DOI:
10.1039/D6RA00934D
(Paper)
RSC Adv., 2026,
16, 22386-22399
Electronic structure engineering of phosphorene through transition metal functionalization for toxic gas detection
Received
3rd February 2026
, Accepted 9th April 2026
First published on 1st May 2026
Abstract
Designing sensitive, selective, and recyclable materials for toxic gas detection is crucial for environmental monitoring and public safety. In this study, density functional theory calculations are employed to systematically investigate the adsorption behavior, electronic structure modulation, charge transfer, optical response, and sensing performance of CO, CO2, H2S, and SO2 on cobalt-doped and cobalt-adsorbed phosphorene. Cobalt doping transforms pristine phosphorene from a direct-bandgap semiconductor (1.06 eV) into an indirect-bandgap semiconductor with a reduced bandgap of 0.83 eV, while cobalt adsorption further narrows the bandgap to 0.42 eV. Across all target gases and studied temperatures, the Co-doped configuration exhibits stronger adsorption and moderate charge transfer, which govern the observed bandgap modulation and sensing response, while the Co-adsorbed system shows excessive charge transfer, leading to near-irreversible adsorption. Among the investigated gases, SO2 and CO show the highest sensing responses, whereas CO2 interacts weakly with both surfaces. Based on literature trends, Co-modified phosphorene is expected to exhibit selective responses toward other common gases, such as NH3 and NO2, with minimal interference from H2O. Recovery-time analysis indicates rapid and reversible desorption under visible-light irradiation for the Co-doped system. In contrast, the Co-adsorbed system exhibits pronounced charge transfer, leading to strongly bound adsorption that is nearly irreversible. These results suggest that Co-doped phosphorene is a promising candidate for gas sensing based on computational predictions.
Introduction
Industries, power plants, and vehicles emit substantial quantities of toxic gases into the atmosphere due to the use of fossil fuels as energy sources.1,2 The release of large volumes of toxic gases, such as CO2, H2S, SO2, NH3, CO, NO, and NO2, contributes to air pollution and poses serious risks to public health.3–5 The World Health Organization has identified air pollution as a “silent killer” responsible for approximately seven million deaths annually, including thousands of children.6,7 Such mortalities are predominantly associated with respiratory pathologies and cardiovascular conditions, including myocardial infarction, stroke, and lung cancer.8 Therefore, the detection of toxic gases is critical for public safety, environmental monitoring, medical diagnostics, agricultural practices, and industrial chemical processes.9 For many industries, identifying hazardous gases, such as NH3 and CO, is essential to ensure compliance with environmental standards and mitigate their harmful impacts.10–12 Recently, gas sensors have become indispensable for applications across multiple domains.13 An ideal gas sensor exhibits characteristics such as high selectivity, high sensitivity, fast recovery time, high precision, high stability, low hysteresis, high repeatability, and high resolution.14,15 Proper material selection is therefore essential to ensure effective gas-sensing performance.16 Materials with two-dimensional (2D) structures, such as graphene, molybdenum disulfide, and other transition metal dichalcogenides (TMDs), have drawn considerable attention from scientists because of their excellent electrical, thermal, and mechanical properties.17,18 These materials offer a high surface-to-volume ratio, high carrier mobility, and advanced synthesis possibilities compared to their one-dimensional (1D) and bulk counterparts.19–21 In addition, 2D materials exhibit significant potential for applications in photovoltaics, photodetectors, nanoelectronics, light emitters, drug delivery, and gas sensors.22 Graphene possesses exceptional electron mobility and a high surface area relative to its volume, making it well-suited for use in energy-storage devices and gas monitoring.23 However, π–π electron coupling in graphene slows charge transfer and reduces sensitivity to gas molecules. Additionally, the zero-bandgap nature of graphene limits its applicability in gas sensors. Consequently, exploring 2D materials with tunable bandgaps and enhanced gas-sensing capabilities remains an important research direction, and appropriately engineered bandgaps constitute a critical area of ongoing research.24 In 2014, phosphorene was successfully fabricated from bulk black phosphorus using mechanical exfoliation.25 Phosphorene, a single atomic layer derived from black phosphorus, is one of the four known allotropes of phosphorus.26 It is a novel 2D semiconductor material characterized by a puckered honeycomb structure with an orthorhombic symmetry.27 Phosphorene has a direct bandgap that varies with the number of layers, ranging from 0.33 to 1.50 eV.28–30 Furthermore, its charge-carrier mobility is approximately 1000 cm2 V−1 s−1, which is higher than that of MoS2.30 The surface-to-volume ratio of phosphorene is higher than that of other 2D materials, providing distinct advantages for gas-sensing applications.31 Phosphorene finds diverse applications in batteries, transistors, solar cells, drug-delivery systems, quantum dot synthesis, and gas sensing.32 Gas molecules generally exhibit higher adsorption energies on phosphorene than on graphene and other 2D materials.33 Its high surface-to-volume ratio and strong chemical reactivity make it highly suitable for gas-sensing applications.34 Previous studies have demonstrated the potential of phosphorene as an effective gas-sensing material. Despite notable progress, understanding the interactions between toxic gases, volatile organic compounds (VOCs), and phosphorene remains an active area of research. First-principles studies have explored the adsorption characteristics of various gas molecules on phosphorene, including rare gases,35 methane on doped systems,30 and toxic species such as phosgene.36 The effect of metal-atom substitution on CO adsorption has also been investigated,37 while pristine phosphorene has been proposed as a promising gas sensor for the selective detection of SF6 decomposition products.38 In addition, the sensitivity of phosphorene has been widely reported, although the recovery dynamics remain less explored,39 and fundamental studies on physisorption have established a baseline for small-molecule interactions.40 In addition, the role of metal adatoms has been examined. However, these studies are generally limited to single gas molecules or a single type of surface modification, and even in multi-gas investigations, different functionalization strategies are rarely compared under identical computational conditions. For instance, Lei S. et al.43 and Sun X. et al.37 focused on CO adsorption using substitutional doping, while Kou L. et al.39 considered pristine phosphorene; Cai Y. et al.40 examined physisorption without functionalization. As a result, a consistent understanding of how different modification approaches influence sensing behavior is still lacking.
In this work, a systematic comparison between Co-doped and Co-adsorbed phosphorene is performed for multiple toxic gases (CO, CO2, H2S, and SO2) within a unified DFT framework. These gases are common atmospheric pollutants and have been frequently investigated in previous studies on phosphorene and other 2D materials.41,42 This direct, side-by-side comparison of substitutional doping and adatom adsorption under identical conditions has not been systematically addressed in previous studies. Such an approach enables clear identification of how the mode of cobalt incorporation governs adsorption strength, charge transfer, and electronic response. Furthermore, beyond adsorption energetics, this study integrates electronic structure, optical response, charge transfer, sensing characteristics, and recovery behavior within a single framework, allowing the simultaneous evaluation of sensitivity, selectivity, and reversibility. Therefore, the novelty of this work lies not merely in extending the range of gases but in establishing a comparative and mechanistic understanding of cobalt-modified phosphorene, providing more reliable design guidelines for high-performance gas sensors.
Computational methods and material parameters
Co-doped and Co-adsorbed phosphorene monolayers were investigated for the adsorption of different toxic gases using DFT simulations.43 For each gas molecule, several initial adsorption configurations, including different orientations and adsorption sites, were considered. Full structural optimization was performed, and the most stable configuration with the lowest total energy was selected for further analysis. The adsorption and sensing of toxic gases were examined using the Spanish Initiative for Electronic Simulations with Thousands of Atoms (SIESTA) computational method.14 A 3 × 2 phosphorene supercell containing 24 P atoms was constructed in Materials Studio, with lattice parameters of a = 3.63 Å and b = 4.29 Å.
A 12 Å vacuum slab was introduced to eliminate periodic interactions.44 The generalized gradient approximation (GGA) with the revised Perdew–Burke–Ernzerhof (RPBE) exchange–correlation functional was employed for structural optimization. Geometry optimizations were performed using Monkhorst–Pack k-point meshes of 3 × 3 × 1 and 6 × 6 × 1. A real-space grid cutoff of 198 Ry (≈2.7 keV electron kinetic energy) was used in the SIESTA calculations. The computational setup employed norm-conserving pseudopotentials in a fully nonlocal semicore form and a double-zeta basis set to describe the valence orbitals. The computational parameters, including k-point sampling, energy cutoff, and supercell size, were selected based on well-established convergence-tested settings reported in previous phosphorene-based studies, ensuring reliable adsorption energies and electronic properties.45–47
The 3 × 2 supercell provides sufficient separation between periodic images, minimizing artificial interactions between Co atoms and adsorbed gas molecules. The Co concentration (∼4 at%) was chosen to represent experimentally relevant doping levels while avoiding significant Co–Co interactions. Such doping levels are feasible in two-dimensional materials and can be achieved via substitutional incorporation during growth (e.g., chemical vapor deposition) or post-synthetic functionalization, including adsorption, followed by annealing or defect-assisted doping. Pristine phosphorene is known to degrade under ambient conditions due to oxidation in the presence of oxygen and moisture; previous studies have shown that transition metal doping and surface functionalization can enhance its chemical stability. Additionally, encapsulation techniques have been demonstrated to significantly improve its environmental stability. Therefore, the chosen model is both computationally practical and experimentally relevant under controlled conditions.39,48
All calculations in this work were performed using a non-spin-polarized approach. While cobalt is a magnetic transition metal and its spin polarization may influence the electronic structure through Co 3d states, previous studies on transition-metal-doped phosphorene have shown that key properties, such as adsorption energies, charge transfer, and relative sensing trends, remain qualitatively consistent.14,43 Therefore, the non-spin-polarized framework adopted here is sufficient for comparative analysis. However, spin-polarized calculations could further refine the understanding of magnetic interactions and will be explored in future investigations.
Calculation of adsorption energy
Determining the adsorption energy is essential for understanding the interaction mechanisms of toxic gases with phosphorene. The adsorption energy provides insight into the interaction strength, thermal stability, and the feasibility of experimental realization. In this study, the interaction energies of gas molecules on Co-doped and Co-adsorbed phosphorene were computed using eqn (1) and (2).30,35,49–51| | |
ECo-dopedads = E(Co-doped+gas) − ECo-doped − Egas
| (1) |
| | |
ECo-adsorbedads = E(Co-adsorbed+gas) − ECo-adsorbed − Egas
| (2) |
In eqn (1), ECo-dopedads represents the adsorption energy of the Co-doped system and E(Co-doped+gas), ECo-doped, and Egas correspond to the total energies of the gas-adsorbed doped system, the isolated Co-doped phosphorene, and the isolated gas molecule, respectively. Similarly, in eqn (2), ECo-adsorbedads denotes the adsorption energy of the Co-adsorbed system and E(Co-adsorbed+gas), ECo-adsorbed, and Egas represent the total energies of the gas-adsorbed Co-adsorbed system, the isolated Co-adsorbed phosphorene, and the isolated gas molecule, respectively. All adsorption energies reported in the tables and text have been carefully checked and are consistently reported using the negative sign convention, where a negative adsorption energy indicates exothermic (thermodynamically favorable) adsorption.51
Results and discussion
Pristine, Co-doped, and Co-adsorbed phosphorene structures
In the optimized phosphorene structure, each phosphorus atom is covalently bonded to three adjacent phosphorus atoms, resulting in a puckered, honeycomb-like framework similar to graphene but with intrinsic wrinkles.52 Upon the substitution of a Co atom for a phosphorus atom, the system fully relaxes. The doping concentration of a single Co atom in a phosphorene monolayer supercell containing 24 phosphorus atoms corresponds to approximately 4%. For Co adsorption on phosphorene, two main adsorption sites are possible: directly above a phosphorus atom or at the hollow site of the honeycomb structure, approximately 2.60 Å above the center. The hollow site is energetically more favorable because it allows interactions with three neighboring phosphorus atoms. This substitution and adsorption significantly affect the overall crystal lattice, inducing local deformations in bond lengths and bond angles, which are influenced by the presence of the dopant. All geometries reached their respective minimum energy states during optimization, indicating structural stability. Table 1 presents the calculated bond lengths and angles obtained from the relaxed structures of the pristine, Co-doped, and Co-adsorbed phosphorene (Fig. 1).
Table 1 Bond lengths (d1 and d2) and bond angles (θ1 and θ2) of the pristine, Co-doped, and Co-adsorbed phosphorene
| Dopants |
d1 (Å) |
d2 (Å) |
θ1 (°) |
θ2 (°) |
| Pristine (this work) |
2.281 |
2.263 |
101.9 |
95.6 |
| Pristine14 |
2.262 |
2.241 |
101.6 |
96.6 |
| Co-doped (this work) |
2.207 |
2.233 |
94.1 |
85.5 |
| Co-doped43 |
— |
2.232 |
— |
— |
| Co-adsorbed |
2.180 |
2.257 |
113.8 |
117.4 |
 |
| | Fig. 1 Top and side views of the phosphorene-based systems: (a) pristine phosphorene, (b) Co-doped phosphorene and (c) Co-adsorbed phosphorene. Phosphorus atoms are shown in orange and cobalt atoms in blue. Bond lengths are indicated in Å. The figure highlights the structural modifications due to Co doping and adsorption. | |
In the pristine phosphorene, the P–P bond lengths d1 and d2 are 2.280 Å along the armchair direction and 2.263 Å along the zigzag direction, respectively, while the corresponding bond angles θ1 and θ2 are 101.0° and 95.6°, respectively. For the Co-doped phosphorene, the P–P bond lengths are reduced to 2.207 Å in the armchair direction and slightly increased to 2.233 Å in the zigzag direction, whereas the bond angles θ1 and θ2 decrease to 94.1° and 85.5°, respectively. In the Co-adsorbed structure, the Co–P bond length is 2.098 Å, and the P–P bond length is 2.257 Å. The corresponding bond angles θ1 and θ2 increase significantly to 113.8° and 117.4°, respectively.
The bond lengths and angles for the pristine, Co-doped, and Co-adsorbed phosphorene are plotted in Fig. 2. These results are consistent with those reported in the literature.25,53 These variations in bond lengths and angles induced by Co doping and adsorption reflect localized geometric distortions in the phosphorene lattice. These distortions enhance surface reactivity and promote the formation of favorable active sites for toxic-gas adsorption.
 |
| | Fig. 2 Comparison of the structural parameters of the pristine, Co-doped, and Co-adsorbed phosphorene. The figure shows variations in bond lengths (d1 and d2, in Å) and bond angles (θ1 and θ2, in degrees) for the three systems. The results illustrate the structural distortions in the phosphorene lattice induced by Co doping and adsorption. | |
Electronic structure and properties
In the next step, the electronic band structure and PDOS of the pristine, Co-doped, and Co-adsorbed phosphorene systems were plotted to investigate their electronic behavior, and the results are shown in Fig. 3(a–f). As evidenced by the bandgap widths shown in Fig. 3(a), the pristine phosphorene exhibited a direct bandgap of 1.06 eV, which is in good agreement with previously reported values in the literature.54,55 The substitution of a Co atom in phosphorene decreases the bandgap from 1.06 eV to 0.83 eV (Fig. 3(c)). This transformation is attributed to the hybridization between the Co 3d orbitals and the P 3p states, which perturbs the band edges and modifies the carrier transport properties owing to the formation of new energy states near the Fermi level. Calculations confirmed that the pristine phosphorene is a direct bandgap material; however, Co doping changes the bandgap from direct to indirect, which strongly influences optical absorption. These results imply that Co doping increases the responsiveness of phosphorene for gas detection, a trend also observed in previous studies.25,43 With Co adsorption, the bandgap of the Co-adsorbed system decreases to 0.42 eV (Fig. 3(e)). The PDOS confirmed significant alterations in the electronic states of the doped phosphorene compared to its pristine counterpart. Furthermore, substituting a Co atom into the pristine phosphorene resulted in a notable distortion in the PDOS near the Fermi level. This deformation is attributable to the emergence of an additional electronic band above the Fermi level compared to the pristine phosphorene. Phosphorus 3p states made the most significant contribution to the valence band near the Fermi level. Strong hybridization between the Co 3d and P 3p states introduced additional states, modifying the density of states near the valence region. The Co 3d states contributed significantly to the lower edge of the conduction band. Hybridization between the Co 3d, P 3p, and P 3s states led to the redistribution of electronic states, influencing charge transport. Additional peaks in the PDOS arose because of the strong interactions between these orbitals, which affected the overall electronic structure.
 |
| | Fig. 3 Band structures (left) and projected density of states (PDOS, right) of the (a and b) pristine, (c and d) Co-doped, and (e and f) Co-adsorbed phosphorene. The Fermi level (EF = 0 eV) is shown by the dashed blue line. Co incorporation reduces the bandgap from 1.06 eV to 0.83 eV and 0.42 eV. | |
Optical absorption and conductivity
The optical absorption spectra of the pristine, Co-doped, and Co-adsorbed phosphorene, with characteristic absorption peaks at 1.06, 0.83, and 0.42 eV, respectively, are presented in Fig. 4. These spectra provide insight into the electronic structure and bandgap features of the material. The high density of states near the Fermi level facilitates electron transitions to the conduction band upon photon absorption, which directly influences the optoelectronic behavior relevant for gas sensing applications. Incorporation of Co into pristine phosphorene enhances the optical absorption and broadens the absorption spectrum. Importantly, the absorption peaks correspond to photon energies capable of promoting electron excitation under visible light. This suggests that light illumination can assist in the desorption of gas molecules from the phosphorene surface, thereby improving sensor recovery and reversibility.
 |
| | Fig. 4 Optical absorption (right) and optical conductivity (left) of the pristine, Co-doped, and Co-adsorbed phosphorene. The x-axis represents the photon energy (eV), while the y-axis shows the absorption coefficient (α × 105 cm−1) and optical conductivity (σ × 105 Ω−1 cm−1), respectively. Black, red, and blue lines indicate the pristine, Co-doped, and Co-adsorbed systems, respectively. | |
The optical conductivities (σ) of the pristine, Co-doped, and Co-adsorbed phosphorene were evaluated using eqn (3) to assess the charge-transport efficiency.49,56
| |
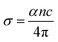 | (3) |
Here,
c,
n, and
α denote the speed of light, refractive index, and absorption coefficient, respectively. According to
eqn (3), optical conductivity depends directly on the absorption coefficient, reflecting the material's response to incident photons. As shown in the right panel of
Fig. 4, Co incorporation enhances the optical conductivity of phosphorene, indicating more efficient charge transport under illumination. Such an improvement can facilitate electron-transfer processes that promote the light-assisted desorption of adsorbed gas molecules, thereby enhancing the overall performance of phosphorene-based sensors.
Overall, the increased optical absorption and conductivity of the Co-doped and Co-adsorbed phosphorene are not only indicative of improved optoelectronic behavior but also play a crucial role in enabling visible-light-assisted recovery in gas sensing applications.
Adsorption and electronic properties of gases on the Co-doped and Co-adsorbed phosphorene
To comprehensively assess the sensing performance of cobalt-modified phosphorene, we conducted a comparative analysis of CO, CO2, H2S, and SO2 on both the Co-doped and Co-adsorbed phosphorene systems. The optimized adsorption configurations are shown in Fig. 5, whereas the corresponding electronic properties, including the band structures and projected density of states (PDOS), are presented in Fig. 6 and 7. Upon adsorption, all gas molecules interact with the Co-modified phosphorene surface, inducing notable changes in both structural and electronic properties. Generally, the distance between the gas molecules and the Co atom decreases after structural optimization, confirming the stability of adsorption. However, the interaction strength varies significantly depending on the type of gas molecule and the modification strategy. Among all the gases, CO exhibits the strongest interaction in both systems. In the Co-doped phosphorene, the distance between the CO molecule and the Co atom decreases from 2.549 Å to 1.779 Å, accompanied by an increase in the Co–P bond lengths and minor variations in bond angles, indicating slight structural deformation. In the Co-adsorbed system, the interaction is even stronger, with the distance further reduced to 1.762 Å and more pronounced changes in the P–P and Co–P bond lengths. This strong interaction is consistent with the calculated adsorption energies of −1.20 eV and −4.00 eV for Co-doped and Co-adsorbed systems, respectively. By contrast, CO2 exhibits a relatively weak interaction, particularly in the Co-doped system. The distance between the oxygen atom and Co decreases slightly from 2.808 Å to 2.402 Å, with minimal structural distortion, which corresponds to a low adsorption energy of −0.42 eV. In the Co-adsorbed phosphorene system, however, the interaction strengthens, as evidenced by the reduction in the Co–C and Co–O distances to approximately 2.0 Å and an increased adsorption energy of −1.9 eV. For H2S adsorption, a moderate interaction is observed in the Co-doped system, where the S–Co distance decreases from 2.549 Å to 2.310 Å, accompanied by slight changes in bond lengths and angles. In the Co-adsorbed system, adsorption is stronger, with the S–Co distance further reduced to 2.172 Å and a higher adsorption energy of −2.60 eV observed. Similarly, SO2 demonstrates a relatively strong interaction with both systems. In Co-doped phosphorene, the S–Co distance decreases from 2.680 Å to 2.129 Å, accompanied by noticeable bond distortions. In the Co-adsorbed system, the interaction is stronger, with a shorter distance of 2.05 Å and more significant structural modifications. The adsorption energy is −0.55 eV for Co-doped and −3.10 eV for Co-adsorbed phosphorene, confirming stronger binding in the latter.
 |
| | Fig. 5 Optimized adsorption configurations of CO, CO2, H2S, and SO2 on phosphorene. Panels (a–d) correspond to CO, CO2, H2S, and SO2, respectively. For each gas, the left panel shows the Co-doped phosphorene, and the right panel shows the Co-adsorbed phosphorene. In each panel, the top row represents the top view, and the bottom row represents the side view of the adsorbed gas molecule. Color scheme: P (purple), Co (blue), O (red), C (gray), S (yellow), and H (white). | |
 |
| | Fig. 6 Electronic band structures of the gas-adsorbed Co-modified phosphorene for the CO, CO2, H2S, and SO2. Panels (a, b), (c, d), (e, f), and (g, h) correspond to CO, CO2, H2S, and SO2, respectively. Panels (a, c, e and g) represent the Co-doped phosphorene, while (b, d, f and h) represent the Co-adsorbed phosphorene. The dashed horizontal line indicates the Fermi level (EF = 0 eV). | |
 |
| | Fig. 7 Projected density of states (PDOS) of the gas-adsorbed Co-modified phosphorene for CO, CO2, H2S, and SO2. Panels (a, b), (c, d), (e, f), and (g, h) correspond to the CO, CO2, H2S, and SO2 adsorptions, respectively. Panels (a, c, e and g) represent the Co-doped phosphorene, while panels (b, d, f and h) represent the Co-adsorbed phosphorene. The dashed vertical line indicates the Fermi level (EF = 0 eV). The PDOS demonstrates significant hybridization between the Co 3d states and the gas molecule orbitals near the Fermi level, providing insights into the electronic origin of the sensing behavior. | |
The calculated band structures and projected density of states (PDOS) reveal that gas adsorption significantly alters the bandgap of phosphorene, which is a key parameter governing sensing performance, as it directly affects electrical conductivity. In Co-doped phosphorene, CO adsorption reduces the bandgap from 0.83 eV to 0.50 eV, while H2S and SO2 lower it to 0.63 eV and 0.60 eV, respectively. By contrast, CO2 slightly increases the bandgap to 0.87 eV, reflecting weaker electronic coupling. A more pronounced electronic response is observed in the Co-adsorbed phosphorene system, where the bandgap decreases from 0.42 eV to 0.30 eV (CO), 0.34 eV (CO2), 0.25 eV (H2S), and 0.29 eV (SO2). This consistent bandgap narrowing indicates enhanced electrical conductivity and stronger sensitivity toward gas adsorption. The PDOS results (Fig. 7(a–h)) further support these findings, showing increased hybridization between the orbitals of the gas molecules and the Co atom near the Fermi level. The adsorption energies and corresponding bandgap values are listed in Table 3. This effect is particularly strong for CO, H2S, and SO2 adsorption, especially in the Co-adsorbed system, indicating stronger electronic coupling. To further clarify the microscopic origin of these electronic changes and their impact on sensing behavior, a Mulliken charge population analysis was performed.
Charge transfer analysis (Mulliken population analysis)
To elucidate the interaction mechanisms between gas molecules and the Co-modified phosphorene, Mulliken charge population analysis was performed. This method provides quantitative insight into both the direction and magnitude of electron transfer, which is crucial for understanding and optimizing gas-sensing performance. Positive values of charge transfer indicate electron donation from the gas molecule to the phosphorene surface, whereas negative values indicate electron withdrawal from the surface by the molecule. The Mulliken charge transfer (ΔQ) is calculated using eqn (4).| | |
ΔQdoped = Qisolatedgas − Qadsorbedgas
| (4) |
Here, Qisolatedgas represents the Mulliken charge of the isolated gas molecule, while Qadsorbedgas denotes the Mulliken charge after adsorption on the phosphorene surface. Positive ΔQ values indicate electron donation from the gas molecule to phosphorene, whereas negative values indicate electron withdrawal from phosphorene by the gas molecule. The analysis reveals that CO and H2S primarily act as electron donors, transferring electrons to the phosphorene substrate. In contrast, SO2 behaves as an electron acceptor, withdrawing electrons from the surface. CO2 exhibits negligible charge transfer, indicating weak interaction with both the Co-doped and Co-adsorbed phosphorene. The calculated charge transfer values are summarized in Table 2. A comparison of the two modification strategies highlights distinct differences. The Co-adsorbed phosphorene system exhibits significantly higher charge transfer than the Co-doped system. This behavior is consistent with its larger adsorption energies and more pronounced bandgap modulation, suggesting stronger orbital hybridization with gas molecules. However, the strong charge transfer in the Co-adsorbed phosphorene results in very strong binding and near-irreversible adsorption, which limits its suitability for reusable gas sensors. In contrast, Co-doped phosphorene demonstrates moderate charge transfer, offering a favorable balance between interaction strength and reversibility. This ensures detectable electronic responses while maintaining rapid recovery, making Co-doped phosphorene a promising candidate for practical gas-sensing applications.
Table 2 Mulliken charge transfer (ΔQ) for gas adsorption on the Co-modified phosphorene
| Gas |
ΔQ (Co-doped, e) |
Nature |
ΔQ (Co-adsorbed, e) |
Nature |
| CO |
+0.18 to + 0.25 |
Donor |
+0.40 to +0.55 |
Strong donor |
| CO2 |
∼±0.05 |
Weak interaction |
+0.15 to +0.25 |
Moderate donor |
| H2S |
+0.08 to +0.12 |
Weak donor |
+0.25 to +0.35 |
Moderate donor |
| SO2 |
−0.15 to −0.22 |
Acceptor |
−0.30 to −0.45 |
Strong acceptor |
Table 3 Adsorption (Eads) and bandgap (Eg) energies of gas molecules adsorbed on Co-doped and Co-adsorbed phosphorene
| Gas molecule |
Eads (eV) |
Eg (eV) |
Gas molecule |
Eads (eV) |
Eg (eV) |
| Co-doped-phosp |
— |
0.83 |
Co-adsorbed-phosp |
— |
0.42 |
| CO |
−1.20 |
0.50 |
CO |
−4.00 |
0.3 |
| CO2 |
−0.42 |
0.87 |
CO2 |
−1.90 |
0.34 |
| H2S |
−0.17 |
0.63 |
H2S |
−2.60 |
0.25 |
| SO2 |
−0.55 |
0.60 |
SO2 |
−3.10 |
0.29 |
These findings highlight the critical role of Co modification in tuning charge transfer and gas-sensing behavior, providing a theoretical basis for selecting materials with optimal sensitivity and reversibility. Overall, the sensing performance of the two systems shows distinct trends. While Co-adsorbed phosphorene exhibits stronger adsorption energies and larger charge transfer, this leads to excessively strong binding and limited recovery, making it less suitable for reusable sensing applications. Conversely, Co-doped phosphorene demonstrates moderate charge transfer combined with pronounced bandgap modulation, resulting in high electronic sensitivity and faster recovery. Therefore, Co-doped phosphorene is more suitable for practical gas-sensing applications, whereas Co-adsorbed phosphorene may be better suited for gas capture or storage.
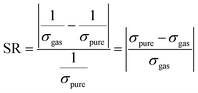
Sensing response (SR)
The sensing response of a gas sensor is defined as the relative change in the electrical conductivity of Co-doped phosphorene and Co-adsorbed phosphorene upon gas adsorption. It is a crucial parameter for evaluating the effectiveness of the sensor in detecting commonly encountered gas species. Determining the sensing response of a material for each gas molecule remains challenging in computational studies. We note that the sensing response estimated from eqn (5) and (6) is based on a simplified Arrhenius-type relation, where bandgap changes are used as a proxy for conductivity.57 This approach neglects factors such as carrier concentration, scattering, and transport effects, and therefore provides qualitative trends only. Moreover, explicit transport calculations (e.g., NEGF or I–V characteristics) are not included, so the predicted responses should be interpreted cautiously.| |
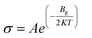 | (5) |
| |
 | (6) |
In the above equations, σ, Bg, A, T, and K denote the conductivity, bandgap energy, a constant, temperature, and the Boltzmann constant, respectively.
The sensing responses of the Co-doped and Co-adsorbed phosphorene toward SO2, H2S, CO2, and CO exhibit a clear dependence on temperature, with noticeable differences between the two configurations. Across all gases and temperature ranges, the Co-doped system shows relatively higher sensing responses than the Co-adsorbed counterpart, suggesting comparatively stronger interactions with the target molecules (Fig. 8). In both systems, the sensing response decreases as the temperature increases from 300 K to 500 K, which can be attributed to enhanced thermal agitation that weakens adsorption strength and reduces charge transfer. Among the gases studied, SO2 and CO show the highest responses, whereas CO2 exhibits the lowest, particularly in the Co-adsorbed configuration. The difference between doping and adsorption is more evident for CO2 and H2S, where the doped system retains comparatively higher responses at elevated temperatures. Overall, these results suggest that both modification strategies improve the sensing characteristics of phosphorene, with Co doping showing comparatively better potential under varying thermal conditions. The temperature-dependent sensing responses are presented in Table 4.
 |
| | Fig. 8 Temperature-dependent sensing responses (%) of the Co-doped and Co-adsorbed phosphorene toward SO2, H2S, CO2, and CO. Co doping shows higher and more stable responses, while all responses decrease with increasing temperature. | |
Table 4 Sensing response of Co-doped and adsorbed phosphorene toward toxic gases at different temperatures
| Temp. (K) |
Co-doped-phosp |
Co-adsorbed-phosp |
| SO2 |
H2S |
CO2 |
CO |
SO2 |
H2S |
CO2 |
CO |
| 300 |
−0.988 |
−0.978 |
−1.160 |
−0.998 |
−0.919 |
−0.962 |
−0.787 |
−0.901 |
| 350 |
−0.977 |
−0.963 |
−0.941 |
−0.995 |
−0.884 |
−0.940 |
−0.734 |
−0.863 |
| 400 |
−0.964 |
−0.945 |
−0.786 |
−0.991 |
−0.848 |
−0.915 |
−0.686 |
−0.824 |
| 450 |
−0.948 |
−0.924 |
−0.675 |
−0.985 |
−0.812 |
−0.888 |
−0.643 |
−0.787 |
| 500 |
−0.930 |
−0.901 |
−0.591 |
−0.978 |
−0.778 |
−0.860 |
−0.604 |
−0.751 |
Recovery time
Recovery times, defined as the period required for gas desorption, were estimated using a simplified Arrhenius-type model with a fixed attempt frequency of 1012 s−1, a standard approximation in computational studies of gas adsorption.58,59 Although this approach neglects surface-specific vibrational modes and dynamic effects, it provides a reasonable first-order estimate of desorption kinetics. Furthermore, visible-light irradiation can enhance desorption by supplying additional energy to overcome adsorption barriers, suggesting that actual recovery times may be shorter than those predicted by the thermal model. The near-irreversible adsorption on the Co-adsorbed surface may indicate potential applications in pollutant capture.
In this work, the recovery time is estimated using a Van't Hoff–Arrhenius-type expression.60–62
| |
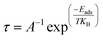 | (7) |
Here,
τ,
A,
Eads,
T, and
KB represent the recovery time, frequency (10
12 s
−1 for visible light), adsorption energy, temperature, and Boltzmann constant, respectively.
It should be noted that this approach is highly simplified. The chosen pre-exponential factor is approximate, and the model neglects important effects, such as multiple desorption pathways, photon-assisted processes, surface defects, and realistic experimental conditions. Therefore, the calculated recovery times should be interpreted only in terms of order-of-magnitude trends, rather than as quantitative predictions. The estimated recovery times are summarized in Table 5. For the Co-doped system, relatively short recovery times are obtained for SO2, H2S, and CO2, indicating that these gases can desorb readily, even at room temperature. In contrast, CO shows comparatively longer recovery times, suggesting stronger binding and the need for elevated temperatures for efficient desorption. These results indicate a balance between adsorption strength and reversibility, which is desirable for sensing applications. In contrast, the Co-adsorbed system exhibits extremely large recovery times across all gases and temperatures. Rather than representing physically meaningful timescales, these values indicate that desorption is effectively negligible under practical conditions. This suggests that gas adsorption on Co-adsorbed phosphorene is essentially irreversible within experimental timescales, limiting its applicability for reusable sensing devices. However, such a strong binding may be advantageous for applications such as gas capture and catalytic processes.
Table 5 Estimated recovery times (s) based on the Arrhenius model. Values indicate order-of-magnitude trends rather than precise predictions
| Temp. (K) |
Co-doped-phosp |
Co-adsorbed-phosp |
| SO2 |
H2S |
CO2 |
CO |
SO2 |
H2S |
CO2 |
CO |
| 300 |
1.74 × 10−3 |
7.18 × 10−10 |
1.14 × 10−5 |
1.44 × 108 |
2.51 × 1038 |
4.78 × 1031 |
8.31 × 1019 |
6.91 × 1051 |
| 350 |
8.31 × 10−5 |
2.80 × 10−10 |
1.12 × 10−6 |
1.90 × 105 |
1.58 × 1031 |
2.75 × 1025 |
2.29 × 1015 |
5.24 × 1042 |
| 400 |
8.51 × 10−6 |
1.39 × 10−10 |
1.96 × 10−7 |
1.32 × 103 |
6.31 × 1025 |
5.75 × 1020 |
8.71 × 1011 |
7.58 × 1035 |
| 450 |
1.44 × 10−6 |
8.02 × 10−11 |
5.06 × 10−8 |
2.75 × 101 |
3.98 × 1021 |
1.32 × 1017 |
1.90 × 109 |
3.63 × 1030 |
| 500 |
3.50 × 10−7 |
5.17 × 10−11 |
1.71 × 10−8 |
1.25 |
1.74 × 1018 |
1.61 × 1014 |
1.42 × 107 |
2.01 × 1026 |
Conclusion
In this work, the adsorption and sensing behaviors of toxic gases (CO, CO2, H2S, and SO2) on Co-doped and Co-adsorbed phosphorene were systematically investigated using first-principles density functional theory calculations. Cobalt doping significantly modifies the electronic structure of phosphorene, with moderate charge transfer from gas molecules inducing bandgap changes and enhanced optical absorption, which together contribute to improved and reversible gas-sensing performance. By comparison, excessive charge transfer in the Co-adsorbed system results in stronger adsorption and near-irreversible binding, limiting reusability but indicating potential for gas capture and catalytic applications. Among the studied gases, SO2 and CO exhibit the strongest sensing responses in both configurations, while CO2 interacts most weakly, particularly on the Co-adsorbed surface. The difference between doping and adsorption is most pronounced for CO2 and H2S, with the Co-doped system maintaining sensitivity and thermal stability even at elevated temperatures. Recovery-time analysis indicates that Co-doped phosphorene allows efficient desorption under visible-light irradiation, with rapid recovery of SO2, H2S, and CO2 at room temperature and moderate recovery of CO at higher temperatures. By contrast, adsorption on Co-adsorbed phosphorene is effectively irreversible under practical conditions, limiting its suitability for reusable sensing, while highlighting applications in pollutant capture, storage, or catalysis. Overall, Co-doped phosphorene achieves a favorable balance of adsorption strength, tunable electronic response, and recovery efficiency, demonstrating its potential as a versatile platform for toxic gas sensing applications.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data presented in this work are exclusively generated from first-principles simulations performed using the SIESTA package. They do not originate from any external crystallographic database or previously published reference sources. Instead, all results are obtained directly from computational modeling and are reported in the form of figures and tables within the manuscript.
Acknowledgements
This work was supported by the Korea Institute of Industrial Technology and Korea Planning & Evaluation Institute of Industrial Technology (Project Number: 1761002860, RS-2023-00255479).
References
- M. Mittal and A. Kumar, Carbon nanotube (CNT) gas sensors for emissions from fossil fuel burning, Sens. Actuators, B, 2014, 203, 349–362 CrossRef CAS
 .
. - R. Rai, M. Rajput, M. Agrawal and S. Agrawal, Gaseous air pollutants: a review on current and future trends of emissions and impact on agriculture, J. Sci. Res., 2011, 55(771), 1 Search PubMed .
- S. Ma, L. Su, L. Jin, J. Su and Y. Jin, A first-principles insight into Pd-doped MoSe2 monolayer: A toxic gas scavenger, Phys. Lett. A, 2019, 383(30), 125868 CrossRef CAS .
- S. G. Chatterjee, S. Chatterjee, A. K. Ray and A. K. Chakraborty, Graphene–metal oxide nanohybrids for toxic gas sensor: a review, Sens. Actuators, B, 2015, 221, 1170–1181 CrossRef .
- M. D. Mohammadi, H. Louis, U. G. Chukwu, S. Bhowmick, M. E. Rasaki and G. Biskos, Gas-Phase Interaction of CO, CO2, H2S, NH3, NO, NO2, and SO2 with Zn12O12 and Zn24 Atomic Clusters, ACS Omega, 2023, 8(23), 20621 CrossRef PubMed .
- S. G. John, Attention of Nigerian newspapers to air pollution, Air Qual., Atmos. Health, 2020, 1–10 Search PubMed .
- M. Méndez, M. G. Merayo and M. Núñez, Machine learning algorithms to forecast air quality: a survey, Artif. Intell. Rev., 2023, 1–36 Search PubMed .
- C. Varghese, L. HG and S. Kalikeri, Understanding the Status of Important Criteria Air Pollutants and Its Health Effects–A Review, Int. J. Health Allied Sci., 2022, 11(1), 5 Search PubMed .
- M. A. H. Khan, M. V. Rao and Q. Li, Recent advances in electrochemical sensors for detecting toxic gases: NO2, SO2 and H2S, Sensors, 2019, 19(4), 905 CrossRef PubMed .
- A. J. Tomassoni, R. N. French and F. G. Walter, Toxic industrial chemicals and chemical weapons: exposure, identification, and management by syndrome, Emerg. Med. Clin., 2015, 33(1), 13–36 CrossRef PubMed .
- Z. D. Leve, E. I. Iwuoha and N. Ross, The Synergistic Properties and Gas Sensing Performance of Functionalized Graphene-Based Sensors, Materials, 2022, 15(4), 1326 CrossRef CAS PubMed .
- S. S. Varghese, S. H. Varghese, S. Swaminathan, K. K. Singh and V. Mittal, Two-dimensional materials for sensing: graphene and beyond, Electronics, 2015, 4(3), 651–687 CrossRef CAS .
- G. W. Hunter, S. Akbar, S. Bhansali, M. Daniele, P. D. Erb, K. Johnson, C.-C. Liu, D. Miller, O. Oralkan and P. J. Hesketh, Editors’ choice—critical review—a critical review of solid state gas sensors, J. Electrochem. Soc., 2020, 167(3), 037570 CrossRef CAS .
- J. Prasongkit, V. Shukla, A. Grigoriev, R. Ahuja and V. Amornkitbamrung, Ultrahigh-sensitive gas sensors based on doped phosphorene: A first-principles investigation, Appl. Surf. Sci., 2019, 497, 143660 CrossRef CAS .
- T. Kaewmaraya, L. Ngamwongwan, P. Moontragoon, A. Karton and T. Hussain, Drastic improvement in gas-sensing characteristics of phosphorene nanosheets under vacancy defects and elemental functionalization, J. Phys. Chem. C, 2018, 122(35), 20186–20193 CrossRef CAS .
- V. S. Bhati, M. Kumar and R. Banerjee, Gas sensing performance of 2D nanomaterials/metal oxide nanocomposites: A review, J. Mater. Chem. C, 2021, 9(28), 8776–8808 RSC .
- H. G. Ali, K. Khan, M. B. Hanif, M. Z. Khan, I. Hussain, M. S. Javed, H. A. AL-bonsrulah, M. Mosiałek, M. Fichtner and M. Motola, Advancements in two-dimensional materials as anodes for lithium-ion batteries: Exploring composition-structure-property relationships emerging trends, and future perspective, J. Energy Storage, 2023, 73, 108980 CrossRef .
- M. Xu, T. Liang, M. Shi and H. Chen, Graphene-like two-dimensional
materials, Chem. Rev., 2013, 113(5), 3766–3798 CrossRef CAS PubMed .
- N. Sultana, A. Degg, S. Upadhyaya, T. Nilges and N. S. Sarma, Synthesis, modification, and application of black phosphorus, few-layer black phosphorus (FLBP), and phosphorene: a detailed review, Mater. Adv., 2022, 3(14), 5557–5574 RSC .
- S. Ray, First-principles study of MoS2, phosphorene and graphene based single electron transistor for gas sensing applications, Sens. Actuators, B, 2016, 222, 492–498 CrossRef CAS .
- S.-J. Choi and I.-D. Kim, Recent developments in 2D nanomaterials for chemiresistive-type gas sensors, Electron. Mater. Lett., 2018, 14, 221–260 CrossRef CAS .
- R. Gusmao, Z. Sofer and M. Pumera, Black phosphorus rediscovered: from bulk material to monolayers, Angew. Chem., Int. Ed., 2017, 56(28), 8052–8072 CrossRef CAS PubMed .
- L. Ji, P. Meduri, V. Agubra, X. Xiao and M. Alcoutlabi, Graphene-based nanocomposites for energy storage, Adv. Energy Mater., 2016, 6(16), 1502159 CrossRef .
- G.-X. Chen, R.-X. Wang, D.-D. Wang, H.-X. Li, S. Liu and J.-M. Zhang, First-principles study of CO and NO adsorption on pristine and transition metal doped blue phosphorene, Vacuum, 2020, 179, 109503 CrossRef CAS .
- A. Hashmi and J. Hong, Transition metal doped phosphorene: first-principles study, J. Phys. Chem. C, 2015, 119(17), 9198–9204 CrossRef CAS .
- H. Wang, X. Li, Z. Liu and J. Yang, ψ-Phosphorene: a new allotrope of phosphorene, Phys. Chem. Chem. Phys., 2017, 19(3), 2402–2408 RSC .
- S. H. Mir, Exploring the electronic, charge transport and lattice dynamic properties of two-dimensional phosphorene, Phys. B, 2019, 572, 88–93 CrossRef CAS .
- S. Sardarzadeh, J. Karamdel and P. Nayebi, Adsorption of SO2, H2S, NH3, PH3, and AsH3 Gas Molecules on Pristine Armchair Phosphorene Nanoribbon: A First-Principles Study, Phys. Status Solidi B, 2020, 257(9), 2000120 CrossRef CAS .
- K. Wang, H. Wang, M. Zhang, Y. Liu and W. Zhao, Electrical properties of phosphorene systems doped with fourth-period elements, Appl. Phys. Lett., 2018, 112(20), 202101 CrossRef .
- S. Yang, Z. Wang, X. Dai, J. Xiao, M. Long and T. Chen, First-principles study of gas molecule adsorption on C-doped zigzag phosphorene nanoribbons, Coatings, 2019, 9(11), 763 CrossRef CAS .
- A. K. Tareen, K. Khan, S. Rehman, M. Iqbal, J. Yu, Z. Zhou, J. Yin and H. Zhang, Recent development in emerging phosphorene based novel materials: Progress, challenges, prospects and their fascinating sensing applications, Prog. Solid State Chem., 2022, 65, 100336 CrossRef CAS .
- G. R. Bhimanapati, Z. Lin, V. Meunier, Y. Jung, J. Cha, S. Das, D. Xiao, Y. Son, M. S. Strano and V. R. Cooper, Recent advances in two-dimensional materials beyond graphene, ACS Nano, 2015, 9(12), 11509–11539 CrossRef CAS PubMed .
- V. V. Kulish, O. I. Malyi, C. Persson and P. Wu, Adsorption of metal adatoms on single-layer phosphorene, Phys. Chem. Chem. Phys., 2015, 17(2), 992–1000 RSC .
- M. Donarelli and L. Ottaviano, 2D materials for gas sensing applications: A review on graphene oxide, MoS2, WS2 and phosphorene, Sensors, 2018, 18(11), 3638 CrossRef PubMed .
- J. Hu, L. Zhao, J. Du and G. Jiang, Adsorption of rare gases on pristine and doped phosphorene, Appl. Surf. Sci., 2020, 504, 144326 CrossRef CAS .
- R. Bhuvaneswari and R. Chandiramouli, First-principles investigation on detection of phosgene gas molecules using phosphorene nanosheet device, Chem. Phys. Lett., 2019, 717, 99–106 CrossRef CAS .
- X. Sun, S. Luan, H. Shen and S. Lei, Effect of metal doping on carbon monoxide adsorption on phosphorene: a first-principles study, Superlattices Microstruct., 2018, 124, 168–175 CrossRef CAS .
- A.-J. Yang, D.-W. Wang, X.-H. Wang, J.-F. Chu, P.-L. Lv, Y. Liu and M.-Z. Rong, Phosphorene: A promising candidate for highly sensitive and selective SF 6 decomposition gas sensors, IEEE Electron Device Lett., 2017, 38(7), 963–966 CAS .
- L. Kou, T. Frauenheim and C. Chen, Phosphorene as a superior gas sensor: selective adsorption and distinct I–V response, J. Phys. Chem. Lett., 2014, 5(15), 2675–2681 CrossRef CAS PubMed .
- Y. Cai, Q. Ke, G. Zhang and Y.-W. Zhang, Energetics, charge transfer, and magnetism of small molecules physisorbed on phosphorene, J. Phys. Chem. C, 2015, 119(6), 3102–3110 CrossRef CAS .
- M. Doust Mohammadi, H. Louis, U. G. Chukwu, S. Bhowmick, M. E. Rasaki and G. Biskos, Gas-phase interaction of CO, CO2, H2S, NH3, NO, NO2, and SO2 with Zn12O12 and Zn24 atomic clusters, ACS Omega, 2023, 8(23), 20621–20633 CrossRef CAS PubMed .
- M. Doust Mohammadi, H. Louis, U. Chukwu, S. Bhowmick, M. Rasaki and G. Biskos, Gas-phase interaction of CO, CO2, H2S, NH3, NO, NO2, and SO2 with Zn12O12 and Zn24 atomic clusters, ACS Omega, 2023, 8, 20621–20633 CrossRef CAS PubMed .
- S. Lei, S. Luan and H. Yu, Co-doped phosphorene: Enhanced sensitivity of CO gas sensing, Int. J. Mod. Phys. B, 2018, 32(06), 1850068 CrossRef CAS .
- L. Xiao, Q. Zheng, S. Luo, Y. Ying, R. Zhou, S. Zhou, X. Li, X. Ye, Z. Yu and Q. Xu, Pd-intercalated black phosphorus: An efficient electrocatalyst for CO2 reduction, Sci. Adv., 2024, 10(25), eadn2707 CrossRef CAS PubMed .
- Y. Luo, C. Ren, S. Wang, S. Li, P. Zhang, J. Yu, M. Sun, Z. Sun and W. Tang, Adsorption of transition metals on black phosphorene: a first-principles study, Nanoscale Res. Lett., 2018, 13(1), 282 CrossRef PubMed .
- J. Zhao, Y. Jiang, H. Fan, M. Liu, O. Zhuo, X. Wang, Q. Wu, L. Yang, Y. Ma and Z. Hu, Porous 3D few-layer graphene-like carbon for ultrahigh-power supercapacitors with well-defined structure-performance relationship, Adv. Mater., 2017, 29(11), 1604569 CrossRef PubMed .
- L. Kou, Y. Ma, X. Tan, T. Frauenheim, A. Du and S. Smith, Structural and electronic properties of layered arsenic and antimony arsenide, J. Phys. Chem. C, 2015, 119(12), 6918–6922 Search PubMed .
- J. D. Wood, S. A. Wells, D. Jariwala, K.-S. Chen, E. Cho, V. K. Sangwan, X. Liu, L. J. Lauhon, T. J. Marks and M. C. Hersam, Effective passivation of exfoliated black phosphorus transistors against ambient degradation, Nano Lett., 2014, 14(12), 6964–6970 Search PubMed .
- S. A. Mian, A. Hussain, A. Basit, G. Rahman, E. Ahmed and J. Jang, Molecular modeling and simulation of transition metal–doped molybdenum disulfide biomarkers in exhaled gases for early detection of lung cancer, J. Mol. Model., 2023, 29(8), 225 CrossRef CAS PubMed .
- A. I. Ayesh, H2S and SO2 adsorption on Cu doped MoSe2: DFT investigation, Phys. Lett. A, 2022, 422, 127798 Search PubMed .
- S. Ahmad, A. Hussain, S. A. Mian, G. Rahman, S. Ali and J. Jang, Sensing and conversion of carbon dioxide to methanol using Ag-decorated zinc oxide nanocatalyst, Mater. Adv., 2024, 5(3), 1119–1129 RSC .
- M. Vanni, Functionalization of Black Phosphorus with Inorganic Reagents, 2021 Search PubMed .
- H. Sun, Y. Shang, Y. Yang and M. Guo, Realization of N-Type Semiconducting of Phosphorene through Surface Metal Doping and Work Function Study, J. Nanomater., 2018, 2018(1), 6863890 Search PubMed .
- M. Pica and R. D'Amato, Chemistry of phosphorene: synthesis, functionalization and biomedical applications in an update review, Inorganics, 2020, 8(4), 29 CrossRef CAS .
- U. O. Abu, Synthesis, Property Tuning and Degradation Studies of Phosphorene-Based Materials, 2023 Search PubMed .
- S. A. Mian, S. U. Khan, A. Hussain, A. Rauf, E. Ahmed and J. Jang, Molecular Modelling of Optical Biosensor Phosphorene-Thioguanine for Optimal Drug Delivery in Leukemia Treatment, Cancers, 2022, 14(3), 545 CrossRef CAS PubMed .
- S. Ahmad, S. A. Mian, A. Hussain, E. Ahmed, M. S. Khan, G. Rahman and J. Jang, Environmental carbonaceous aerosols conversion into methanol using a zinc-decorated ZnO nanocage, Mater. Chem. Phys., 2025, 339, 130648 CrossRef CAS .
- D. Xiao, Z. Jin, G. Sheng, L. Chen, X. Xiao, T. Shan, J. Wang, R. Navik, J. Xu and L. Zhou, Single crystals of purely organic free-standing two-dimensional woven polymer networks, Nat. Chem., 2024, 16(11), 1906–1914 Search PubMed .
- B. Liu, H. Wu and I. P. Parkin, Gaseous photocatalytic oxidation of formic acid over TiO2: A comparison between the charge carrier transfer and light-assisted mars–van krevelen pathways, J. Phys. Chem. C, 2019, 123(36), 22261–22272 Search PubMed .
- E. Zuluaga-Hernandez, M. Mora-Ramos, E. Flórez and J. Correa, Adsorption of nitrogen-based gases on different layers of blue phosphorene oxides, J. Mater. Sci., 2021, 56(28), 15824–15843 Search PubMed .
- M. Ghashghaee and M. Ghambarian, Defect engineering and zinc oxide doping of black phosphorene for nitrogen dioxide capture and detection: quantum-chemical calculations, Appl. Surf. Sci., 2020, 523, 146527 Search PubMed .
- R. Shahriar, O. Hassan and M. K. Alam, Adsorption of gas molecules on buckled GaAs monolayer: a first-principles study, RSC Adv., 2022, 12(26), 16732–16744 Search PubMed .
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a and
Joonkyung Jang
*a and
Joonkyung Jang










