBJ method in this work are smaller than those obtained with the GGA-PBE method (see Table 8). For perovskite systems containing transition metals (B' = Ti, Zr) and heavier elements (B″ = Sn, Ge), similar band-gap differences are often discussed in the literature in relation to spin–orbit coupling (SOC), the sensitivity of pseudopotential treatment (semi-core layer) and TB-mBJ parameterization in systems with semi-localized d electrons.15,16 In the present study, scalar relativistic effects were included in all calculations, whereas explicit spin–orbit coupling (SOC) was not considered. Therefore, the reported band gaps and band-edge transitions reflect scalar-relativistic DFT results. While SOC may slightly modify the conduction band levels, it is not explicitly included in the present scalar-relativistic calculations, the overall qualitative trends and direct-to-indirect transitions observed upon structural distortion are expected to remain valid. Analysis of the band structure and density of states further indicates that the structural distortion from Fmm to P21/c alters the local bonding environment, including octahedral tilting and variations in B–O–B bond angles. These distortions modify the orbital overlap and hybridization near the band edges, which in turn induces the observed direct-to-indirect band gap transitions. Specifically, the conduction band minimum is dominated by the d orbitals of the B′ elements, while the valence band maximum is primarily contributed by the 2p orbitals of oxygen. The relative energies of these orbitals are affected by the degree of octahedral distortion, leading to the observed shifts in the band edges.
Table 8 Band gap values for Sr2B′B″O6 under four calculation methods (Δ and Δ(+U)) denote band gap differences under TB-mBJ and PBE algorithms, respectively; TB-mBJ+U and PBE+U denote differences under TB-mBJ+U and PBE+U algorithms; U values: Ti: 3.5, Zr: 3.0, Sn: 3.5, Ge: 0
| Material |
SG |
PBE |
TB-mBJ |
Δ |
PBE+U |
TB-mBJ+U |
Δ(+U) |
| Sr2TiSnO6 |
Fmm |
3.05 |
2.47 |
−0.59 |
3.27 |
2.70 |
−0.57 |
| P21/c |
2.91 |
2.63 |
−0.28 |
3.13 |
2.88 |
−0.26 |
| Sr2TiGeO6 |
Fmm |
2.70 |
2.14 |
−0.56 |
2.95 |
2.40 |
−0.55 |
| P21/c |
2.70 |
2.14 |
−0.56 |
2.95 |
2.40 |
−0.55 |
| Sr2ZrSnO6 |
Fmm |
3.95 |
3.56 |
−0.39 |
3.96 |
3.85 |
−0.11 |
| P21/c |
3.94 |
3.75 |
−0.20 |
4.17 |
4.09 |
−0.08 |
| Sr2ZrGeO6 |
Fmm |
4.10 |
3.08 |
−1.02 |
4.10 |
3.41 |
−0.69 |
| P21/c |
3.90 |
3.19 |
−0.72 |
3.90 |
3.51 |
−0.39 |
Given the absence of reported experimental band gap data for Sr2B′B″O6 (B′ = Ti, Zr; B″ = Sn, Ge), we conducted preliminary predictions based on similar oxide materials of the Ba2TiB″O6 type (Fig. 6 and 7). The band gap range for oxide materials containing Sn4+ and Ge4+ is approximately 2 to 4 electron volts. Substituting Ti4+ with Zr4+ is expected to increase the band gap, a prediction consistent with our experimental results.17 Based on the band structure calculations for Sr2TiGeO6 (shown in Fig. 8 and Fig. 9), we performed further analysis. The results show that under the Fmm space group structure, Sr2TiGeO6 consistently maintains a direct band gap, indicating significant potential for optoelectronic applications. This is because electronic transitions can occur without phonon assistance, leading to strong light absorption capabilities. However, upon transitioning to the P21/c space group, the band gap size increases compared to the original structure. Furthermore, under the TB-mBJ+U calculation method, the band gap nature changes from direct to indirect. Except for Sr2TiGeO6, the other three materials exhibit a transition from direct to indirect band gaps upon transformation to the P21/c space group, regardless of computational method. This indicates that structural distortion significantly influences the band edge states, though the effect is relatively weaker for Sr2TiGeO6. Furthermore, using the TB-mBJ method as an example, we analyzed the density of states distribution for the materials. The results show that, regardless of the space group, the valence band maximum (VBM) is primarily contributed by the 2p orbitals of the X element (oxygen's 2p orbitals), while the conduction band minimum (CBM) is dominated by the d orbitals of the Y element (where the B′ elements are Ti and Zr). These observations indicate that structural distortion significantly influences the band edge states through modification of orbital overlap and hybridization. The analysis also suggests a hole-dominated valence-edge character, indicating a qualitative p-type tendency in electronic behavior, while recognizing that definitive carrier type determination would require further transport calculations.
 |
| | Fig. 6 Electronic band structure of Sr2TiGeO6 in the Fmm space group calculated using different methods: (a) PBE, (b) PBE+U, (c) TB-mBJ, (d) TB-mBJ+U. The comparison illustrates the effect of exchange–correlation functionals and Hubbard U on band-gap estimation. | |
 |
| | Fig. 7 Electronic band structure of Sr2TiGeO6 in the P21/c space group calculated using different methods: (a) PBE, (b) PBE+U, (c) TB-mBJ, (d) TB-mBJ+U. Structural distortion induces modifications in band dispersion and band-gap character. | |
 |
| | Fig. 8 Density of states (DOS) of Sr2TiGeO6 in the Fmm space group. The valence band is mainly dominated by O 2p states, while the conduction band is primarily contributed by Ti 3d orbitals. | |
 |
| | Fig. 9 Density of states (DOS) of Sr2TiGeO6 in the P21/c space group. The redistribution of orbital contributions reflects the influence of structural distortion on electronic structure. | |
 |
| | Fig. 10 Band structures and corresponding dominant orbital-resolved density of states of Sr2TiGeO6 calculated using the TB-mBJ method. (a and b) Fmm phase; (c and d) P21/c phase. The results illustrate the orbital origin of band-edge states and their evolution with symmetry lowering. | |
3.4 Optical properties
The dielectric response of any material can be studied using its dielectric function, which can be expressed as:| | |
ε (ω) = ε1(ω) + iε2(ω)
| (4) |
whereε1 (ω), ε2 (ω) correspond to the real and imaginary parts of the dielectric function, respectively. Under Fmm space group symmetry, the real and imaginary parts of the dielectric function for the four materials as a function of photon energy are shown in Fig. 11(a) and (b). For the compound Sr2TiGeO6 (Sr2TiSnO6), the real part of the dielectric function gradually increases with energy, reaching a peak at 3.28 eV (3.53 eV), followed by a decrease at 3.80 eV (4.07 eV). Additionally, for the compound Sr2TiGeO6(Sr2TiSnO6), the peaks of ε1 (ω) are observed at 9.67 and 9.59, respectively Sr2TiGeO6 and Sr2TiSnO6. The real parts of the static dielectric function for the compound ε1 (0) were 5.20 and 4.89, respectively. For the compound Sr2ZrGeO6(Sr2ZrSnO6), the real part of the dielectric function gradually increases to 4.54 (5.59) eV, then decreases to 5.11 (6.08) eV, with peak values at 10.05 (9.49) eV. The static real part of the dielectric function ε1 (0) is 4.60 and 4.25, respectively. Fig. 11(b) shows that the ε2 (ω) of Sr2B′B″O6 curve exhibits a pronounced upward trend across the 2–4 eV range, with values in this energy interval closely matching the corresponding band gap values after Hubbard U correction. Fig. 11(c) and (d) respectively display the Sr2B′B″O6 changes in the real part σ1 and imaginary part σ2 of the photocurrent with photon energy. Similar to the imaginary part of the dielectric function, the real part of the photocurrent conductivity also exhibits a pronounced upward trend between 2 and 4 eV. Specifically, Sr2TiGeO6 and Sr2TiSnO6 reaches peak values of 527.28 S cm−1 and 507.99 S cm−1 within the 6–8 eV range, while Sr2ZrGeO6 and Sr2ZrSnO6 achieves peak values of 478.17 S cm−1 and 555.64 S cm−1 within the 4–6 eV range. Within the 4–8 eV range, the material exhibits high conductivity, reflecting its strong photoelectric response in the ultraviolet band. Similarly, in Fig. 12(a) and (b), the optical absorption coefficient and extinction coefficient ε2 (ω) curves of Sr2B′B″O6 both show a significant increase in the 2–4 eV region, indicating similar absorption edge characteristics for the material. Specifically, the extinction coefficient in Fig. 12(b) approaches zero below 3 eV, indicating excellent transparency for this material class. As shown in Fig. 12(a), the optical absorption coefficient of Sr2B′B″O6 remains consistently high between 4-12 eV, exceeding that of 5 × 105 cm−1, with peaks exceeding 106 cm−1, further confirming its outstanding ultraviolet light absorption performance. Furthermore, the static refractive index n(0) of these materials ranges between 2 and 2.5. Combined with their near-zero extinction coefficient in the visible region, this confirms their status as high-refractive-index transparent media. Their reflectance gradually increases from a static value of 0.1–0.2 to a peak of approximately 0.3 as the extinction coefficient k rises. Notably, after elemental substitution from Ti → Zr and Ge → Sn, both B′-based and B″-based materials exhibit a blue shift in their optical properties.
 |
| | Fig. 11 Optical properties of Sr2B′B″O6 in the Fmm space group: (a) real part of dielectric function, (b) imaginary part of dielectric function, (c) real part of photoconductivity, and (d) imaginary part of photoconductivity. The spectra indicate strong ultraviolet response and visible transparency. | |
 |
| | Fig. 12 Optical constants of Sr2B′B″O6 in the Fmm space group: (a) absorption coefficient, (b) extinction coefficient, (c) refractive index, and (d) reflectivity. The results highlight optical response characteristics across different energy regions. | |
For the low-symmetry space group P21/c, this paper selects the representative Sr2TiGeO6 for analysis. As shown in Fig. 13(a), the peak values of Sr2TiGeO6 along the XX, YY, and ZZ directions increase to 3.29 eV, 3.23 eV, and 3.29 eV, respectively, before decreasing to approximately 3.77 eV. Additionally, during this process, the real part peaks of the dielectric function for Sr2TiGeO6 reached 9.62, 9.53, and 9.70 in the XX, YY, and ZZ directions, respectively. Meanwhile, the real part of the static dielectric functionε1 (0) remained stable at approximately 5.21. The curve ε2 (ω) exhibited a pronounced upward trend in the 2–3 eV range, corresponding to the variation in Sr2TiGeO6 for the Fmm space group (shown in Fig. 13(b)). Fig. 13(c) and (d) respectively show the trends and average values of the real part σ1 and imaginary part σ2 of the photoconductivity Sr2TiGeO6 along the XX, YY, and ZZ directions as a function of photon energy. Similar to the imaginary part of the dielectric function, both the real and imaginary parts of the photoconductivity exhibit a significant upward trend in the 2–3 eV range. The real part of the photoconductivity reaches peak values of 526.53 S cm−1, 508.93 S cm−1, and 525.79 S cm−1 in the 7–8 eV range, respectively. Similar to Fmm space group materials, this class of materials exhibits strong photoresponse capabilities in the ultraviolet region. Furthermore, in Fig. 14(a) and (b), the optical absorption coefficient and extinction coefficient ε2 (ω)of Sr2TiGeO6 curves both exhibit a significant increase in the 2–3 eV range, indicating consistency in their absorption edges. Notably, in Fig. 10(b), the extinction coefficient remains near zero below 3 eV, demonstrating that these materials retain excellent transparency even after transforming into the P21/c space group. As shown in Fig. 10(a),Sr2TiGeO6 rapidly increases to above 5 × 105 cm−1 within the 3–4 eV range and remains substantially above 5 × 105 cm−1, with peaks reaching the 106 cm−1 magnitude. This demonstrates that even after transforming into the P21/c space group, the material exhibits exceptionally strong light absorption properties in the ultraviolet band. Meanwhile, the static refractive index n(0) of Sr2TiGeO6 is approximately 2.25. Combined with its near-zero extinction coefficient k in the visible region, this confirms the material remains a high-refractive-index transparent medium. Concurrently, as the extinction coefficient k increases, its reflectance rises from approximately 0.15 in the static state to a maximum of about 0.3
 |
| | Fig. 13 Optical properties of Sr2TiGeO6 in the P21/c space group along the XX, YY, and ZZ directions and their average values: (a) real part of photoconductivity, (b) imaginary part of photoconductivity, (c) real part of dielectric function, and (d) imaginary part of dielectric function. | |
 |
| | Fig. 14 Direction-dependent optical constants of Sr2TiGeO6 in the P21/c space group along the XX, YY, and ZZ directions and their average values: (a) absorption coefficient, (b) extinction coefficient, (c) refractive index, and (d) reflectivity. The results reveal optical anisotropy induced by symmetry lowering. | |
Sr2B′B″O6 Comprehensive analysis indicates that the transition from the Fmm space group to the P21/c space group has a minor impact on optical properties. Furthermore, under the P21/c space group, optical properties exhibit high consistency in the XX, YY, and ZZ directions. This material exhibits high-refractive-index transparency in the visible spectrum while demonstrating significant photoelectric response and light absorption in the ultraviolet range. Consequently, it holds clear application potential in visible-light-transparent optical components and ultraviolet detectors.
3.5 Thermoelectric properties
To evaluate a material's thermoelectric performance, parameters such as the Seebeck coefficient, electrical conductivity, and thermal conductivity are typically measured to calculate the key thermoelectric figure of merit (ZT). During thermoelectric performance calculations, accurate band structures significantly influence the effective mass of carriers and the Seebeck coefficient. Therefore, this study employed BoltzTrap software to perform relevant calculations based on self-consistent field (SCF) data obtained via the TB-mBJ+U method,18 yielding thermoelectric parameters of Sr2B′B″O6 including the Seebeck coefficient and electrical conductivity (σ) etc. Ultimately, this paper focuses on analyzing the material's thermoelectric performance at room temperature (300 K). All thermoelectric transport calculations were carried out at 300 K under the constant relaxation time approximation (CRTA). Therefore, the obtained transport coefficients and ZT values reflect the behavior at 300 K only.
To facilitate research and incorporate appropriate relaxation times (τ values), this paper considers perovskite-type oxides where site A consists of similar elements and transition metal–oxygen octahedra form the basic structural unit. These perovskites exhibit carrier transport primarily influenced by similar lattice vibrations and electron-phonon scattering mechanisms. Relevant literature on oxides such as Sr2TiO3 is referenced18–20. The relaxation time was set to τ = 1.5 × 10−14 and τ = 1 × 10−14 s. Within the CRTA framework, these relaxation times are assumed constants and may introduce uncertainty in the absolute values of transport coefficients. In the Fmm space group, Sr2B′B″O6 the Seebeck coefficient exhibited a gradual decrease from approximately 500 µV K−1 to about 130 µV K−1 within the optimal ZT value carrier concentration range (1 × 1019 to 1 × 1021 cm−3). This phenomenon indicates that at low carrier concentrations, the material resides in the non-degenerate semiconductor region. As the doping concentration increases, it enters the degenerate region, causing the Fermi level to shift toward the conduction band or valence band, resulting in a decrease in the Seebeck coefficient. This aligns with the expected behavior predicted by the Pisarenko relation. Concurrently, within this concentration range, the electrical conductivity σ exhibits a change with respect to the logarithm of carrier concentration n at τ = 1 × 10−14 s, it rises from approximately 1500–2000 S m−1 to 120000–150000 S m−1, while at τ = 1.5 × 10−14, it changes from approximately 2000–2300 S m−1 to 190000–200000 S m−1. Regarding the power factor PF, at τ = 1.5 × 10−14 s, it is approximately 3.5 µW cm−1 K−2 at low carrier concentrations. As carrier concentration increases, the power factor gradually rises and approaches a high-concentration plateau of approximately 32 µW cm−1 K−2. Therefore, the peak power factor PF can be estimated at approximately 32 µW cm−1 K−2. For τ = 1.5 × 10−14 s, the power factor is about 5 µW cm−1 K−2 at low carrier concentrations. As carrier concentration increases, the power factor gradually rises and approaches a high-concentration plateau of approximately 45 µW cm−1 K−2. The peak power factor PF can be estimated within the range of 37 to 45 µW cm−1 K−2,21 indicating that under reasonable τ conditions, this material can achieve or approach practical-scale power factor levels in highly doped regions. Finally, within the carrier concentration range of 1 × 1019 to 1 × 1021 cm−3, the thermoelectric figure of merit ZT of Sr2B′B″O6 remains near its optimal value. The electronic thermal conductivity ke was calculated using BoltzTrap software with τ = 1.5 × 10−14 and τ = 1 × 10−14 s; results indicate values ranging from approximately 8 to 20. The lattice thermal conductivity was referenced from Ba0.2Sr1.8TiCoO6 at 300 K (kl = 0.11), as shares similarities in crystal structure type and transition metal–oxygen framework. It should be noted that the lattice thermal conductivity is not explicitly calculated for the present systems but estimated from a similar oxide, which introduces additional uncertainty in the ZT evaluation. Additionally, mass differences in A-site elements enhance phonon scattering and reduce lattice thermal conductivity.22 As shown in Fig. 11 and 12, the optimal ZT value is approximately 0.7 regardless of whether τ = 1.5 × 10−14 or τ = 1 × 10−14 s. This optimal ZT value should be regarded as a comparative, qualitative trend at 300 K rather than a quantitatively predictive result. This indicates that this class of materials exhibits high Seebeck coefficients and promising thermoelectric performance even at low doping levels or in undoped states.20 However, the enhancement of thermoelectric performance is constrained by the inability to significantly improve it through simple increases in carrier concentration.
When the material's space group is transformed to P21/c, within the carrier concentration (n) region corresponding to the maximum ZT value (1 × 1019 to 1 × 1021 cm−3), the Seebeck coefficient of Sr2B′B″O6 decreases from approximately 550 µV K−1 to about 170 µV K−1, while that of Sr2TiGeO6 slightly decreases from approximately 500 µV K−1 to about 130 µV K−1. Within this concentration range, The conductivity σ varies logarithmically with carrier concentration. At τ = 1 × 10−14 s, it increases from approximately 800–1600 S m−1 to 70000–160000 S m−1. At τ = 1.5 × 10−14 s, it rises from approximately 1500–2500 S m−1 to 110000–250000 S m−1. Additionally, at τ = 1 × 10−14 s, the power factor (PF) ranges from approximately 2.5 to 5 µW cm−1 K−2 at low carrier concentrations. As carrier concentration increases, PF gradually rises, reaching a plateau of approximately 21 to 35 µW cm−1 K−2 at high concentrations. The peak power factor PF is approximately 21 to 35 µW cm−1 K−2. At τ = 1.5 × 10−14 s, the power factor PF ranges from about 4 to approximately 7 µW cm−1 K−2 at low carrier concentrations. As carrier concentration increases, PF gradually rises and reaches a plateau at high concentrations, ranging from about 32 to approximately 52 µW cm−1 K−2. The peak power factor PF ranges from approximately 32 to 52 µW cm−1 K−2, demonstrating that under reasonable τ, this material in the P21/c space group can achieve or approach practical-scale power factors in highly doped regions. Finally, within the carrier concentration range of 1 × 1019 to 1 × 1021 cm−3, as shown in Fig. 13 and 14, the Sr2B′B″O6 under P21/c space group exhibits an optimal ZT value around 0.7–0.8 regardless of whether τ = 1.5 × 10−14 s or τ = 1 × 10−14 s. It continues to demonstrate a high Seebeck coefficient and promising thermoelectric performance potential. The reported ZT values at 300 K provide a comparative, qualitative assessment of the material's thermoelectric behavior, highlighting trends rather than exact predictions (Fig. 15 and 16).
 |
| | Fig. 15 Thermoelectric properties of Sr2B′B″O6 in the Fmm space group at 300 K with relaxation time τ = 1 × 10−14 s: (a) Seebeck coefficient, (b) electrical conductivity, (c) power factor, and (d) figure of merit ZT as functions of carrier concentration. | |
 |
| | Fig. 16 Thermoelectric properties of Sr2B′B″O6 in the Fmm space group at 300 K with relaxation time τ = 1.5 × 10−14 s: (a) Seebeck coefficient, (b) electrical conductivity, (c) power factor, and (d) figure of merit ZT as functions of carrier concentration. | |
Overall, the transition from the Fmm space group to the P21/c space group has little effect on the thermoelectric properties of Sr2B′B″O6. Additionally, the B′ and B″ elements do not significantly influence the material's thermoelectric performance and do not exhibit a dominant role (Fig. 17 and 18).
 |
| | Fig. 17 Thermoelectric properties of Sr2B′B″O6 in the P21/c space group at 300 K with relaxation time τ = 1 × 10−14 s: (a) Seebeck coefficient, (b) electrical conductivity, (c) power factor, and (d) figure of merit ZT as functions of carrier concentration. | |
 |
| | Fig. 18 Thermoelectric properties of Sr2B′B″O6 in the P21/c space group at 300 K with relaxation time τ = 1.5 × 10−14 s: (a) Seebeck coefficient, (b) electrical conductivity, (c) power factor, and (d) figure of merit ZT as functions of carrier concentration. | |
4. Conclusions
We have conducted a comprehensive first-principles study of Sr2B′B″O6 (B′ = Ti, Zr; B″ = Sn, Ge) double perovskite oxides to clarify the respective roles of crystal symmetry and B-site chemistry. All compositions are predicted to be stable within the present 300 K AIMD window, with the distorted P21/c phase energetically favored over the cubic Fmm structure. Mechanical analysis indicates stable elastic behavior in both phases, with symmetry lowering generally promoting ductility and modifying bonding characteristics.
Electronic structure calculations suggest that structural distortion plays a decisive role in band-edge reconstruction, frequently inducing a direct-to-indirect band-gap transition and band-gap enlargement. In contrast, optical properties are largely intrinsic to the chemical framework, exhibiting strong ultraviolet absorption and visible transparency with only minor symmetry dependence. B-site substitution is predicted to primarily govern the blue shift of the absorption edge. Thermoelectric transport properties show limited sensitivity to symmetry transition under the assumptions used in this study.
Overall, this study indicate that symmetry lowering can substantially modify electronic structure while having comparatively modest influence on optical and thermoelectric responses. Compared to prior first-principles studies on double perovskites, this work systematically examines eight Sr2B′B″O6 compositions across two distinct space groups (Fmm and P21/c), providing a unified analysis of thermodynamic, mechanical, electronic, optical, and thermoelectric properties. The novelty lies in the explicit correlation between crystal symmetry, B-site chemistry, and multi-property responses, offering a comprehensive structure–property map that has not been previously reported. These findings provide qualitative insight into structure–property relationships in double perovskite oxides and may guide symmetry-informed materials design, while experimental validation is required to confirm the predicted trends.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper entitled “Symmetry-dependent electronic reconstruction and intrinsic ultraviolet response in Sr2B′B″O6 (B′ = Ti, Zr; B″ = Sn, Ge) double perovskite oxides: a first-principles study”.
Data availability
The data supporting this study are available from the corresponding author upon reasonable request.
Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d6ra01242f.
Acknowledgements
This research received no external funding.
References
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, Organometal Halide Perovskites as Visible-Light Sensitizers for Photovoltaic Cells, J. Am. Chem. Soc., 2009, 131, 6050–6051, DOI:10.1021/ja809598r.
- S. Vasala and M. Karppinen, A2B′B″O6 perovskites: A review, Prog. Solid State Chem., 2015, 43, 1–36, DOI:10.1016/j.progsolidstchem.2014.08.001.
- J. M. Rondinelli and C. J. Fennie, Octahedral Rotation-Induced Ferroelectricity in Cation Ordered Perovskites, Adv. Mater., 2012, 24, 1961–1968, DOI:10.1002/adma.201104674.
- Y. Wang, B. Baldassarri, J. Shen, J. He and C. Wolverton, Landscape of Thermodynamic stabilities of A2BB'O6 compounds, Chem. Mater., 2024, 36, 6816–6830, DOI:10.1021/acs.chemmater.4c00576.
- N. Rahman, M. Husain, W. Ullah, A. Azzouz-Rached, H. Albalawi, Z. Bayhan and S. A. Alsalhi, Comprehensive analysis of structural, mechanical, optoelectronic, and thermodynamic properties of Ba2XBiO6 (X = Y, La) double perovskites using density functional theory, Phys. Scr., 2024, 99, 095984, DOI:10.1088/1402-4896/ad6e30.
- G. B. G. de Souzaa, V. B. Nascimentoa, R. de Paivac and A. M. Rappeb, Thermoelectric properties of M2AgAlBr6 (M = K, Rb,Cs) double perovskites: a first principles study, Solid State Commun., 2024, 378, 115399, DOI:10.1016/j.ssc.2023.115399.
- H. Karwasara, K. C. Bhamub, S. G. Kangc, A. K. Kushwahad, D. P. Raie, S. Sappatif, J. Sahariyag and A. Soni, Ab-initio investigations for structural, mechanical, optoelectronic, and thermoelectric properties of Ba2SbXO6 (X=Nb, Ta) compounds, J. Alloys Compd., 2022, 893, 162332, DOI:10.1016/j.jallcom.2021.162332.
- C. Ezeakunne, B. Lamichhane and S. Kattel, Integrating density functional theory with machine learning for enhanced band gap prediction in metal oxides, Phys. Chem. Chem. Phys., 2025, 27, 5338–5358, 10.1039/D4CP03397C.
- Q. Li, S. Li and J. Xiao, First-principles study on the mechanical and electronic properties of energetic molecular perovskites AM(ClO4)3 (A = C6H14N22+, C4H12N22+C6H14O22+; M = Na+, K+), RSC Adv., 2022, 12, 24647–24653, 10.1039/D2RA03407G.
- N. Pandech, K. Sarasamak and S. Limpijumnong, Elastic properties of perovskite ATiO3 (A = Be, Mg, Ca, Sr, and Ba) and PbBO3 (B = Ti, Zr, and Hf): First principles calculations, J. Appl. Phys., 2015, 117, 174108, DOI:10.1063/1.4919837.
- E. K. Mahmoud, A. A. Farghali, S. I. El-dek and M. Taha, Structural stabilities, mechanical and thermodynamic properties of chalcogenide perovskite ABS3 (A=Li, Na, K, Rb, Cs; B=Si, Ge, Sn) from first-principles study, Eur. Phys. J. Plus, 2022, 137, 1006, DOI:10.1140/epjp/s13360-022-03211-7.
- S. A. Shah, M. Husain, N. Rahman, N. Sfina, M. Elhadi, V. Tirth, A. Alotaibi and A. Khan, Revealing the Structural, Elastic, Electronic, and Optical Properties of K2ScCuCl6 and K2YCuCl6: An In-Depth Exploration Using Density Functional Theory, ACS Omega, 2024, 9, 16860–16867, DOI:10.1021/acsomega.4c01923.
- D. S. P. Tanner, M. A. Caro, S. Schulz and E. P. O'Reilly, Hybrid functional study of nonlinear elasticity and internal strain in zinc-blende III-V materials, Phys. Rev. Mater., 2019, 3, 013604, DOI:10.1103/PhysRevMaterials.3.013604.
- F. I. H. Alias, M. H. Ridzwan, M. K. Yaakob, C. W. Loy and Z. Mohamed, Structural, electronic and optical studies of Sr2NiTeO6 double perovskite by first-principle DFT-LDA + U calculation, J. Mater. Res. Technol., 2022, 18, 1623–1630, DOI:10.1016/j.jmrt.2022.03.017.
- B. Traoré, G. Bouder, W. Lafargue-Dit-Hauret, X. Rocquefelte, C. Katan, F. Tran and M. Kepenekian, Efficient and accurate calculation of band gaps of halide perovskites with the Tran-Blaha modified Becke-Johnson potential, Phys. Rev. B, 2019, 99, 035139, DOI:10.1103/PhysRevB.99.035139.
- H. Jiang, Band gaps from the Tran-Blaha modified Becke-Johnson approach: A systematic investigation, J. Chem. Phys., 2013, 138, 134115, DOI:10.1063/1.4798706.
- Z. U. Rehman and Z. Lin, Optoelectronic characteristics and stability evaluation of Ba2TiMxO6 (Mx = Ge, Sn, Se, Te) p-type semiconductors as candidates for functional layers in optoelectronic devices, J. Mater. Chem., 2025, 13, 8151–8168, 10.1039/D5TC00036J.
- S. K. Singh, J. A. Abraham, A. S. Alofi, A. Srivastava, K. L. Meena, B. Alshahrani, R. Sharma and A. J. A. Moayad, Physical, optoelectronic and thermoelectric characteristics of double perovskite (Sr2ScBiO6) for green energy technology using ab initio computations, RSC Adv., 2023, 13, 35145–35160, 10.1039/D3RA05397K.
- M. Sajjad, Q. Mahmood, N. Singh and J. A. Larsson, Ultralow Lattice Thermal Conductivity in Double Perovskite Cs2PtI6: A Promising Thermoelectric Material, ACS Appl. Energy Mater., 2020, 3, 11293–11299, DOI:10.1021/acsaem.0c02236.
- G. Ding, G. Gao and K. Yao, High-efficient thermoelectric materials: The case of orthorhombic IV-VI compounds, Sci. Rep., 2015, 5, 9567, DOI:10.1038/srep09567.
- K. Guo, F. Yang, T. Weng, J. Chen, J. Zhang, J. Luo, H. Li, G. Rao and J. Zhao, The Electrical and Thermal Transport Properties of La-Doped SrTiO3 with Sc2O3 Composite, Materials, 2021, 14, 6279, DOI:10.3390/ma14216279.
- T. Maiti, M. Saxena and P. Roy, Double perovskite (Sr2B′B″O6) oxides for high-temperature thermoelectric power generation—A review, J. Mater. Res., 2019, 34, 107–125, DOI:10.1557/jmr.2018.376.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Yanan Zhangbc,
Songrui Weia,
Xiaoning Guan
a,
Yanan Zhangbc,
Songrui Weia,
Xiaoning Guan



















