DOI:
10.1039/D6RA01015F
(Paper)
RSC Adv., 2026,
16, 27495-27503
Grading schemes of VGO hydrotreating catalysts according to the classification of their active sites
Received
5th February 2026
, Accepted 3rd April 2026
First published on 26th May 2026
Abstract
Based on the difference in the degree of sulfidization, hydrogenation active sites can be divided into Type-I and Type-II. In this study, one concept of catalyst grading design based on Type-I and Type-II active sites is proposed. Two NiMo/γ-Al2O3-supported hydrotreating catalysts with Type-I and Type-II active sites were prepared. Based on characterization results, theoretical models of the active sites were constructed, and their catalytic characteristics were predicted by DFT calculations. The catalysts were then evaluated by acridine model compounds and vacuum gas oil systems under four grading schemes. The DFT calculations and hydrogenation experiments results show that Type-I active sites excel in the hydrogenation saturation of lateral aromatic rings, while Type-II active sites preferentially promote the hydrogenolysis of heteroatom–carbon bonds. The catalyst dominated by Type-I active site exhibits high activity at low temperatures; the nitrogen content of the product hydrotreated at 340 °C by the single Type-I catalyst is approximately 68.4% of that hydrotreated by the single Type-II catalyst. However, the Type-II catalyst performs better at high temperatures; the nitrogen content of the product hydrotreated at 370 °C by the Type-II catalyst can be reduced to 1.6 µg g−1, which is only 28.1% of that obtained by the Type-I catalyst. Among the four schemes of grading catalysts combining Type-I and Type-II active sites, the optimal one is to place the catalysts with Type-I active sites first, followed by the Type-II catalyst, which has the lowest nitrogen content of 2.9 µg g−1.
1 Introduction
Hydrotreatment is a current industrialized process for clean fuel production in the refining industry.1–3 The feedstocks of hydrotreating units are complex mixtures composed of hydrocarbons, sulphur compounds, nitrogen compounds and other impurities.4–6 Competitive adsorption and reaction among various reactants seriously influence the activity and selectivity of hydrotreating catalysts.7–11 On the other hand, the interior of a hydrogenation reactor can be considered a thermally isolated system, and the axial temperature rise is considerable, which affects the performance of the catalysts.12,13 Catalyst grading provides one of the effective methods for achieving deep conversion. The classic gradation is divided by series function zones, which consist of catalysts with specific activity and selectivity.14–16
Albemarle Corporation has developed the STAX™ grading technology, which divides the hydrotreating reactor into zones for direct hydrodesulphurization (HDS), sufficient hydrodenitrogenation (HDN) and deep hydrogenation saturation. The strategy of the STAX™ technology is to sequentially convert target reactants to avoid reaction inhibition, in which the catalysts array also fits for the axial temperature variation of the reactor.17–19 SINOPEC Dalian Research Institute of Petroleum and Petrochemicals (DRIPP) has proposed a novel catalyst concept grading scheme, which aims to convert all the target reactants synchronously. The key strategy is loading the hydrotreating catalysts with varying activities successively in a fixed-bed reactor to efficiently achieve deep HDS, deep HDN and moderate hydrodearomatization (HDA) through functional zoning.20
Although current grading schemes have achieved positive industrial application results, these grading methods are usually sorted according to the specific functions of catalysts, such as HDS first, then HDN, and finally HDA. In fact, the conversion of each reactant molecule in the reactor involves multiple steps. One scientific and effective grading method should match the conversion progress of the molecules with the corresponding catalysts possessing the appropriate functions.
In this study, hydrogenation catalyst grading schemes based on the classification of active sites are established. Generally, Type-I active sites exhibit strong interactions with the support, and the presence of Mo–O–Al or W–O–Al chemical bonds at the interface between the support and the active metal can significantly impair the hydrotreating activity of the catalysts. Therefore, hydrotreating catalysts with Type-I and Type-II active sites are prepared and characterized, followed by the establishment of the theoretical models of the active sites and the prediction of catalytic characteristics through DFT calculations. Furthermore, several grading schemes are proposed and comparatively evaluated through small-scale hydrogenation devices to determine the optimal grading sequence.
2 Experiments and calculations
2.1 Preparation of hydrotreating catalysts
In this study, a commercial γ-Al2O3 support produced by SINOPEC (brand number is FF-76-ZT) was chosen as the support for the hydrotreating catalysts. The properties of the γ-Al2O3 are listed in Table 1.
Table 1 Properties of γ-Al2O3 used in this study
| Items |
Values |
| Stacking density/kg m−3 |
525.37 ± 0.42 |
| Specific surface area/m2 g−1 |
309.63 ± 0.70 |
| Pore volume/cm3 g−1 |
0.798 ± 0.012 |
| Crushing strength/N cm−1 |
183.63 ± 0.83 |
Nickel and molybdenum were chosen as the active metals to load onto the support by an impregnation method.15,21 The specific synthesis steps are as follows: for the first type of NiMo hydrotreating catalyst (Cat-I), ammonium heptamolybdate ((NH4)6Mo7O24·4H2O) was selected as the Mo source and nickel nitrate (Ni(NO3)2·6H2O) as the Ni source. The impregnation solution (J-1) consisted of 0.36 mol L−1 (NH4)6Mo7O24·4H2O, 0.6 mol L−1 Ni(NO3)2·6H2O and 0.5 mol L−1 ammonia (NH3·H2O). Then, 100 g of FF-76-ZT and 100 mL of J-1 were separately weighed and impregnated under the condition of pH ranging from 10.0 to 11.0. After the process, it was dried at 120 °C for 4.0 hours and then calcined at 500 °C for 4.0 hours. Cat-I can be obtained in this way.
For the second type of NiMo hydrotreating catalyst (Cat-II), molybdenum trioxide (MoO3) was selected as the Mo source and nickel citrate (C12H10Ni3O14) as the Ni source. The impregnation solution (J-2) consisted of 2.5 mol L−1 MoO3, 0.6 mol L−1 C12H10Ni3O14 and 1.0 mol L−1 phosphoric acid (H3PO4). Similarly, 100 g of FF-76-ZT and 100 mL of J-2 were separately weighed and impregnated under the condition of pH ranging from 1.0 to 2.0. After the process, it only needed to be dried at 150 °C for 6.0 hours to obtain Cat-II.
2.2 Characterization of the catalyst
X-ray photoelectron spectroscopy (XPS) characterization of the sulfurized catalysts was performed using a Shimadzu AXIS SPURA+ spectrometer with Al Kα (Eb = 1486.6 eV) as the X-ray source.
Transmission electron microscopy (TEM) images of the sulphide catalysts were obtained using a JEOL JEM-2200FS transmission electron microscope. Morphological data of the active sites were analyzed based on 60 images. The average stacking layer number and the average length of the active site were calculated by the following formulae:22
| |
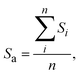 | (1) |
where
n means the number of active sites counted in the images, and
Si means the stack layer of a certain active site.
| |
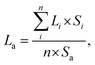 | (2) |
where
n means the number of active sites counted in the images, and
Li means the length of a certain active site.
2.3 Modeling and computational methods
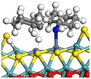
To better understand the catalytic characteristics of Type-I and Type-II active sites, two Ni–Mo–S models with different sulfurized degrees were constructed. The fully sulfurized Ni–Mo–S active site, which represents the Type-II active site (denoted as Ni-Mo-SS), was established as shown in Fig. 1 according to relevant reports.23–27 On the Ni–Mo-edge, the nickel coverage is 50%. The Ni atom is in square-planar coordination with four S atoms, and the normal direction of the square plane is exposed without S atoms.28–31 On the other hand, the insufficient sulfurized Ni–Mo–S active site (denoted as Ni-Mo-SO), which represents the Type-I active site, is derived from Ni-Mo-SS. In detail, the inner sulphur atoms are replaced by oxygen atoms. In the optimized Ni-Mo-SO nanocluster, the distance between adjacent molybdenum atoms shortens from 3.2 nm to 2.8 nm, whereas the atomic configuration at the edge does not change obviously.
 |
| | Fig. 1 Atomic structure of the model active sites of (a) Ni-Mo-SS and (b) Ni-Mo-SO. | |
2.4 Computational methods
The calculations were conducted using the DMol3 code with numerical atomic basis functions. The exchange–correlation functional was the revised Perdew–Burke–Ernzerhof functional belonging to the generalized gradient approximation.32,33 The basis set was a double numerical plus polarization basis (DNP).34
The complete linear synchronous transit (LST) and quadratic synchronous transit (QST) methods were used to find the transition state, and the nudged elastic band (NEB) method was used to confirm the transition state. The D2 correction method was used to calculate the dispersion force.35,36 Other calculation details and parameters are listed in Table 2.
Table 2 Detailed calculation parameters
| Calculating items |
Parameters |
| Electronic treatment |
Orbital cut off |
5.0 Å |
| Thermal smearing |
5 × 10−4 Ha |
| Convergence tolerance |
Self-consistent field density convergence (SCF) |
2 × 10−5 |
| Binding energy tolerance |
2 × 10−5 Ha |
| Force tolerance |
4 × 10−3 Ha Å−1 |
| D2 correction37 |
Exchange–correlation dependent factor s6 |
1.0 |
| Damping coefficient d |
20.0 |
2.5 HDN performance
The hydrotreating performances of various catalysts were evaluated in a series-connected small-scale fixed-bed reactors. The feedstocks for the experiments included a model probe system and vacuum gas oil. The model probe system contained acridine as the probe and decalin as the solvent. The composition and properties of the model probe system are shown in Table 3, and the properties of the VGO feedstock are listed in Table 4. The reaction flowchart is shown in Fig. 2. 100 mL of catalyst and 100 mL of quartz sand (as an inert diluent) were evenly loaded into each fixed-bed reactor. Before introducing the feedstock, two catalysts were sulfurized by cyclohexane with 4.0% mass fraction dimethyl-disulfides at 6.0 MPa under a hydrogen atmosphere, and the gas flow rate was fixed at 200 mL min−1. Meanwhile, the sulphide solution was pumped into the reactor at a rate of 100 mL h−1 and the temperature rose to 200 °C at the rate of 2.0 °C min−1 and maintained for 4 hours, then rose to 350 °C at the rate of 2.0 °C min−1 and maintained for 6 hours.
Table 3 Composition and properties of the model system
| Items |
Mass fraction of acridine/% |
Mass fraction of decalin/% |
Nitrogen content/µg g−1 |
| Values |
1.3 |
98.7 |
1015 |
Table 4 Properties of the VGO feedstock
| Items |
Density/g cm−3 |
Nitrogen content/µg g−1 |
IBP/°C |
EBP/°C |
BMCI |
| Values |
0.901 |
989.6 |
385.6 |
538.1 |
35.92 |
 |
| | Fig. 2 Reaction flowchart of the series-connected reactors. | |
After sulfidation is completed, the two feedstocks are separately pumped into the series-connected small-scale fixed-bed reactors. The reaction conditions for the model system and VGO are listed in Table 5.
Table 5 Reaction conditions for the model system and VGO
| Feedstock |
Pressure/MPa |
LHSV/h−1 for single reactor |
Gas–oil ratio v/v |
Temperature/°C |
| Model system |
15.0 |
2.0 |
1000 |
340–370 |
| VGO |
10.0 |
3.0 |
600 |
320–350 |
3 Results and discussion
3.1 Properties of catalysts
The TEM images of the two sulfurized Cat-I and Cat-II catalysts are shown in Fig. 3. The related statistical analyses of the average slab length and stacking layers of NiMoS active sites are summarized in Table 6. It can be concluded that the active sites of Cat-Ⅰ are shorter and have relatively low stacking layers, consisting mainly of single layer. In contrast, the ones of Cat-Ⅱ are longer and have higher stacking layers, with an average stacking layer number and average slab length of only 3.3 and 6.4 nm, respectively. The characterization results indicate that the Cat-I catalyst mainly contains type-I active sites, while the Cat-II catalyst mainly has type-II active sites.
 |
| | Fig. 3 TEM images of (a) Cat-I and (b) Cat-II. | |
Table 6 Morphology of the active sites
| Catalyst |
Average stacking layer |
Average length/nm |
| Cat-I |
1.4 |
5.5 |
| Cat-II |
3.3 |
6.4 |
To further analyze the distributions of active metals on the surface of two types of sulfurized catalysts, the XPS spectra of the Mo 3d orbitals are shown in Fig. 4. The percentage of Mo species in the +4 valence state is approximately 59.3%, whereas that of Mo species in the +6 valence state is approximately 40.7% for sulfurized Cat-I. On the other hand, the percentage of Mo species in the +4 valence state is up to 71.1%, whereas that of Mo species in the +6 valence state is only 28.9% for sulfurized Cat-II. It indicates that the Mo species in Cat-I remain insufficiently sulfurized compared to those in Cat-II.
 |
| | Fig. 4 XPS spectra of the 3d region for (a) Cat-I and (b) Cat-II. | |
3.2 Theoretical analysis
3.2.1 Effects of oxygen on charge distribution and molecular orbitals. The oxygen atoms in the Ni-Mo-SO active site first change the charge distribution at the Ni–Mo edge and the properties of the unoccupied orbitals (as shown in Table 7). According to the calculation results, the inner oxygen atoms with strong electronegativity attract a positive charge from the surface. In addition, the LUMO eigenvalues of Ni-Mo-SO are 0.82 eV lower than those of Ni-Mo-SS, which may further influence the adsorption and catalytic characteristics of the active site.
Table 7 Charge distribution and LUMO properties
| Active sites |
Atoms |
Averaged charge/e |
LUMO |
Eigenvalues/eV |
| Type-II |
Mo |
+0.31 |
 |
−4.37 |
| Ni |
−0.10 |
| S |
−0.19 |
| Type-I |
Mo |
+0.35 |
 |
−5.19 |
| Ni |
−0.08 |
| S |
−0.16 |
3.2.2 Effects of oxygen on hydrogen activation and transfer. Hydrogen dissociation and activation are one of the key functions of the hydrogenation active site in hydrotreating catalysts. Hydrogen dissociation on the Ni–Mo edge can occur on different atom pairs. The details of the hydrogen dissociation on Ni-Mo-SS and Ni-Mo-SO are shown in Table 8. In general, the hydrogen dissociation on the Ni–Mo edge is an endothermic process. On the Ni-Mo-SS active site, the reaction energy for hydrogen dissociation exceeds +45 kJ mol−1, and the activation energy is even up to +120 kJ mol−1. On the other hand, on the Ni-Mo-SO active site, the reaction energy for hydrogen dissociation is no more than 40 kJ mol−1. For the Mo–S atom pairs, the reaction energy is only +19.70 kJ mol−1, and the activation energy is 50 kJ mol−1 lower than that on Ni-Mo-SS. Therefore, it could be predicted that hydrogen dissociation on Ni-Mo-SO is more favorable than on Ni-Mo-SS.
Table 8 Hydrogen dissociation on Ni-Mo-SS and Ni-Mo-SO
| Active sites |
Location |
Morphology |
Dissociation energy/kJ mol−1 |
Transition state |
Activation energy/kJ mol−1 |
| Ni-Mo-SS |
Ni–Ni |
 |
+53.12 |
 |
+139.33 |
| Ni-Mo-SS |
Mo–S |
 |
+46.34 |
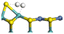 |
+125.07 |
| Ni-Mo-SO |
Ni–Ni |
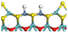 |
+38.93 |
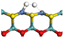 |
+104.96 |
| Ni-Mo-SO |
Mo–S |
 |
+19.70 |
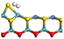 |
+72.53 |
The hydrogen transfer is another key process in hydrogenation reactions. Active hydrogen migrates along the edge of the active site to increase the possibility of contact the reactants. The details of hydrogen transfer are listed in Table 9. According to the calculation results, the hydrogen transfer on the Ni-Mo-SS active site requires lower energy, and the activation energy is no more than 32 kJ mol−1. The hydrogen transition energy on the Ni-Mo-SO active site is similar to that on the Ni-Mo-SS, whereas the activation energy of the former is approximately 10–20 kJ mol−1 higher than that of the latter. The energy changes and the activation barriers for hydrogen transfer are relatively smaller than those for hydrogen dissociation, indicating that the inhibition of hydrogen transfer caused by oxygen is less effective than the promotion of hydrogen activation.
Table 9 Hydrogen transfer on Ni-Mo-SS and Ni-Mo-SO
| Active sites |
Transfer route |
Transition state |
Transition energy/kJ mol−1 |
Activation energy/kJ mol−1 |
| Ni-Mo-SS |
Mo–Ni |
 |
+11.82 |
+26.50 |
| Ni–Ni |
 |
+2.24 |
+23.62 |
| S–Mo |
 |
−10.90 |
+31.96 |
| Ni-Mo-SO |
Mo–Ni |
 |
+13.76 |
+50.98 |
| Ni–Ni |
 |
−1.72 |
+43.93 |
| S–Mo |
 |
−10.45 |
+52.87 |
3.2.3 Effects of oxygen on the adsorption of the reactant. Among various reactant molecules, basic nitrogen compounds with multiple aromatic rings exhibit the strongest polarity and adsorption capacity. In this study, acridine and its fully saturated intermediate (saturated by fourteen hydrogen atoms, denoted as THA for short) were used as probes to investigate the adsorption performance of the active sites (as listed in Table 10). According to the calculation results, the adsorption could be considered as charge transfer from acridine to the active site, and the adsorption bond is formed between the nitrogen atom and the metal atom. When the nitrogen atom of acridine coordinates with a Mo atom, the adsorption energy is approximately 25 kJ mol−1 higher than that for coordination with a Ni atom. The adsorption energy and charge transfer of acridine on the Ni-Mo-SO active site are both higher than that on the corresponding metal atom of Ni-Mo-SS. As the lateral cycloalkane rings of the THA provide additional volume and steric hindrance, the nitrogen atom cannot fully bond with the semi-exposed Mo atom. The optimal adsorption form for THA is N–Ni configuration. In this case, the adsorption energy of THA on Ni-Mo-SO is still approximately 25 kJ mol−1 higher than that on Ni-Mo-SS, and there is also charge transfer involved. According to the calculation results, it can be concluded that inner oxygen in the Ni-Mo-SO active site strengthens the adsorption ability toward electron-rich compounds.
Table 10 Adsorption of nitrogen compounds on Ni-Mo-SS and Ni-Mo-SO
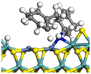
3.2.4 Effects of oxygen on hydrogenation saturation and hydrogenolysis. The hydrogenation saturation of an aromatic ring is a complicated process involving multiple elementary reactions including repeated hydrogen activation and hydrogen transfer, whereas the rate-controlling step is the first active hydrogen bond with the aromatic carbon accompanied by the destruction of the conjugated system.39 In this study, the elementary reaction of acridine saturation by the first active hydrogen on Ni-Mo-SS and Ni-Mo-SO was calculated. Based on previous calculations of hydrogen activation and reactant adsorption, the hydrogenation saturation process of the lateral aromatic ring of acridine on the active site was calculated (as shown in Table 11). The rational position for hydrogen attack is the Ni atom near the aromatic ring, where active hydrogen can arrive by rapidly diffusing along the Ni–Mo edge. The reaction energies for hydrogenation saturation on both active sites are close to 30 kJ mol−1, and the activation energies are no more than 100 kJ mol−1. Compared with the dissociation and transfer process of hydrogen, the generation of high-energy active hydrogen is the elementary reaction with the highest activation energy on the Ni-Mo-SS active site, and this step should be considered the rate-controlling step for the hydrogenation saturation of the aromatic ring. For the Ni-Mo-SO active site, although the activation energy of certain hydrogen transfer pathways is relatively high, which hinders the approach of active hydrogen to the aromatic ring, inner oxygen strengthens the ability of hydrogen dissociation, effectively reducing the activation energy of the rate-controlling steps in the overall reaction process.
Table 11 Hydrogenation saturation of acridine on Ni-Mo-SS and Ni-Mo-SO
| Reaction |
 |
| Active sites |
Type-II |
Type-I |
| Transition state |
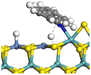 |
 |
| Reaction energy/kJ mol−1 |
+26.45 |
+34.66 |
| Activation energy/kJ mol−1 |
+74.57 |
+96.82 |
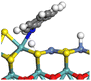
The hydrogenolysis of heteroatom–carbon bonds is another core function of the active site. The C–N bond is generally considered as the typical stubborn bond in feedstocks. The cleavage of the C–N bond in THA was calculated, and the details are listed in Table 12. At the transition state, the cleavage of the C–N bond generates two radical species, namely, a nitrogen-centered radical and an α-carbon-centered radical. Active hydrogen preferentially bonds with the α-carbon to lower the bonding energy of the system; thus, the coordination state of the nitrogen radical largely determines the reaction barrier of this elementary reaction. Notably, the nitrogen atom can coordinate with the Mo atom at the edge of the Ni-Mo-SS active site. In that way, the nitrogen atom achieves a triply coordinated state, and the bonding energy at the transition state effectively reduces. However, in the Ni-Mo-SO active site, the inner Mo–O bond is shorter than the outer Mo–S bond, resulting in a certain curvature and stretch on the Ni–Mo-edge. In other words, the distance and angle between Ni and Mo atoms are not favorable for simultaneous coordination with the nitrogen atom. Therefore, the reaction barrier for C–N bond cleavage on Ni-Mo-SO is much higher than that on Ni-Mo-SS, indicating that inner oxygen restricts hydrogenolysis activity by reducing the planarity of the edge.
Table 12 C–N bond cleavage of THA on Ni-Mo-SS and Ni-Mo-SO
| Reaction |
 |
| Active sites |
Type-II |
Type-I |
| Transition state |
 |
 |
| Reaction energy/kJ mol−1 |
+35.96 |
+52.11 |
| Activation energy/kJ mol−1 |
+157.74 |
+186.90 |
In summary, inner oxygen in the Ni-Mo-SO active site changes the properties of the unoccupied orbitals and the charge distributions on the Ni–Mo edge. Furthermore, these changes enhance the hydrogen dissociation, hydrogen activation and reactant adsorption while hindering hydrogen transfer and reducing the planarity of the active edge. The hydrogenation saturation of lateral aromatic rings becomes more favorable, whereas the hydrogenolysis of heteroatom-containing bonds becomes more difficult on the oxygen-substituted active sites.
3.3 Hydrogenation experiments
In the theoretical section, the effects of sulfidation on the catalytic properties of the active sites have been discussed. The calculation results predict that Type-I active sites exhibit superior ability to saturate lateral aromatic rings at lower temperatures due to their lower hydrogen dissociation activation energy and stable adsorption capacity. In contrast, Type-II active sites require overcoming their higher hydrogen dissociation energy barrier to achieve efficient HDN, which necessitates relatively stringent reaction conditions. The performances of catalysts with Type-I and Type-II active sites demonstrate distinct differences, which warrant further investigation into their grading order. For this purpose, hydrotreating performance tests were conducted in series-connected fixed-bed reactors using VGO as the feedstock. The grading schemes are listed in Table 13.
Table 13 Catalyst grading scheme
| Grading scheme |
A |
B |
C |
D |
| Reactor 1 (100 mL) |
Cat-I |
Cat-II |
Cat-I |
Cat-II |
| Reactor 2 (100 mL) |
Cat-I |
Cat-II |
Cat-II |
Cat-I |
First, the performance of the single catalyst at different temperatures was investigated. The temperatures of the two reactors were identical, and only the products at sample port 2 were collected. The results of the hydrotreating performance of the single catalysts are listed in Tables 14 and 15. As shown in Tables 14 and 15, the single Cat-I exhibits better hydrogenation saturation and HDN activities than the single Cat-II at low temperatures. In the model system, the nitrogen content of the product obtained with Cat-I is lower than that obtained with Cat-I at 320 °C and 330 °C. However, when the reaction temperature exceeds 340 °C, the trend of nitrogen content in the product reverses. At 350 °C, the nitrogen content of the product obtained with Cat-I is approximately four times that obtained with Cat-II. In the VGO hydrotreating system, at 340 °C, the nitrogen content of the product obtained with Cat-I is only approximately 68.4% of that obtained with Cat-II. The bureau of mines correlation index (BMCI) and density of the Cat-I product are both lower than those of the Cat-II product as well. As the reaction temperature rises, the activity of Cat-II, which contains more Type-II active sites, increases faster than that of Cat-I. When the temperature approaches 370 °C, the nitrogen content of Cat-I product is 5.3 ± 0.5 µg g−1, whereas that of Cat-II even reduces to 1.7 ± 0.3 µg g−1. Meanwhile, the BMCI and density of the Cat-II product reduce to 22.0 and 0.852 g cm−3, respectively. Combining these results with DFT calculations revealed that Cat-I exhibits superior lateral aromatic rings saturation capacity and a certain hydrogenolysis ability for C–N bonds at lower temperatures, while the Ni-Mo-SS active sites in Cat-II struggle to increase the overall HDN rate due to their weaker saturation capacity of lateral aromatic rings. However, the lateral aromatic ring saturation capability of Ni-Mo-SS in Cat-II increases rapidly at higher temperatures, resulting in superior HDN performance. The activation energy required for direct C–N bond cleavage is significantly higher than that for the hydrogenation saturation of lateral aromatic rings, which results from the conjugated effect between the nitrogen heterocyclic ring and aromatic rings in polyaromatic nitrogen compounds.38,39 Therefore, the saturation capacity of lateral aromatic rings significantly restricts the rates of HDN. As lateral aromatic rings progressively saturate, the conjugated structure is gradually disrupted, which makes the hydrogenation saturation rate of lateral aromatic rings the primary rate-limiting step in HDN.40
Table 14 Model system hydrogenation performance of Cat-I and Cat-II
| Scheme |
A |
B |
| Temperature of reactor-1/°C |
320 |
330 |
340 |
350 |
320 |
330 |
340 |
350 |
| Temperature of reactor-2/°C |
320 |
330 |
340 |
350 |
320 |
330 |
340 |
350 |
| Nitrogen content/µg g−1 |
86.6 ± 0.5 |
42.8 ± 0.4 |
23.9 ± 0.4 |
6.7 ± 0.2 |
156.2 ± 0.8 |
49.5 ± 0.9 |
10.5 ± 0.2 |
1.9 ± 0.3 |
Table 15 VGO hydrogenation performance of Cat-I and Cat-II
| Scheme |
A |
B |
| Temperature of reactor-1/°C |
340 |
350 |
360 |
370 |
340 |
350 |
360 |
370 |
| Temperature of reactor-2/°C |
340 |
350 |
360 |
370 |
340 |
350 |
360 |
370 |
| Density/g cm−3 |
0.873 |
0.870 |
0.867 |
0.862 |
0.879 |
0.874 |
0.863 |
0.852 |
| Nitrogen content/µg g−1 |
78.9 ± 1.1 |
40.1 ± 0.7 |
21.0 ± 0.9 |
5.3 ± 0.5 |
115.3 ± 1.3 |
57.1 ± 0.8 |
19.8 ± 0.7 |
1.7 ± 0.3 |
| BMCI |
28.6 |
26.3 |
24.4 |
23.1 |
29.9 |
27.1 |
24.3 |
22.0 |
The performance of the grading schemes involving two different catalysts was evaluated in two reactors operated at different temperatures. The products were collected at both sample ports 1 and 2. The grading schemes and hydrogenation performance results are listed in Tables 16 and 17. Compared with the single-catalyst results, the effects of catalyst gradation show a trend of differentiation. For the first grading scheme (Cat-I + Cat-II), both of the catalysts perform high hydrogenation activities, which presents the optimum performance among the four schemes with a product nitrogen content of only 2.6 ± 0.1 µg g−1 for the model system and 3.4 ± 0.3 µg g−1 for the VGO. By contrast, the reverse grading scheme leads to the worst performance, with a nitrogen content of 10 µg g−1 in the product of VGO, exceeding the basic nitrogen content requirement for feedstock in the following hydrocracking process. The comprehensive advantages of the first grading scheme via the key indicators of the hydrogenation products are briefly summarized in Table 18. It can be concluded that the grading scheme with Type-I catalyst in the front and Type-II catalyst in the back exhibits the best HDN performance for both the model system and VGO and the best HDA ability for VGO feedstock.
Table 16 Model system hydrogenation performance of the grading schemes
| Grading scheme |
Type-I |
Type-II |
Type-I |
Type-II |
| Type-I |
Type-II |
Type-II |
Type-I |
| Temperature of reactor-1/°C |
320 |
| Temperature of reactor-2/°C |
350 |
| Sample of reactor-1 |
| Nitrogen content/µg g−1 |
218 ± 4.1 |
367 ± 3.5 |
213 ± 4.0 |
364 ± 6.3 |
| Sample of reactor-2 |
| Nitrogen content/µg g−1 |
8.8 ± 0.4 |
6.9 ± 0.2 |
2.6 ± 0.1 |
12.8 ± 0.5 |
Table 17 VGO hydrogenation performance of the grading schemes
| Grading scheme |
Type-I |
Type-II |
Type-I |
Type-II |
| Type-I |
Type-II |
Type-II |
Type-I |
| Temperature of reactor-1/°C |
340 |
| Temperature of reactor-2/°C |
370 |
| Sample of reactor-1 |
| Density/g cm−3 |
0.878 |
0.882 |
0.877 |
0.883 |
| Nitrogen content/µg g−1 |
169 ± 3.4 |
301 ± 5.2 |
165 ± 1.4 |
308 ± 6.7 |
| BMCI |
29.0 |
30.9 |
29.3 |
31.1 |
| Sample of reactor-2 |
| Density/g cm−3 |
0.863 |
0.862 |
0.858 |
0.867 |
| Nitrogen content/µg g−1 |
6.9 ± 0.4 |
4.7 ± 0.2 |
3.4 ± 0.3 |
10.1 ± 0.7 |
| BMCI |
26.4 |
25.8 |
24.1 |
28.8 |
Table 18 Brief comparison of the grading schemes
| Grading scheme |
Front |
Type-I |
Type-II |
Type-I |
Type-II |
| Back |
Type-II |
Type-I |
| Nitrogen content for model system/µg g−1 |
8.8 ± 0.4 |
6.9 ± 0.2 |
2.6 ± 0.1 |
12.8 ± 0.5 |
| Nitrogen content for VGO/µg g−1 |
6.9 ± 0.4 |
4.7 ± 0.2 |
3.4 ± 0.3 |
10.1 ± 0.7 |
| BMCI for VGO product |
26.4 |
25.8 |
24.1 |
28.8 |
| Comprehensive effect ranking |
3rd |
2nd |
1st |
4th |
The results of the grading schemes can be attributed to differences in temperature sensitivity and the synergies in hydrocarbon molecule conversion. The single-catalyst hydrogenation evaluation reveals that Cat-I exhibits higher activities at lower temperatures, whereas Cat-II performs better at the higher temperatures. The positive sequence grading matches the temperature distribution in thermally insulated industrial reactors. On the other hand, theoretical calculations predict that Type-I active sites preferred hydrogenation saturation of lateral aromatic rings, whereas Type-II active sites excel in the hydrogenolysis of obstructed heteroatom–carbon bonds. The sequence grading matches the conversion process of the complicated polycyclic molecules.
4 Conclusions
The theoretical calculations and hydrogenation experiments indicate that the type of hydrogen active sites on the catalyst could be controlled by the preparation method. Impregnation under high pH values and the high-temperature calcination led to Type-I active sites with a low sulfidation degree, while impregnation under low pH values and low-temperature drying led to Type-II active sites with a high sulfidation degree. The theoretical calculations predict that Type-I active sites enhance the activity of hydrogen dissociation and reactant adsorption, which is helpful for the hydrogenation saturation of lateral aromatic rings. Type-II active sites promote hydrogen transfer and the ability to bind radical atoms, which contributes to the hydrogenolysis of obstructed heteroatom–carbon bonds. The hydrogenation experiments support that catalysts dominated by Type-I active sites perform better at low temperatures, whereas those dominated by Type-II active sites perform better at high temperatures. The optimal grading scheme is to place the catalysts with Type-I active sites in the front and those with Type-II in the back.
Author contributions
Qianmin Jiang: conceptualization, methodology, writing—original draft, and writing—review and editing. Sijia Ding: methodology, software, and writing—original draft. Shuandi Hou: validation and supervision. Zhentao Chen: writing—review and editing, resources, funding acquisition, and supervision.
Conflicts of interest
There are no conflicts to declare.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article.
Acknowledgements
The authors acknowledge the support by the National Natural Science Foundation of China (NSFC) (no. 22278430 and 21878329).
References
- S. Liu and H. Bai, Contemp. Chem. Ind., 2023, 52, 2237–2242 Search PubMed.
- M. Liu, D. Zhao, L. Zhang and S. Zhao, Fuel Process. Technol., 2017, 158, 238–246 Search PubMed.
- S. Vedachalam and A. K. Dalai, Catal. Today, 2023, 407, 165–171 CrossRef CAS.
- P. Becker, B. Celse, D. Guillaume, H. Dulot and V. Costa, Fuel, 2015, 139, 133–143 CrossRef CAS.
- P. Misra, S. Badoga, A. K. Dalai and J. Adjaye, Fuel, 2018, 226, 127–136 CrossRef CAS.
- D. Soogund, P. Lecour, A. Daudin, B. Guichard, C. Legens, C. Lamonier and E. Payen, Appl. Catal., B, 2010, 98, 39–48 CrossRef CAS.
- C. Boscagli, C. Yang, A. Welle, W. Wang, S. Behrens, K. Raffelt and J.-D. Grunwaldt, Appl. Catal., A, 2017, 544, 161–172 CrossRef CAS.
- J. Horáček, U. Akhmetzyanova, L. Skuhrovcová, Z. Tišler and H. d. P. Carmona, Appl. Catal., B, 2020, 263, 118328 CrossRef.
- J. Janssens, Y. Xu, G. Witte, A. D. v. Langeveld, S. T. Sie and J. A. Moulijn, Fuel, 1998, 77, 1367–1375 Search PubMed.
- Y. Dong, H. Zhao, Z. Liu, M. Yang, Z. Zhang, T. Zhu and H. Cheng, RSC Adv., 2020, 10, 11039–11045 RSC.
- I. Vázquez-Garrido, A. López-Benítez, G. Berhault and A. Guevara-Lara, Fuel, 2019, 236, 55–64 Search PubMed.
- B. Chu, N. Zhang, X. Zhai, X. Chen and Y. Cheng, J. Energy Chem., 2014, 23, 593–600 Search PubMed.
- Y. Zhang, F. Liu, W. Chen, W. Han, W. Zhai, Y. Lu and M. Li, Fuel, 2024, 356, 129640 Search PubMed.
- L. Qi, C. Peng, Z. Cheng and Z. Zhou, Fuel, 2023, 351, 128941 CrossRef CAS.
- Z. Cao, X. Zhang, C. Xu, X. Huang, Z. Wu, C. Peng and A. Duan, J. Energy Chem., 2021, 52, 41–50 Search PubMed.
- J. Ge, P. Zhang, F. Sun, Z. Xie, Z. Wu and B. Liu, Pet. Sci., 2023, 20, 3798–3806 CrossRef CAS.
- S. Mayo and B. Leliveld, in NPRA Annual Meeting, AM-09-14, San Antonio, USA, 2009 Search PubMed.
- S. Mayo, K. Vogt and B. Leliveld, in NPRA Annual Meeting, AM-10-170, Phoenix, Arizona, USA, 2010 Search PubMed.
- S. Mayo, in NPRA Annual Meeting, AM-11-23, San Antonio, USA, 2011 Search PubMed.
- S. Fan, C. Peng, H. Pang, Z. Wu and Z. Wang, Acta Pet. Sin., 2022, 38, 1194–1204 CAS.
- Z. Cao, S. Ding, R. Guo, Z. Chen, J. Yu, Z. Wu, D. Gao, A. Duan, H. Liu and X. Zhang, Appl. Catal., B, 2024, 343, 123536 CrossRef CAS.
- Z. Cao, R. Guo, P. Du, J. Mei, X. Zhang, C. Xu, J. Liu, G. Jiang, H. Li and A. Duan, Chem. Eng. Sci., 2020, 213, 115415 CrossRef CAS.
- S. Ding, S. Jiang, Y. Zhou, Q. Wei and W. Zhou, J. Catal., 2017, 345, 24–38 CrossRef CAS.
- S. Ding, Y. Zhou, Q. Wei, S. Jiang and W. Zhou, Catal. Today, 2018, 305, 12 CrossRef.
- S. Ding, A. Li, S. Jiang, Y. Zhou, Q. Wei, W. Zhou, Y. Huang, Q. Yang and T. Fan, Fuel, 2019, 237, 429–441 CrossRef CAS.
- J. Kibsgaard, J. Lauritsen, E. Laegsgaard, B. Clausen, H. Topsoe and F. Besenbacher, J. Am. Chem. Soc., 2006, 128, 13950 CrossRef CAS PubMed.
- Y. Zhu, Q. M. Ramasse, M. Brorson, P. G. Moses, L. P. Hansen, C. F. Kisielowski and S. Helveg, Angew. Chem., Int. Ed., 2014, 53, 10827 CrossRef.
- Y. Yi, X. Jin, L. Wang, Q. Zhang, G. Xiong and C. Liang, Catal. Today, 2011, 175, 460–466 CrossRef CAS.
- E. Krebs, A. Daudin and P. Raybaud, Oil Gas Sci. Technol., 2009, 64, 707–718 CrossRef CAS.
- S. Cristol, J. Paul, C. Schovsbo, E. Veilly and E. Payen, J. Catal., 2006, 239, 145–153 CrossRef CAS.
- S. Humbert, G. Izzet and P. Raybaud, J. Catal., 2016, 333, 78–93 CrossRef CAS.
- Y. Inada and H. Orita, J. Comput. Chem., 2010, 29, 225–232 CrossRef PubMed.
- B. Delley, J. Chem. Phys., 1982, 76, 1949–1960 CrossRef CAS.
- E. C. Anota and G. H. Cocoletzi, Phys. E, 2014, 56, 134–140 CrossRef.
- S. Grimme, J. Comput. Chem., 2010, 27, 1787–1799 CrossRef PubMed.
- S. Grimme, Wiley Interdiscip. Rev.:Comput. Mol. Sci., 2011, 1, 211–228 CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Ding, S. Peng, Z. Yan, J. Wang, S. Jiang and Z. Yang, Pet. Sci., 2022, 19, 339–344 CrossRef CAS.
- S. Ding, S. Jiang, Z. Yang, S. Peng and Q. Jiang, Chem. Ind. Eng. Prog., 2024, 43, 2436–2448 CAS.
- H. Farag, M. Kishida and H. Al-Megren, Appl. Catal., A, 2014, 469, 173–182 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Sijia Ding
a,
Sijia Ding




































