
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic palladium coordination cages formed by self-assembly of extended tetra(nitrile)cavitands
Zsolt Csók *ab,
Leonidas-Dimitrios Syntrivanisc,
Daniel Häussingerd and
László Kollár*ab
*ab,
Leonidas-Dimitrios Syntrivanisc,
Daniel Häussingerd and
László Kollár*ab
aDepartment of Inorganic Chemistry, University of Pécs and János Szentágothai Science Center, P.O. Box 266, H-7624 Pécs, Hungary. E-mail: zscsok@gamma.ttk.pte.hu
bHUN-REN-PTE Selective Chemical Syntheses Research Group, Ifjúság u. 6, H-7624 Pécs, Hungary. E-mail: kollar@gamma.ttk.pte.hu
cDepartment of Chemistry, University of Colorado, Boulder, Colorado 80309, USA
dNMR Laboratory, Department of Chemistry, University of Basel, St. Johanns Ring 19, CH-4056 Basel, Switzerland
First published on 11th February 2026
Abstract
Two extended tetra(4-cyanophenylethynyl)cavitands with methylene (6S) and ethylene (6L) bridges were synthesized for coordination-driven self-assembly. Reaction with Pd(dppp)(OTf)2 in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio afforded discrete, symmetric coordination cages (8S and 8L), characterized by NMR, IR and DOSY. The assemblies exhibit dynamic behavior that is dependent on concentration, with intermediate species identified through NMR titration studies. No cage formation was observed with Pt(II) or Ni(II) analogues, highlighting the importance of the kinetic lability of Pd(II) species, the size of the metal center and the geometry of the dppp ligand.
:4 ratio afforded discrete, symmetric coordination cages (8S and 8L), characterized by NMR, IR and DOSY. The assemblies exhibit dynamic behavior that is dependent on concentration, with intermediate species identified through NMR titration studies. No cage formation was observed with Pt(II) or Ni(II) analogues, highlighting the importance of the kinetic lability of Pd(II) species, the size of the metal center and the geometry of the dppp ligand.
Introduction
Resorcin[4]arenes and related cavitands constitute well-established scaffolds for the design and synthesis of large-scale molecular containers.1,2 Once a functionally tailored molecular subunit is accessible, self-assembly provides a powerful strategy for constructing increasingly complex nanoscale architectures.3 Initially, self-assembly through hydrogen bonding was employed to link two or more cavitand-based subunits, typically bearing N–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functionalities within self-complementary motifs such as ureas, imides or carboxamides.4–9
O functionalities within self-complementary motifs such as ureas, imides or carboxamides.4–9
Self-assembly through highly directional metal–ligand coordination bonds offers another opportunity for the preparation of nanoscale molecular containers.10 Dalcanale and co-workers reported the synthesis and host–guest properties of coordination cages assembled from tetradentate cavitand ligands and square-planar Pd(II) and Pt(II) complexes, such as M(dppp)(OTf)2 (M = Pt, Pd; dppp = 1,3-bis(diphenylphosphino)propane) (Scheme 1a).11–15 The efficiency of the self-assembly process is governed by several factors, including: (a) the size of the chelate ring in the square-planar metal complex, (b) the intrinsic properties of the metal centre, and (c) the degree of preorganization within the cavitand scaffold.
 | ||
| Scheme 1 Representative examples of coordination cages. | ||
This concept was further advanced by Kobayashi et al., who demonstrated the formation of both homo- and heterocavitand cages under thermodynamic or kinetic control (Scheme 1b and c).16–18 Hong and co-workers reported the synthesis of self-assembled nanocapsules constructed from tetra(pyridyl)cavitands and M(dppp)(OTf)2 complexes (M = Pt, Pd).19,20 Haino et al. prepared an octadentate cavitand bearing four bipyridyl ligands, which coordinates four silver(I) cations in a tetrahedral geometry, resulting in the self-assembly of a discrete coordination cage.21 Additionally, hybrid supramolecular capsules were synthesized through the controlled self-assembly of two precisely designed cavitand subunits that are held together by multiple hydrogen and metal–ligand coordination bonds.22–24
In recent years, we have developed a novel family of enlarged cavitands, including a tetra(4-cyanophenylethynyl)cavitand designed for applications in nonlinear optics.25–28 This compound may enable access to the missing member of the coordination cages derived from tetra(nitrile)cavitands. In view of the limited solubility of the compound in common organic solvents, we also sought to improve solubility by appending long alkyl chains to the lower rim of the cavitand scaffold. In this study, we report the coordination chemistry of two tetra(4-cyanophenylethynyl)cavitands with a series of square-planar cis-metal triflate complexes, including Pd(dppp)(OTf)2. The enlarged lateral portals characteristic of this cavitand family may impose constraints on their efficacy as host molecules, as previously demonstrated for a related hydrogen-bonded bimolecular capsule.29 Consequently, encapsulation studies were not pursued for these new coordination cages, as their design does not favor stable guest inclusion.
Results and discussion
Synthesis of new tetra(4-cyanophenylethynyl)cavitands
Tetra(4-cyanophenylethynyl)cavitand (6S), featuring methylene bridges and methyl ‘feet’, was synthesized following established procedures.28 The acid-catalyzed condensation between resorcinol and aldehyde can yield various tetrameric resorcin[4]arene isomers exhibiting different conformations (cone, boat, chair, or diamond) as well as different relative configurations of the bridging alkyl/aryl groups (rccc, rcct, rctt, rtct).30–33 Under standard conditions, the condensation of 2-methylresorcinol with undecanal produced a mixture of isomeric macrocycles that proved inseparable by preparative chromatography. Consequently, this isomeric mixture was used directly in subsequent reactions without further purification. The major component (1L) was identified in the 1H NMR spectrum by: (a) singlets corresponding to both aromatic and phenolic protons, and (b) a triplet associated with the bridging methine protons. This NMR pattern is consistent with a cone conformation featuring all four decyl ‘feet’ in axial positions (rccc stereoisomer). Subsequently, tetra(iodo)cavitand 4L was synthesized from this mixture in three consecutive steps, without isolating the intermediates. Despite extensive purification efforts, these intermediates (2L, 3L) remained contaminated with various isomers.First, tetra(methyl)resorcin[4]arene 1L was bridged using ethylene di(p-toluenesulfonate) to afford tetra(methyl)cavitand 2L (Scheme 2). This intermediate was then selectively brominated with N-bromosuccinimide (NBS) in the presence of 1,1′-azobis(cyclohexanecarbonitrile) (ACHN) as a radical initiator to yield tetra(bromomethyl)cavitand 3L. Subsequent reaction of 3L with an excess of 4-iodophenol in the presence of sodium hydroxide furnished tetra(iodo)cavitand 4L. Notably, cavitand 4L was isolated in pure cone conformation with an overall 67% yield across the three-step sequence. Next, trimethylsilylethynyl groups were introduced at the upper rim of the cavitand scaffold under standard Sonogashira coupling conditions. Removal of the trimethylsilyl protecting groups using tetrabutylammonium fluoride trihydrate (TBAF·3H2O) provided the corresponding tetra(ethynyl)cavitand 5L. Finally, a second Sonogashira coupling of 5L with 4-iodobenzonitrile, in the presence of Pd(OAc)2/PPh3/CuI in situ catalytic system, enabled the installation of a third row of aromatic panels functionalized with nitrile groups.34
 | ||
| Scheme 2 Synthesis of extended tetra(4-cyanophenylethynyl)cavitands 6S and 6L. | ||
Cavitands 4L–6L were fully characterized using standard techniques, including 1H and 13C NMR spectroscopy as well as ESI-MS. Notably, cavitand 6L – featuring ethylene bridges and decyl ‘feet’ – exhibited broad NMR signals in CD2Cl2 at room temperature, attributable to rapid interconversion between two equivalent flattened C2v cone conformers.11,35 Furthermore, as anticipated, 6L displayed significantly enhanced solubility relative to 6S.
Coordination chemistry of the extended tetra(4-cyanophenyl-ethynyl)cavitands 6S and 6L
New coordination compounds were obtained in 80–85% yields by reacting tetra(4-cyanophenylethynyl)cavitands 6S or 6L with Pd(dppp)(OTf)2 in a 2:4 molar ratio at room temperature in CH2Cl2. The 1H, 13C, and 31P NMR spectra of the resulting main products indicate the formation of highly symmetric species in CD2Cl2 (Fig. 1d and e; see also SI). In the case of the complex derived from 6L, the 1H NMR signals appear as very broad resonances likely due to dynamic equilibria and increased conformational flexibility of the ethylene-bridged cavitand. In contrast, the signals for the methylene-bridged cavitand 6S remain sharp under identical conditions. Upon coordination, the 1H signals for both cavitand scaffolds experience a slight upfield shift of approximately 0.02 ppm. A subset of aromatic protons of the Pd(dppp) fragment appear at 7.72 ppm (0.1 ppm downfield compared to uncoordinated Pd(dppp)(OTf)2), while the P–CH2 signals shift downfield by 0.14 ppm, resonating at 2.88 ppm after complexation. Because of severe spectral overlap in the aromatic region, the aromatic protons adjacent to the cyano groups could not be assigned unambiguously. In both complexes, the 31P resonance was shifted upfield by approximately 3 ppm upon coordination. However, a minor signal corresponding to an unidentified side product appeared at 6.6 ppm, accounting for approximately 10% of the mixture in both cases. The triflate counterions produced a singlet at −78.5 ppm in the 19F NMR spectra, indicating that all triflate anions remain outside the cavitand cavity.11 Owing to the limited solubility of 8S in CD2Cl2, detailed NMR studies, including diffusion-ordered spectroscopy (DOSY), were carried out on the more soluble analogue 8L. Interestingly, the IR spectra revealed the presence of both uncoordinated (ca. 2210 cm−1) and coordinated (ca. 2270 cm−1) nitrile stretching bands for both complexes (see Fig. S17 and S23 in the SI).
 | ||
| Fig. 1 1H NMR spectra (500 MHz, CD2Cl2) of (a) 6S; (b) 6L; (c) Pd(dppp)(OTf)2; (d) 8S and (e) 8L. Asterisks (*) indicate residual CD2Cl2 and minor impurities, including water. | ||
Variable-temperature 1H NMR spectra of compound 8L were recorded between 238 K and 388 K in CDCl2–CDCl2 (Fig. S19 in the SI). Upon increasing the temperature, the signals shifted gradually downfield, the broad resonances partially sharpened and began to display fine structure, consistent with an increased exchange rate between closely related sub-conformations of compound 8L. Unfortunately, the compound decomposed above 388 K before reaching fast-exchange conditions for all resonances. At lower temperatures, the resonances broadened even further, and coalescence was observed for several resonances (e.g. at 3.8 and 4.2 ppm). However, the signals did not resolve into sharp peaks in the slow-exchange regime even at 238 K; further cooling was prevented by freezing of the solvent.
The DOSY experiments were carefully set up to ensure a negligible temperature gradient, thereby providing reliable diffusion coefficients (see Experimental). In 8L, the signals arising from both the cavitand skeleton and the Pd(dppp) moieties diffuse together as a single molecular entity. DOSY measurements in CDCl2–CDCl2 (3.4 mM, 298 K) yielded average diffusion coefficients of 8.06 × 10−11 m2 s−1 (range: 7.55 to 8.71 × 10−11 m2 s−1) for the free tetradentate ligand 6L and 6.45 × 10−11 m2 s−1 (range: 6.15 to 6.74 × 10−11 m2 s−1) for the coordination complex 8L, respectively (Fig. S11 and S21 in the SI). The observed decrease in diffusion coefficient upon complexation reflects the formation of a larger, more slowly diffusing entity. The magnitude of this change is consistent with reported values for similar cavitand-based monomer/dimer systems.21,29,36,37 Derived from the Stokes–Einstein equation, the cubed ratio of the two diffusion coefficients corresponds to the inverse ratio of their molar volumes.38 On this basis, the volume of complex 8L was calculated to be 1.95 times that of 6L. These results rule out intramolecular complexation, where coordination would involve two adjacent nitrile groups on the same cavitand, and instead support the formation of an intermolecularly bridged self-assembly.
NMR titration experiments were performed to investigate the formation of the coordination complex derived from cavitand 6L. Upon addition of 0.5 equiv. of Pd(dppp)(OTf)2 to a 3 mM CD2Cl2 solution of 6L, diagnostic downfield 1H and upfield 31P NMR signals emerged, indicating complex formation (Fig. S24 and S25 in the SI). Further stepwise additions of the Pd precursor (up to 4 equiv.) led to gradual upfield shifts of the characteristic proton resonances, while the 31P signal shifted downfield. For comparison, mono- and bis-benzonitrile coordination complexes were also synthesized as reference compounds. The 31P NMR signal of Pd(dppp)(PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N)(OTf)2 appears at 15.5 ppm in CD2Cl2, closely matching the resonance observed at a 6L/[Pd] molar ratio of 2:8. Similarly, the 31P resonance of Pd(dppp)(PhCN)2(OTf)2 is found at 14.5 ppm in CD2Cl2, nearly identical to the signal observed at a 6L/[Pd] ratio of 2:4 and corresponding to the resonance of the isolated coordination complex 8L. This behavior contrasts with that of smaller coordination cages, where the cage was the sole product regardless of the stoichiometric excess of either component (Scheme 1a).11 Furthermore, the formation of the cage complex 8L from the reaction of 6L with Pd(dppp)(OTf)2 in a 2:4 molar ratio exhibits concentration dependence: the equilibrium shifts toward cage formation at higher concentrations (Fig. S26 in the SI).
N)(OTf)2 appears at 15.5 ppm in CD2Cl2, closely matching the resonance observed at a 6L/[Pd] molar ratio of 2:8. Similarly, the 31P resonance of Pd(dppp)(PhCN)2(OTf)2 is found at 14.5 ppm in CD2Cl2, nearly identical to the signal observed at a 6L/[Pd] ratio of 2:4 and corresponding to the resonance of the isolated coordination complex 8L. This behavior contrasts with that of smaller coordination cages, where the cage was the sole product regardless of the stoichiometric excess of either component (Scheme 1a).11 Furthermore, the formation of the cage complex 8L from the reaction of 6L with Pd(dppp)(OTf)2 in a 2:4 molar ratio exhibits concentration dependence: the equilibrium shifts toward cage formation at higher concentrations (Fig. S26 in the SI).
Together, these findings support the conclusion that cavitand 6L and Pd(dppp)(OTf)2 self-assemble at a 2:4 molar ratio to form coordination cage 8L in CD2Cl2 and in CDCl2–CDCl2 (Scheme 3). Formation of this highly symmetric D4h cage structure requires that the tetra(4-cyanophenylethynyl)cavitand subunits adopt a C4v symmetry cone conformer, in which all four nitriles have the same angle with respect to the C4 symmetry axis.11 NMR titration experiments indicate a dynamic equilibrium between free cavitand 6L, coordination cage 8L, and an intermediate species 7L, in which only one nitrile group coordinates to the palladium center. The intermediate is presumed to exist in a state of rapid equilibrium between two equivalent C2v flattened cone conformers. The pronounced line broadening observed in the NMR spectra suggests rapid exchange between all these species on the NMR timescale. In contrast, the faster temporal resolution of IR spectroscopy enables differentiation between uncoordinated (ca. 2210 cm−1) and coordinated nitrile groups (ca. 2270 cm−1). Moreover, the absence of detectable peaks for either 8S or 8L by MALDI and ESI-TOF mass spectrometry suggests that the nitrile–palladium coordination bonds are comparatively weak and dynamic in nature when contrasted with earlier coordination cages.11–13,16,17
 | ||
| Scheme 3 Dynamic behavior of the cavitand-based palladium coordination cages 8S and 8L. | ||
Attempts to form coordination cages by reacting tetra(4-cyanophenylethynyl)cavitands 6S or 6L with other square-planar cis-metal triflate complexes – such as Pd(dppe)(OTf)2 (dppe = 1,2-bis(diphenylphosphino)ethane), Pt(dppp)(OTf)2, cis-Pt(PEt3)2(OTf)2, Ni(dppe)(OTf)2, Ni(dppp)(OTf)2 and Ni(dppf)(OTf)2 (dppf = 1,1′-bis(diphenylphosphino)ferrocene) – were unsuccessful. In the case of the nickel-based complexes, the tetranitrile ligands were recovered unreacted, suggesting no significant coordination occurred. Reactions involving the platinum complexes resulted in the formation of multiple unidentified species, indicating non-selective or uncontrolled coordination. When these reactions were carried out at elevated temperature (85 °C) in tetrachloroethane, insoluble materials precipitated from the reaction mixtures, consistent with the formation of oligomeric or polymeric species rather than discrete cages.
Both the size of the chelate ring and the nature of the metal center exert a significant influence on cage formation. In good agreement with previous findings,11 successful self-assembly into a cage structure was observed only with dppp, whose P–M–P bite angle (approximately 90°) closely matches the ideal geometry for square-planar coordination. The same study found that self-assembly of the ethylene-bridged tetra(nitrile)cavitands proceeds only with Pt(dppp)(OTf)2, as the Pd(II) analogues proved ineffective under the same conditions (Scheme 1a). In contrast, on the enlarged cavitand platform presented here, the desired cage could not be obtained using the Pt(dppp)(OTf)2 precursor, either under standard conditions or at elevated temperatures. This limitation can be attributed to the greater kinetic inertness of platinum compared to palladium, which may hinder the reversibility and dynamic ‘error–correction processes’ necessary for thermodynamically controlled self-assembly. Furthermore, the complete lack of reactivity observed with the nickel complexes is likely due to the inherently weak coordination between nitrile groups and nickel(II), preventing effective complex formation.
Conclusions
In this study, we synthesized two extended tetra(4-cyanophenylethynyl)cavitands, 6S and 6L, featuring rigid methylene and more flexible ethylene bridging units, respectively. Their stepwise functionalization enabled the installation of terminal nitrile groups suitable for coordination-driven self-assembly. Coordination experiments with Pd(dppp)(OTf)2 in a 2:4 molar ratio led to the formation of discrete, symmetric coordination cages (8S and 8L), as evidenced by NMR spectroscopy, DOSY measurements and IR analysis. The assembly process is dynamic and reversible, with NMR titration studies revealing the presence of intermediates and a concentration-dependent equilibrium between the free ligand and the final cage structure. Notably, cage formation was not observed with Pt(II) or Ni(II) analogues, emphasizing the essential role of both the geometric compatibility of the dppp ligand and the kinetic lability of the Pd(II) metal center. These findings may further advance the design of cavitand-based coordination architectures and highlight the utility of nitrile–metal interactions in dynamic supramolecular assemblies.
Experimental
General experimental methods
All reagents and organometallic precursors were purchased from Merck and used without further purifications. Square-planar cis-metal triflate complexes (ref. 11) and cavitands 1S–6S (ref. 25–28) were synthesized according to literature procedures; their NMR spectra were identical to those previously reported. 1H, 31P and 13C NMR spectra were recorded at 500, 202 and 125 MHz, respectively. Chemical shifts (δ) are reported in parts per million (ppm) downfield from the solvent residual peaks: CDCl3 (7.27 ppm for 1H, 77.0 ppm for 13C), CD2Cl2 (5.32 ppm for 1H, 53.84 ppm for 13C) and DMSO-d6 (2.50 ppm for 1H, 39.51 ppm for 13C). High-resolution mass spectra were obtained using ESI-TOF technique on a Bruker maXis 4G mass spectrometer.Self-diffusion measurements were performed with the bipolar gradient pulse sequence39 using a Bruker Avance III NMR spectrometer operating at 600 MHz proton frequency. The instrument was equipped with a 5 mm broadband direct observe BBFO probe with a shielded z-gradient coil and a GAB gradient amplifier (10 A, maximum gradient strength 52.5 G cm−1). The diffusion experiments were performed at 298 K and the gradient strength was calibrated using a Shigemi tube filled with H2O to a height of 4.0 mm and imaging this water cylinder.40 The diffusion experiments were performed by varying the gradient strength between 5% and 95% of the maximum strength in 16 single experiments using an acquisition time of 2 s and 16 scans, while keeping the diffusion times and gradient lengths constant. The diffusion time was set to 90 ms (big delta), and a gradient duration of 1.5 ms was applied (little delta). The intensity decrease of the signal of interest was determined and fitted with a Bruker t1/t2 software package suitable for DOSY experiments, which is included in the Topspin 3.6 software package.41 Variable temperature experiments of 8L in CDCl2–CDCl2 were performed in the range of 238 to 388 K on a broadband inverse detection probe (BBI) equipped with a shielded z-gradient. The temperature was calibrated using a methanol standard for low and a glycerol sample for high temperatures, showing accuracy within ±0.2 K.
Synthetic procedures
:1 ethanol/water mixture until the washings were neutral (pH ≈ 7). The crude solid was triturated with hot methanol (100 mL) and dried under reduced pressure to yield the title compound 1L as a peach-colored solid (18.5 g, 83%). Mp 230–235 °C. νmax(KBr): 1098, 1165, 1263, 1337, 1441, 1477, 1612, 2853, 2924, 3389 cm−1. Major isomer: 1H NMR (500 MHz, DMSO-d6) δ 0.85 (12H, t, J = 7.0 Hz, CH3), 1.10–1.36 (64H, m, (CH2)8), 1.94 (12H, s, ArCH3), 2.17 (8H, m, CH2CH), 4.18 (4H, m, CHCH2), 7.19 (4H, s, ArH), 8.64 (8H, s, OH). 13C{1H} NMR (500 MHz, DMSO-d6) δ 10.0 (CH3), 13.8 (ArCH3), 22.2 (CH2), 28.0 (CH2), 28.9 (CH2), 29.1 (CH2), 29.29 (CH2), 29.31 (CH2), 29.4 (CH2), 31.4 (CH2), 33.5 (CHCH2), 34.2 (CH2CH), 111.6 (Ar), 120.1 (Ar), 124.3 (Ar), 149.2 (Ar). HRMS (ESI-TOF) m/z: [M + H]+ calcd for C72H113O8 1105.8430; found: 1105.8453.Tetra(methyl)cavitand 2L was dissolved in benzene (200 mL) and added to N-bromosuccinimide (15.60 g, 87.46 mmol) and 1,1′-azobis(cyclohexanecarbonitrile) (1.34 g, 5.47 mmol) under an argon atmosphere. The reaction mixture was stirred at 80 °C for 2 days. After cooling to rt, the upper liquid layer was removed by decantation, and the solvent was evaporated under reduced pressure. The residue was dissolved in CHCl3 (200 mL) and washed with 1 M NaOH (3 × 200 mL). The combined aqueous layers were extracted with another portion of CHCl3 (50 mL). The combined organic phases were washed with water (100 mL), dried over anhydrous MgSO4 and concentrated under vacuum to afford intermediate 3L as a brown, viscous oil. This material was used in the subsequent step without further purification. Major component: 1H NMR (CDCl3, 500 MHz): δ 0.88 (12H, t, J = 7.0 Hz, CH3), 1.15–1.39 (64H, m, (CH2)8), 2.05 (8H, m, CH2CH), 4.17 (8H, m, inner of OCH2CH2O), 4.63 (8H, m, outer of OCH2CH2O), 4.76 (8H, s, ArCH2Br), 5.20 (4H, t, J = 8.1 Hz, CHCH2), 7.43 (4H, s, Ar).
To a solution of 4-iodophenol (12.10 g, 54.66 mmol) in THF (50 mL) was added aqueous NaOH (1.825 M, 30 mL), and the mixture was stirred at rt for 30 minutes. This solution was then added dropwise to a solution of tetra(bromomethyl)cavitand 3L in THF (100 mL). The resulting mixture was refluxed at 70 °C for 16 hours, then cooled to rt. The reaction mixture was washed with 1 M NaOH (100 mL) and CHCl3 (100 mL). The organic layer was further washed with 1 M NaOH (2 × 100 mL), and the combined aqueous layers were extracted with another portion of CHCl3 (50 mL). The combined organic phases were dried over anhydrous MgSO4 and concentrated under reduced pressure. The resulting residue was triturated with methanol (60 mL), and the solid that precipitated was collected and dried in vacuo to afford the title compound 4L as a yellow solid (7.60 g, combined yield: 67%). Mp 103–105 °C. νmax(KBr): 818, 995, 1061, 1096, 1234, 1466, 1485, 1584, 2853, 2924 cm−1; 1H NMR (CDCl3, 500 MHz): δ 0.89 (12H, t, J = 7.0 Hz, CH3), 1.18–1.43 (64H, m, (CH2)8), 2.14 (8H, m, CH2CH), 3.75 (8H, m, inner of OCH2CH2O), 4.21 (8H, m, outer of OCH2CH2O), 5.04 (8H, s, ArCH2O), 5.30 (4H, t, J = 8.0 Hz, CHCH2), 6.66 (8H, d, J = 9.0 Hz, Ar), 7.50 (4H, s, Ar), 7.57 (8H, d, J = 9.0 Hz, Ar). 13C{1H} NMR (CDCl3, 125 MHz): δ 14.2 (CH3), 22.7 (CH2), 27.9 (CH2), 29.4 (CH2), 29.6 (CH2), 29.68 (CH2), 29.69 (CH2), 29.72 (CH2), 31.9 (CH2), 33.9 (CHCH2), 34.4 (CH2CH), 61.1 (ArCH2O), 73.5 (OCH2CH2O), 83.3 (Ar), 116.8 (Ar), 123.6 (Ar), 125.3 (Ar), 135.9 (Ar), 138.5 (Ar), 153.8 (Ar), 158.0 (Ar). HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C104H132O12I4Na 2103.5790; found: 2103.5776.
To a solution of the tetra(trimethylsilylethynyl)cavitand intermediate in THF (80 mL) was added tetrabutylammonium fluoride trihydrate (3.14 g, 9.944 mmol). The reaction mixture was stirred at rt for 30 minutes. The solvent was then removed under reduced pressure, and the residue was dissolved in CH2Cl2 (60 mL). This solution was washed sequentially with 10% aqueous HCl (60 mL) and water (2 × 100 mL). The organic layer was dried over anhydrous MgSO4, filtered and concentrated under reduced pressure. The crude product was triturated with methanol (30 mL), the resulting solid was collected and dried in vacuo to afford the title compound 5L as a brown solid (2.7 g, 75%). Mp 100–103 °C. νmax(KBr): 831, 997, 1063, 1096, 1169, 1238, 1287, 1466, 1506, 1605, 2106, 2853, 2924, 3287 cm−1.1H NMR (CDCl3, 500 MHz): δ 0.90 (12H, t, J = 7.1 Hz, CH3), 1.20–1.43 (64H, m, (CH2)8), 2.15 (8H, m, CH2CH), 3.00 (4H, s, CCH), 3.77 (8H, m, inner of OCH2CH2O), 4.23 (8H, m, outer of OCH2CH2O), 5.09 (8H, s, ArCH2O), 5.32 (4H, t, J = 8.1 Hz, CHCH2), 6.83 (8H, d, J = 8.9 Hz, Ar), 7.43 (8H, d, J = 8.9 Hz, Ar), 7.52 (4H, s, Ar). 13C{1H} NMR (CDCl3, 125 MHz): δ 14.1 (CH3), 22.7 (CH2), 27.9 (CH2), 29.4 (CH2), 29.5 (CH2), 29.63 (CH2), 29.69 (CH2), 29.73 (CH2), 31.9 (CH2), 34.0 (CHCH2), 34.5 (CH2CH), 61.1 (ArCH2O), 73.5 (OCH2CH2O), 76.1 (CC), 83.3 (CC), 114.4 (Ar), 114.9 (Ar), 123.6 (Ar), 125.3 (Ar), 133.8 (Ar), 135.9 (Ar), 153.9 (Ar), 158.5 (Ar). HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C112H136O12Na 1695.9924; found: 1695.9917.
N), 2853, 2924 cm−1.1H NMR (CDCl3, 500 MHz): δ 0.90 (12H, t, J = 7.1 Hz, CH3), 1.15–1.45 (64H, m, (CH2)8), 2.16 (8H, m, CH2CH), 3.79 (8H, m, inner of OCH2CH2O), 4.25 (8H, m, outer of OCH2CH2O), 5.13 (8H, s, ArCH2O), 5.33 (4H, t, J = 8.0 Hz, CHCH2), 6.90 (8H, d, J = 8.8 Hz, Ar), 7.49 (8H, d, J = 8.8 Hz, Ar), 7.52–7.64 (20H, m, Ar). 1H NMR (CD2Cl2, 500 MHz): δ 0.89 (12H, t, J = 7.0 Hz, CH3), 1.10–1.42 (64H, m, (CH2)8), 2.17 (8H, m, CH2CH), 3.76 (8H, m, inner of OCH2CH2O), 4.22 (8H, m, outer of OCH2CH2O), 5.11 (8H, s, ArCH2O), 5.30 (4H, t, J = 7.9 Hz, CHCH2), 6.92 (8H, d, J = 8.6 Hz, Ar), 7.48 (8H, d, J = 8.6 Hz, Ar), 7.52–7.66 (20H, m, Ar). 13C{1H} NMR (CDCl3, 125 MHz): δ 14.1 (CH3), 22.7 (CH2), 27.9 (CH2), 29.4 (CH2), 29.6 (CH2), 29.69 (CH2), 29.70 (CH2), 29.72 (CH2), 31.9 (CH2), 34.0 (CHCH2), 34.4 (CH2CH), 61.3 (ArCH2O), 73.5 (OCH2CH2O), 87.0 (CC), 93.6 (CC), 111.2, 114.7, 114.9, 118.5, 123.4, 125.5, 128.4, 131.8, 132.0, 133.5, 136.0, 153.9, 158.8. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C140H148N4O12Na 2100.0986; found: 2100.0987.N), 2268 (coordinated CN), 2943 cm−1.1H NMR (CD2Cl2, 500 MHz): δ 1.85 (24H, d, J = 7.4 Hz, CH3CH), 2.33 (8H, br m, PCH2CH2CH2P), 2.88 (16H, br s, PCH2CH2CH2P), 4.49 (8H, d, J = 7.3 Hz, inner of OCH2O), 4.98 (16H, s, ArCH2O), 5.07 (8H, q, J = 7.4 Hz, CHCH3), 5.69 (8H, d, J = 7.3 Hz, outer of OCH2O), 6.85 (16H, d, J = 8.8 Hz, Ar), 7.30–7.55 (104H, m, Ar), 7.72 (32H, m, Ar). 13C{1H} NMR (CD2Cl2, 125 MHz): δ 16.3 (CH3CH), 18.8 (PCH2CH2CH2P), 22.5 (d, J(C–P) = 45 Hz, PCH2CH2CH2P), 31.8 (CHCH3), 61.2 (ArCH2O), 87.3 (CC), 95.2 (CC), 100.5 (OCH2O), 108.7, 114.9, 115.2, 120.9, 121.0 (q, J(C–F) = 320 Hz), 121.9, 122.9, 124.6, 125.1, 130.1 (m), 132.1, 133.3, 133.4, 133.8 (m), 133.9, 139.4, 154.4, 159.9. 31P NMR (CD2Cl2, 202 MHz): δ 14.2 (s). 19F NMR (CD2Cl2, 564 MHz): δ −78.6 (s).N), 2273 (coordinated CN), 2853, 2924 cm−1. 1H NMR (CD2Cl2, 500 MHz): δ 0.88 (24H, br t, J = 7.0 Hz, CH3), 1.10–1.42 (128H, br m, (CH2)8), 2.16 (16H, br m, CH2CH), 2.32 (8H, br m, PCH2CH2CH2P), 2.88 (16H, br s, PCH2CH2CH2P), 3.76 (16H, br m, inner of OCH2CH2O), 4.20 (16H, br m, outer of OCH2CH2O), 5.10 (16H, br s, ArCH2O), 5.29 (8H, br m, CHCH2), 6.91 (16H, br d, J = 8.5 Hz, Ar), 7.34–7.60 (104H, br m, Ar), 7.71 (32H, br s, Ar). 13C{1H} NMR (CD2Cl2, 125 MHz): δ 14.3 (CH3), 18.7 (PCH2CH2CH2P), 22.7 (d, J(C–P) = 40 Hz, PCH2CH2CH2P), 23.1, 28.4, 29.8, 30.1 (double intensity, CH2), 30.2 (double intensity, CH2), 32.4, 34.5 (CHCH2), 35.0 (CH2CH), 61.7 (ArCH2O), 73.9 (OCH2CH2O), 87.3 (CC), 95.3 (CC), 108.9, 115.0, 115.4, 124.0, 124.6, 125.0, 128.2, 128.7, 129.7, 130.1, 132.1, 132.8, 133.3, 133.8, 136.4, 154.4, 159.7. 31P NMR (CD2Cl2, 202 MHz): δ 14.5 (br s). 19F NMR (CD2Cl2, 564 MHz): δ −78.5 (s).Conflicts of interest
The authors declare no conflicts of interest.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: NMR and IR spectra of the synthesized compounds, along with additional experimental details. See DOI: https://doi.org/10.1039/d6ra00433d.Acknowledgements
Project no. TKP2021-EGA-17 has been implemented with the support provided by the Ministry of Culture and Innovation of Hungary from the National Research, Development and Innovation Fund, financed under the TKP2021 funding scheme. We would like to thank Dr Luis Escobar (Institute of Chemical Research of Catalonia, Tarragona, Spain) for some HRMS measurements.Notes and references
- P. Timmerman, W. Verboom and D. N. Reinhoudt, Tetrahedron, 1996, 52, 2663–2704 CrossRef CAS
.
- D. J. Cram and J. M. Cram, Container Molecules and their Guests, Monographs in Supramolecular Chemistry, ed. J. F. Stoddart, Royal Society of Chemistry, Cambridge, 1994, Vol. 4 Search PubMed
- K. Kobayashi and M. Yamanaka, Chem. Soc. Rev., 2015, 44, 449–466 RSC
- T. Heinz, D. M. Rudkevich and J. Rebek Jr., Nature, 1998, 394, 764–766 CrossRef CAS
- S. Ma, D. M. Rudkevich and J. Rebek Jr., J. Am. Chem. Soc., 1998, 120, 4977–4981 CrossRef CAS
- M. H. K. Ebbing, M.-J. Villa, J.-M. Valpuesta, P. Prados and J. de Mendoza, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4962–4966 CrossRef CAS PubMed
- H.-J. Choi, Y. S. Park, C. S. Cho, K. Koh, S.-H. Kim and K. Paek, Org. Lett., 2004, 6, 4431–4433 CrossRef CAS PubMed
- Y. S. Park, S. Seo, E.-H. Kim and K. Paek, Org. Lett., 2011, 13, 5904–5907 CrossRef CAS PubMed
- S. Merget, L. Catti, S. Zev, D. T. Major, N. Trapp and K. Tiefenbacher, Chem. – Eur. J., 2011, 27, 4447–4453 CrossRef PubMed
- R. Pinalli, F. Boccini and E. Dalcanale, Isr. J. Chem., 2011, 51, 781–797 CrossRef CAS
- F. Fochi, P. Jacopozzi, E. Wegelius, K. Rissanen, P. Cozzini, E. Marastoni, E. Fisicaro, P. Manini, R. Fokkens and E. Dalcanale, J. Am. Chem. Soc., 2001, 123, 7539–7552 CrossRef CAS PubMed
- N. Cuminetti, M. H. K. Ebbing, P. Prados, J. de Mendoza and E. Dalcanale, Tetrahedron Lett., 2001, 42, 527–530 CrossRef CAS
- D. Zuccaccia, L. Pirondini, R. Pinalli, E. Dalcanale and A. Macchioni, J. Am. Chem. Soc., 2005, 127, 7025–7032 CrossRef CAS PubMed
- R. Pinalli, V. Cristini, V. Sottili, S. Geremia, M. Campagnolo, A. Caneschi and E. Dalcanale, J. Am. Chem. Soc., 2004, 126, 6516–6517 Search PubMed
- L. Pirondini, D. Bonifazi, B. Cantadori, P. Braiuca, M. Campagnolo, R. De Zorzi, S. Geremia, F. Diederich and E. Dalcanale, Tetrahedron, 2006, 62, 2008–2015 CrossRef CAS
- K. Kobayashi, Y. Yamada, M. Yamanaka, Y. Sei and K. Yamaguchi, J. Am. Chem. Soc., 2004, 126, 13896–13897 CrossRef CAS PubMed
- M. Yamanaka, Y. Yamada, Y. Sei, K. Yamaguchi and K. Kobayashi, J. Am. Chem. Soc., 2006, 128, 1531–1539 CrossRef CAS PubMed
- M. Nakamura, Y. Tsukamoto, T. Ueta, Y. Sei, T. Fukushima, K. Yoza and K. Kobayashi, Chem. – Asian J., 2020, 15, 2218–2230 CrossRef CAS PubMed
- S. J. Park and J.-I. Hong, Chem. Commun., 2001, 1554–1555 RSC
- S. J. Park, D. M. Shin, S. Sakamoto, K. Yamaguchi, Y. K. Chung, M. S. Lah and J.-I. Hong, Chem. – Eur. J., 2005, 11, 235–241 CrossRef PubMed
- T. Haino, M. Kobayashi, M. Chikaraishi and Y. Fukazawa, Chem. Commun., 2005, 2321–2323 RSC
- M. Yamanaka, N. Toyoda and K. Kobayashi, J. Am. Chem. Soc., 2009, 131, 9880–9881 CrossRef CAS PubMed
- M. Yamanaka, M. Kawaharada, Y. Nito, H. Tayaka and K. Kobayashi, J. Am. Chem. Soc., 2011, 133, 16650–16656 CrossRef CAS PubMed
- Y. Nito, H. Adachi, N. Toyoda, H. Takaya, K. Kobayashi and M. Yamanaka, Chem. – Asian J., 2014, 9, 1076–1082 CrossRef CAS PubMed
- Z. Csók, T. Kégl, L. Párkányi, Á. Varga, S. Kunsági-Máté and L. Kollár, Supramol. Chem., 2011, 23, 710–719 CrossRef
- Z. Csók, A. Takátsy and L. Kollár, Tetrahedron, 2012, 68, 2657–2661 CrossRef
- Z. Csók, T. Kégl, Y. Li, R. Skoda-Földes, L. Kiss, S. Kunsági-Máté, M. H. Todd and L. Kollár, Tetrahedron, 2013, 69, 8186–8190 CrossRef
- Z. Csók, P. Szuroczki, L. Kollár, H. M. Ngo, I. Ledoux-Rak, N. A. M. S. Caturello and R. Q. Albuquerque, J. Phys. Chem. C, 2015, 119, 12608–12615 CrossRef
- Z. Csók, L.-D. Syntrivanis, N. A. M. S. Caturello, R. Q. Albuquerque and L. Kollár, Tetrahedron, 2025, 185, 134807 CrossRef
- A. G. S. Högberg, J. Org. Chem., 1980, 45, 4498–4500 CrossRef
- L. Abis, E. Dalcanale, A. Du Vosel and S. Spera, J. Org. Chem., 1988, 53, 5475–5749 CrossRef CAS
- L. M. Tunstad, J. A. Tucker, E. Dalcanale, J. Weiser, J. A. Bryant, J. C. Sherman, R. C. Helgeson, C. B. Knobler and D. J. Cram, J. Org. Chem., 1989, 54, 1305–1312 CrossRef CAS
- O. Middel, W. Verboom, R. Hulst, H. Kooijman, A. L. Spek and D. N. Reinhoudt, J. Org. Chem., 1998, 63, 8259–8265 CrossRef CAS
- Using 4-iodopyridine, 4-iodoaniline and 4-iodobenzoic acid in the second Sonogashira coupling, we attempted to incorporate alternative building blocks into the cavitand scaffold. However, these reactions failed to yield the desired products under various reaction conditions.
- D. J. Cram, S. Karbach, H.-E. Kim, C. B. Knobler, E. F. Maverick, J. L. Ericson and R. C. Helgeson, J. Am. Chem. Soc., 1988, 110, 2229–2237 CrossRef CAS
- J. N. Smith, C. Ennis and N. T. Lucas, Chem. Sci., 2021, 12, 11858–11863 RSC
- I. Horin, T. Adiri, Y. Zafrani and Y. Cohen, Org. Lett., 2018, 20, 3958–3961 CrossRef CAS PubMed
- From the Einstein–Stokes equation:
 , where D is the translational diffusion coefficient, r is the hydrodynamic radius
, where D is the translational diffusion coefficient, r is the hydrodynamic radius 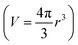 , k is the Boltzmann constant, T is the absolute temperature and η is the viscosity of the solvent; the following expression for the molar volume ratio is obtained:
, k is the Boltzmann constant, T is the absolute temperature and η is the viscosity of the solvent; the following expression for the molar volume ratio is obtained: 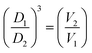 .
. - D. H. Wu, A. D. Chen and C. S. Johnson, J. Magn. Reson., Ser. A, 1995, 115, 260–264 CrossRef CAS
- M. Holz and H. Weingärtner, J. Magn. Reson., 1991, 92, 115–125 CAS
- XWINNMR, Bruker Analytik GmbH, Software Dept., Rheinstetten, Germany Search PubMed



| This journal is © The Royal Society of Chemistry 2026 |