DOI:
10.1039/D6RA00870D
(Paper)
RSC Adv., 2026,
16, 23770-23782
A DFT analysis of structural and electronic modulation of Cs2AgBiX6 (X = Cl, Br) via A-site NH4+ substitution for photovoltaic applications
Received
1st February 2026
, Accepted 1st April 2026
First published on 6th May 2026
Abstract
To address environmental pollution and sustainable energy challenges, lead-free Ag–Bi double perovskites Cs2AgBiX6 (X = Cl, Br) and their ammonium-substituted variants CsNH4AgBiX6 and (NH4)2AgBiX6 are investigated using first-principles FP-LAPW calculations within density functional theory. Ammonium incorporation slightly reduces lattice size while enhancing structural flexibility. Band-structure analysis (GGA, SOC, hybrid-PBE) shows decreasing band gaps with NH4 doping from 2.52 eV to 2.09 eV, with the CBM dominated by Bi states and the VBM by halide p states. Effective mass calculations indicate high carrier mobility due to the low effective mass of (NH4)2AgBiX6 (X = Cl, Br) compared to Cs-based double perovskites, which results in  values that are between 0.524 and 0.939 eV, and
values that are between 0.524 and 0.939 eV, and 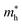 values that are between 1.2 and 1.645 eV. The stability of these compounds is confirmed through mechanical (Cij), formation of enthalpy ΔHf and Goldschmidt tolerance factor (τG) analyses. The elastic constants confirm the mechanically stable and ductile nature of these materials. Furthermore, ab initio molecular dynamics simulations and phonon band-structure calculations have been performed and confirm the stability of the materials. Optical properties reveal stronger light absorption (∼45 × 104 cm−1 in the visible region) and an enhanced dielectric response after NH4+ substitution. Band-edge alignment analysis supports the potential for photocatalytic water splitting, while SLME analysis identifies (NH4)2AgBiBr6 (ηmax = 6.47%) as the most promising photovoltaic absorber. Overall, A-site ammonium engineering effectively tunes the structural, electronic, optical, and photocatalytic properties of Ag–Bi double perovskites for energy applications.
values that are between 1.2 and 1.645 eV. The stability of these compounds is confirmed through mechanical (Cij), formation of enthalpy ΔHf and Goldschmidt tolerance factor (τG) analyses. The elastic constants confirm the mechanically stable and ductile nature of these materials. Furthermore, ab initio molecular dynamics simulations and phonon band-structure calculations have been performed and confirm the stability of the materials. Optical properties reveal stronger light absorption (∼45 × 104 cm−1 in the visible region) and an enhanced dielectric response after NH4+ substitution. Band-edge alignment analysis supports the potential for photocatalytic water splitting, while SLME analysis identifies (NH4)2AgBiBr6 (ηmax = 6.47%) as the most promising photovoltaic absorber. Overall, A-site ammonium engineering effectively tunes the structural, electronic, optical, and photocatalytic properties of Ag–Bi double perovskites for energy applications.
1 Introduction
Humans are currently dealing with a number of environmental issues, including hazardous waste, toxic air pollution and polluted groundwater. Moreover, organic pollutants have received significant attention. Many industries, including textiles, food, printing, cosmetics, and dyeing, mostly use dyes for their products. These dyes can emit significant amounts of organic-based pollutants into the water and soil, threatening both the environment and human health.1 The pressing need for alternate energy sources is highlighted by the present energy and environmental crises. The abundance, affordability, and environmental friendliness of hydrogen make it a tempting alternative; nonetheless, its widespread usage requires improvements in hydrogen storage and transportation technologies.2–6 Photocatalytic technologies outperform traditional methods by removing organic pollutants in the parts per billion (ppb) range from both air and water in a nonselective, cost-effective process that operates at ambient pressure and room temperature, and does not create polycyclic molecules.7 Photocatalysis is a photoinduced process that is accelerated by the presence of a catalyst. Three essential phases comprise the photocatalytic process: (1) charge carriers produced by photolysis; (2) charge-carrier separation and diffusion to the outermost layer of the photocatalyst; and (3) redox processes occurring on the surface of the catalyst.8 Photocatalysts are typically solid semiconductors that are chemically and biologically stable, light-absorbing, non-toxic, and low in cost.9 The development of photocatalytic devices that function efficiently under visible light is urgently needed. A number of researchers have worked to develop semiconductor-based photocatalysts with reduced band gaps, enabling absorption in the visible region; these are commonly known as second-generation photocatalysts.10 Semiconductors powered by visible light, such as Cu2O, WO3 and Fe2O3, have been used as photocatalysts because of their easy preparation, simple structure, high photosensitivity, and low toxicity.11 On the other hand, hybrid halide perovskites have attracted significant attention in photovoltaic research and development due to the rapid increase in their solar-cell power conversion efficiency, which has risen from approximately 3.8% to 23.7% over the past decade.12 Thus, it is assumed that technology associated with perovskites may be cheaper and more efficient compared to other silicon (Si)-based materials.13 Additionally, perovskites have the capability to be utilized in a vast range of optoelectronic applications, beyond their most prominent use in photovoltaics, because these materials have some remarkable physical characteristics.14–17 Innovative perovskite hydride materials have become attractive options for applications involving the storage of hydrogen. The hydrides BeXH3 (X = Pd, Ag, and Cd) and Mg2XrH6 (X = V, Cr) provide tremendous potential for advancements in energy technology.18,19 Materials based on perovskites and antiperovskites have become appealing options for solid-state hydrogen storage systems.20–23 However, the hybrid halide perovskites still face various unresolved challenges in large-scale commercial applications, as these materials are structurally unstable and easily degrade upon exposure to heat, air and moisture, and they often contain toxic lead (Pb), which is hazardous to the environment. The problem of poor perovskite stability may be addressed using numerous techniques, such as carbon encapsulation, altering the composition of the perovskite, and the incorporation of hydrophobic molecules.24 Nowadays, organic–inorganic hybrid double perovskites have received enormous focus in photovoltaic optoelectronic research because these materials are believed to have the potential to sufficiently resolve both toxicity and instability concerns.25 By modifying the B′ and B″ combinations in double perovskites A2B′B″X6, these can potentially provide a better and more effective replacement for Pb-containing perovskites in photovoltaics as well as optoelectronics. For hybrid functions such as spintronics, ferroelectricity, thermoelectricity, and optoelectronics, perovskites have been extensively researched.26 The band gap provides a basis for a compound’s optical properties, showing significant absorption and conduction in high-energy spectra, while allowing transparency for low-energy photons.27 The majority of the reported organic–inorganic hybrid double perovskites exhibit low PCE and are unsuitable for use in solar cells because they either have a large electronic band gap or low photoconductivity and absorption.28–30 According to recent studies, it may be possible to adjust and improve the characteristics of double perovskites by substituting organic MA or FA for the A-site element (Cs).31–33 Similarly, a DFT analysis of ASbCuX6 (A = Cs2/organics, X = Cl/Br/I) reveals indirect band gaps of 1–2 eV, high absorption/photoconductivity, and superior properties in Cs2SbCuI6, emphasizing the significance of cation engineering for stability and performance tuning of Pb-free photovoltaics.34 Halide-based double perovskites also show promising results for photocatalytic and photovoltaic applications using ab initio calculations.35–37 Electronic structure simulations reveal an indirect band gap with variable semiconducting characteristics for these compounds.38 Following this trend of successful A-site cation engineering, similar engineering was performed in this study for lead-free double perovskites Cs2AgBiX6 (X = Cl, Br) by partially and fully substituting the inorganic Cs+ cation with the molecular ammonium (NH4+) ion. For each halide composition, four modified structures CsNH4AgBiCl6, (NH4)2AgBiCl6, CsNH4AgBiBr6 and (NH4)2AgBiBr6 were constructed to systematically examine the influence of isovalent molecular-cation incorporation on the structural, electronic, and optoelectronic properties.
2 Computational details
To perform DFT-based computations, the WIEN2k code with the FP-LAPW method was employed.39 In order to analyze the structural, electrical, optical, photocatalytic and elastic properties, we utilized the more precise WC-GGA generalized gradient approximation as the exchange–correlation potential.40 To increase the accuracy of the determined band gaps, the modified Becke–Johnson (mBJ) potential was applied on top of the WC-GGA functional.41,42 Here, however, TB-mBJ has been used in place of HSE06 (VASP code), as it produces comparable results.43 To control plane-wave expansion based on muffin-tin radius (RMT) and maximum k-vector (kmax), RMTkmax = 7.0 was applied and −6.0 Ryd selected as a cutoff energy. The core-valence separation energy utilized in our plane-wave computations is denoted by the cutoff energy of −6.0 Ryd. This value guarantees proper handling of the core and valence states and is selected based on conventional procedures for the materials under study.44 The accuracy of DFT calculations is strongly affected by two key parameters: the number of k-points used for Brillouin zone (BZ) integration and the kinetic energy cutoff, which determines the number of plane waves in the basis set.45 Within the muffin-tin radius, the sphere has a charge density of Gmax = 20 and an angular momentum of lmax = 10. To define the structure, a denser k-mesh of 12 × 12 × 12 was used, while a denser k-mesh of 16 × 16 × 16 was used to determine the optical and electronic properties.
3 Results and discussion
3.1 Structural properties
Generally, double-perovskite compounds possess a cubic crystal structure that belongs to the Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group (number 225). Typically, double perovskites are described by the general formula A2B′B″X6. The crystal structures of Cs-containing inorganic double perovskites, i.e., Cs2AgBiCl6 and Cs2AgBiBr6 are shown in Fig. 1. Once the inorganic perovskite crystal structure was produced, it was transformed into a 1 × 1 × 1 primitive supercell, and afterwards four Cs+ cations were fully replaced by molecular ammonium (NH4+) ions. The six model structures of double perovskites under consideration, namely, Cs2AgBiCl6, CsNH4AgBiCl6, (NH4)2AgBiCl6, Cs2AgBiBr6, CsNH4AgBiBr6 and (NH4)2AgBiBr6, were put through geometry optimization to identify their optimum ground-state structures. Their structural properties, such as optimized lattice constant, density, and bond lengths are listed in Table 1. Initially, an optimization technique was used to reduce the energy to the ground-state level. As a starting point, we took the experimental lattice parameters of Cs2AgBiCl6 and Cs2AgBiBr6, which are 10.77 Å and 11.27 Å.29 The optimized lattice constants for all studied compounds ranged from 10.67 Å to 11.12 Å. The investigated lattice parameters are seen to decrease as a result of the A-site alteration. In crystalline materials, this is a common occurrence where one atom is replaced by a smaller atom, which decreases the unit cell volume. Similarly, A-site substitution induces measurable changes in N–H, Ag–(Cl/Br) and Bi–(Cl/Br) bond lengths, and the overall density of all materials, as displayed in Table 1, indicating lattice distortion and enhanced structural flexibility, which underpin the observed modulation of the electronic properties.
m space group (number 225). Typically, double perovskites are described by the general formula A2B′B″X6. The crystal structures of Cs-containing inorganic double perovskites, i.e., Cs2AgBiCl6 and Cs2AgBiBr6 are shown in Fig. 1. Once the inorganic perovskite crystal structure was produced, it was transformed into a 1 × 1 × 1 primitive supercell, and afterwards four Cs+ cations were fully replaced by molecular ammonium (NH4+) ions. The six model structures of double perovskites under consideration, namely, Cs2AgBiCl6, CsNH4AgBiCl6, (NH4)2AgBiCl6, Cs2AgBiBr6, CsNH4AgBiBr6 and (NH4)2AgBiBr6, were put through geometry optimization to identify their optimum ground-state structures. Their structural properties, such as optimized lattice constant, density, and bond lengths are listed in Table 1. Initially, an optimization technique was used to reduce the energy to the ground-state level. As a starting point, we took the experimental lattice parameters of Cs2AgBiCl6 and Cs2AgBiBr6, which are 10.77 Å and 11.27 Å.29 The optimized lattice constants for all studied compounds ranged from 10.67 Å to 11.12 Å. The investigated lattice parameters are seen to decrease as a result of the A-site alteration. In crystalline materials, this is a common occurrence where one atom is replaced by a smaller atom, which decreases the unit cell volume. Similarly, A-site substitution induces measurable changes in N–H, Ag–(Cl/Br) and Bi–(Cl/Br) bond lengths, and the overall density of all materials, as displayed in Table 1, indicating lattice distortion and enhanced structural flexibility, which underpin the observed modulation of the electronic properties.
 |
| | Fig. 1 Crystal structures of (a) Cs2AgBiX6, (b) CsNH4AgBiX6 and (c) (NH4)2AgBiX6 (where X = Cl, Br). | |
Table 1 Calculated lattice constant a (Å), Goldschmidt tolerance factor τG, octahedral factor µ, enthalpy of formation ΔHf (eV per atom), density ρ (g cm−3), and bond lengths d (Å) of AAgBiX6 (where A = Cs2, CsNH4, (NH4)2; and X = Cl, Br)
| Material |
a (Å) |
τG |
µ |
ΔHf |
ρ (g cm−3) |
d (Å) |
| N–H |
Ag–Cl/Br |
Bi–Cl/Br |
| Cs2AgBiCl6 |
10.67, 10.77 (exp)29 |
0.923 |
0.53 |
−1.55 |
4.34 |
— |
2.643 |
2.694 |
| CsNH4AgBiCl6 |
10.64 |
1.008 |
0.53 |
−1.42 |
4.75 |
1.036 |
2.635 |
2.685 |
| (NH4)2AgBiCl6 |
10.59 |
1.092 |
0.53 |
−1.28 |
4.20 |
1.037 |
2.617 |
2.681 |
| Cs2AgBiBr6 |
11.19, 11.27 (exp)29 |
0.913 |
0.49 |
−1.80 |
4.22 |
— |
2.756 |
2.839 |
| CsNH4AgBiBr6 |
11.15 |
0.993 |
0.49 |
−1.70 |
4.52 |
1.036 |
2.748 |
2.831 |
| (NH4)2AgBiBr6 |
11.12 |
1.072 |
0.49 |
−1.66 |
4.02 |
1.037 |
2.733 |
2.826 |
The Goldschmidt tolerance factor τG (ref. 46) serves as a primary indicator of the structural stability of perovskite materials. Specifically, it is particularly useful for assessing the crystalline stability of compounds in which two cations (monovalent and trivalent) coexist at the B‑site of the structure. This tolerance factor (τG) and octahedral values µ can be determined using the relations
| |
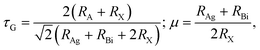 | (1) |
where
RA,
RAg,
RBi and
RX (X = Cl and Br) represent the ionic radii of Cs (or NH
4), Ag, Bi and X = Cl/Br, respectively. The ionic radii used for Cs, NH
4, Ag, Bi, Cl, and Br were 1.02, 1.46, 1.15, 1.03, 1.81, and 1.96 Å, respectively.
47 Previous statistical analyses of all existing halide perovskites have shown that
τG ranges from 0.81 to 1.11, while the optimal range for
µ is 0.42–0.75. If the material’s
τG value does not lie within the given range, it will be considered unstable. The calculated values of
τG given in
Table 1 for the studied compounds satisfy these stability criteria.
In addition, thermal stability can be quantified by calculating the formation energy (ΔHf) of the crystal,23 which is given as
| |
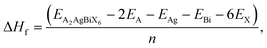 | (2) |
where
n represents the total number of atoms,
EA2AgBiX6 represents the total energy, and
EA,
EAg,
EBi, and
EX represent the energies of Cs/NH
4, Ag, Bi, and X = Cl/Br atoms, respectively. These values can be used for calculating Δ
Hf as given in
Table 1. If the values of Δ
Hf are negative, this proves the thermodynamic stability of the material. These energy parameters define the molecular structure and bonds within the crystal, providing valuable insights into its stability. Differences in stability and energy may arise due to lattice strain, ionic sizes, and bonding strengths affected by the halide ions (Br, Cl) in each material.
3.2 Electronic properties
The band structure effectively describes the physical properties of solids, such as resistivity and optical behavior. This information can also be utilized to design solar cells and transistors. For this reason, we calculated each compound’s band structure. The band gaps of all the compounds computed with GGA, SOC, and hybrid+SOC are listed in Table 2. First, we computed the band gaps of all studied compounds using GGA and obtained values ranging from 1.17 to 1.65 eV. A small reduction in the band gap is observed upon A-site substitution, the same trend that was observed in a DFT study of A-site substitution in Sb–Cu analogs ASbCuX6.34 Then, in order to obtain band gap values that were closer to the experimental band gaps, we employed SOC and then the hybrid PBE, as reported in Table 2. SOC reduced the band gaps, and subsequent use of the hybrid functional led to band gap values of 2.52, 2.44, 2.42, 2.12, 2.12 and 2.09 eV for Cs2AgBiCl6, CsNH4AgBiCl6, (NH4)2AgBiCl6, Cs2AgBiBr6, CsNH4AgBiBr6 and (NH4)2AgBiBr6, respectively. The anticipated values of the band gap for the examined double perovskites are displayed in a bar diagram in Fig. 2 and the band structures along with the density of states (DOS) are shown in Fig. 3. The findings imply that the band gaps decreased upon A-site cation engineering, proving the materials’ potential for photovoltaic applications. To determine the atoms’ contribution (Cs, N, H, Ag, Bi, Br and Cl) to the VBM and CBM, we have computed the total density of states (TDOS) of all considered materials, as given in Fig. 3. It is evident that the VBM is fully contributed by Cl and Br atoms in Cl-based and Br-based compounds, respectively. On the other hand, the CBM is contributed mainly by the Bi atom in all compounds except (NH4)2AgBiBr6, where the CBM is mainly contributed by the Br atom. To determine which orbitals of Cl, Br, Bi and Br play a role in the VBM and CBM, we also computed the partial density of states (PDOS), as highlighted in Fig. 3. Cl-(p,d), Cl-p, Cl-d, Br-(p,d), Br-d and Br-d orbitals contribute to the VBM and Bi-(p,d), Bi-d, Bi-(p,d), Bi-(p,d), Bi(p,d) and Br-d orbitals contribute to the CBM in Cs2AgBiCl6, CsNH4AgBiCl6, (NH4)2AgBiCl6, Cs2AgBiBr6, CsNH4AgBiBr6 and (NH4)2AgBiBr6 compounds, respectively.
Table 2 Band gap values of all studied compounds
| System |
Eg (eV) |
| GGA |
SOC |
Hybrid-PBE |
| Cs2AgBiCl6 |
1.65 |
1.32 |
2.52–2.77(exp)29 |
| CsNH4AgBiCl6 |
1.61 |
1.34 |
2.44 |
| (NH4)2AgBiCl6 |
1.58 |
1.33 |
2.42 |
| Cs2AgBiBr6 |
1.20 |
0.96 |
2.12–2.19(exp)29 |
| CsNH4AgBiBr6 |
1.20 |
0.98 |
2.11 |
| (NH4)2AgBiBr6 |
1.17 |
0.95 |
2.09 |
 |
| | Fig. 2 Band gap values of AAgBiX6 (where A = Cs2, CsNH4, (NH4)2; and X = Cl, Br) hybrid double perovskites obtained using the hybrid PBE functional. | |
 |
| | Fig. 3 Band structure and TDOS of (a) Cs2AgBiCl6, (b) Cs2AgBiBr6 (c) CsNH4AgBiCl6, (d) CsNH4AgBiBr6, (e) (NH4)2AgBiCl6 and (f) (NH4)2AgBiBr6 computed using the hybrid PBE scheme. | |
Large carrier mobility is crucial for capable electronic and optoelectronic devices.48 The mobility of charge carriers is higher for a smaller effective mass. Charge carriers basically fall into two categories, i.e., electrons and holes. The electron’s effective mass 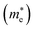 and the hole’s effective mass
and the hole’s effective mass  for all six studied compounds computed using the hybrid PBE functional are given in Table 3. It is clear that the
for all six studied compounds computed using the hybrid PBE functional are given in Table 3. It is clear that the  values are between 0.524 and 0.939 eV, which are lower than that of Si (1.09), hence it is anticipated that the carrier mobility of all six materials exceeds that of Si.49 A smaller effective mass results in higher carrier mobility. Due its value of
values are between 0.524 and 0.939 eV, which are lower than that of Si (1.09), hence it is anticipated that the carrier mobility of all six materials exceeds that of Si.49 A smaller effective mass results in higher carrier mobility. Due its value of  being the smallest (0.52 eV), the compound (NH4)2AgBiBr6 has the greatest carrier mobility among all the compounds, therefore this material is highly sought after for advanced optoelectronic and electronic applications.
being the smallest (0.52 eV), the compound (NH4)2AgBiBr6 has the greatest carrier mobility among all the compounds, therefore this material is highly sought after for advanced optoelectronic and electronic applications.
Table 3 Computed effective mass (m*/mo) of halide double perovskites AAgBiX6 (where A = Cs2, CsNH4, (NH4)2; and X = Cl, Br)
| Material |
m*/mo |
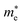
|

|
| Cs2AgBiCl6 |
0.695 |
1.619 |
| CsNH4AgBiCl6 |
0.589 |
1.644 |
| (NH4)2AgBiCl6 |
0.581 |
1.645 |
| Cs2AgBiBr6 |
0.939 |
1.344 |
| CsNH4AgBiBr6 |
0.704 |
1.201 |
| (NH4)2AgBiBr6 |
0.524 |
1.509 |
3.3 Elastic properties
Studying the elastic characteristics of solids is crucial because it provides a better understanding of how materials react to tiny mechanical stresses.50 Elastic constants quantify the material’s response to external forces, and provide information on their structural stability. For cubic crystals, there are three basic elastic parameters, namely, C11, C12, and C44. By applying a series of small, symmetry-adapted deformations to the equilibrium structures, the finite-strain approach was used to determine the elastic constants.51 The calculated elastic constants for all the species are displayed in Table 4 and represented graphically in Fig. 4(a–h). These materials satisfy the Born requirements for cubic crystal stability, specifically C11 > 0, C44 > 0, C11 + 2C12 > 0, C11 − C12 > 0 and C12 < B < C11.52 This demonstrates that these materials consistently exhibit elastic stability when deformed. The fundamental mechanical properties of the materials under study were also computed using established relationships and elastic constants. The elastic constants listed in Table 4 were employed to analyze the mechanical behavior of the investigated cubic hybrid double perovskites AAgBiX6 (A = Cs2, NH4Cs, (NH4)2 and X = Cl, Br). From the computed elastic constants C11, C12 and C44, several fundamental mechanical parameters were derived, including the bulk modulus (B), shear moduli (GV, GR and GH), Young’s modulus (Y), Pugh’s ratio (B/GH), Poisson’s ratio (ν), elastic anisotropy factor (A), Kleinman parameter (ξ), and melting temperature (Tm).53,54 The materials are shown to be mechanically stable by analyzing their mechanical properties, which include the elastic constants, the bulk modulus B, the shear modulus G, Young’s modulus Y, and Poisson’s ratio ν.55 These parameters are essential for evaluating the suitability of materials for structural and optoelectronic applications. The bulk modulus (B), which measures resistance to volume compression, ranges from 32.08 to 38.31 GPa. Among the studied systems, CsNH4AgBiCl6 exhibits the highest bulk modulus, indicating stronger resistance to hydrostatic deformation, whereas (NH4)2AgBiBr6 shows the lowest value, reflecting increased compressibility. This trend is consistent with the softer lattice induced by NH4+ substitution and the larger ionic radius of Br compared to Cl. The shear modulus, calculated using the Voigt, Reuss and Hill approximations, provides insight into the resistance against shape deformation. The Hill shear modulus (GH), which represents a realistic average of Voigt’s shear modulus (GV) and Reuss’s shear modulus (GR), varies between 8.29 and 20.87 GPa. The highest shear rigidity is observed for CsNH4AgBiBr6, while (NH4)2AgBiCl6 exhibits the lowest rigidity. This variation reflects differences in bonding strength and internal structural relaxation caused by cation substitution. Young’s modulus (Y), which quantifies stiffness, follows a similar trend and lies in the range 31.10–52.66 GPa. Compounds with higher Y values are mechanically stiffer and more resistant to elastic deformation.56 The relatively moderate Young’s modulus values indicate that these materials possess intermediate stiffness, suitable for flexible optoelectronic and photovoltaic device applications. The ductile or brittle nature of the materials was assessed using Pugh’s ratio (B/GH).57 According to the Pugh criterion, materials with B/GH > 1.75 are ductile, while those with lower values are brittle.58,59 As shown in Table 4 and Fig. 4(c, g), most compounds exhibit B/GH > values significantly larger than 1.75, suggesting ductile behavior. The Pugh ratio is higher in Cl-based double perovskites compared with Br-based counterparts, and upon incorporation of the ammonium cation NH4 ion in Cs2AgBiX6 (where X = Cl, Br), its value decreases but the ductile nature is retained. Cauchy pressure (C′ = C12 − C44) provides information about the nature of chemical bonding.60–63 Positive Cauchy pressure values generally indicate metallic or ionic bonding, while negative values imply directional or covalent bonding. The calculated Cauchy pressures reveal both positive and near-zero values, indicating a mixed ionic–covalent bonding character, with increased covalency in compounds exhibiting lower Cauchy pressure values. The Poisson’s ratio (ν) values, which reflect the degree of volume change under uniaxial stress, range from 0.25 to 0.34 for the studied compounds. These values suggest that the materials are mechanically stable and moderately compressible, consistent with predominantly ionic bonding with partial covalent contributions. Elastic anisotropy, quantified using the anisotropy factor (A), deviates significantly from unity for all compounds, confirming that these materials are elastically anisotropic. Such anisotropy is an important consideration for practical applications, as it influences crack formation and mechanical reliability under external stress. The Kleinman parameter (ξ), which characterizes the balance between bond stretching and bond bending under strain, lies in the range 0.427–0.456. These values indicate that lattice deformation in these hybrid double perovskites is primarily governed by bond-bending mechanisms, with slight enhancement of bond stretching upon NH4+ substitution. Finally, the estimated melting temperatures (Tm), derived from the elastic constant C11, fall between 927 and 967 K, indicating moderate thermal stability suitable for high-temperature optoelectronic and photovoltaic applications. Overall, the positive values of the shear constant (C′) and compliance with stability criteria confirm that all studied compounds are mechanically stable, ductile to moderately brittle, and suitable for practical device integration.
Table 4 Computed elastic properties i.e., elastic constants (C11, C12 and C44), Young’s modulus (Y), Voigt’s shear modulus (GV), Hill’s shear modulus (GH), Reuss’s shear modulus (GR), bulk modulus (B), Pugh’s ratio (B/GH), shear constant (C′), Lames coefficient (ν), Cauchy pressure (C″), Kleinman parameter (ξ), melting temperature (Tm) and anisotropy constant (A) of hybrid double perovskites AAgBiX6 (A = Cs2, CsNH4, (NH4)2 and X = Cl, Br)
| Computed property |
Cs2AgBiCl6 |
CsNH4AgBiCl6 |
(NH4)2AgBiCl6 |
Cs2AgBiBr6 |
CsNH4AgBiBr6 |
(NH4)2AgBiBr6 |
| C11 (GPa) |
69.137 |
70.112 |
65.311 |
67.871 |
67.868 |
63.289 |
| C12 (GPa) |
19.596 |
22.412 |
17.312 |
18.073 |
19.001 |
16.473 |
| C44 (GPa) |
4.8274 |
16.224 |
3.280 |
6.731 |
18.805 |
4.774 |
| 3C44 (GPa) |
14.482 |
48.672 |
9.840 |
20.194 |
56.415 |
14.322 |
| GV (GPa) |
12.804 |
19.274 |
11.567 |
13.998 |
21.036 |
−12.227 |
| GR (GPa) |
7.120 |
18.603 |
5.010 |
9.505 |
20.699 |
7.004 |
| GH (GPa) |
9.96 |
18.938 |
8.288 |
11.752 |
20.868 |
9.615 |
| Y (GPa) |
34.353 |
49.519 |
31.103 |
37.013 |
52.664 |
32.547 |
| B (GPa) |
36.109 |
38.312 |
33.311 |
34.672 |
35.356 |
32.078 |
| B/GH |
3.624 |
2.022 |
4.018 |
2.950 |
1.694 |
3.330 |
| C′ (GPa) |
24.77 |
23.850 |
23.999 |
24.898 |
24.384 |
23.407 |
| C″ (GPa) |
14.768 |
6.188 |
14.032 |
11.341 |
0.295 |
11.699 |
| ν |
0.341 |
0.284 |
0.344 |
0.322 |
0.251 |
0.330 |
| A |
0.194 |
0.680 |
0.136 |
0.270 |
0.771 |
0.203 |
| ξ |
0.432 |
0.456 |
0.427 |
0.439 |
0.448 |
0.430 |
| Tm (K) |
961.6 |
967.4 |
939.0 |
954.2 |
954.2 |
927.1 |
 |
| | Fig. 4 Elastic properties from (a–d) AAgBiCl6 and (e–h) AAgBiBr6 (A = Cs2, CsNH4, (NH4)2). | |
3.4 Optical properties
Determining the optical characteristics of novel crystals is crucial for predicting their potential performance in photovoltaic and optoelectronic devices. The computed optical properties, such as the dielectric function, optical absorption, reflectivity, conductivity, refractive index and extinction coefficient, for the selected double perovskites are depicted in Fig. 6(a, b), 7(a–d) and 8(a–d). The real and imaginary parts of the dielectric function describe how a material responds to an external electromagnetic field, especially light. In DFT/optical studies, the dielectric function can be expressed as ε(ω) = ε1(ω) + iε2(ω), where ε1 represents the real part of the dielectric constant, while ε2 represents the imaginary part.64 Table 5 summarizes the static optical parameters, namely, the static dielectric constant ε1(0), static reflectivity, R(0), and static refractive index n(0), for all studied halide double perovskites. An increase in the values of ε1(0), R(0) and n(0) from 3.81 to 5.15, 0.10 to 0.15 and 1.86 to 2.97, respectively, is observed upon partial and full A-site substitution with NH4/ This indicates increased dielectric screening and polarizability, indicating stronger light–matter interaction and improved optoelectronic performance. In order to observe the increasing or decreasing trend of the dielectric constant due to the partial and full A-site substitution with NH4, the calculated dielectric constants of the compounds studied here are presented as a bar chart in Fig. 5. For the selected materials, the trend is the reverse of the change in the value of the electronic band gap. The dielectric constant increases upon the substitution of Cs with NH4 and Cl with Br. These results also indicate that the high dielectric constant of perovskites with narrow electronic band gaps is crucial for the development of materials with the optimum characteristics for photovoltaic applications. The imaginary part of the dielectric function, ε2(ω), as shown in Fig. 6(a and b), indicates the optical absorption behavior of Ag–Bi double perovskites. In all compounds, ε2(ω) is negligible in the infrared region, confirming their semiconducting nature, while the absorption onset in the visible region corresponds to the optical band gap. Substitution of Cs with NH4 and Cl with Br causes a red-shift and enhancement of the ε2(ω) peaks due to increased orbital hybridization and band-gap narrowing. Consequently, NH4- and Br-rich compounds exhibit stronger visible-light absorption, making them more suitable for optoelectronic and photovoltaic applications. From Fig. 7 and 8, it is clear that the optical spectra show a weak response in the infrared region, confirming the semiconducting nature of all studied double perovskites. In the visible region, a rapid increase in the extinction coefficient, absorption coefficient and optical conductivity marks the onset of interband transitions. NH4 substitution enhances peak intensities and slightly red-shifts the spectra, indicating stronger light–matter interaction and improved dielectric screening. In the UV region, the optical response becomes strongest, with high absorption coefficients of 50.4 × 104 cm−1, 56.6 × 104 cm−1, 59.8 × 104 cm−1, 56.1 × 104 cm−1, 58.6 × 104 cm−1 and 61.4 × 104 cm−1 for Cs2AgBiCl6, CsNH4AgBiCl6, (NH4)2AgBiCl6, Cs2AgBiBr6, CsNH4AgBiBr6 and (NH4)2AgBiBr6, respectively, along with increased conductivity, while the reflectivity remains moderate. Meanwhile, in the visible region, absorption decreases but remains appreciable, with values of 35.2 × 104 cm−1, 29.4 × 104 cm−1, 23.6 × 104 cm−1, 52.1 × 104 cm−1, 47.7 × 104 cm−1 and 45.2 × 104 cm−1 for Cs2AgBiCl6, CsNH4AgBiCl6, (NH4)2AgBiCl6, Cs2AgBiBr6, CsNH4AgBiBr6 and (NH4)2AgBiBr6, respectively. Overall, partial to full NH4 substitution increases optical absorption, making these materials more promising for optoelectronic and photovoltaic applications. This comprehensive optical analysis demonstrates that NH4- and Br-rich Ag–Bi double perovskites possess a superior optical response and light-harvesting capability, making them suitable for future photovoltaic and optoelectronic applications.
Table 5 Static optical parameters of AAgBiX6 (A = Cs2, CsNH4, (NH4)2 and X = Cl, Br)
| Parameters |
ε1(0) |
R(0) |
n(0) |
| Cs2AgBiCl6 |
3.81 |
0.10 |
1.86 |
| CsNH4AgBiCl6 |
4.03 |
0.12 |
1.99 |
| (NH4)2AgBiCl6 |
4.19 |
0.13 |
2.02 |
| Cs2AgBiBr6 |
4.72 |
0.13 |
2.86 |
| CsNH4AgBiBr6 |
4.92 |
0.14 |
2.91 |
| (NH4)2AgBiBr6 |
5.15 |
0.15 |
2.97 |
 |
| | Fig. 5 Dielectric constants of double perovskites AAgBiX6 (A = Cs2, CsNH4, (NH4)2 and X = Cl, Br) calculated using a hybrid functional. | |
 |
| | Fig. 6 (a) Real part ε1 and (b) imaginary part ε2 of the dielectric constant of AAgBiX6 (A = Cs2, CsNH4, (NH4)2 and X = Cl, Br). | |
 |
| | Fig. 7 (a) Optical constants (n, k), (b) optical conductivity σ(ω), (c) absorption coefficient α(ω) and (d) reflectivity R(ω) of AAgBiCl6 (A = Cs2, CsNH4, (NH4)2). | |
 |
| | Fig. 8 (a) Optical constants (n, k), (b) optical conductivity σ(ω), (c) absorption coefficient α(ω) and (d) reflectivity R(ω) of AAgBiBr6 (A = Cs2, CsNH4, (NH4)2). | |
3.5 Photocatalytic properties
Using semiconductor materials as photocatalysts to drive redox processes, photocatalytic water splitting is a sustainable technique that uses solar energy to produce hydrogen (H2).36,37 With appropriate indirect band gap semiconductors, water can be dissociated to produce hydrogen using solar energy.65,66 Thus, clean, sustainable energy can be produced through the photolysis of water.67,68 Reduction of water occurs via electrons, and oxidation occurs via holes during the photocatalytic process.69 For this process, the oxidation (reduction) potential of 1.23 eV must be lower (higher) than the valence and conduction bands for any material under consideration.70 Fig. 9 displays the oxidation potential of −4.44 eV and the reduction potential of −5.67 eV. The Fermi level is set at −4.4 eV to find the band-edge positions of the VB and CB with respect to standard oxidation.71,72 The pH of the water can have an impact on the conventional redox potentials. The redox potential at different pH values can be calculated using a reported formula:73 in addition to EO2/H2O = 5.67 eV + pH × 0.059 eV, EH+/H2 = 4.44 eV + pH × 0.059 eV. For pH = 0 (typical aqueous acid conditions for redox reactions), water reduction and oxidation have standard potentials of 4.44 eV and 5.67 eV, respectively.74 The WC-GGA+mBJ functional was used to estimate the valence band (VB) and conduction band (CB) potentials for all studied compounds. From Fig. 9(a and b) it is clear that all of the materials display favorable reactions for both oxidation and reduction, except CsNH4AgBiCl6 and (NH4)2AgBiCl6, which exhibit favorable reactions only for water reduction to generate hydrogen. Thus, it is concluded that all the species can be utilized for extensive solar hydrogen production.
 |
| | Fig. 9 Photocatalytic properties of (a) AAgBiCl6 and (b) AAgBiBr6 (A = Cs2, CsNH4, (NH4)2). | |
3.6 Photovoltaic performance assessment via SLME formalism
The spectroscopic limited maximum efficiency (SLME) simulation model assesses the power conversion efficiency of solar cells. To evaluate a solar cell’s power conversion efficiency, the enlarged Shockley–Queisser (SQ) model can be utilized.75,76 SLME efficiency exceeds the SQ limit when the absorption spectrum and film thickness are taken into account. SLME has been competently utilized for Si, chalcogenides, perovskites, etc., since it was developed.77–79 The maximum theoretical efficiency is defined as
The maximum output power density, Pmax, is obtained from the voltage–current (V–I) characteristics of the solar cell, while Pin is the incident solar power under the AM1.5G spectrum, taken as 100.3 mW cm−2. In the SLME framework, Pmax is calculated from the J–V characteristics of the device:
Pmax = JV = V[JSC − J0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(eV/kT − 1)], exp(eV/kT − 1)], |
where
T,
e,
V,
JSC,
J0 and
k are the device temperature, elementary charge, voltage across the absorber, short-circuit current density, reverse-saturation current density and Boltzmann’s constant, respectively. The material’s permitted band gap, global solar spectra (AM-1.5G), and absorption spectra calculated using DFT are the basic input parameters utilized to assess SLME.
80 The computed SLME results for all studied double perovskites are shown in
Fig. 10. The energy gap, temperature and material thickness have a profound impact on the SLME. We can see that the SLME increases moderately with material thickness and eventually reaches a constant value, as displayed in
Fig. 10(a and b). By following standard SLME analysis, compared to other compounds, (NH
4)
2AgBiBr
6 has the greatest SLME efficiency of 2.96% for a 0.5-µm-thick layer. This makes (NH
4)
2AgBiBr
6 a promising compound for solar-cell applications using a 500-nm-thick layer. The same compound has a maximum efficiency of 6.67%.
 |
| | Fig. 10 SLME efficiency vs. thickness for (a) AAgBiCl6 and (b) AAgBiBr6 (A = Cs2, CsNH4, (NH4)2 and X = Cl, Br). | |
3.7 Thermal and vibrational stability
We have calculated the thermal stability (see Fig. 11) and phonon band structure (see Fig. 12) of all studied compounds. To check thermal stability, ab initio molecular dynamics simulations (AIMD)81 simulations were performed at a temperature of 300 K for a total time of 5 ps with a time interval of 1 fs, as shown in Fig. 11(a–f). It is clear that all materials maintain their cubic symmetry without any structural distortion. To further confirm the vibrational stability, we calculate the phonon band structures for all compounds, as shown in Fig. 12(a–f). We use the supercell approach (2 × 2 × 2 supercell), as implemented in the PHONOPY package,82 to perform the relevant frozen-phonon calculations. All structures are dynamically stable and exhibit no known negative frequencies, which also validates the elastic results.
 |
| | Fig. 11 Ab initio molecular dynamics calculations of the thermal stability of AAgBiX6 (A = Cs2, CsNH4, (NH4)2 and X = Cl, Br). | |
 |
| | Fig. 12 Phonon band structures of (a–c) AAgBiCVl6 and (d–f) AAgBiBr6 (A = Cs2, CsNH4, (NH4)2). | |
4 Conclusions
A detailed DFT study has been carried out to explore the impact of partial and full A-site substitution of Cs+ with NH4+ in Pb-free halide double perovskites Cs2AgBiX6 (X = Cl, Br). Structural analysis reveals that ammonium incorporation induces lattice contraction, bond-length modulation, and enhanced structural flexibility without compromising mechanical stability. Electronic structure calculations demonstrate a systematic narrowing of the band gap upon NH4+ substitution, bringing the band gaps into an optimal range for visible-light-driven applications. Density of states and effective mass analyses indicate favorable charge transport characteristics, with (NH4)2AgBiBr6 exhibiting the lowest electron effective mass and highest predicted carrier mobility. The stability of these compounds is confirmed through mechanical (Cij), formation of enthalpy ΔHf and Goldschmidt tolerance factor (τG) analyses. Elastic-property calculations confirm that all studied compounds satisfy the Born stability criteria and predominantly exhibit ductile behavior with moderate stiffness and elastic anisotropy, making them suitable for device integration. Optical investigations reveal enhanced dielectric constants, stronger absorption coefficients, and red-shifted optical responses in NH4- and Br-rich compositions, indicating improved light-harvesting capability. Band-edge alignment analysis confirms that most compounds possess appropriate conduction and valence band positions for photocatalytic water splitting, highlighting their potential for solar hydrogen production. Finally, SLME calculations identify (NH4)2AgBiBr6 as the most promising photovoltaic absorber among the studied systems, achieving the highest theoretical efficiency at practical film thicknesses. The thermal (AIMD) and dynamical (phonon) calculations confirm the stability of the studied compounds. Overall, this study demonstrates that the ammonium-based A-site cation engineering scheme is an effective and viable strategy to enhance the structural, electronic, optical, photocatalytic, and photovoltaic performance of Ag–Bi-based double perovskites. The results provide valuable theoretical guidance for the design of stable, non-toxic, and multifunctional perovskite materials for next-generation optoelectronic, photovoltaic, and photocatalytic applications.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
Data will be made available on request.
Funding
This research was funded by Ongoing Research Funding program – Research Chairs (ORF-RC-2025-5532), King Saud University, Riyadh, Saudi Arabia.
Acknowledgements
The authors extend their appreciation to Ongoing Research Funding program – Research Chairs (ORF-RC-2025-5532), King Saud University, Riyadh, Saudi Arabia. The author Y. Saeed would like to thank the Higher Education Commission (HEC) of Pakistan for providing a grant under NRPU-15844.
Notes and references
- S. Garcia-Segura and E. Brillas, Applied photoelectrocatalysis on the degradation of organic pollutants in wastewaters, J. Photochem. Photobiol., C, 2017, 31, 1–35 CrossRef CAS.
- Y. Didi, Z. E. Fatouaki, R. Touti, R. Ahfir, A. Tahiri, M. Naji, M. Idiri and A. Rjeb, First principles computational study of MgX3H8 (X = V and Fe) for hydrogen storage applications, Int. J. Hydrogen Energy, 2025, 133, 140–151 CrossRef CAS.
- Y. Didi, A. Tahiri, H. Naïli, M. B. Camara, M. Naji, R. Ahfir and R. Touti, First-principles investigation of Li3XH8 (X = Al, Ti, and Zr) complex hydrides for hydrogen storage applications, J. Phys. Chem. Solids, 2026, 209, 113274 CrossRef CAS.
- O. Eddahmani, M. Hadhoud, A. Tahiri, A. O. Tayebi Hassani and R. Touti, DFT-based study of NaYH3 and NaWH3 perovskite hydrides: structural, mechanical, electronic, and optical insights for hydrogen storage, Next Mater., 2025, 9, 101150 CrossRef CAS.
- M. Taleb, Y. Didi, O. T. H. Abdallah, R. Touti and A. Tahiri, Computational exploration of XV3H8 (X = Li and Na) hydrides for hydrogen storage applications, Chem. Phys., 2026, 600, 112916 CrossRef CAS.
- E. H. Akarchaou, et al., Computational analysis of X2MgTiH6 (X =Li, Na, and K) double perovskite hydride materials for hydrogen storage applications, Int. J. Hydrogen Energy, 2025, 161, 150644 CrossRef CAS.
- G. Liu, J. Ji, H. Huang, R. Xie, Q. Feng, Y. Shu, Y. Zhan, R. Fang, M. He, S. Liu, X. Ye and D. Y. C. Leung, UV/H2O2: An efficient aqueous advanced oxidation process for VOCs removal, Chem. Eng. J., 2017, 324, 44–50 CrossRef CAS.
- A. Mills and S. Le Hunte, An overview of semiconductor photocatalysis, J. Photochem. Photobiol., A, 1997, 108, 1–35 CrossRef CAS.
- A. Kubacka, M. Fernández-García and G. Colón, Advanced nanoarchitectures for solar photocatalytic applications, Chem. Rev., 2012, 112, 1555–1614 CrossRef CAS PubMed.
- N. Quici, M. L. Vera, H. Choi, G. L. Puma, D. D. Dionysiou, M. I. Litter and H. Destaillats, Effect of key parameters on the photocatalytic oxidation of toluene at low concentrations in air under 254+185 nm UV irradiation, Appl. Catal., B, 2010, 95, 312–319 CrossRef CAS.
- F. Wang, Y. T. Zhao, F. Feng, C. L. Li, F. Cao and W. F. Shangguan, Fabrication and shape evolution of petal-like Cu2O nanocrystal toward enhanced photoactivity and stability for hydrogen generation under visible light irradiation, J. Alloys Compd., 2016, 688, 632–638 CrossRef CAS.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, Organometal halide perovskites as visible-light sensitizers for photovoltaic cells, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- W. Zhang, G. E. Eperon and H. J. Snaith, Metal halide perovskites for energy applications, Nat. Energy, 2016, 1, 16048 CrossRef CAS.
- M. Roknuzzaman, C. Zhang, K. Ostrikov, A. Du, H. Wang, L. Wang and T. Tesfamichael, Electronic and optical properties of lead-free hybrid double perovskites for photo voltaic and optoelectronic applications, Sci. Rep., 2019, 9, 718 CrossRef PubMed.
- O. M. Bakr and O. F. Mohammed, Powering up perovskite photoresponse, Science, 2017, 355, 1260–1261 CrossRef CAS PubMed.
- Y. Yang and J. You, Make perovskite solar cells stable, Nature, 2017, 544, 155–156 CrossRef CAS PubMed.
- W.-J. Yin, T. Shi and Y. Yan, Unique properties of halide perovskites as possible origins of the superior solar cell performance, Adv. Mater., 2014, 26, 4653–4658 CrossRef CAS PubMed.
- H. Benaali, Y. Didi, A. Tahiri, H. Fatihi, R. Tobuti and M. Naji, An ab-initio study of physical properties of BeXH3 (X = Pd, Ag, and Cd) perovskites hydrides for hydrogen storage applications, Int. J. Hydrogen Energy, 2025, 180, 151745 CrossRef CAS.
- R. Oualaid, Y. El bid, N. El Biaze, R. Markazi, K. El-moudenib, M. Bouzelmad and A. Aboulkassim, Study of Mechanical, Optical, Electrical and Structural properties of Magnesium-Based double perovskites Mg2CrH6 (X= V, Cr) for hydrogen storage applications using DFT, Solid State Commun., 2025, 404, 116102 Search PubMed.
- A. E. Mekkaouy, A. Tahiri, S. Chtita and R. Touti, First principles computational study of X2CaTiH6 (X = Li, and Na) for hydrogen storage applications, Int. J. Hydrogen Energy, 2025, 164, 150723 CrossRef.
- A. Meziany, M. Essami, S. M. Aboufaris El Alaoui, Y. Didi, M. Lazrak, R. Touti, A. Tahiri and M. Naji, Toward lightweight solid-state hydrogen storage: computational investigation of potassium antiperovskites, Phys. Status Solidi A, 2026, 223, e202500168 CrossRef CAS.
- A. N. Khan, N. U. Khan, M. Kaleem, M. Tanzeel, A. Nasir, A. Hosen, A. Akremi and I. Boukhris, Lead-free X2MgGeI6 (X = Rb, Cs) double perovskites for multi-functional energy applications: a DFT and SCAPS-1D perspective, Solid State Sci., 2025, 168, 108049 CrossRef CAS.
- M. Kaleem, M. M. A. Iqbal and A. N. Khan, Stability and hydrogen storage performance of Na2LiXH6 (X = Zr, V, Cr) double perovskite hydrides via DFT and AIMD, RSC Adv., 2026, 16, 995–1007 RSC.
- P. Zhang, J. Yang and S. H. Wei, Manipulation of cation combinations and configurations of halide double perovskites for solar cell absorbers, J. Mater. Chem. A, 2018, 6, 1809–1815 Search PubMed.
- Z. Xiao, K. Z. Du, W. Meng, J. Wang, D. B. Mitzi and Y. Yan, Intrinsic instability of Cs2In (I)M(III)X6 (M = Bi, Sb; X = Halogen) double perovskites: a combined density functional theory and experimental study, J. Am. Chem. Soc., 2017, 139, 6054–6057 CrossRef CAS PubMed.
- A. EL Mekkaouy, I. J. Idrissi, S. Chtita, A. Jabar, M. Naji, A. Tahiri and R. Touti, Investigation of the multifunctional properties of X2TiF5 (X = Mg, Ca, Sr) for use in optoelectronics and radiation shielding”, Mater. Today Chem., 2025, 47, 102813 CrossRef CAS.
- T. Karafi, E. M. Hrida, M. Idiri, Y. Didi, A. Tahiri, R. Touti and M. Naji, DFT-Based Ab Initio Calculations of Structural, Electronic, Mechanical, and Optical Properties of Ga-based Fluoroperovskite GaXF3 (X = Ca and Sr), ChemistrySelect, 2025, 10, e202404985 Search PubMed.
- M. R. Filip, S. Hillman, A. A. Haghighirad, H. J. Snaith and F. Giustino, Band gaps of the lead-free halide double perovskites Cs2BiAgCl6 and Cs2BiAgBr6 from theory and experiment, J. Phys. Chem. Lett., 2016, 7, 2579–2585 CrossRef CAS PubMed.
- E. T. McClure, M. R. Ball, W. Windl and P. M. Woodward, Cs2AgBiX6 (X = Br, Cl): new visible light absorbing, lead-free halide perovskite semiconductors, Chem. Mater., 2016, 28, 1348–1354 CrossRef CAS.
- Z. Deng, F. Wei, S. Sun, G. Kieslich, A. K. Cheetham and P. D. Bristowe, Exploring the properties of lead-free hybrid double perovskites using a combined computational-experimental approach, J. Mater. Chem. A, 2016, 4, 12025–12029 RSC.
- M. Roknuzzaman, K. Ostrikov, H. Wang, A. Du and T. Tesfamichael, Towards lead-free perovskite photovoltaics and optoelectronics by ab-initio simulations, Sci. Rep., 2017, 7, 14025 CrossRef PubMed.
- M. Roknuzzaman, K. Ostrikov, K. Chandula Wasalathilake, C. Yan, H. Wang and T. Tesfamichael, Insight into lead-free organic-inorganic hybrid perovskites for photovoltaics and optoelectronics: a first-principles study, Org. Electron., 2018, 59, 99–106 CrossRef CAS.
- M. Roknuzzaman, C. Zhang, K. Ostrikov, A. Du, H. Wang, L. Wang and T. Tesfamichael, Electronic and optical properties of lead-free hybrid double perovskites for photovoltaic and optoelectronic applications, Sci. Rep., 2019, 9, 718 Search PubMed.
- M. Roknuzzaman, J. A. Alarco, H. Wang and K. K. Ostrikov, Structural, electronic and optical properties of lead-free antimony-copper based hybrid double perovskites for photovoltaics and optoelectronics by first principles calculations, Comput. Mater. Sci., 2021, 186, 110009 CrossRef CAS.
- M. A. Ullah, M. Kaleem, A. Nasir, Z. Sarfraz, M. M. A. Iqbal, M. Rizwan, K. N. Riaz and M. Tanzeel, An approach towards next-generation hydrogen storage: a DFT study on A2LiTiH6 (A = K, Ca) perovskite hydrides, RSC Adv., 2025, 15, 38714 RSC.
- M. M. Asif Iqbal, M. Abaid Ullah, M. Kaleem and A. N. Khan, Quantum chemical investigation of A2LiBiI6 perovskites with Na, K, and Rb for photocatalytic water-splitting application, npj Clean Energy, 2026, 2, 1 CrossRef.
- A. N. Khan, M. Kaleem, N. U. Khan, A. Nasir, A. Khan and M. Z. Abbasi, Multi-functional DFT and SCAPS-1D analysis of lead-free Z2MgGeI6 (Z = Na, K) double perovskites for optoelectronic, photo-catalytic, and photovoltaic applications, Sol. Energy Mater. Sol. Cells, 2026, 294, 113922 CrossRef CAS.
- A. Moutaouaffiq, A. El Mekkaouy, Y. Didi, A. Tahiri, A. Rjeb and R. Touti, Holmium doping effects on the structural and functional properties of BaTiO3: Combined sol-gel and first-principles investigation, Ceram. Int., 2026, 52, 12862–12880 CrossRef CAS.
- P. Blaha, K. Schwarz, F. Tran, R. Laskowsk, G. K. H. Madsen and L. D. Marks, WIEN2k: An APW+lo program for calculating the properties of solids, J. Chem. Phys., 2020, 152, 074101 CrossRef CAS PubMed.
- Z. Wu and R. E. Cohen, Generalized gradient approximation made more accurate for solids, Phys. Rev. B:Condens. Matter Mater. Phys., 2006, 73, 235116 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple”, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- E. Engel and S. H. Vosko, Exact exchange-only potentials and the virial relation as microscopic criteria for generalized gradient approximations, Phys. Rev. B:Condens. Matter Mater. Phys., 1993, 47, 13164 Search PubMed.
- Y. Liu, I. J. Cleveland, M. N. Tran and E. S. Aydil, Stability of the halide double perovskite Cs2AgInBr6, J. Phys. Chem. Lett., 2023, 14, 3000–3006 CrossRef CAS PubMed.
- A. N. Khan, N. U. Khan, A. K. Alqorashi, H. U. Shah, A. Khan, Z. khan and A. Hosen, First-principles study of lead-free double perovskites Cs2MgGeBr6 and Rb2MgGeBr6 for energy applications, Phys. B, 2025, 717, 417848 CrossRef CAS.
- F. Tran and P. Blaha, Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential, Phys. Rev. Lett., 2009, 102, 226401 Search PubMed.
- V. M. Goldschmidt, Die gesetze der krystallochemie, Naturwissenschatfen, 1926, 14, 477–485 CrossRef CAS.
- W. Travis, E. N. K. Glover, H. Bronstein, D. O. Scanlon and R. G. Palgrave, On the application of the tolerance factor to inorganic and hybrid halide perovskites: a revised system, Chem. Sci., 2016, 7, 4548 RSC.
- Y. Liu, X. Duan, Y. Huang and X. Duan, Two-dimensional transistors beyond graphene and TMDCs, Chem. Soc. Rev., 2018, 47, 6388–6409 RSC.
- M. A. Green, Intrinsic concentration, effective densities of states, and effective mass in silicon, J. Appl. Phys., 1990, 67, 2944–2954 CrossRef CAS.
- M. Abdellaoui, R. Touti, A. El Mekkaouy, E. H. Akarchaou, L. Talha, A. Tahiri, M. Filali and S. Chtita, Ab initio study of the structural, electronic, and optical properties of fluoro-double perovskites A2SbAgF6 (A = Li, K, Rb) for photovoltaic and optoelectronic applications, Next Mater., 2026, 10, 101548 Search PubMed.
- N. Gaidi, A. EL Mekkaouy, L. Talha, M. Filali, S. Chtita and R. Touti, First-principles insights into lead-free double perovskites X2SbAgBr6 (X = Li, Na, K) for sustainable optoelectronic applications, Sci. Afr., 2026, 31, e03189 CAS.
- S. Chaba Mouna, M. Radjai, M. A. Rahman, A. Bouhemadou, A. Abdullah, D. Houatis, D. Allali, S. S. Essaoud and H. Allaf, Physical properties of Be-based fluoroperovskite compounds XBeF3 (X = K, Rb): a first-principles study, J. Phys.: Condens. Matter, 2024, 36, 055701 CrossRef PubMed.
- L. D. Whalley, J. M. Frost, Y.-K. Jung and A. Walsh, Perspective: Theory and simulation of hybrid halide perovskites, J. Chem. Phys., 2017, 146, 220901 CrossRef PubMed.
- W. J. Yin, J. H. Yang, J. Kang, Y. Yan and S. H. Wei, Halide perovskite materials for solar cells: a theoretical review, J. Mater. Chem. A, 2015, 3, 8926–8942 RSC.
- A. Moutaouaffiq, A. El Mekkaouy, H. Benaali, E. H. Akarchaou, N. El Gaidi, A. K. Alanazi, A. Tahiri and R. Touti, Exploring the multifunctional properties of Sr2BiXO6 (X = La, Y) double perovskites via DFT: A path toward photocatalytic applications, J. Phys. Chem. Solids, 2026, 209, 113276 CrossRef CAS.
- Z. Ali, A. Razzaq, S. M. Ali, M. U. Saeed, H. O. Alansary, I. M. Moussa, M. A. El-Sheikh, A. U. R. Bacha and Y. saeed, A dft study of structural, electronic, optical, and thermoelectric properties of TMX (TM = Mo and W; X= N, P, and As) compounds, J. Electron. Mater., 2024, 53, 3834–3847 Search PubMed.
- S. F. Pugh, Relations between the elastic moduli and the plastic properties of polycrystalline pure metals, Phil. Mag., 1954, 45, 823–843 CAS.
- D. G. Isaak, E. K. Graham, J. D. Bass and H. Wang, The elastic properties of single-crystal fayalite as determined by dynamical measurement techniques, Pure Appl. Geophys., 1993, 141, 393–414 CrossRef.
- A. N. Khan, S. Rabhi, N. U. Khan, S. A. Ansari, S. Sadaf and M. W. Alam, Harnessing solar energy with lead-free Tl2BPI6 (B = Cs, Rb) double perovskites for photocatalytic water splitting, Ceram. Int., 2025, 51, 59579–59589 CrossRef CAS.
- N. Korozlu, K. Colakoglu, E. Deligoz and G. Surucu, First-principles study of structural, elastic, lattice dynamical and thermodynamical properties of GdX (X = Bi, Sb), Philos. Mag., 2010, 90, 1833–1852 CrossRef CAS.
- G. Pagare, S. S. Chouhan, P. Soni, S. P. Sanyal and M. Rajagopalan, First principles study of structural, electronic and elastic properties of lutetium mono-pnictides, Comput. Mater. Sci., 2010, 50, 538–544 CrossRef CAS.
- D. G. Pettifor, Theoretical predictions of structure and related properties of intermetallics, Mater. Sci. Technol., 1992, 8, 345 CrossRef CAS.
- M. H. Rubel, M. Mozahar Ali, M. S. Ali, R. Parvin, M. M. Rahaman, K. M. Hossain, M. I. Hossain, A. K. M. A. Islam and N. Kumada, First-principles study: Structural, mechanical, electronic and thermodynamic properties of simple-cubic-perovskite (Ba0.62K0.38)(Bi0.92Mg0.08)O3, Solid State Commun., 2019, 288, 22–27 CrossRef CAS.
- M. A. Khan, A. Kashyap, A. K. Solanki, T. Nautiyal and S. Auluck, Interband optical properties of Ni3Al, Phys. Rev. B:Condens. Matter Mater. Phys., 1993, 48, 16974 CrossRef CAS PubMed.
- K. Maeda and K. Domen, Photocatalytic water splitting: recent progress and future challenges, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- R. M. Navarro
Yerga, M. C. Alvarez Galvan, F. Del Valle, J. A. Villoria de la Mano and J. L. Fierro, Water splitting on semiconductor catalysts under visible-light irradiation, ChemSusChem, 2009, 2, 471–485 CrossRef PubMed.
- F. E. Osterloh, Inorganic materials as catalysts for photochemical splitting of water, Chem. Mater., 2008, 20, 35–54 CrossRef CAS.
- X. Hu, G. Li and J. C. Yu, Design, fabrication, and modification of nanostructured semiconductor materials for environmental and energy applications, Langmuir, 2010, 26, 3031–3039 Search PubMed.
- V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Splitting water with cobalt, Angew. Chem., Int. Ed., 2011, 50, 7238–7266 CrossRef CAS PubMed.
- A. Kudo, Photocatalysis and solar hydrogen production, Pure Appl. Chem., 2007, 79, 1917–1927 CrossRef CAS.
- J. Liu, X. Fu, S. Chen and Y. Zhu, Electronic structure and optical properties of AgP3PO4 photocatalyst calculated by hybrid density functional method, Appl. Phys. Lett., 2011, 99, 191903 CrossRef.
- F. Mouhat and F. X. Coudert, Necessary and sufficient elastic stability conditions in various crystal systems, Phys. Rev. B:Condens. Matter Mater. Phys., 2014, 90, 224104 CrossRef.
- K. D. Pham, First-principles prediction of electronic, mechanical, transport and optical properties of the silicane/Ga2SSe heterostructure, RSC Adv., 2022, 12, 31935–31942 RSC.
- Z. Huang, C. He, X. Qi, H. Yang, W. Liu, X. Wei, X. Peng and J. Zhong, Band structure engineering of monolayer MoS2 on h-BN: first-principles calculations, J. Phys. D: Appl. Phys., 2014, 47, 075301 CrossRef CAS.
- L. Yu and A. Zunger, Identification of potential photovoltaic absorbers based on first-principles spectroscopic screening of materials, Phys. Rev. Lett., 2012, 108, 068701 Search PubMed.
- W. Shockley and H. J. Queisser, Detailed balance limit of efficiency of p–n junction solar cells, J. Appl. Phys., 1961, 32, 510 CrossRef CAS.
- I. H. Lee, J. Lee, Y. J. Oh, S. Kim and K. J. Chang, Computational search for direct band gap silicon crystals, Phys. Rev. B:Condens. Matter Mater. Phys., 2014, 90, 115209 CrossRef.
- W.-J. Yin, T. Shi and Y. Yan, Unique properties of halide perovskites as possible origins of the superior solar cell performance, Adv. Mater., 2014, 26, 4653–4658 CrossRef CAS PubMed.
- W.-J. Yin, J.-H. Yang, J. Kang, Y. Yan and S.-H. Wei, Halide perovskite materials for solar cells: a theoretical review, J. Mater. Chem. A, 2015, 3, 8926–8942 RSC.
- R. M. Abraham, J. Alvarez-Muniz, C. A. Arguelles, A. Ariga, T. Bostan, M. Bustamante and A. Camming, Tau neutrinos in the next decade form GeV to EeV, J. Phys. G: Nucl. Part. Phys., 2022, 49, 110501 CrossRef CAS.
- R. Yuan, J. A. Napoli, C. Yan, O. Marsalek, T. E. Markl and M. D. Fayer, Tracking Aqueous Proton Transfer by Two-Dimensional Infrared Spectroscopy and ab Initio Molecular Dynamics Simulations, ACS Cent. Sci., 2019, 5, 1269–1277 CrossRef CAS PubMed.
- A. Togo, L. Chaput, T. Tadano and I. Tanaka, Implementation strategies in phonopy and phono3py, J. Phys.:Condens. Matter, 2023, 35, 353001 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Hosam O. Elansaryc,
Ihab Mohamed Moussad,
Sohail Mumtaz
b,
Hosam O. Elansaryc,
Ihab Mohamed Moussad,
Sohail Mumtaz values that are between 0.524 and 0.939 eV, and
values that are between 0.524 and 0.939 eV, and  values that are between 1.2 and 1.645 eV. The stability of these compounds is confirmed through mechanical (Cij), formation of enthalpy ΔHf and Goldschmidt tolerance factor (τG) analyses. The elastic constants confirm the mechanically stable and ductile nature of these materials. Furthermore, ab initio molecular dynamics simulations and phonon band-structure calculations have been performed and confirm the stability of the materials. Optical properties reveal stronger light absorption (∼45 × 104 cm−1 in the visible region) and an enhanced dielectric response after NH4+ substitution. Band-edge alignment analysis supports the potential for photocatalytic water splitting, while SLME analysis identifies (NH4)2AgBiBr6 (ηmax = 6.47%) as the most promising photovoltaic absorber. Overall, A-site ammonium engineering effectively tunes the structural, electronic, optical, and photocatalytic properties of Ag–Bi double perovskites for energy applications.
values that are between 1.2 and 1.645 eV. The stability of these compounds is confirmed through mechanical (Cij), formation of enthalpy ΔHf and Goldschmidt tolerance factor (τG) analyses. The elastic constants confirm the mechanically stable and ductile nature of these materials. Furthermore, ab initio molecular dynamics simulations and phonon band-structure calculations have been performed and confirm the stability of the materials. Optical properties reveal stronger light absorption (∼45 × 104 cm−1 in the visible region) and an enhanced dielectric response after NH4+ substitution. Band-edge alignment analysis supports the potential for photocatalytic water splitting, while SLME analysis identifies (NH4)2AgBiBr6 (ηmax = 6.47%) as the most promising photovoltaic absorber. Overall, A-site ammonium engineering effectively tunes the structural, electronic, optical, and photocatalytic properties of Ag–Bi double perovskites for energy applications.




 and the hole’s effective mass
and the hole’s effective mass  for all six studied compounds computed using the hybrid PBE functional are given in Table 3. It is clear that the
for all six studied compounds computed using the hybrid PBE functional are given in Table 3. It is clear that the  values are between 0.524 and 0.939 eV, which are lower than that of Si (1.09), hence it is anticipated that the carrier mobility of all six materials exceeds that of Si.49 A smaller effective mass results in higher carrier mobility. Due its value of
values are between 0.524 and 0.939 eV, which are lower than that of Si (1.09), hence it is anticipated that the carrier mobility of all six materials exceeds that of Si.49 A smaller effective mass results in higher carrier mobility. Due its value of  being the smallest (0.52 eV), the compound (NH4)2AgBiBr6 has the greatest carrier mobility among all the compounds, therefore this material is highly sought after for advanced optoelectronic and electronic applications.
being the smallest (0.52 eV), the compound (NH4)2AgBiBr6 has the greatest carrier mobility among all the compounds, therefore this material is highly sought after for advanced optoelectronic and electronic applications.










