DOI:
10.1039/D6RA01178K
(Paper)
RSC Adv., 2026,
16, 17450-17461
Harnessing the neutral Ca(NO3)2 electrolyte in CaMnO3 perovskite systems for calcium-ion supercapacitors
Received
10th February 2026
, Accepted 16th March 2026
First published on 31st March 2026
Abstract
The need for sustainable and high-performance energy storage technologies has intensified the research interest in calcium-ion supercapacitors, driven by the abundance, low cost, and favourable redox properties of calcium. In this study, we present a CaMnO3 perovskite electrode system operating in a neutral Ca(NO3)2 electrolyte, exhibiting outstanding electrochemical characteristics. The optimized CaMnO3 electrode achieves a specific capacitance of 258.6 F g−1 at 0.5 A g−1 within a wide potential window of 1.9 V, achieving unprecedented performance for this material. It retains 72.6% of its initial capacitance even after 1500 charge–discharge cycles, with 100% coulombic efficiency. A CMO‖CMO symmetric device assembled using this electrode delivers a specific capacitance of 76.1 F g−1, an energy density of 3.8 W h kg−1, and a power density of 1190 W kg−1, emphasizing its potential for high-power applications. To demonstrate the practical application of the fabricated device, both red and yellow LEDs were illuminated. Electrochemical analysis indicates that the superior performance results from the synergistic interaction between the CaMnO3 perovskite structure and the Ca2+-based neutral electrolyte, enabling efficient charge storage through combined electrical double-layer capacitance and pseudocapacitance. This work establishes a new benchmark for calcium-ion supercapacitors and provides valuable insights into ion-intercalation and surface-redox behaviour within perovskite–electrolyte interfaces. These findings pave the way for safe, sustainable, and cost-effective energy storage solutions.
1. Introduction
The global energy framework is shifting from relying on fossil fuels towards embracing clean and renewable resources like hydro, wind and solar energies. Effective and easily accessible energy storage systems are expected to accelerate this process.1 Lithium-ion batteries (LIBs) represent the forefront of technology utilized in portable electronics, electric vehicles, and hybrid electric vehicles, among other applications.2 Nonetheless, the limited availability of lithium resources and their uneven geographical distribution are contributing to the increased costs associated with lithium metal and lithium-ion batteries.3 There is a vital need for alternative, cost-effective energy storage technologies that utilize materials abundant on Earth for large-scale applications.4,5
The energy storage systems utilizing multivalent metal ions are regarded as promising candidates because they offer double or triple electron exchange per charge carrier, which could result in enhanced volumetric and gravimetric capacities.6,7 According to previous reports, non-noble metal oxides, like copper, manganese, and cobalt oxide, show potential as supercapacitor electrode materials.8–11 Mixed metal oxides have been identified as effective supercapacitor electrodes, besides pure metal oxides.12 Among several transition metal oxides, manganese oxide stands out as one of the most promising electrodes for supercapacitor applications. This is primarily due to its wide potential window, high theoretical specific capacitance, eco-friendliness, low cost, and high abundance. However, the main limitations are its poor cyclic stability and low conductivity.13,14 There has been a growing requirement for superior energy storage materials, which has led to the exploration of potential options, such as magnesium and calcium-based energy storage systems, which can possibly deliver double the energy density of lithium batteries by providing two electrons per atom. Multivalent-ion batteries are gaining research attention as a safer, low-cost, environmental friendly, and abundant alternative to lithium-ion batteries.15–20 Substituting organic electrolytes with water also enables the utilization of the complete pH spectrum and a diverse array of ionic solutions, including H2SO4 at low pH, Na2SO4 at neutral pH, and NaOH and KOH under alkaline conditions.21 A calcium-ion battery based on aqueous electrolyte was introduced in 2017 by Gheytani et al.19 Compared with its Mg ion-based counterpart, the Ca ion offered faster electrode kinetics and the benefit of a multivalent-ion battery. The initial version of the cell exhibited excellent efficiency and consistent capacity at both high and low current rates (40 mA h g−1); following cycling, it retained 88% of its capacity and had an average coulombic efficiency of 99%.19 Thereafter, a perovskite-type CaMnO3 anode material for lithium-ion storage was reported by Chang et al. The synthesized CaMnO3 demonstrated excellent rate capability as well as stable cycling performance at a current density of 0.2 A g−1, achieving a specific capacity of 225.4 mA h g−1 after 90 cycles. Furthermore, under an extreme temperature of 0 °C, it was capable of achieving 175.2 mA h g−1 capacity even after 300 cycles at a current density of 0.2 A g−1.22 By synthesizing and electrochemically evaluating CaMnO3−δ perovskite as a pseudocapacitor material, Forslund and Pender advanced our understanding of how complex metal oxides can be tuned to enhance their capacity to store charge through anion intercalation by utilizing previously reported trends in catalytic and capacitive behaviour.23 The application of calcium-based mixed metal oxides as supercapacitor electrodes was reported by Ali et al. in 2015. They synthesised CaMnO3 via coprecipitation method (calcined at 900 °C), which was electrochemically tested in 5 M KOH; the specific capacitance value obtained was 227 F g−1 at 0.25 A g−1.12,24 In 2023, Kanagarajan et al. prepared calcium manganese oxide (CMO) by the co-precipitation method at two different temperatures (700 °C and 800 °C) and investigated its suitability for supercapacitor application. The CV and the GCD curves of CMO (800 °C) showed a maximum specific capacitance of 247 F g−1 at the scan rate of 5 mV s−1 and 327 F g−1 at a current density of 1 A g−1.13 In 2024, CaMnO3−δ perovskite was prepared by the sol–gel method by Abdel-Khalek et al., which showed a specific capacitance of only 7.5 F g−1 at a current density of 1 A g−1.25 As mentioned in Table 1, supercapacitor applications of calcium-based metal oxides were all carried out in an alkaline medium (KOH).
Table 1 Previously reported CaMnO3-based calcium-ion supercapacitors
| Electrode material |
Electrolyte |
Voltage window (V) |
Specific capacitance |
Current collector |
Reference |
| CaMnO3 |
5 M KOH |
0–0.5 |
227 F g−1 at 0.25 A g−1 |
Nickel foam |
12 (2015) |
| CaMnO3 |
5 M KOH |
0–0.4 |
327 F g−1 at 1 A g−1 |
Nickel foam |
13 (2023) |
| CaMnO3 |
6 M KOH |
0–0.5 |
7 F g−1 at 1 A g−1 |
Nickel foam |
26 (2024) |
| CaMnO3 |
1 M KOH |
−0.5–0.7 |
191 F g−1 at 0.5 A g−1 |
Graphite sheet |
This work |
| CaMnO3 |
0.5 M Ca(NO3)2 |
−0.4–1.5 |
258 F g−1 at 0.5 A g−1 |
Graphite sheet |
This work |
The investigation on the electrochemical redox behaviour of perovskite CaMnO3 as a host for calcium-ion intercalation was introduced in 2020 by Pathreeker et al. using an electrolyte of 0.5 M calcium tetrafluoroborate (Ca(BF4)2) in acetonitrile.26
In this work, we synthesised CaMnO3 by a simple hydrothermal method, using calcium nitrate tetrahydrate (Ca(NO3)2·4H2O) and manganese nitrate tetrahydrate (Mn(NO3)2·4H2O), followed by the calcination of the synthesised product to obtain CaMnO3. Using a neutral electrolyte, i.e., 0.5 M Ca(NO3)2, the electrochemical studies were performed. This study demonstrates CaMnO3 as a potential electrode material that requires additional exploration regarding its function.
2. Results and discussion
2.1 Structural and morphological analysis
The XRD (X-ray diffraction) pattern of the as-prepared CaMnO3 is shown in Fig. 1. The diffraction peaks located at 2θ values of 23.8°, 26.7°, 33.9°, 41.9°, 48.8°, 55.02°, 56.4°, 60.8°, 71.5°, and 81.5° correspond to the (101), (111), (121), (112), (202), (222), (311), (321), (400) and (161) planes of orthorhombic CaMnO3, respectively (JCPDS card no. 00-050-1746). The obtained diffraction confirms the formation of the CaMnO3 material, with minor low-intensity peaks corresponding to Ca2MnO4, a member of the layered perovskite Ruddlesden Popper family.
 |
| | Fig. 1 XRD pattern of the synthesised CaMnO3, where  indicates the Ca2MnO4 phase and “Δ” indicates CaO. indicates the Ca2MnO4 phase and “Δ” indicates CaO. | |
The morphology and elemental composition of the synthesized sample were analysed using Scanning Electron Microscopy (SEM). SEM images (Fig. 2a–c) show an agglomerated structure, which appeared like an irregular spongy-like texture. Notably, the presence of some well-defined cubical particles was also observed. The absence of impurity from Fig. 2d of EDX confirms the purity and homogeneity of the material. Overall, the SEM and EDX mapping results indicate the successful synthesis of a nanostructured perovskite-type CaMnO3 metal oxide (Table S3), offering great potential for enhanced electrochemical performance.
 |
| | Fig. 2 (a–c) SEM images of CaMnO3. (d) EDX (energy-dispersive X-ray spectroscopy) analysis of CaMnO3 with atomic percentages of Ca, Mn and O. (e–h) Elemental mapping images of Ca, Mn and O, respectively. | |
Fig. 3a and b show the transmission electron microscopy (TEM) image of the CaMnO3 perovskite sample. The brightfield image shows the presence of aggregated nanoparticles. Fig. 3c–e show the HRTEM (High-Resolution TEM) images of the CaMnO3 perovskite sample. These images displays the lattice fringes, which indicate the high crystallinity of the CaMnO3 perovskite sample.22 The d-spacings between the lattice fringes were measured to be 0.264 nm, 0.225 nm and 0.333 nm, corresponding to the (121), (112) and (111) lattice planes of CaMnO3 with an orthorhombic structure, respectively. The obtained d-spacings are in good agreement with the d-values estimated from the XRD analysis. Fig. 3f shows the SAED pattern of the CaMnO3 perovskite sample. This pattern confirms the polycrystallinity of the sample.
 |
| | Fig. 3 (a and b) Transmission electron microscopy (TEM) images of the CaMnO3 perovskite sample. (c–e) High-resolution TEM (HRTEM) images of CaMnO3 depicting the lattice spacings. (f) Selected area electron diffraction (SAED) pattern of the CaMnO3 sample exhibiting the rings of corresponding diffraction planes. | |
Fig. S1 shows the X-ray photoelectron spectroscopy (XPS) survey spectra of Ca 2p, Mn 2p, and O 1s in the CaMnO3 perovskite sample. The survey spectra indicated the presence of elements, i.e. Ca, Mn, and O, in the CaMnO3 perovskite sample without any other impurity. The calcium core level spectra in Fig. 4a show the Ca 2p3/2 peak at 347.8 eV and the Ca 2p1/2 peak at 351.1 eV, which match the expected splitting of 3.3 eV for Ca2+. The additional peak at 345.8 eV, along with the carbonate signal at C 1s survey spectra indicated surface CaCO3 (∼288 eV) due to adventitious carbon. The Mn 2p high-resolution spectra show the characteristic peaks of Mn 2p3/2 and Mn 2p1/2 at 642.2 eV and 653.9 eV (Fig. 4b), respectively, with a spin–orbit splitting of 11.7 eV, characteristic of the existence of Mn4+ ions in the sample.28,29 The O 1s high-resolution spectra in Fig. 4c depict two peaks located at 529.9 eV and 531.5 eV, corresponding to the lattice oxygen and surface oxygen, respectively.30–32
 |
| | Fig. 4 XPS spectra of CaMnO3 illustrating the core-level binding energies for (a) Ca 2p, (b) Mn 2p and (c) O 1s. | |
The existence of surface oxygen species at a higher binding energy of 531.5 eV indicates the electron-deficient surface of this sample, which facilitates the redox activity in the oxygen carriers.27,33–35
2.2 Electrochemical characterization
The electrochemical properties of CaMnO3, including their redox behaviour, operational potential window, and specific capacitance, were systematically evaluated using cyclic voltammetry (CV), galvanostatic charge–discharge (GCD), and electrochemical impedance spectroscopy (EIS) methods.
The CV curves of the CaMnO3 materials at different scan rates (5, 10, 25, 50, 75 and 100 mV s−1) are shown in Fig. 5a. The CV curves demonstrate a linear increase in the current with an increase in scan rate. As the scan rate increases, the area beneath the curve increases, indicating rapid ion diffusion and effective charge-storage characteristics of the electrode materials through the electrochemical process. A perceptible redox peak can be seen in the lower scan rates in the CV curves, due to the slower voltage sweep, which provides adequate time for redox reactions to take place, thus indicating better reversibility.36 There are subtle anodic and cathodic peaks at 0.58 and −0.12 V, respectively, indicating the presence of reversible redox reactions (Fig. S2). These are likely associated with the redox transitions of Mn, particularly Mn3+ ↔ Mn4+, which are common in manganese-based oxides.
 |
| | Fig. 5 (a) CV curves of CaMnO3 in the potential window of −0.4–1.5 V at different scan rates (5–100 mV s−1). (b) Specific capacitance calculated from the CV curves in the potential window of −0.4–1.5 V. | |
However, there is a slight deviation from the ideal rectangular shape at higher scan rates due to the fast voltage change, which limits the time available for redox processes, leading to a current response mainly governed by surface-limited capacitive effects. The specific capacitance was calculated from the CV curve using eqn (1):
| |
 | (1) |
where
Cs is the specific capacitance in F g
−1, ∫
i![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
d
v is the integral area under the CV curves,
m is the active mass of the electrode material (mg),
s is the sweep rate (mV s
−1), and
V is the potential window. The estimated specific capacitance values of the CaMnO
3 material from the CV curves are mentioned in Table S1. CaMnO
3 showed a specific capacitance of 208 F g
−1 at 5 mV s
−1. It is seen that as the scan rate increases, the specific capacitance decreases (
Fig. 5b). This is due to the faster ion transport at elevated scan rates, which results in the limitation of ion transport, promoting the formation of a complete electrical double layer at the electrode interface.
37
GCD curves of CaMnO3 at various specific currents are shown in Fig. 6a. A deviation from the linear behaviour indicates the pseudocapacitive behaviour resulting from the redox reactions, as seen in the CV patterns of the synthesised CaMnO3. The specific capacitance is calculated from the GCD curve using eqn (2):
| |
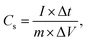 | (2) |
where
Cs,
I, Δ
t,
m, and Δ
V are the specific capacitance, current applied, discharge time, active mass of electrode material, and potential window, respectively. The specific capacitance values calculated were 258.6 F g
−1 and 244 F g
−1 at different specific currents of 0.5 A g
−1 and 1 A g
−1, respectively. Further, the rate performance of CaMnO
3 is tabulated in Table S2. From
Fig. 6b, it is clear that as the applied current increases, the specific capacitance decreases. This could be linked to the time required for the electrolyte ions to reach the electrode surface. At a low specific current, ions require more time to reach the electrode surface, resulting in high specific capacitance. On the contrary, at a high specific current, these ions have less time to access the electrode surface, leading to a lower specific capacitance.
 |
| | Fig. 6 (a) GCD curves of CaMnO3 in the potential window of −0.4–1.5 V at different specific currents varying from 0.5–12 A g−1. (b) Rate performance of CaMnO3 at different specific currents varying from 0.5–12 A g−1. (c) Long-term cycling performance of CaMnO3 in a 0.5 M Ca(NO3)2 electrolyte at 5 A g−1 for 1500 cycles (inset: up to 3000 cycles). (d) Nyquist plots of CaMnO3 before and after electrochemical cycling. | |
To benchmark the electrochemical performance of CaMnO3, additional measurements were performed in an alkaline 1 M KOH electrolyte. The electrochemical behaviour was evaluated within a potential window of −0.5–0.7 V, as shown in Fig. S3a. Galvanostatic charge–discharge measurements were performed at various specific currents ranging from 0.5–10 A g−1 (Fig. S3b). The CaMnO3 electrode delivered a specific capacitance of 191 F g−1 at 0.5 A g−1 within this potential window. Notably, the synthesized CaMnO3 retained a capacitance of 64 F g−1 even at a high specific current of 10 A g−1 (Fig. S3c). To the best of our knowledge, such performance of CaMnO3 in 1 M KOH has not been widely reported and has been included in Table 1. Unlike KOH, which contains K+ ions, Ca(NO3)2 provides divalent Ca2+ ions that potentially contributed to enhanced charge storage. Within this neutral electrolyte system, the CaMnO3 electrode exhibits better electrochemical performance, delivering a specific capacitance of 258 F g−1 at a specific current of 0.5 A g−1 over a wide potential window of −0.4–1.5 V. The broader potential window and higher capacitance highlight the advantages of utilizing a divalent Ca2+-based neutral electrolyte, which aligns with the main objective of developing an efficient calcium-ion supercapacitor.
The cycling of the CaMnO3 electrode was performed at 5 A g−1; it is observed that after 1500 continuous charge–discharge cycles, CaMnO3 showed 100% coulombic efficiency (Fig. S4), but the capacitance faded to 76% of its initial capacitance, as shown in Fig. 6c. This loss of specific capacitance can also be attributed to the mechanical expansion of the electrode material that takes place during these charging and discharging cycles, as well as the minor dissolution of electrode materials that occur during the cycling process, which has been described in the proposed mechanism in Section 2.2.1.38,39 This can be due to the increase in internal resistance.
To further analyse the kinetics of electrochemical reactions and the conductive properties of CaMnO3 in the 0.5 M Ca(NO3)2 electrolyte, Electrochemical impedance spectroscopy (EIS) measurements were carried out. The results from EIS demonstrated a decrease in electrochemical performance following cycling, as shown in Fig. 6d. A subtle shift to the right in the Nyquist plot within the high-frequency range indicates a rise in the solution resistance, which may be attributed to the degradation at the electrode–electrolyte interface. The expansion of the semicircle in the mid-frequency region indicates an increased charge-transfer resistance, potentially due to structural changes. To further analyse the internal kinetics, the diffusion coefficient and ionic conductivity of the three electrode system were calculated from EIS using the following equations:
| |
 | (3) |
| |
 | (4) |
| |
 | (5) |
where
Z′ is the real part of the resistance (Ω),
R1 is the solution resistance,
Rct is the charge-transfer resistance,
ω is the angular frequency,
R is the universal gas constant (J mol
−1 K
−1),
T is absolute temperature (K),
A is the electrode surface area (cm
2), n is the number of electrons involved in redox reaction,
F is the Faraday constant,
C is the molar concentration (mol cm
−3),
σ is the Warburg diffusion factor, and
D is the diffusion coefficient. Fig. S5 shows the linear fitting results. The linear fitted line gives the slope values of 30.9 and 7.34 for the pristine and cycled electrodes, respectively. The diffusion coefficient values are calculated using
eqn (4) and are found to be 1.43 × 10
−10 and 2.54 × 10
−9 cm
2 s
−1 for the pristine and cycled electrodes, respectively, which shows that the diffusion has increased after cycling. While the diffusion coefficient has increased, the corresponding ionic conductivity increased from 2.69 × 10
−7 S cm
−1 to 4.77 × 10
−7 S cm
−1. This suggests that cycling enhanced the ion transport, likely due to the improved electrolyte penetration despite an increase in charge-transfer resistance.
In order to evaluate the charge-storage contribution of CaMnO3 in the 0.5 M Ca(NO3)2 electrolyte, the power law was used:
where
i is current,
v is the scan rate and a and
b are adjustable parameters. When
b is closer to 1, the charge storage is dominated by capacitive behaviour, whereas when
b is closer to 0.5, the electrochemical process will be diffusion-controlled.
Fig. 7a shows the logarithm plots of the cyclic voltametric current corresponding to the cathodic peaks of CaMnO
3 in a 0.5 M Ca(NO
3)
2 electrolyte, at a scan rate ranging from 5 to 100 mV s
−1. The cathodic
b value obtained was 0.86, which is close to 1, indicating that the charge storage is predominantly governed by capacitive behaviour.
 |
| | Fig. 7 (a) Dual logarithmic plot of the current vs. scan rate of the CaMnO3 electrode derived from the cathodic peak current at 1.2 V, with the slope corresponding to the calculated ‘b’ value. (b) Capacitive-diffusive contribution bar diagram of the CaMnO3 electrode at the potential of 1.2 V. (c) Capacitive contribution of CaMnO3 in the 0.5 M Ca(NO3)2 electrolyte from the CV analysis performed at 5 mV s−1. | |
Dunn's method was used to differentiate and quantify the capacitive and diffusive behaviour within the electrode material by the following equation:
where
i is the current response,
v is the scan rate and
k1 and
k2 are the constants that correspond to the capacitive contribution and diffusive contribution, respectively. From
Fig. 7b, it can be seen that as the scan rate increases, the capacitive contribution increases. At lower scan rates, the ions have enough time to diffuse to the electrode and undergo the redox process, giving a distinct peak, but as soon as the scan rate increases, the ions do not have enough time to diffuse; thus, capacitive charge storage dominates, giving a more rectangular cyclic voltammogram that can be clearly seen in
Fig. 7c.
2.2.1 Post-cycling analysis. In order to evaluate the structural and morphological properties of the cycled electrodes, post-cycling analysis of the CaMnO3 electrode was performed in the three-electrode configuration at 5 A g−1. We have compared the SEM images with those of the pristine CaMnO3 electrode. Fig. 8a–d shows the SEM images of the CaMnO3 electrodes before and after 3000 cycles. The capacitance fading of the CaMnO3 electrodes after electrochemical cycling in the three-electrode configuration is proven from the significant morphological changes of the cycled electrode (Fig. 8d–f), as compared with the pristine electrode (Fig. 8a–c). SEM images were taken before and after cycling to understand how the morphology changed after the cycling. The SEM images of the pristine electrode in Fig. 8a–c show that the electrode is composed of nanoscale CaMnO3 particles, which are uniformly distributed. The cycled electrode in Fig. 8d–f appears to have cracks; this degradation can hinder the cycling stability of the material.
 |
| | Fig. 8 (a–c) SEM images of the CaMnO3 electrode before cycling in the three-electrode configuration. (d–f) SEM images of the CaMnO3 electrode after 3000 continuous cycles at a specific current of 5 A g−1 in the three-electrode configuration. | |
Fig. 9 shows the XRD patterns of the electrode before and after charge–discharge cycling in an aqueous Ca(NO3)2 electrolyte. The pristine electrode exhibits diffraction peaks corresponding to CaMnO3, along with dominant reflections from the graphite sheet at 26.4° and 54.6°, and a weak diffraction peak at ∼42°. After cycling, additional diffraction peaks appeared at 18.3° and 33.9°, which are attributed to the formation of MnOOH. The overlap of the MnOOH and graphite peaks at ∼24.9° is also observed. These reflections are in good agreement with the standard JCPDS pattern of MnOOH (PDF No. 01-088-0649), indicating the formation of an MnOOH phase during electrochemical cycling. Furthermore, the intensity of the weak graphite reflection at ∼42° decreased. In the pristine electrodes, the particles are preferentially oriented with their basal planes to the current collector, resulting in high-intensity XRD reflections. After prolonged cycling, the orientation of the particles changes, which would influence the XRD reflections.40 During cycling, manganese undergoes reversible Mn4+/Mn3+ redox reactions, while the overall electrochemical performance is dominated by capacitive behaviour arising from the surface adsorption and desorption of Ca2+ ions. So, from the above understanding, the mechanism can be proposed as follows:
| (CaMnO3)surface + Ca2+ + e− ↔ (CaMnO3–Ca2+)surface |
 |
| | Fig. 9 X-ray diffraction pattern of the CaMnO3 electrode before and after electrochemical cycling in 0.5 M Ca(NO3)2 in the three-electrode configuration (peaks at ∼26° and ∼55° arise from the current collector (graphite)). The  peaks are indexed to MnOOH (JCPDS 01-088-0649). peaks are indexed to MnOOH (JCPDS 01-088-0649). | |
The reversible redox reaction of Mn at the surface can be proposed as follows:
Some of the Mn3+ species react with the aqueous electrolyte, form MnOOH on the surface of the electrode, and continue to participate in the charge–discharge process. The mechanism can be written as follows:
The FTIR (Fourier transform infrared spectroscopy) spectra of the CaMnO3-coated graphite electrode before cycling are shown in Fig. S7. The spectrum shows characteristic Mn–O and Mn–O–Mn vibrations of the basic MnO6 octahedra in the 400–900 cm−1 region, confirming the presence of the CaMnO3 perovskite phase.41 After electrochemical cycling, additional broad O–H stretching bands (∼3200–3600 cm−1) and a distinct –OH bending vibration near ∼1626 cm−1 emerge.42 The appearance of a peak at ∼736 cm−1 after electrochemical cycling is attributed to the ν4 in-plane bending vibration of nitrate ions originating from the Ca(NO3)2 electrolyte.43 These spectra indicate the electrochemically induced formation of MnOOH on the surface of CaMnO3, while the presence of Mn–O lattice vibrations suggests that the CaMnO3 core structure is retained.
2.3 Performance of the CaMnO3 symmetric supercapacitor
The two-electrode studies were done by fabricating a coin cell supercapacitor device with Whatman glass fibre filter paper dipped in a 0.5 M aqueous Ca(NO3)2 electrolyte solution sandwiched between CaMnO3 electrodes with an active mass of 1.32 mg cm−2 each. The CVs were measured within a stable voltage of 0–1.2 V, and further increase in the voltage window led to oxygen evolution. The CV curve shown in Fig. 10a has a rectangular shape, characteristic of the electrical double-layer capacitor, with no distinct redox peaks. It can also be seen that there is no significant change in the shape of the CV curves when the scan rate increases, indicating a reversible electrochemical reaction. The galvanostatic charge–discharge was carried out at a specific current of 0.1 A g−1 to evaluate the capacitive performance (Fig. 10b) of the assembled CaMnO3 symmetric supercapacitor device. The device attained a specific capacitance of 76.1 F g−1 with a stable operating voltage window of 1.2 V. The specific capacitance of the symmetric supercapacitor was calculated using the following equation:44–49| |
 | (8) |
where I is the current, m is the sum of the masses of active materials on both electrodes, Δt is the discharge time and ΔV is the total potential window. Fig. 10c and d show the rate performance of the symmetric device at different specific currents, varying from 0.1–2 A g−1. The cycling of the aqueous CMO‖CMO symmetric supercapacitor was performed at a specific current of 0.25 A g−1, at a stable voltage of 0–1.2 V. An initial value of 49.3 F g−1 was obtained, which then dropped to 65% of the initial value after 3000 continuous cycles, as shown in Fig. 10f. The device exhibited 100% coulombic efficiency even after 3000 continuous charge–discharge cycles, as shown in Fig. 10f. To our knowledge, this study is the first to report on a symmetric supercapacitor utilizing CaMnO3 as the electrode material for calcium-ion energy storage, highlighting its novelty and potential for alternative energy storage systems.
 |
| | Fig. 10 (a) Cyclic voltammograms of the CaMnO3 symmetric supercapacitor in the potential window of 0–1.2 V at various scan rates (5–100 mV s−1). (b) GCD analysis performed at 0.1 A g−1 specific current in the voltage window of 0–1.2 V. (c) GCD analysis performed at different specific currents (0.1–1 A g−1). (d) Rate performance of the CaMnO3 symmetric supercapacitor in the voltage window of 0–1.2 V. (e) Ragone plot of the fabricated CaMnO3 symmetric supercapacitor. (f) Capacitance retention and coulombic efficiency of the fabricated CaMnO3 symmetric supercapacitor. | |
The energy density and power density of the device were calculated using the equations below:50,51
| |
 | (9) |
| |
 | (10) |
where
Cs is the Specific capacitance, Δ
V is the voltage window, and Δ
t is the discharge time. An energy density of 3.8 W h kg
−1 and a high-power density of 1190 W kg
−1 were obtained for the fabricated symmetric supercapacitor device, as shown in the Ragone plot in
Fig. 10e. These results are comparable with those of the reported symmetrical MnO
2/RGO//MnO
2/RGO and RGO//RGO devices fabricated by Haojie Fei, which had energy densities of 2.1 W h kg
−1 and 1.9 W h kg
−1, respectively.
52 Daraghmeh
et al. synthesised a carbon-nanofibre-based symmetric supercapacitor, which showed a maximum specific energy of 2.3 W h kg
−1 at a specific power of 197 W kg
−1, comparable with the above mentioned results.
53 Maity
et al. synthesised a nitrogen-functionalized covalent triazine-based framework for a supercapacitor, which exhibited 7 W h kg
−1 and a power density of 200 W kg
−1.
54 N. Sudhan
et al. synthesised carbon from rice straw for symmetrical aqueous cells, which exhibited a high energy density of 7.8 W h kg
−1 at a power density of 150.2 W kg
−1, comparable with the results in this work.
55
To further analyse the kinetics of the symmetric CaMnO3-based perovskite supercapacitor device, electrochemical impedance spectroscopy analysis was done. The cycled symmetric supercapacitor exhibited a progressive decline in electrochemical performance with the increase in resistance components, as observed in the post-cycling Nyquist plot (Fig. 11). After extended cycling, both the ohmic resistance (Rs) and charge-transfer resistance (Rct) increased, indicating higher diffusion resistance, all of which hinder rapid ionic transport to the electroactive sites. The diffusion coefficient was calculated from Fig. S6; using eqn (3) and (4). A decrease from 4.29 × 10−11 cm2 s−1 to 1.37 × 10−11 cm2 s−1 was observed after electrochemical cycling. Similarly the ionic conductivity decreased from 8 × 10−8 to 2.68 × 10−8 S cm−1 for the pristine and cycled CMO‖CMO symmetric supercapacitor, respectively. The noticeable rightward shift in the high-frequency Nyquist region reflects the higher charge-transfer resistance due to surface layer formation on both electrodes and the combined Warburg contribution, which limit the ion transfer and hinder the gradual capacity fading from an initial 49.3 F g−1 to 65% retention after 3000 cycles. These results demonstrate that, while both the three-electrode and two-electrode configurations experience minor interfacial resistance increases, the three-electrode setup maintains higher ionic transport and better capacitance retention compared with the two-electrode symmetric cell, along with the cumulative degradation of interface kinetics and ion accessibility.
 |
| | Fig. 11 Electrochemical impedance spectroscopy analysis of the fabricated CaMnO3 symmetric supercapacitor device before and after electrochemical cycling; the inset shows an enlarged view of the high-frequency region. | |
To further analyse the practical application of the aqueous symmetric CMO‖CMO supercapacitor device, red and yellow LEDs were illuminated by connecting two charged supercapacitors (after charging up to 1.2 V). Fig. 12a and b demonstrate the glowing red and yellow LEDs powered by the aqueous CMO‖CMO symmetric supercapacitor. Thus, this study highlights the practical implications of the symmetric perovskite-type CaMnO3-based aqueous supercapacitor.
 |
| | Fig. 12 Practical utility of the aqueous CMO‖CMO symmetric supercapacitor device for illuminating a (a) red LED and (b) yellow LED. | |
3. Experimental section
3.1 Materials
Calcium nitrate tetrahydrate, manganese nitrate tetrahydrate, urea, N-methyl-2-pyrrolidone (NMP), PVDF (poly(vinylidene difluoride)) binder, and Super P carbon were used as received. A graphite sheet (thickness = ∼381 microns) was used as the substrate.
3.2 Synthesis of CaMnO3
The synthesis of the CaMnO3 powder was carried out by the hydrothermal method, as shown in Fig. 13. First, 5.02 g of calcium nitrate tetrahydrate and 4.72 g of manganese nitrate tetrahydrate were dissolved separately in 20 mL of deionized water. Then, these solutions were stirred together for 30 minutes. 2.4 g of urea was dissolved in 20 mL of deionised water and added dropwise to the above solution as a chelating agent to form a homogenous solution. This solution was then transferred to a 100 mL Teflon-lined stainless-steel autoclave, which was then kept in a hot-air oven at 150 °C for 12 hours, after which the precipitate formed was evaporated at 80 °C. Finally, the obtained product was annealed at 500 °C for 3 hours in a tubular furnace, followed by calcination at 900 °C for 12 h to obtain CaMnO3 powder.
 |
| | Fig. 13 Synthesis of the CaMnO3 perovskite material by a simple hydrothermal method, followed by calcination at 900 °C. | |
3.3 Electrode preparation and electrochemical testing
The working electrode was prepared by using the active material (CaMnO3), Super P carbon, and PVDF as binder and NMP as solvent. The above materials were taken in a weight ratio of 80:10:10 and ground in a mortar; NMP was added dropwise to get a homogenous mixture of the slurry. The slurry was then brush-coated onto a graphite sheet and dried at 100 °C in an oven overnight. The area of the electrode was 1 cm2, and the active mass on the electrode was 1.2 mg cm−2. The electrochemical capacitive performance of CaMnO3 was characterized by cyclic voltammetry (CV), galvanostatic charge–discharge (GCD) and electrochemical impedance spectroscopy (EIS) analyses in a 0.5 M Ca(NO3)2 aqueous electrolyte using Bio-Logic BCS-810. The measurements were conducted in a three-electrode setup using a CaMnO3-coated graphite sheet as the working electrode, a platinum electrode as the counter electrode and Ag/AgCl as the reference electrode. The CV measurements of CaMnO3 were performed within a potential window of −0.4 to 1.5 V at different scan rates (5, 10, 25, 50, 75 and 100 mV s−1). The GCD measurements of CaMnO3 were performed at different specific currents within the potential window of −0.4 to 1.5 V. EIS measurements of CaMnO3 were performed over the frequency range of 100 kHz to 10 mHz, with an AC amplitude of 10 mV.
4. Conclusion
In summary, a high-performance calcium-ion supercapacitor based on CaMnO3 perovskite electrode has been successfully developed using a neutral Ca(NO3)2 electrolyte. The optimized CaMnO3 electrode exhibits a specific capacitance of 258.6 F g−1 at 0.5 A g−1 and excellent cycling stability, retaining 72.6% of its initial capacitance after 1500 charge–discharge cycles with 100% coulombic efficiency. The as-assembled CMO‖CMO symmetric device delivered a specific capacitance of 76.1 F g−1, an energy density of 3.8 W h kg−1, and a power density of 1190 W kg−1 within a stable 1.2 V operating window. The aqueous CMO‖CMO symmetric supercapacitor maintained 65% of its initial capacitance even after 3000 continuous cycles. The practical application of the symmetric supercapacitor device was demonstrated by powering red and yellow LEDs. This remarkable performance surpasses most previously reported perovskite-type CaMnO3-based supercapacitors, making this supercapacitor a highly promising candidate for next-generation sustainable and high-power energy storage applications.
Conflicts of interest
The authors declare no conflicts of interest.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d6ra01178k.
Acknowledgements
The authors would like to thank the SRM Institute of Science and Technology for providing the research facilities, including the SRM-SCIF and the Nanotechnology Research Centre (NRC) for the structural measurements.
References
- W. Cai, C. Yan, Y. X. Yao, L. Xu, R. Xu, L. L. Jiang, J. Q. Huang and Q. Zhang, Small Struct., 2020, 1, 2000010, DOI:10.1002/sstr.202000010.
- H. Li, Z. Wang, L. Chen and X. Huang, Adv. Mater., 2009, 21, 4593–4607, DOI:10.1002/adma.200901710.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367, DOI:10.1038/35104644.
- Y. Liang, H. Dong, D. Aurbach and Y. Yao, Nat. Energy, 2020, 5, 646–656, DOI:10.1038/s41560-020-0655-0.
- M. C. Lin, M. Gong, B. Lu, Y. Wu, D. Y. Wang, M. Guan, M. Angell, C. Chen, J. Yang, B. J. Hwang and H. Dai, Nature, 2015, 520, 325–328, DOI:10.1038/nature14340.
- M. E. Arroyo-de Dompablo, A. Ponrouch, P. Johansson and M. R. Palacín, Chem. Rev., 2020, 120, 6331–6357, DOI:10.1021/acs.chemrev.9b00339.
- J. Hao, B. Li, X. Li, X. Zeng, S. Zhang, F. Yang, S. Liu, D. Li, C. Wu and Z. Guo, Adv. Mater., 2020, 32, 2003021, DOI:10.1002/adma.202003021.
- Y. Li, K. Huang, Z. Yao, S. Liu and X. Qing, Electrochim. Acta, 2011, 56, 2140–2144, DOI:10.1016/j.electacta.2010.11.074.
- Y. Li, S. Chang, X. Liu, J. Huang, J. Yin, G. Wang and D. Cao, Electrochim. Acta, 2012, 85, 393–398, DOI:10.1016/j.electacta.2012.07.127.
- G. A. M. Ali, L. L. Tan, R. Jose, M. M. Yusoff and K. F. Chong, Mater. Res. Bull., 2014, 60, 5–9, DOI:10.1016/j.materresbull.2014.08.008.
- I. I. Misnon, R. A. Aziz, N. K. M. Zain, B. Vidhyadharan, S. G. Krishnan and R. Jose, Mater. Res. Bull., 2014, 57, 221–230, DOI:10.1016/j.materresbull.2014.05.044.
- G. A. M. Ali, O. A. G. Wahba, A. M. Hassan, O. A. Fouad and K. F. Chong, Ceram. Int., 2015, 41, 8230–8234, DOI:10.1016/j.ceramint.2015.02.100.
- K. Kanagarajan, R. Velayutham and S. Vembu, Appl. Phys. A:Mater. Sci. Process., 2023, 129, 560, DOI:10.1007/s00339-023-06835-3.
- X. Li, X. Li, G. Wang, X. Wang and J. Ji, J. Mater. Chem. A, 2013, 1, 10103–10106, 10.1039/C3TA11727H.
- J. Muldoon, C. B. Bucur and T. Gregory, Chem. Rev., 2014, 114, 11683–11720, DOI:10.1021/cr500049y.
- A. L. Lipson, B. Pan, S. H. Lapidus, C. Liao, J. T. Vaughey and B. J. Ingram, Chem. Mater., 2015, 27, 8442–8447, DOI:10.1021/acs.chemmater.5b04027.
- S. Wu, F. Zhang and Y. Tang, Adv. Sci., 2018, 5, 1701082, DOI:10.1002/advs.201701082.
- R. Y. Wang, B. Shyam, K. H. Stone, J. N. Weker, M. Pasta, H. W. Lee, M. F. Toney and Y. Cui, Adv. Energy Mater., 2015, 5, 1401869, DOI:10.1002/aenm.201401869.
- S. Gheytani, Y. Liang, F. Wu, Y. Jing, H. Dong, K. K. Rao, X. Chi, F. Fang and Y. Yao, Adv. Sci., 2017, 4, 1700465, DOI:10.1002/advs.201700465.
- B. Ji, H. He, W. Yao and Y. Tang, Adv. Mater., 2021, 33, 2005501, DOI:10.1002/adma.202005501.
- F. Wang, S. Xiao, Y. Hou, C. Hu, L. Liu and Y. Wu, RSC Adv., 2013, 3, 13059–13084, 10.1039/C3RA23466E.
- L. Chang, J. Li, Z. Le, P. Nie, Y. Guo, H. Wang, T. Xu and X. Xue, J. Colloid Interface Sci., 2021, 584, 698–705, DOI:10.1016/j.jcis.2020.04.014.
- R. P. Forslund, J. Pender, C. T. Alexander, K. P. Johnston and K. J. Stevenson, J. Mater. Chem. A, 2019, 7, 21222–21231, 10.1039/C9TA06094D.
- H. Abd El-Wahab, A. M. A. Hassan, O. A. Fouad, O. Abd and E.-G. Wahba, Egypt. J. Chem., 2011, 54, 205–222 CrossRef.
- E. K. Abdel-Khalek, E. A. Mohamed and Y. A. M. Ismail, J. Solgel Sci. Technol., 2025, 113, 461–472, DOI:10.1007/s10971-024-06632-1.
- S. Pathreeker, S. Reed, P. Chando and I. D. Hosein, J. Electroanal. Chem., 2020, 874, 114453, DOI:10.1016/j.jelechem.2020.114453.
- N. Yu, M. M. Nair and N. Mahinpey, Can. J. Chem. Eng., 2019, 97, 2131–2136, DOI:10.1002/cjce.23495.
- A. A. Nechikott, A. Williams, P. Ragupathy and P. K. Nayak, ACS Appl. Energy Mater., 2025, 8, 13215–13230, DOI:10.1021/acsaem.5c00996.
- C. Pan, H. Gu and L. Dong, J. Power Sources, 2016, 303, 175–181, DOI:10.1016/j.jpowsour.2015.11.002.
- T. Wang, X. Qian, D. Yue, X. Yan, H. Yamashita and Y. Zhao, Chem. Eng. J., 2020, 398, 125368, DOI:10.1016/j.cej.2020.125638.
- D. Yan, P. Yan, S. Cheng, J. Chen, R. Zhuo, J. Feng and G. Zhang, Cryst. Growth Des., 2009, 9, 218–222, DOI:10.1021/cg800312u.
- L. Chang, J. Li, Z. Le, P. Nie, Y. Guo, H. Wang, T. Xu and X. Xue, J. Colloid Interface Sci., 2021, 584, 698–705, DOI:10.1016/j.jcis.2020.04.014.
- N. Yu, M. M. Nair and N. Mahinpey, Can. J. Chem. Eng., 2019, 97, 2131–2136, DOI:10.1002/cjce.23495.
- B. Bulfin, J. Vieten, D. E. Starr, A. Azarpira, C. Zachäus, M. Hävecker, K. Skorupska, M. Schmücker, M. Roeb and C. Sattler, J. Mater. Chem. A, 2017, 5, 7912–7919, 10.1039/c7ta00822h.
- E. K. Abdel-Khalek, E. A. Mohamed and Y. A. M. Ismail, J. Solgel Sci. Technol., 2025, 113, 461–472, DOI:10.1007/s10971-024-06632-1.
- J. Yan, Z. Fan, W. Sun, G. Ning, T. Wei, Q. Zhang, R. Zhang, L. Zhi and F. Wei, Adv. Funct. Mater., 2012, 22, 2632–2641, DOI:10.1002/adfm.201102839.
- A. A. Mohamad, Inorg. Chem. Commun., 2025, 172, 113677, DOI:10.1016/j.inoche.2024.113677.
- R. Kumar, P. Rai and A. Sharma, RSC Adv., 2016, 6, 3815–3822, 10.1039/c5ra20331g.
- Y. He, W. Chen, X. Li, Z. Zhang, J. Fu, C. Zhao and E. Xie, ACS Nano, 2013, 7, 174–182, DOI:10.1021/nn304833s.
- D. Aurbach, B. Markovsky, A. Rodkin, M. Cojocaru, E. Levi and H.-J. Kim, Electrochim. Acta, 2002, 47, 1899–1911, DOI:10.1016/S0013-4686(02)00013-0.
- B. Zhang, C. Li, S. Liu, L. Zhuang, W. Zhang, L. Huang, Z. Jia, D. Chen and X. Li, RSC Adv., 2024, 14, 36181–36192, 10.1039/D4RA06407K.
- J. Wang, J. G. Wang, H. Liu, C. Wei and F. Kang, J. Mater. Chem. A, 2019, 7, 13727–13735, 10.1039/C9TA03541A.
- K. Al-Amin, M. Kawsar, M. T. R. B. Mamun and M. S. Hossain, Nanoscale Adv., 2025, 7, 6677–6702, 10.1039/d5na00522a.
- H. Wang, H. Yi, X. Chen and X. Wang, J. Mater. Chem. A, 2014, 2, 3223–3230, 10.1039/C3TA15046A.
- A. Noori, M. F. El-Kady, M. S. Rahmanifar, R. B. Kaner and M. F. Mousavi, Chem. Soc. Rev., 2019, 48, 1272–1341, 10.1039/C8CS00581H.
- S. Sundriyal, V. Shrivastav, H. Kaur, S. Mishra and A. Deep, ACS Omega, 2018, 3, 17348–17358, DOI:10.1021/acsomega.8b02065.
- M. K. Sahoo and G. R. Rao, Nanoscale Adv., 2021, 3, 5417–5429, 10.1039/D1NA00261A.
- S. Roldán, D. Barreda, M. Granda, R. Menéndez, R. Santamaría and C. Blanco, Phys. Chem. Chem. Phys., 2015, 17, 1084–1092, 10.1039/C4CP05124F.
- M. D. Stoller and R. S. Ruoff, Energy Environ. Sci., 2010, 3, 1294–1301, 10.1039/C0EE00074D.
- J. Yu, N. Fu, J. Zhao, R. Liu, F. Li, Y. Du and Z. Yang, ACS Omega, 2019, 4, 15904–15911, DOI:10.1021/acsomega.9b01916.
- S. Sundriyal, H. Kaur, S. K. Bhardwaj, S. Mishra, K. H. Kim and A. Deep, Coord. Chem. Rev., 2018, 369, 15–38, DOI:10.1016/j.ccr.2018.04.018.
- H. Fei, N. Saha, N. Kazantseva, R. Moucka, Q. Cheng and P. Saha, Materials, 2017, 10, 1251, DOI:10.3390/ma10111251.
- A. Daraghmeh, S. Hussain, I. Saadeddin, L. Servera, E. Xuriguera, A. Cornet and A. Cirera, Nanoscale Res. Lett., 2017, 12, 615, DOI:10.1186/s11671-017-2415-z.
- A. Maity, M. Siebels, A. Jana, M. Eswaran, R. Dhanusuraman, C. Janiak and A. Bhunia, ChemSusChem, 2024, 17, e202401716, DOI:10.1002/cssc.202401716.
- N. Sudhan, K. Subramani, M. Karnan, N. Ilayaraja and M. Sathish, Energy Fuels, 2017, 31, 977–985, DOI:10.1021/acs.energyfuels.6b01829.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence and
Helen Annal Therese
and
Helen Annal Therese
 indicates the Ca2MnO4 phase and “Δ” indicates CaO.
indicates the Ca2MnO4 phase and “Δ” indicates CaO.












 peaks are indexed to MnOOH (JCPDS 01-088-0649).
peaks are indexed to MnOOH (JCPDS 01-088-0649).





