DOI:
10.1039/D5RA10073A
(Review Article)
RSC Adv., 2026,
16, 19038-19078
NHC-catalyzed asymmetric synthesis of natural products and pharmaceutical drugs
Received
29th December 2025
, Accepted 11th March 2026
First published on 13th April 2026
Abstract
Asymmetric processes catalyzed by N-heterocyclic carbenes (NHCs) have become extremely important as strategic tools for the efficient synthesis of bioactive molecular scaffolds. Over the past few years, these catalysts have been widely employed for the total synthesis of pharmaceutical drugs and structurally distinct natural products. This review provides an integrated survey of the asymmetric formal and total synthesis of biologically active natural products and medicinally important compounds using triazolium, imidazolium, and thiazolium-based NHC catalysts from 2016 onwards. This review covers the asymmetric synthesis of different classes of natural products, such as alkaloids, terpenoids, lignans, polyketides, pentadecaketides, and flavonoids, along with pharmaceutical drugs, such as NSAIDs, antidepressants, antibacterial and analgesic agents, by harnessing NHC catalysis.
1 Introduction
N-Heterocyclic carbenes (NHCs) are defined as singlet carbenes, having a divalent carbon atom in their ring structure that is linked to one or more nitrogen atoms to form a heterocyclic scaffold.1 As carbenes have six electrons in their valence shell, their incompletely filled valence shell and coordinative unsaturation make free carbenes unstable in nature.2,3 Depending on the number of nitrogen atoms in the ring structure, a wide variety of carbene compounds exist with different substituents and ring sizes (Fig. 1).3,4 Because of the easily accessible starting materials, sustainable reaction conditions and ease of use, NHCs have been acknowledged as versatile and efficient organocatalysts for the rapid synthesis of a broad range of cyclic and acyclic compounds of pharmacological and biological significance.5–11 Along with C–C bond formation, C–heteroatom bond formation can also take place by utilizing NHC organocatalysts.5,12–15
 |
| | Fig. 1 Structures of the most widely used NHCs. | |
Since the mid-19th century, chemists have been trying to isolate carbenes, but all attempts have failed.16 Carbenes typically play the role of extremely reactive intermediates and have a short lifespan. However, the characteristics of N-heterocyclic carbenes, where the carbene center is positioned on an N-heterocyclic ring, are distinct.17,18 NHCs continue to intrigue researchers because these compounds are not only employed as organocatalysts3,5,19–22 but also serve as ligands for transition-metal catalysis,23–26 and they are also used in materials science27,28 and nanoparticle applications.29–32 Tschugajeff prepared the first complex of a carbene ligand stabilized by an adjacent heteroatom in 1925.33 The first metal carbene complex was constructed and evaluated by Fischer in 1964.34 In the 1960s, Wanzlick made efforts to prepare and isolate stable NHC, but instead, the corresponding dimer was obtained.35 In 1991, Arduengo et al. isolated and identified the first crystalline NHC, IAd, named 1,3-di(adamantly)imidazole-2-ylidene, which was obtained by the removal of a proton from the 1,3-imidazolium salt (Fig. 2).36
 |
| | Fig. 2 Synthesis and isolation of the first NHC. | |
Different types of NHC-bound intermediates have been generated as a result of organocatalytic reactions. These can be categorized as Breslow intermediates, radical intermediates, azolium intermediates, homoenolate intermediates, azolium enolate intermediates, acyl azolium intermediates and azolium dienolate intermediates.11,37–40 The polarity of carbonyl compounds is inverted when NHC compounds are used as organocatalysts. As a result, a number of nucleophilic addition reactions can take place by utilizing different electrophiles.41–52 Hence, NHCs are utilized in a number of transformations or annulation reactions,53–55 including Stetter,56,57 benzoin,58,59 radical,60,61 Mannich, and Michael reactions, as well as Claisen rearrangements, C–C/C–H activation and cycloaddition reactions.41–52
Natural products play a crucial role in the drug discovery process due to their distinctive and physiologically active scaffolds.62–64 In natural products and pharmaceutical research, chemists have been focused on the synthesis of enantiomeric compounds, and designing drugs with defined stereochemistry is one of the core challenges in organic synthesis. For example, one enantiomer may have beneficial pharmacological features, while the other enantiomer may have unfavorable side effects. Therefore, there can be significant risks associated with using racemic substances as pharmaceutical medications.65 N-Heterocyclic carbenes catalysis is recognized as a potent and adaptable method for the stereoselective synthesis of various compounds,3,5,12,15,19,66–71 and natural products such as seragakinone A,72 clausenamide,73 cassialoin74 and chalcitrin75 have been accessed via NHC catalysis (Fig. 3). NHC catalysis has also been employed to synthesize fragments of natural products76 and pharmaceutics-related compounds.77–79 Over a prolonged timespan, a range of NHC precursors have been constructed for asymmetric organocatalysis. First, Sheehan and Hunneman harnessed precatalyst chiral thiazolium salts for the enantioselective benzoin condensation reaction about half a century ago.80 Then, in 1996, a triazolium-based NHC salt was used as a precatalyst by Enders and co-workers in the Stetter reaction.81 Since then, there has been notable progress in NHC-catalyzed asymmetric synthesis.
 |
| | Fig. 3 Natural products synthesized via NHC catalysis. | |
A number of review articles have been published over many years on the versatile utility of NHCs as catalysts, cooperative catalysts, and ligands.82,83 Zhang et al., in 2025, presented a review on asymmetric axial and planar chirality construction through the use of NHC organocatalysis.84 Another review was published by Li et al. in 2025 on asymmetric electrophilic activation of carbonyl compounds by NHC catalysis.85 In 2024, Cai et al. presented a review on radical reactions carried out by the SET reduction of NHC-derived acyl azoliums.86 In 2020, Que et al. reviewed the advances in NHC catalysis for natural product synthesis.87 Moreover, in 2012, Scheidt and coworkers also summarized the application of NHC catalysis in the total synthesis of natural products.67 Since then, much more progress has been made on the applications of NHC catalysis. Thus, this review provides an overview of various NHC catalysts that have been utilized (Table 1) for the synthesis of different classes of natural products and pharmaceutical drugs, reported since 2016.
Table 1 Summary of the discussed NHC catalysts
| S. no. |
NHC catalyst |
S. no. |
NHC catalyst |
| Triazolium-based NHCs |
| 1 |
 |
2 |
 |
| 3 |
 |
4 |
 |
| 5 |
 |
6 |
 |
| 7 |
 |
8 |
 |
| 9 |
 |
10 |
 |
| 11 |
 |
12 |
 |
| 13 |
 |
14 |
 |
| 15 |
 |
16 |
 |
| 17 |
 |
18 |
 |
| 19 |
 |
20 |
 |
| 21 |
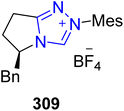 |
22 |
 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| Imidazolium-based NHCs |
| 23 |
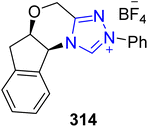
 |
24 |
 |
| 25 |
 |
26 |
 |
| 27 |
 |
28 |
 |
| 29 |
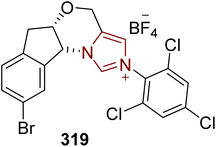 |
30 |
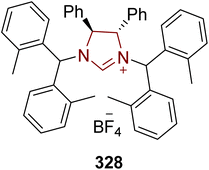 |
|
| Thiazolium-based NHCs |
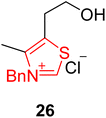
| 31 |
 |
32 |
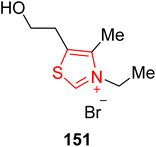 |
|
| Oxazole-based NHC |
| 33 |
 |
2 2- Applications of NHC catalysts in asymmetric synthesis
2.1 Natural products
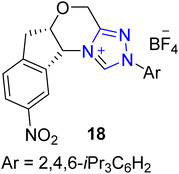
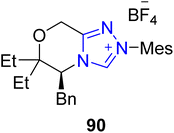
2.1.1 Synthesis of alkaloids. Gelsemoxonine 22, a gelsemium alkaloid, was first extracted from the foliage of Gelsemium elegans Benth., by the research group of Clardy in 1991.88 The original structure of Gelsemoxonine (22) was revised in 2003 by Aimi, based on X-ray crystallographic analysis, as the initial proposed structure was misassigned.89 Gelsemium alkaloids exhibit analgesic, anti tumor and antispasmodic activity; furthermore, they are also used to treat skin ulcers.90–93 In 2016, an asymmetric synthesis of Gelsemoxonine 22 was reported by Zhao et al. by utilizing indanol-triazolium-based NHC 18 as a catalyst. In this synthetic protocol, redox esterification of 6-hydroxy pyranone 16 and enal 17 was observed in the presence of triazolium-based NHC 18 (catalyst) and sodium benzoate (base) at room temperature. As a result, ester 19 was generated with 64% yield and 99% ee after recrystallization. The ester 19 was then reacted with bromomalonate 20 by employing DBU in THF at 0 °C. The resulting compound was allowed to react further with triethyl silane in the presence of trimethylsilyl trifluoromethanesulfonate (TMSOTf) and MeCN to synthesize the key intermediate 21 with 99% ee.94 The fabricated key intermediate 21 had been reported earlier, in 2011, by Shimokawa et al. and used for the synthesis of Gelsemoxonine 22 (Scheme 1).95
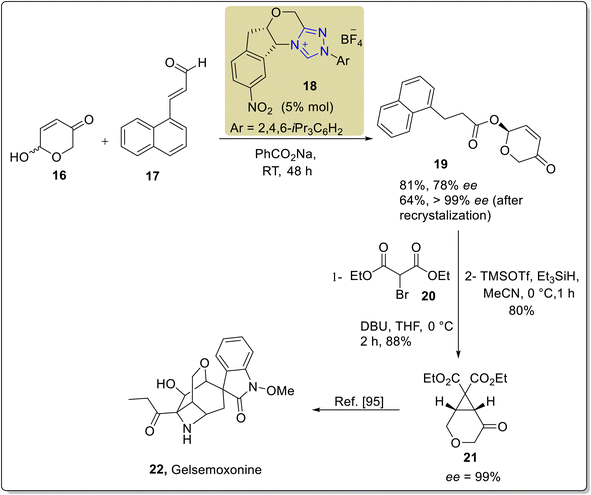 |
| | Scheme 1 Asymmetric synthesis of gelsemoxonine 22. | |
Himalensine A 28, which belongs to the Daphniphyllum class of alkaloids with a trinorcalyciphylline A type skeleton, was first isolated in 2016 by Yue and coworkers from Daphniphyllum himalense, found in the Himalaya Mountains.96–98 In 2017, Dixon and coworkers reported a novel strategy for the enantioselective synthesis of Himalensine A 28 by utilizing NHC 26 as a catalyst via 22-steps. In this unique synthetic protocol, N-Boc protected furan 23 was allowed to undergo intramolecular Diels–Alder furan (IMDAF) cycloaddition reaction in the presence of bifunctional iminophosphorane (BIMP) catalyst to furnish the tricyclic core structure 24. Over a few steps, this tricyclic core structure 24 was converted into the aldehyde moiety 25. The aldehyde 25 was then reacted with thiazolium-based NHC 26 (catalyst) and TEA (base) in ethanol at 60 °C to obtain an intermediate, i.e. the pendant cyclopentenone 27, with 75% yield via Stetter cyclization. An alkene migration of intermediate cyclopentenone 27 generated the enone that underwent further reduction in the presence of Vaska's catalyst [Ir(CO)(PPh3)2Cl] and formic acid at 60 °C to generate (−)-himalensine A 28 in 67% yield (Scheme 2).99
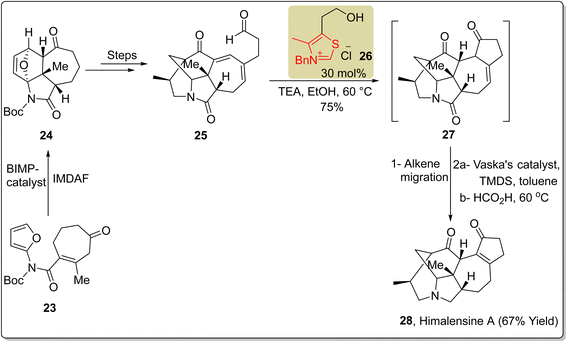 |
| | Scheme 2 Asymmetric synthesis of himalensine A 28. | |
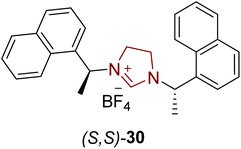
Aurantioclavine 32 is an ergot alkaloid that was first extracted from the Penicillium aurantiovirens in 1981.100 Its tricyclic indole core structure has been utilized as a biosynthetic precursor for the synthesis of complex communesin alkaloids,101,102 which demonstrate cytotoxic activity against blood cancer cells.103 In 2018, Glorius and coworkers introduced an asymmetric approach to synthesize (−)-aurantioclavine 32 by utilizing NHC (S,S)-30 as a ligand, with ruthenium metal catalyst. In their synthetic route, 2 moles of Ru–NHC complex accelerated the asymmetric hydrogenation of 2-oxazolones 29 under 50 bar H2 in cyclohexane and tetrahydrofuran (solvent) at 0 °C to obtain 2-oxazolidinone 31 in 97% yield with 92% enantioselectivity.104 The synthesized key intermediate 2-oxazolidinone 31 was employed by Park et al. in 2016 for the total asymmetric synthesis of aurantioclavine 32 (Scheme 3).105
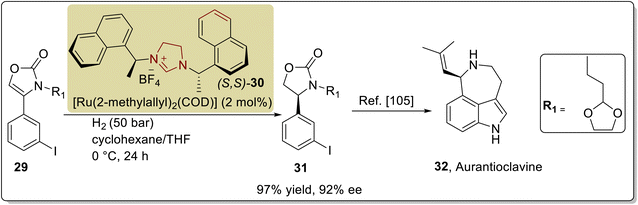 |
| | Scheme 3 Asymmetric synthesis of aurantioclavine 32. | |
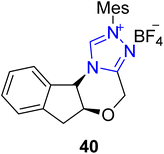
Cruciferane is a naturally occurring alkaloid composed of fused rings of pyrroloindoline and quinazolinone. Cruciferane was first extracted from the non-woody plant Isatis indigotica (Chinese woad) in 2012.106 The desiccated roots and foliage of Isatis indigotica have traditionally been employed to cure influenza, erysipelas, epidemic hepatitis, encephalitis B and carbuncles.107–109 Moreover, it exhibits hepatoprotective and antipyretic activity.110 In 2019, Mhaske and coworkers introduced a novel approach to synthesize the asymmetric deoxy-cruciferane 41 by utilizing the triazolium-based NHC 40 as a catalyst for the intramolecular [3 + 2] annulation reaction. The synthetic protocol commenced with the Heck coupling reaction of methyl acrylate 33 and o-bromoaniline 34 in the presence of Pd(PPh3)2Cl2 and TEA at 100 °C to generate amine 35. After that, unstable benzoxazinone 37 was furnished by the reaction of anthranilic acid 36 and triethyl orthoformate in the presence of p-TsOH. Next, amine 35 and unpurified benzoxazinone 37 were allowed to react to deliver quinazolinone ester 38 in the presence of EDCI in toluene. In the next step, ester 38 was subjected to reduction in the presence of DIBAL-H and THF at −50 °C to synthesize the corresponding alcohol, which further underwent oxidation in crude form by utilizing MnO2 in DCM at room temperature to synthesize the aldehyde 39. Finally, enal 39 was made to undergo intramolecular [3 + 2] cycloaddition reaction with quinazolinone's internal imine in the presence of NHC 40 (catalyst) and K2CO3 (base) in toluene at 65 °C to deliver deoxy-cruciferane 41 in 42% yield with 78:22 enantiomeric ratio (Scheme 4).111
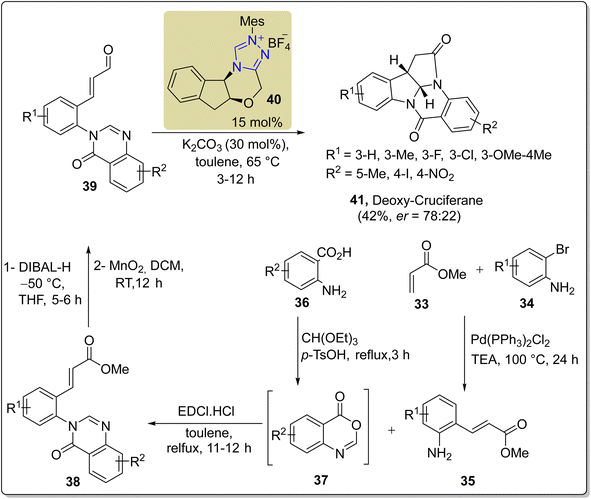 |
| | Scheme 4 Asymmetric synthesis of deoxy-cruciferane 41. | |
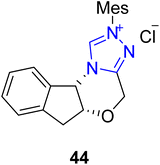
The clausena alkaloids, clausenamide and its derivatives, including neoclausenamide 47, were obtained from the hydrous extract of the Chinese traditional medicinal plant Clausena lansium Skeels in 1988. As a traditional medicine, the water extract of Clausena lansium obtained by decoction is used for the treatment of various diseases, including dermatological diseases and hepatitis.112 Moreover, it also shows a wide range of bioactivities, including hepatoprotective activity, anti-aging activity, anti-ischemic activity and nootropic activity.113–117 Hu et al., in 2019, discovered an efficient approach to synthesize an asymmetric derivative of clausenamide, known as epi-neoclausenamide 47, by utilizing NHC 44 as a catalyst. In this new strategy, α-bromoenal 42 and α-aminoketone 43 were made to undergo [3 + 2] annulation reaction in the presence of triazolium-based NHC 44 (catalyst) derived from the amino indanol core structure and TEA (base) in 1,4-dioxane to synthesize the γ-lactam 45 with 98% ee, 20:1 dr and 84% yield. Next, the Ns group in the γ-lactam 45 was removed by using sodium benzenethiolate, potassium carbonate, dimethyl sulfoxide and acetonitrile, and subsequent methylation was achieved in the presence of methyl iodide and sodium hydride in DMF to synthesize 5-benzoyl-1-methyl-4-phenylpyrrolidin-2-one 46.118 The synthesized compound 46 was reported earlier in 2015 and utilized for the total synthesis of epi-neoclausenamide 47 (Scheme 5).119
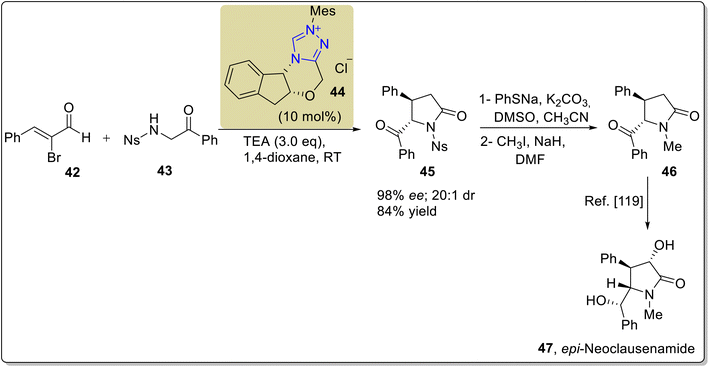 |
| | Scheme 5 Asymmetric synthesis of epi-neoclausenamide 47. | |
Pumiliotoxin B is a dendrobatid alkaloid that was first isolated from Dendrobates pumilio, a poison frog, in 1967. Pumiliotoxin B 54 shows cardiotonic and myotonic activity.120 The marine-sponge-derived triannular guanidine alkaloid netamine C 69 was extracted from Biemna laboutei in 2006 by Kashman and coworkers. Netamine C 69 was inferred to exhibit cytotoxic activity against colon (HT29), breast (MDS-MB-231), and lung (A549) cancer cells.121 In this regard, Hoveyda and coworkers, in 2019, introduced enantioselective routes to generate netamine C 69 and the fragment of pumiliotoxin B 54 by utilizing NHC 50 as a ligand with Cu metal catalyst. For the synthesis of pumiliotoxin B 54, Z-trisubstituted alkenyl-B (pin) 48, monosubstituted alkene 49 and polymethylhydrosilane (PMHS) were reacted to generate 1,5-diene 51 by utilizing NHC 50 (ligand) with copper metal catalyst. Over a few steps, 1,5-diene 51 was converted into compound 52, which was further reacted with LDA in THF to synthesize the alkyne 53.122 The synthesized intermediate, i.e., alkyne 53, had been reported earlier by Lin et al. in 1996, leading toward the synthesis of pumiliotoxin B 54 (Scheme 6).123
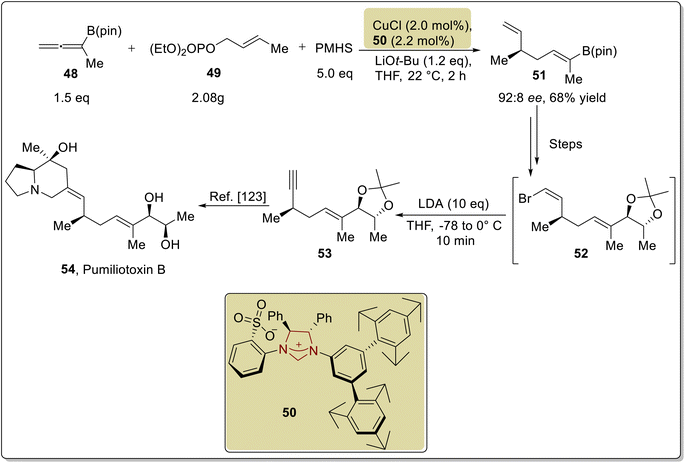 |
| | Scheme 6 Asymmetric synthesis of pumiliotoxins B 54. | |
To synthesize netamine C 69 through a novel synthetic strategy, allylic phosphate 55, allenyl-B(pin) 56 and polymethylhydrosiloxane (PMHS) were reacted in the presence of Cu catalyst with NHC 50 as ligand and LiOt-Bu in THF at 22 °C to furnish 1,5-diene 57. After cross-metathesis of 57 with diol 58 and bis-phosphate 61 separately, in the presence of Ru catalysts 59 and 62, respectively, Z-60 and E-63 allylic phosphates were obtained. Both E- and Z-compounds (60 and 63) produced the same isomer 64 when they underwent reaction with n-hexylzinc bromide by utilizing the Cu–NHC complex (formed in situ from 50). Then, compound 64 was allowed to react with n-BuLi, ClCH2I, Et2O and H2O2 to synthesize the alcohol, which was subsequently converted into the corresponding phosphate 65 by utilizing ClPO(OEt)2, TEA, DMAP, and CH2Cl2. Next, another allylic substitution was made between n-hexylphosphate 65 and allenyl-B(pin) 56 by utilizing a Cu catalyst with NHC 50 and followed by oxidation to furnish the corresponding aldehyde 66. Ring closure of aldehyde 66 through metathesis generated cyclohexenyl intermediate 67, which further underwent a [3 + 2] cycloaddition reaction in the presence of NH2NHBn, HCl and EtOH at 100 °C to generate 68. Finally, netamine C 69 was obtained by catalytic hydrogenation of 68 (Scheme 7).122
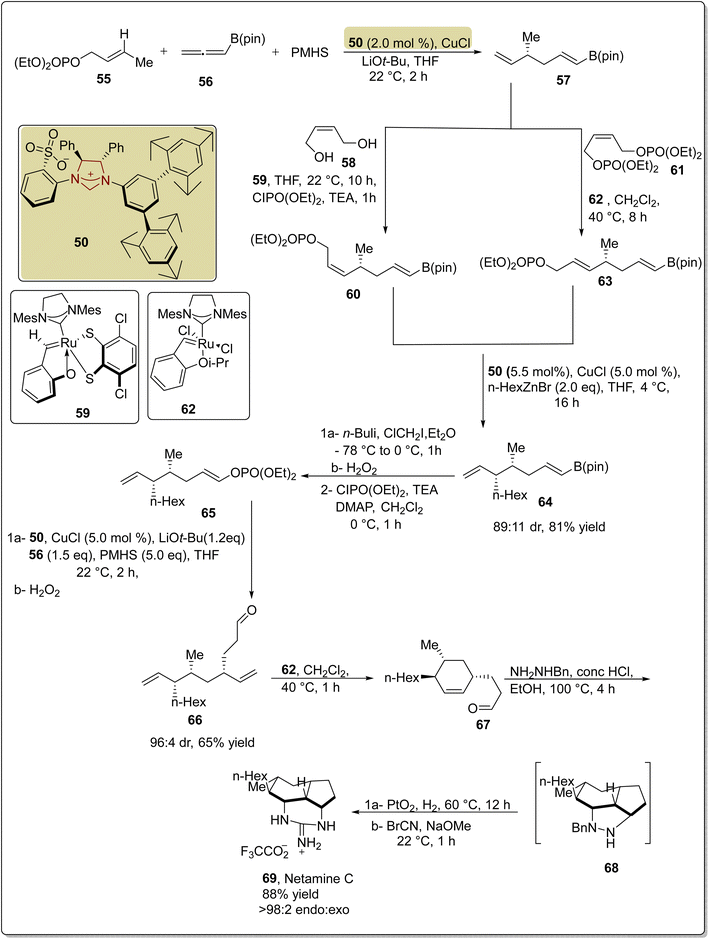 |
| | Scheme 7 Asymmetric synthesis of netamine C 69. | |
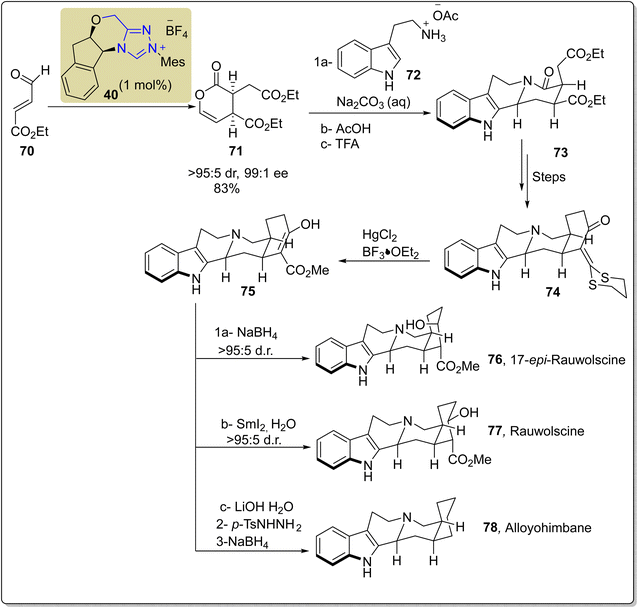
A monoterpenoid indole-based alkaloid, i.e. yohimbine, was first extracted from the outer covering of a tree found in Western Africa named Pausinystalia yohimbe. On the basis of stereochemistry around the D-ring, these alkaloids are further classified into four subfamilies; normal, allo, pseudo and epiallo. Yohimbine alkaloids demonstrate anti-adrenaline, antidiuretic, mydriatic, prototypical α2-receptor antagonistic and serotonin antagonistic activity.124 Scheidt and coworkers, in 2020, reported an enantioselective approach to synthesize the yohimbine alkaloid by utilizing NHC 40 as a catalyst. In this synthetic protocol, enol lactam 71 was obtained with 99:1 ee, >95:5 dr and 83% yield by dimerization of aldehyde 70 by utilizing triazolium-based NHC 40. Next, the enal lactam 71 was made to undergo amidation/N-acyliminium ion cyclization in a three-step sequence. Initially, the enal lactam 71 was treated with tryptamine 72 and Na2CO3 in CH2Cl2; subsequent acidification was done with AcOH, then treatment with TFA favored the cyclization to synthesize compound 73. Over a few steps, compound 73 was converted into α-oxo-ketene dithioacetal 74. In the next step, the α-oxo-ketene dithioacetal 74 was treated with mercury(II) chloride and BF3·OEt2 to generate the main component, i.e. methyl ester 75. Finally, ester 75 was treated in three different ways to produce yohimbine-based natural products. Asymmetric reduction of methyl ester 75 was performed in the presence of NaBH4 to obtain 17-epi-rauwolscine 76. On the other hand, methyl ester 75 was treated with SmI2 and H2O to produce (−)-rauwolscine 77. Finally, decarboxylation of β-ketoester 75 was done by using LiOH·H2O with subsequent Wolff–Kishner deoxygenation to synthesize alloyohimbane 78 (Scheme 8).125
 |
| | Scheme 8 Asymmetric synthesis of yohimbine alkaloids. | |
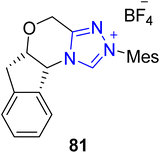
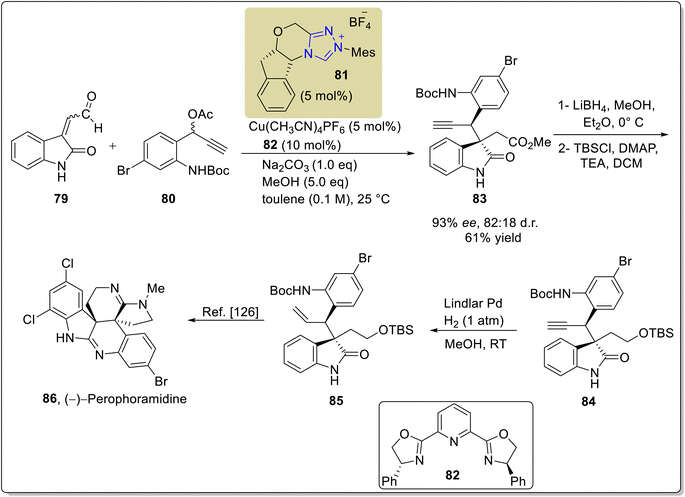
Verbitski et al., in 2001, extracted the polyannular alkaloid perophoramidine 86 for the first time from Perophora namei, an organism found in the Philippines.126 Perophoramidine 86 exhibits cytotoxic activity against the HCT116 colon cancer cell line.127 In 2022, Gong and coworkers reported a novel strategy for the asymmetric synthesis of perophoramidine 86 by exploring Cu/NHC 81 cooperative catalysis. In this new synthetic protocol, enal 79 and propargylic acetate 80 were made to undergo a stereoselective propargylic alkylation reaction in the presence of the copper/NHC 81 dual catalyst, pyridine bisoxazoline 82 (ligand), Na2CO3 and methanol in toluene at 25 °C to obtain oxindole derivative 83 with 61% yield with 93% ee and 82:18 d.r. Next, the oxindole 83 was reduced by employing LiBH4, methanol and diethyl ether at 0 °C to generate the primary alcohol, which was subsequently protected with TBSCl in the presence of DMAP, TEA and DCM. Finally, the TBS-protected oxindole derivative 84 underwent Lindlar reduction to generate the key component, i.e. alkene 85.128 The synthesized alkene 85 had been reported earlier by Trost et al., in 2015, and utilized to furnish asymmetric perophoramidine 86 (Scheme 9).126
 |
| | Scheme 9 Asymmetric synthesis of perophoramidine 86. | |
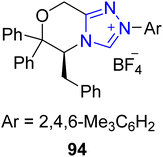
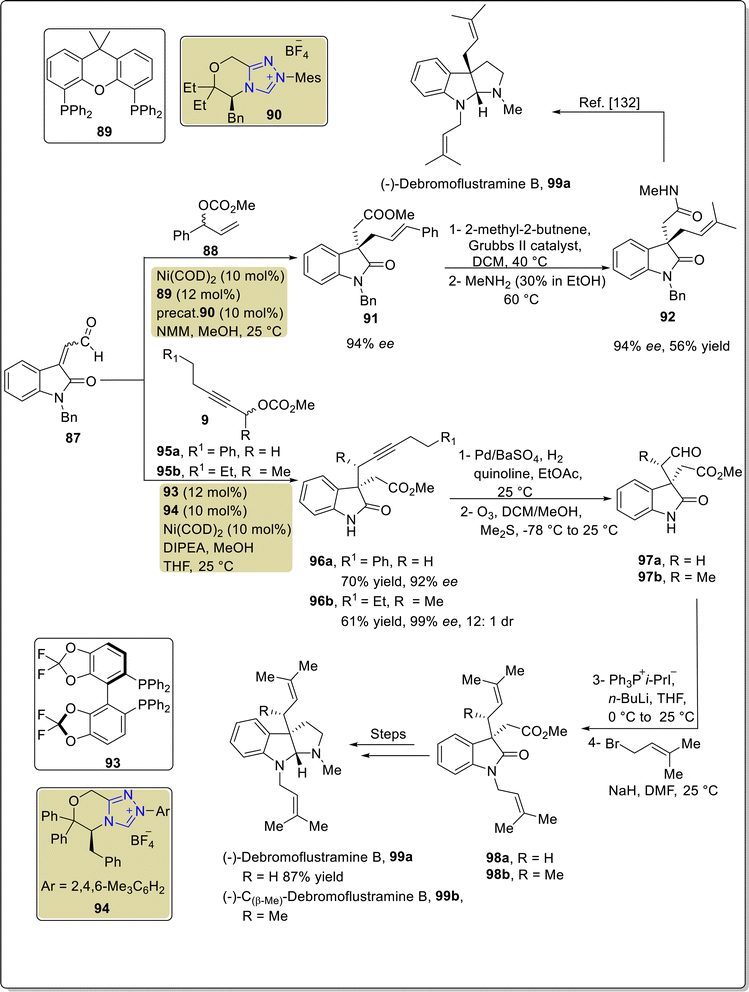
Debromoflustramine B 99a is a pyrrolidinoindoline alkaloid that was first extracted from Flustra foliacea, a marine bryozoan, in 1994. Debromoflustramine B 99a has been found to demonstrate bactericidal activity against methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococci.129 In 2022, Fan et al. introduced an asymmetric approach to generate debromoflustramine B 99a by utilizing Ni–NHC 90 synergistic catalysis. In this synthetic pathway, enal 87 and allylic carbonate 88 were made to undergo allylic alkylation reaction in the presence of Ni(COD)2 catalyst along with NHC 90 (precatalyst), Xantphos 89 (ligand) and N-methylmorpholine (NMM) (base) in methanol at 25 °C to furnish 3,3′-disubstituted oxindole 91 with 94% ee. Next, the cross-metathesis reaction of oxindole 91 with 2-methyl-2-butene, in the presence of Grubbs II catalyst, generated the required alkene, which further underwent amidation reaction with methylamine to obtain methyl amide 92 in 56% yield with 94% ee.130 The synthesized key component, methyl amide 92, had been reported earlier, in 2013, by Miyamoto et al. and utilized for the total synthesis of debromoflustramine B 99a.131 After one year, Peng et al. demonstrated another distinguished and efficient asymmetric approach towards the total synthesis of debromoflustramine B 99a by using Ni–NHC 94 synergistic catalysis. In this synthetic protocol, enal 87 and propargylic carbonate 95a and 95b were allowed to undergo stereochemically divergent propargylation reactions under combined catalysis of Ni–NHC 94 by utilizing diphosphine ligand 93, DIPEA and MeOH, in THF at room temperature to generate asymmetric 3,3′-disubstituted oxindoles 96a and 96b in 70% yield with 92% ee and 61% yield with 99% ee, respectively. The alkyne groups in oxindoles 96a and 96b were allowed to undergo Lindlar reduction, followed by ozone-mediated oxidation, resulting in the synthesis of aldehydes 97a and 97b, respectively. Next, Wittig olefination of aldehydes 97a and 97b was performed by reaction with triphenylisopropylphosphonium iodide to furnish the corresponding compounds, which were further treated with 1-bromo-3-methylbut-2-ene to generate esters 98a and 98b, respectively. Over a few steps, ester 98a was converted into the final natural product, i.e. debromoflustramine B 99a. Synthesis of the methyl analogue of debromoflustramine B, i.e. (−)-C(β-Me)-debromoflustramine B 99b, was also achieved by this research group (Scheme 10).132
 |
| | Scheme 10 Asymmetric synthesis of debromoflustramine B 99a. | |
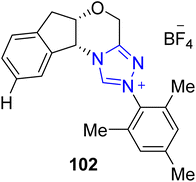
The alkaloid physostigmine 107 was extracted from the poisonous Calabar bean (Physostigma venenosum), a vine native to tropical Africa. Physostigmine 107 demonstrates therapeutic potential against Alzheimer's disease.133 Dutta et al., in 2023, reported an enantioselective approach to the synthesis of esermethole 106 and physostigmine 107 by utilizing NHC 102 as the catalyst. In this unique strategy, acylation of diol 100 was achieved by utilizing benzaldehyde 101 as an acylation source in the presence of NHC 102 as a catalyst, with MnO2 and DABCO in THF at 0 °C to generate the mono-ester 103. The ester 103 was then treated with PPh3, imidazole I2 and THF at 80 °C to furnish its iodo derivative 104. In the next step, iodo derivative 104 was converted into an inseparable mixture by utilizing (TMS)3SiH and azobisisobutyronitrile (AIBN) in toluene at 85 °C, which was subsequently exposed to hydrazine hydrate to furnish a key component, i.e. the asymmetric alcohol 105.134 The synthesized key intermediate 105 had been reported earlier to be utilized for the total synthesis of esermethole 106 and physostigmine 107 (Scheme 11).135,136
 |
| | Scheme 11 Asymmetric synthesis of esermethole 106 and physostigmine 107. | |
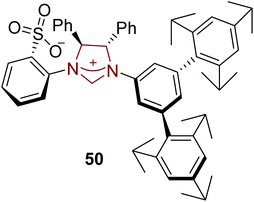
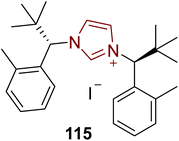
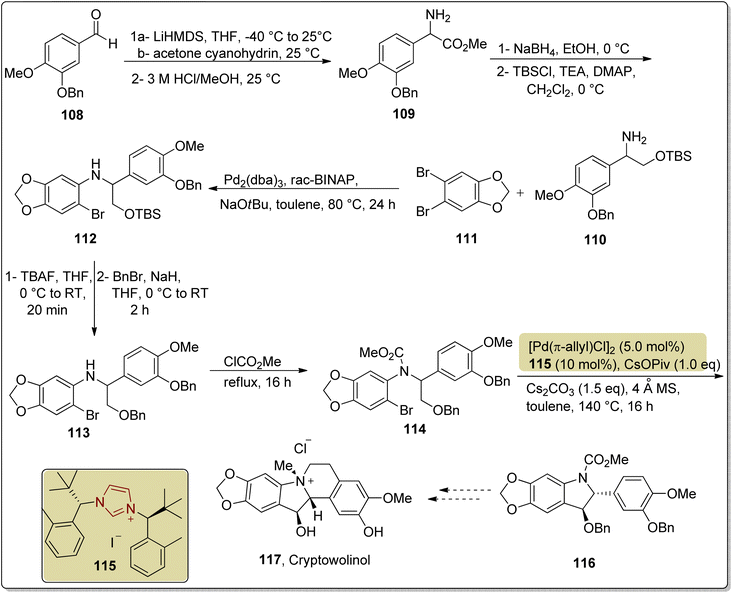
Cryptowolinol 117 is a dibenzopyrrocoline alkaloid that was extracted from the Lauraceae plant Cryptocarya phyllostemon, which is endemic to New Caledonia, in 1989.137–139 Miyakoshi et al., in 2024, reported an enantioselective strategy towards the synthesis of a key component en route to cryptowolinol 117 by utilizing NHC 115 as ligand with Pd-catalyst via parallel kinetic resolution. In this synthetic protocol, benzaldehyde 108 was made to undergo the Strecker reaction in the presence of acetone cyanohydrin to synthesize crude aminonitrile, which subsequently underwent the Pinner reaction in the presence of HCl/MeOH to generate 2-amino ester 109. Reduction of amino ester 109 generated the alcohol, which was then masked with TBSCl to give 110, and further reacted with dibromide 111 via Buchwald-Hartwig N-arylation to synthesize the intermediate 112. Deprotection of the TBS-protected alcohol 112 and subsequent re-protection were done with TBAF and BnBr, respectively, to furnish the benzyl-protected alcohol 113, which was further reacted with methyl chloroformate to protect the amine in order to generate methyl carbamate 114. Next, methyl carbamate 114 was cyclized in the presence of Pd catalyst with NHC 115 as ligand, CsOPiv, Cs2CO3 and toluene at 140 °C to furnish indoline 116, a potential intermediate to obtain cryptowolinol 117. Despite various experimentations, the total synthesis of cryptowolinol 117 was not successful via this approach; however, the significant intermediate 116 was afforded (Scheme 12).139
 |
| | Scheme 12 Asymmetric synthesis of cryptowolinol 117. | |
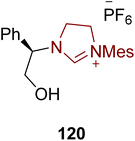
2.1.2 Synthesis of polyketides. In 1974, Hesseltine and colleague extracted equisetin 125 from the secondary metabolites of Fusarium heterosporum, which demonstrates strong antibacterial and HIV-inhibiting activities.140 In this regard, Meng et al. (2016) reported an enantioselective approach to synthesize equisetin 125 by utilizing imidazole-based NHC 120 and CuCl as a catalyst. In this synthetic protocol, dienoate 118 and allenyl-B(pin) 119 were allowed to undergo 1,6-conjugate addition in the presence of NHC 120 and CuCl (as catalyst) with NaOt-Bu and NaOPh in THF at 22 °C to synthesize the enyne 121 in 72% yield with 92:8 ee. Next, the enyne 121 was allowed to undergo regio- and stereo-controlled proto-boryl addition by utilizing compound 122 and CuCl to furnish compound 123. Over a few steps, compound 123 was converted into compound 124, an intermediate141 that was reported earlier, in 2001, by Yuki et al. and utilized for the total synthesis of equisetin 125 (Scheme 13).142
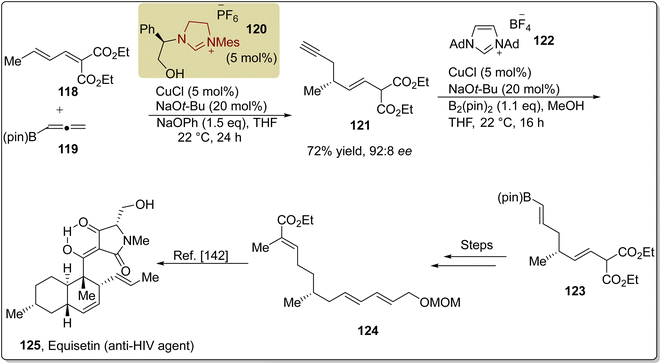 |
| | Scheme 13 Asymmetric synthesis of equisetin 125. | |
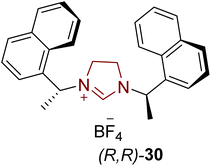
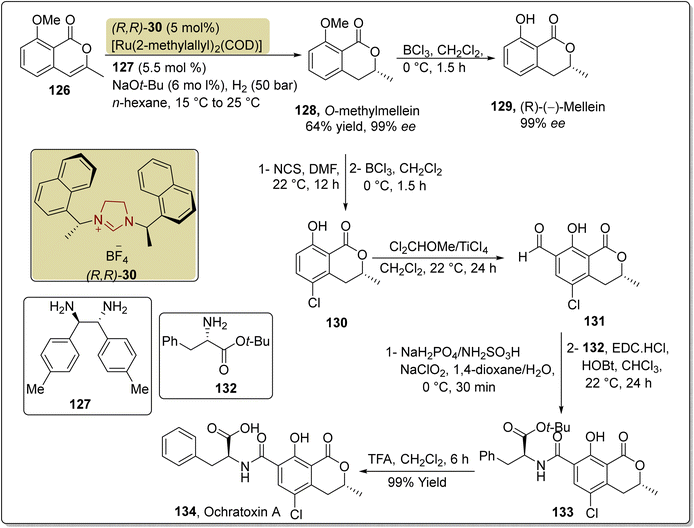
The isocoumarin-based polyketide mellein 129 was first extracted from the fungus Aspergillus melleus in 1933. Mellein 129 demonstrates mycocidal and antibacterial activities.143,144 In 1965, Steyn and fellow researchers isolated ochratoxin A 134 from Aspergillus ochraceus.145 In 2017, Li et al. disclosed an enantioselective route to furnish O-methylmellein 128, mellein 129 and ochratoxin A 134 by harnessing NHC (R,R)-30-diamine-127 as ligand with a ruthenium transition-metal catalyst. In this unique synthetic protocol, 8-methoxy-3-methylisocoumarin 126 was allowed to undergo reduction by utilizing the Ru–NHC (R,R)-(30)-diamine 127 catalytic system in the presence of NaOt-Bu, H2 gas at 50 bar pressure and n-hexane at 15–25 °C to obtain O-methylmellein 128 in 64% yield with 99% ee. Next, O-methylmellein 128 was demasked by employing BCl3 and CH2Cl2 at 0 °C to furnish another natural product, mellein 129, with 99% ee. In order to synthesize ochratoxin A 134, O-methylmellein 128 was treated with N-chlorosuccinimide (NCS) to synthesize its chloro-derivative, which further underwent demethylation in the presence of BCl3 and CH2Cl2 at 0 °C to generate 5-chloromellein 130. Next, chloromellein 130 was made to undergo Rieche formylation in the presence of Cl2CHOMe/TiCl4 to furnish aldehyde 131, which was converted into the corresponding acid via Pinnick oxidation, and subsequently treated with L-phenylalanine 132 to generate the amide 133. Finally, the t-butyl ester group was removed from amide 133 by utilizing TFA and CH2Cl2 to obtain ochratoxin A 134 (Scheme 14).146
 |
| | Scheme 14 Asymmetric synthesis of mellein 129, O-methylmellein 128 and ochratoxin A 134. | |
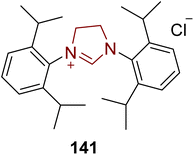
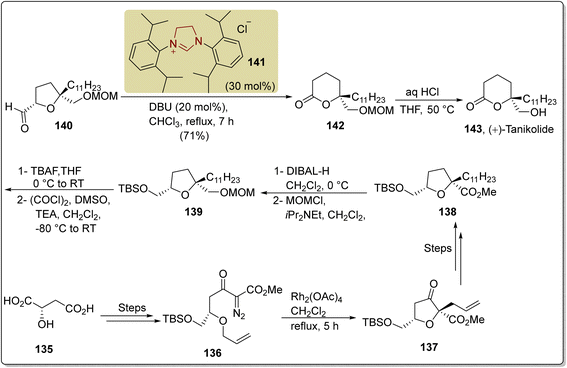
Tanikolide 143 is a polyketide with a lactonic core structure that was isolated from the lipid extract of the marine cyanobacterium Lyngbya majuscula by Gerwick and coworkers in 1999.147 Tanikolide 143 shows molluscicidal, antifungal and narcotic activities.148 In 2018, Yakura and coworkers introduced an asymmetric route to furnish tanikolide 143 by employing NHC 141 as a catalyst. In this synthetic protocol, L-malic acid 135 was transformed into diazoketoester 136 over a number of steps, and subsequently submitted to [2,3]-sigmatropic rearrangement in the presence of dirhodium(II) tetraacetate and refluxing in dichloromethane for 5 hours to synthesize dihydrofuranone 137 in 93% yield. Dihydrofuranone 137 was then converted into the tetrahydrofuran 138 over a few steps. The methyl ester of tetrahydrofuran 138 was then converted into the alcohol by reduction with DIBAL-H, and further protected with methoxy methyl ether to generate compound 139. In the next step, MOM-protected tetrahydrofuran 139 underwent desilylation by utilizing TBAF, and subsequent Swern oxidation furnished the corresponding aldehyde 140. Ring expansion of the aldehyde-containing tetrahydrofuran 140 to the δ-lactone 142 proceeded with 71% yield by employing imidazolium-based NHC 141 as a catalyst with the aid of DBU in chloroform at reflux for 7 hours. Finally, MOM-deprotection of δ-lactone 142 was achieved to furnish tanikolide 143 (Scheme 15).149,150
 |
| | Scheme 15 Asymmetric synthesis of tanikolide 143. | |
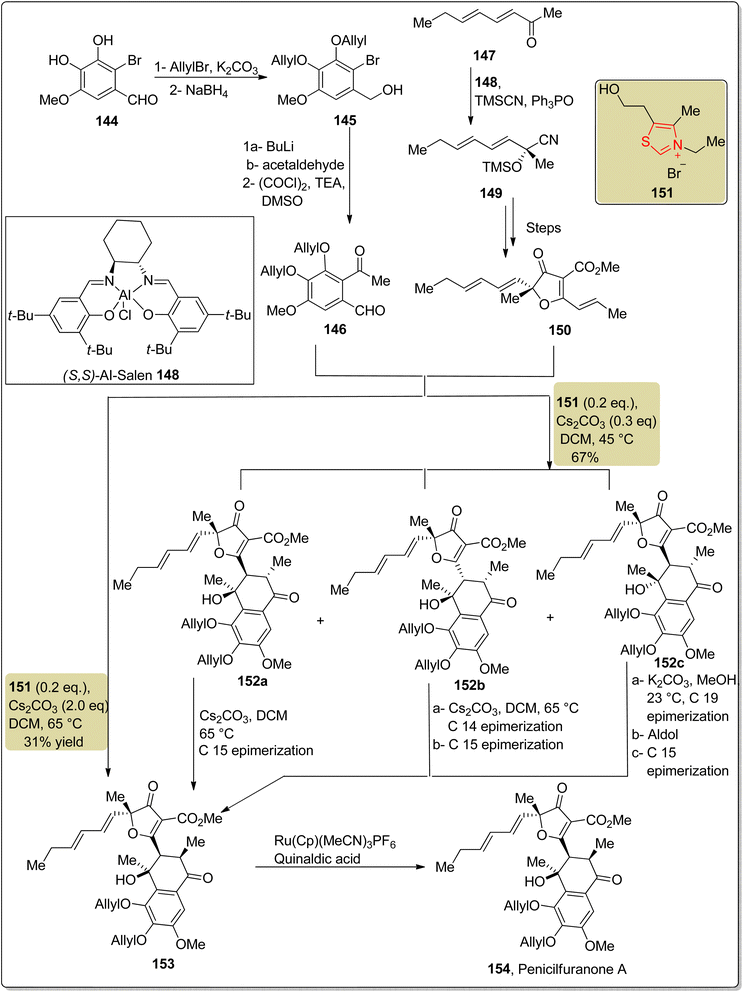
Penicilfuranone A 154, a furancarboxylic acid that belongs to the class of tricyclic aromatic polyketides, was obtained from Penicillium sp. Sh18 that was extracted from the stem of the plant I. eriocalyx var.Laxiflora in 2016 by Pu and fellow researchers.151,152 Penicilfuranone A 154 shows fibrosis attenuating effects in fibrogenic hepatic stellate cells and is considered an important compound to treat hepatic fibrosis. In 2025, Ding et al. reported the first asymmetric approach to the synthesis of penicilfuranone A 154 by employing thiazolium-based NHC 151 as a catalyst. The synthetic protocol commenced with the synthesis of benzaldehyde 146, which was produced in a three-step reaction involving allyl bromide protection, aldehyde reduction to generate alcohol 145 and subsequent Swern oxidation. On the other hand, gregatin A 150 was generated by utilizing Al-salen 148 to catalyze the asymmetric cyanosilylation of methyl ketone 147, yielding intermediate 149. Over a few steps, compound 149 was then converted into gregatin A 150. Next, benzaldehyde 146 and gregatin A 150 were allowed to undergo dimerization in the presence of NHC 151 as a catalyst, 0.3 eq. of Cs2CO3 and DCM at 65 °C to generate three diastereomers, 152a, 152b and 152c, which were subsequently converted into the precursor of penicilfuranone A 153, individually. Benzaldehyde 146 and gregatin A 150 were also converted directly into penicilfuranone A precursor 153 when they were treated with NHC 151, 2 eq. of Cs2CO3 and DCM at 45 °C. Finally, compound 153 was allowed to undergo deallylation in the presence of a ruthenium catalyst to furnish penicilfuranone A 154 (Scheme 16).152
 |
| | Scheme 16 Asymmetric synthesis of penicilfuranone A 154. | |
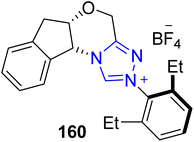
2.1.3 Synthesis of terpenoids. The sesquiterpenoid natural products, marasmane and protoilludane, were isolated from the parasitic basidiomycete fungus species Lactarius vellereus and Armillaria mellea in the 1960s. Protoilludane demonstrates antimicrobial activity, and marasmane shows anti-feedant and wide-spectrum antimicrobial activity. Moreover, mellolite sesquiterpenoid exhibits radiosensitizing and apoptotic activity against various malignant cell lines. Owing to its high pharmaceutical importance, Hovey et al., in 2017, introduced a unified synthetic approach to generate echinocidin D 165 and B 167 (protoilludane), isovelleral 164 (marasmane) and armillaridin 170 (mellolite) natural products by employing NHC 160 as an efficient catalyst. The total synthesis began with the ozonolysis of 1,5-dienoate 155 to generate aldehyde 156, which was subjected to Masamune–Roush modified Horner–Wadsworth–Emmons (HWE) olefination to furnish enone 158. In the next step, enone 158 was subjected to α-iodination and subsequent Arbuzov reaction, in conjunction with reduction, followed by oxidation in the presence of manganese oxide to generate enal 159. Next, enal 159 underwent an annulation reaction by employing triazolium-based NHC 160 (catalyst) to afford lactone 161 with 99:1 er and >20:1 dr. Over a few steps, lactone 161 was converted into aldehyde 162, which was further subjected to intramolecular pinacol reductive coupling to convert it into cis-cyclobutane-diol 163 by employing a vanadium(II)/zinc(II) bimetallic complex generated in situ. After desilylation of cis-cyclobutane-diol 163 by utilizing TASF, echinocidin D 165 (protoilludane) was accessed. On the other hand, when cis-diol 163 was exposed to para-nitrobenzoic acid, cyclopropane was obtained, which further underwent desilylation with TBAF, followed by oxidation with TEMPO to furnish isovelleral 164 (marasmane). In order to synthesize armillaridin 170 (melloide), cis-diol 163 was allowed to undergo reduction by employing sodium triacetoxyborohydride to furnish the trans-diol 166, followed by esterification with orsellinic acid derivative 168 to deliver orsellinate ester 169, with subsequent desilylation with TBAF and oxidation with TEMPO to afford armillaridin 170. Desilylation of trans-diol 166 with TASF afforded echinocidin B 167 (Scheme 17).153
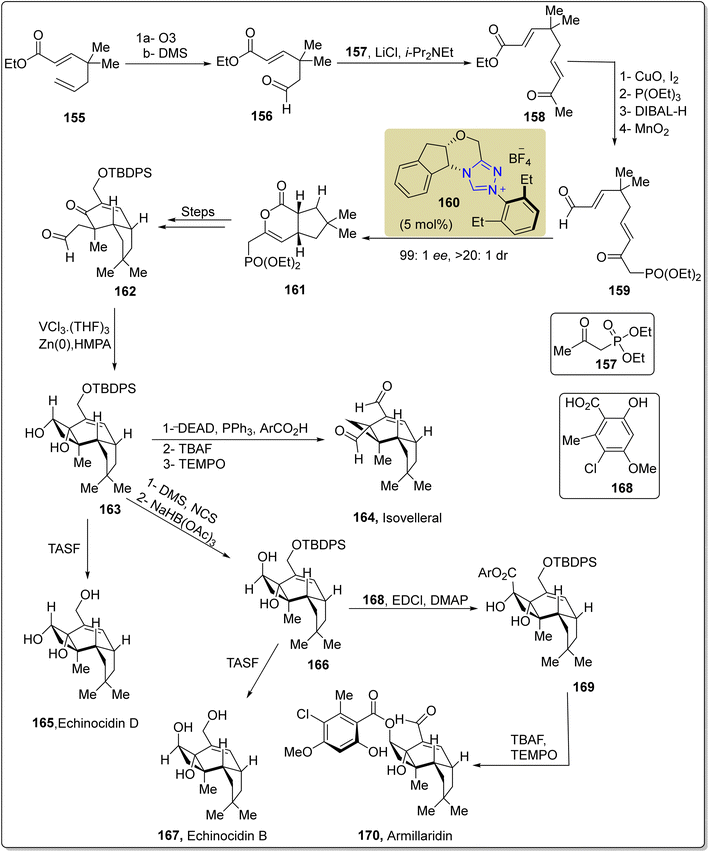 |
| | Scheme 17 Asymmetric synthesis of isovelleral 164, echinocidin D 165 and B 167, and armillaridin 170. | |
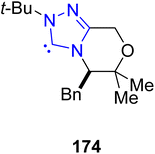
Delta-9-tetrahydrocannabinol (Δ9-THC) 179 is a monoterpenoid natural product extracted in 1964 from the Cannabis sativa L in its trans-form. Δ9-THC 179 demonstrates antiglaucoma, antiemetic, analgesic and antinauseant activities.154,155 In this regard, Ametovski et al., in 2019, reported an enantioselective approach to synthesize normethyl (−)-Δ9-THC 176, (−)-Δ8-THC 178 and (−)-Δ9-THC 179 by utilizing triazolium-based NHC 174 as a catalyst. The total synthesis commenced from the commercially and easily available cinnamate 171, which was converted into acyl fluoride 172 by employing diethylaminosulfur trifluoride (DAST) in CH2Cl2 at 0 °C. In the next step, [4 + 2] annulation reaction was performed between acyl fluoride 172 and cyclobutane 173 by utilizing NHC 174 (catalyst) in DMF/THF at 80 °C to afford β-lactone 175 in 45% yield with 98:2 enantioselectivity and 20:1 diastereoselectivity ratio. Over a few steps, β-lactone 175 was converted into normethyl-(−)-Δ9-THC 176 in 62% yield. Treatment of β-lactone 175 with KCN and MeOH at 0 °C and subsequent oxidation with IBX and EtOAc at 80 °C afforded β-ketoester 177. Next, β-ketoester 177 was transformed into (−)-Δ8-THC 178 in 40% yield via multiple steps. Furthermore, (−)-Δ8-THC was isomerized into (−)-Δ9-THC 179 in two steps by utilizing ZnCl2 and HCl, and then KOC5H11 in toluene (Scheme 18).156
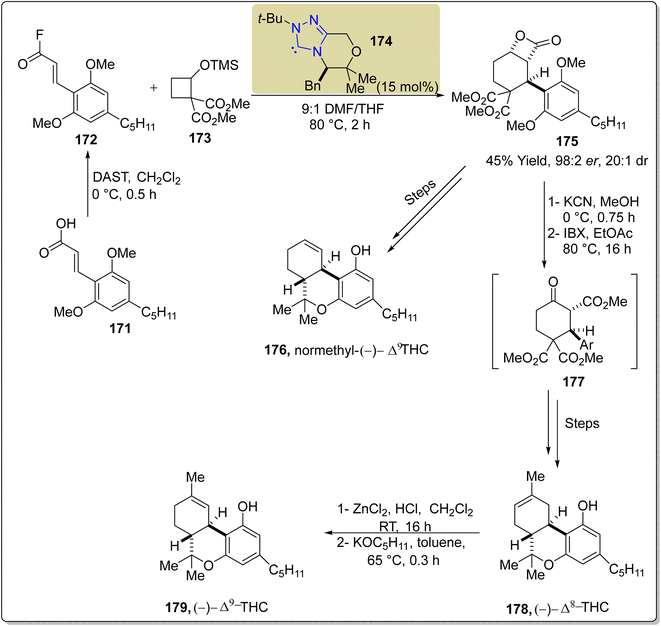 |
| | Scheme 18 Asymmetric synthesis of normethyl (−)-Δ9-THC 176, (−)-Δ8-THC 178 and (−)-Δ9-THC 179. | |
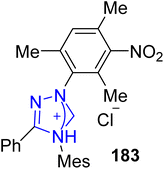
Nepetalactone 185 belongs to the class of iridoid monoterpenoids, which are present in several plants of the genus Nepeta, which includes about 300 species in the Lamiaceae family of mints.157 Nepetalactone 185 is the key component of mosquito repellents, aphid sex pheromones and insects. Moreover, they show euphoric effects in cats. In 2020, Harnying et al. reported an enantiospecific strategy to synthesize nepetalactone 185 by employing triazolium-based NHC 183 as a catalyst. The synthesis began with the allylic oxidation of (S)-citronellol 180 with SeO2/t-BuOOH to obtain an unrefined mixture of 181a and 181b, which subsequently underwent IBX oxidation to deliver 8-oxocitronellal (S)-182. In the next step, oxidative bicyclization took place to convert 8-oxocitronellal (S)-182 into (+)-nepetalactone (+)-185 in 84% yield and 97% enantiomeric excess in the presence of NHC 183 (catalyst) by utilizing quinone 184 (oxidant) and DIPEA (base) in THF. Moreover, (−)-nepetalactone (−)-185 was also furnished in 80% yield and >99% enantiomeric excess via the same synthetic protocol (Scheme 19).158
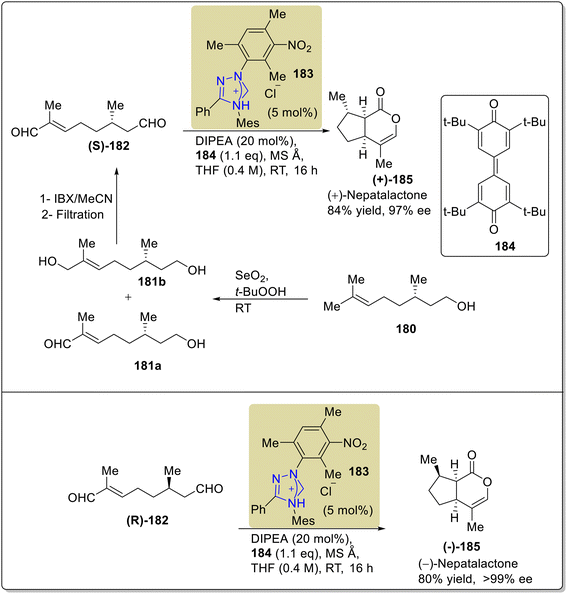 |
| | Scheme 19 Asymmetric synthesis of (+)-nepetalactone (+)-185 and (−)-nepetalactone (−)-185. | |
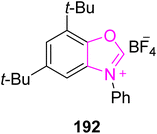
Pseudosinin I 194 and dihydro-ar-turmerone 196 are naturally occurring sesquiterpenoids that were isolated separately from Pseudotsuga sinensis and the wood of Himalayan Cedar, respectively.159–161 Zhang et al., in 2024, reported an enantioselective approach to synthesize pseudosinin I 194, (+)-ar-juvabione 195 and dihydro-ar-turmerone 196 by utilizing oxazole-based NHC 192 as a catalyst. In this synthetic protocol, ketones 186 and 187 were allowed to react with MeCHO and LDA to furnish the corresponding β-hydroxy ketones 188 and 189. In the next step, the β-hydroxy ketones 188 and 189 were subjected to deoxygenative asymmetric reductive cross-coupling with aryl bromides 190 and 191 in the presence of NHC 192 (catalyst) to furnish final natural products, pseudosinin I 194, (+)-ar-juvabione 195 and (−)-dihydro-ar-turmerone 196 (Scheme 20).162
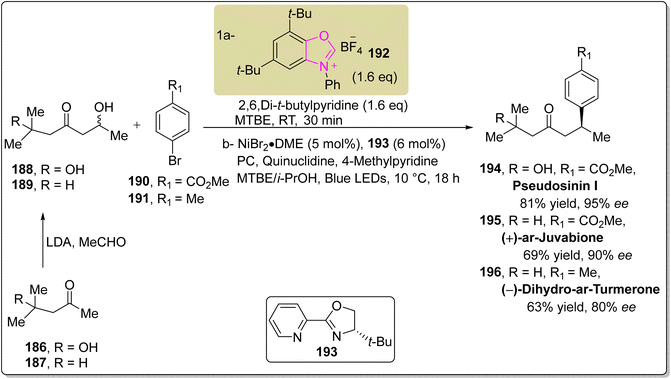 |
| | Scheme 20 Asymmetric synthesis of pseudosinin I 194, (+)-ar-juvabione 195 and (−)-dihydro-ar-turmerone 196. | |
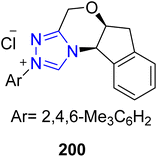
2.1.4 Synthesis of lignans. Hinokinin 204 is a dibenzylbutyrolactone-lignan that was isolated from the heartwood of L. formosana Florin.163 Hinokinin 204 demonstrates antimicrobial, antiinflammatory and regulatory effects on human γ-aminobutyric acid (GABA) translocation activities.164,165 Owing to its high medicinal importance, Singha et al., in 2020, reported the use of NHC 200 and iridium cooperative catalysis to achieve the enantioselective and diastereodivergent synthesis of hinokinin 204 via a [3 + 2] annulation reaction. The total synthesis commenced with [3 + 2] annulation reaction between α-chloro aldehyde 197 and vinylethylene carbonate 198 in the presence of NHC 200/Ir cooperative catalyst by utilizing 1,1′-spirobiindane-7,7′-diol (SPINOL) ligand ent-199 and NMM (base) in toluene to generate trans-lactone 201 in 83% yield with 99% enantiomeric excess. In the next step, dihydroxylation of the double bond and subsequent oxidative cleavage delivered the aldehyde 202. Next, the Nozaki–Hiyama–Kishi reaction was performed between aldehyde 202 and aryl iodide 203, followed by hydrogenolysis utilizing Pd/C (catalyst) to furnish the final compound (−)-hinokinin 204 in 73% yield with 98% enantiomeric excess (Scheme 21).166
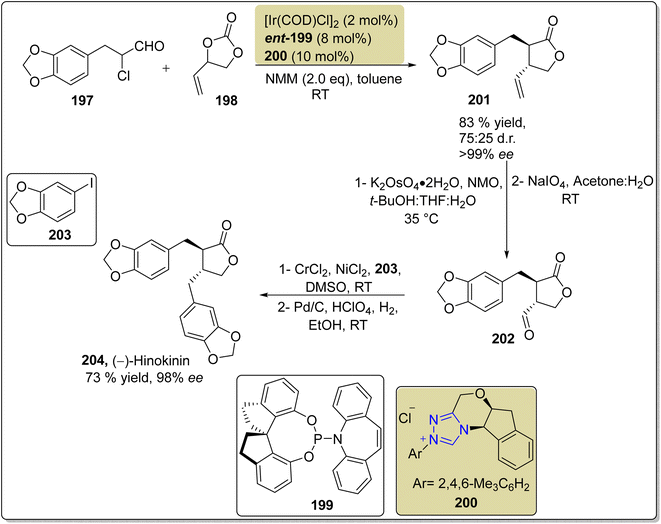 |
| | Scheme 21 Asymmetric synthesis of hinokinin 204. | |
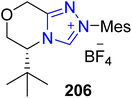
Shi and colleagues isolated a unique polycyclic neolignane named codonopiloneolignanin A 210 in 2016 from the roots of C. pilosula.167 In 2021, Li et al. demonstrated the use of NHC 206 and titanium(IV) cooperative catalysis to deliver asymmetric codonopiloneolignanin A 210 in four steps. The total synthesis commenced with asymmetric dimerization of cinnamaldehyde 205 to furnish cis-cyclopentene 207 in the presence of Ti(IV)/NHC 206 as a cooperative catalyst, TBD (base) and a mixture of isopropanol and acetonitrile (MeCN) (solvent). Next, reduction of the double bond of cis-cyclopentene 207 was achieved by utilizing sodium borohydride (NaBH4) and nickel chloride hexahydrate, with further subsequent reduction using DIBAL to convert the ester intermediate into aldehyde 209. Due to over-reduction, a small amount of alcohol 208 was also formed, which was re-oxidized back to aldehyde 209 by utilizing Dess–Martin periodinane. In the next step, aldehyde 209 was subjected to transannular intramolecular Prins reaction followed by cation-mediated cyclization by utilizing TFA in DCM, and the product was subjected to deprotection to form the final natural product codonopiloneolignanin A 210. Codonopiloneolignanin A 210 was further treated with bromobenzoyl chloride along with TEA in DMAP and DCM at 0 °C to furnish the codonopiloneolignanin A derivative 211 (Scheme 22).168
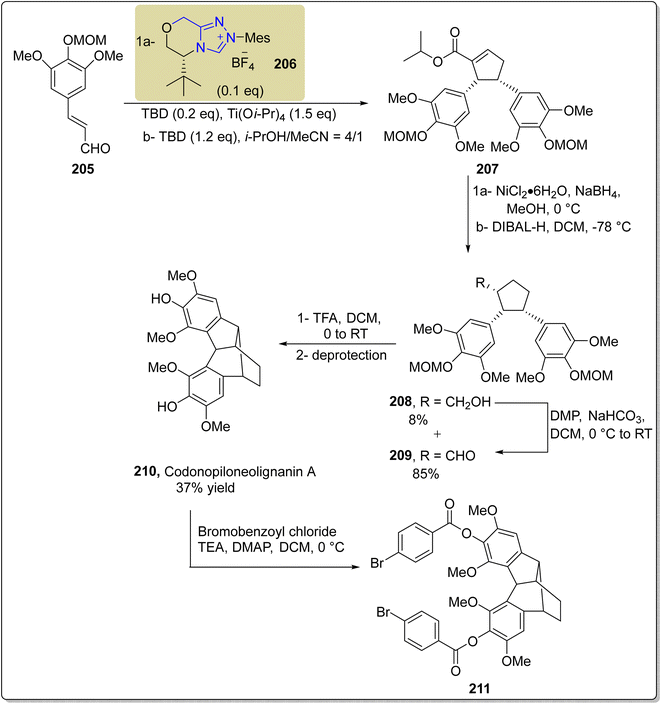 |
| | Scheme 22 Asymmetric synthesis of codonopiloneolignanin A 210. | |
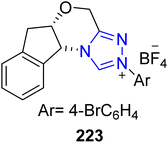
2.1.5 Synthesis of flavonoids. Rotenoids are a significant class of naturally occurring compounds that have been discovered in the Derris and Lonchocarpus species. In 2019, Perveen et al. demonstrated an approach by utilizing NHC 223 as a catalyst that enabled the efficient construction of the cis-fused tetrahydrochromeno[3,4-b]chromene central scaffold of rotenoids. The approach has been utilized for the synthesis of various rotenoids, including 12a-hydroxymunduserone 224 (antitumor activity), tephrosin 226 (pesticides), milletosin 229, 12a-hydroxyrotenone 231, retenone 232 (anticancer activity) and deguelin 227. In order to synthesize naturally occurring rotenoids, three aldehyde modules 213, 215, and 217 and two ketone modules 219 and 221 were first generated. Next, the conjunction of modules 213 and 219 took place, followed by deprotection to generate rac-222, which was further submitted to an intramolecular annulation reaction utilizing NHC 223 as a catalyst with Cs2CO3 and additives A, B, C, D in THF at −20 °C to furnish 12a-hydroxymunduserone 224 in 58% yield with 95.5:4.5 er (Scheme 23). By utilizing similar optimized conditions, tephrosin 226, milletosin 229 and 12a-hydroxyrotenone 231 were also synthesized.169 Next, deguelin 227 from tephrosin 226 and rotenone 232 from 12a-hydroxyrotenone 231 were also achieved by utilizing the method reported by XiaoHui and coworkers (Scheme 24).170
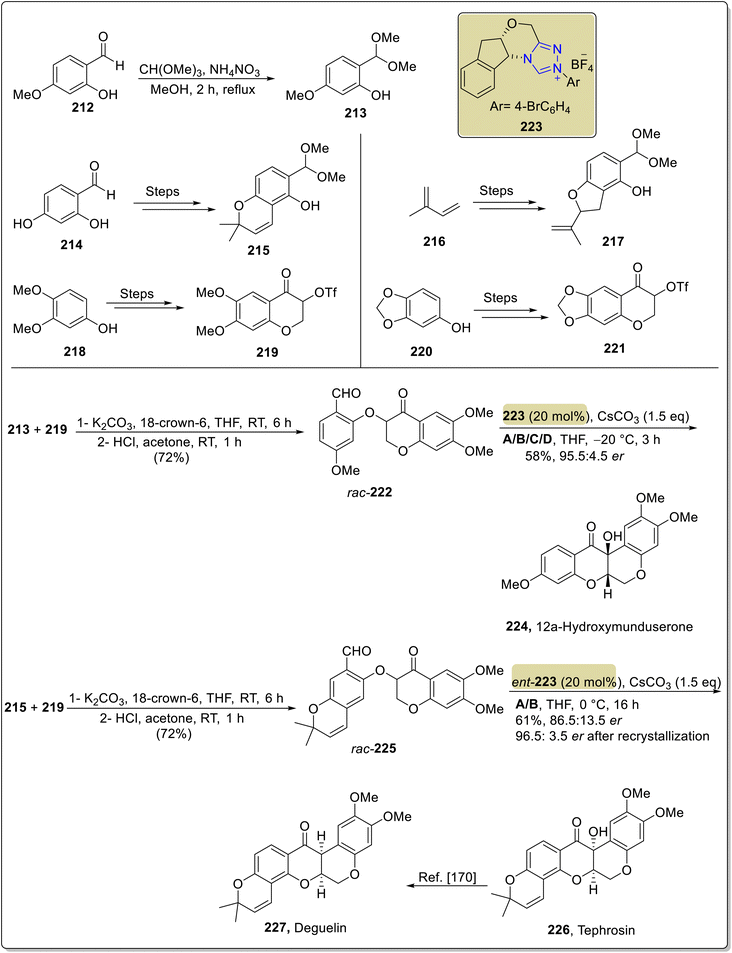 |
| | Scheme 23 Asymmetric synthesis of 12a-hydroxymunduserone 224, tephrosin 226, and deguelin 227. | |
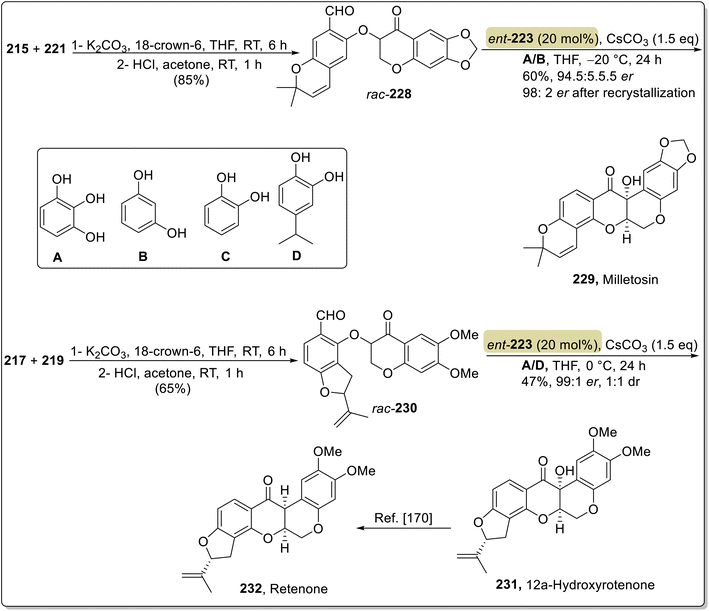 |
| | Scheme 24 Asymmetric synthesis of milletosin 229, 12a-hydroxyrotenone 231 and retenone 232. | |
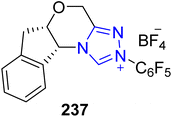
Umehara and co-researchers isolated an isoflavanonol derivative named isodarparvinol B 238 from the duramen of the medicinal plant Dalbergia parviflora, which is native to Thailand.171 Iwai et al., in 2020, demonstrated an enantioselective approach to furnish isodarparvinol B 238 by utilizing NHC 237 as a catalyst. The total synthesis commenced with the bromination of ketone 233 with phenyltrimethylammonium tribromide to generate compound 234. Next, compounds 234 and 235 were subjected to Williamson ether synthesis followed by acetal deprotection to generate aldehyde 236 in 62% yield. In the next step, aldehyde 236 was allowed to undergo intramolecular benzoin reaction in the presence of NHC 237 (catalyst) by utilizing Cs2CO3 in CHCl3 at 40 °C to afford the 4-chromanone, which subsequently underwent hydrogenation to generate (−)-isodarparvinol B 238 in 93% yield with 90% ee (Scheme 25).172
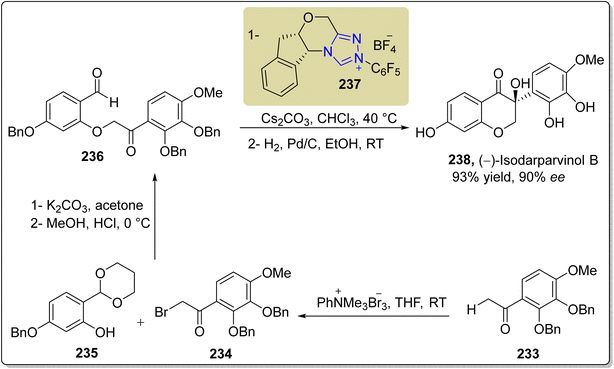 |
| | Scheme 25 Asymmetric synthesis of isodarparvinol B 238. | |
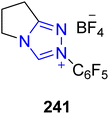
2.1.6 Miscellaneous natural products. D-ribo-Phytosphingosine exhibits cytotoxic activity against a cell line derived from human leukemia and also serves as an indicator of thermal stress in yeast cells. Several analogues of D-ribo-phytosphingosine have been observed to demonstrate antitumor, antiviral and immunostimulatory activities.173 In 2016, Haghshenas et al. reported a diastereoselective strategy to synthesize a derivative of D-ribo-phytosphingosine named D-arabino-phytosphingosine 244 by employing NHC 241 as a catalyst. The total synthesis began with the cross-benzoin reaction between aliphatic aldehyde 239 and N-Boc-protected amino aldehyde 240 in the presence of triazolium-based NHC 241 (catalyst) to furnish a mixture of 242 with 5:1 diastereoselectivity ratio. In the next step, the diastereomeric mixture 242 was reduced with ZnCl2/NaBH4 to generate the amino diols 243 with >20:1 dr. Finally, amino diol 243 was subjected to deprotection to furnish the final natural product D-arabino-phytosphingosine 244 (Scheme 26).174
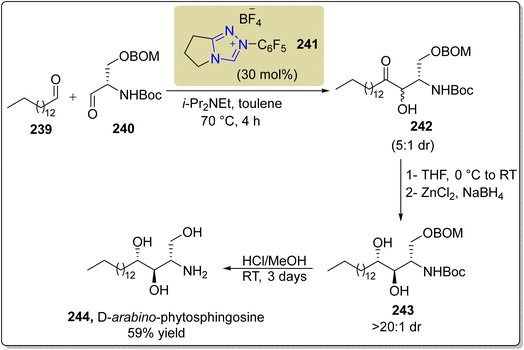 |
| | Scheme 26 Asymmetric synthesis of D-arabino-phytosphingosine 244. | |
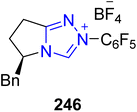
In 2017, Kang et al. reported a cross-benzoin type cyclization reaction by utilizing NHC 246 as a catalyst to furnish epi-inositol hexaacetate 249, muco-inositol hexaacetate 251 and myo-inositol hexaacetate 259. Among them, myo-inositol 259 and its derivatives have been extensively investigated due to their abundance and wide range of biological characteristics.175 myo-Inositol 259 exhibits diverse functionalities, including roles in regulating phosphate levels, ion-channel permeability, metabolic flux, embryonic development and insulin signaling.176 The total synthesis began with the benzoin cyclization reaction of 3,4-O-acetonide 245 masked with TBDPSO groups in the presence of triazolium-based NHC 246 (as a catalyst) with NaOBz (as base) in toluene to furnish a crude product, which was further treated with HF in aq. MeCN at −20 °C to generate the inosose 247. Next, compound 247 was subjected to reduction by utilizing BH3·THF to synthesize compound 248, which was further deprotected with TBAF and then acylated with Ac2O to furnish the final compound epi-inositol hexacetate 249. Parallel to this, compound 247 was also allowed to undergo reduction in the presence of Me4N(AcO)3BH, AcOH and MeCN to synthesize 250, which was further deprotected with TBAF and acylated with Ac2O to furnish another natural product, muco-inositol hexaacetate 251. Next, another unsymmetrical dialdose 252 was allowed to undergo benzoin cyclization reaction under similar conditions to synthesize compounds 253, 254, 255 and 256. In the next step, compound 255 was reduced with BH3·THF to deliver compound 257, which was further treated with MeONa, followed by deprotection with TBAF in conjunction with acylation using Ac2O to synthesize epi-inositol hexaactetate 249. Parallel to this, compound 255 was also protected with TESOTf, then reduced with BuNH2·BH3 to generate compound 258. Finally, compound 258 was treated with MeONa followed by deprotection with TBAF and then subjected to acylation to synthesize myo-inositol hexaacetate 259 (Scheme 27).175
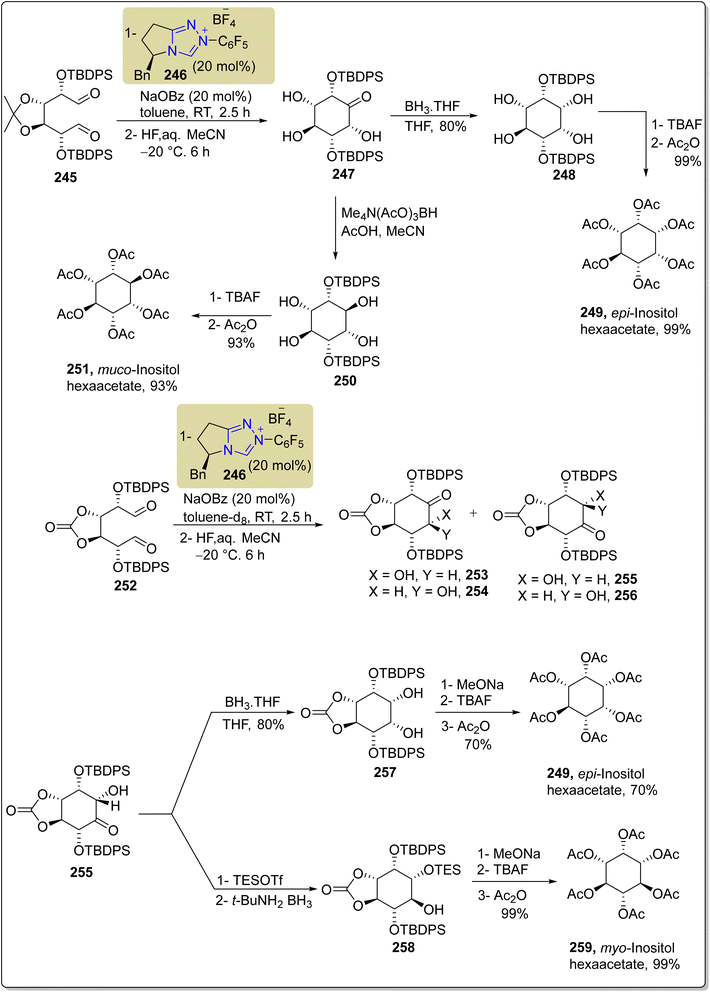 |
| | Scheme 27 Asymmetric synthesis of epi-inositol hexaacetate 249, muco-inositol hexaacetate 251 and myo-inositol hexaacetate 259. | |
Asymmetric, naturally occurring paraconic acids exhibit various biological activities, including anti-HIV-1, antibiotic and antimicrobial activity, depending on their substitution and chiral configuration.177–180 In this regard, Sarkale et al., in 2019, reported a stereodivergent approach to synthesize paraconic acids by utilizing triazolium-based NHC 241 as a catalyst. In this synthetic protocol, alkyl aldehyde 261 and N-benzylmaleimide 260 were allowed to react in the presence of NHC 241 (catalyst) by utilizing DIPEA (base) in toluene to synthesize 3-acylsuccinimide 262. Next, asymmetric transfer hydrogenation (ATH), along with dynamic kinetic resolution (DKR) of 3-acylsuccinimide 262, was performed to generate a diastereomeric mixture of alcohols 265 and 266, which could not be separated, by employing 264 with HCO2H/TEA in DMF at room temperature. In the next step, alcohol 265 was treated with DBU and aq. KOH in 1,4-dioxane at 55 °C to synthesize trans-paraconic acid 267. Parallel to this, alcohol 265 was also subjected to hydrolysis of the imide by employing 5% aqueous KOH, followed by treatment with NaNO2 and Ac2O in acetic acid to furnish cis-paraconic acid 268. Next, the cis-paraconic acid 268 was treated with NaHMDS and MeI in THF to synthesize (−)-phaseolinic acid 269 in 93% yield and (−)-nephromopsinic acid 270 in 96% yield. When ent-267 was treated under similar conditions, nephrosteranic acid 274 was obtained in 89% yield, and (+)-roccellaric acid 275 was obtained in 88% yield. (−)-Methylenolactocin 271 was furnished from 267 upon treatment with MeOMgOCO2Me in DMF and then with CH2O, AcOH, AcONa and PhNHMe. Moreover, when ent-267 was treated under similar conditions, (+)-nephrosterinic acid 272 and (+)-protolichesterinic acid 273 were obtained in 56% and 53% yield, respectively (Scheme 28).181
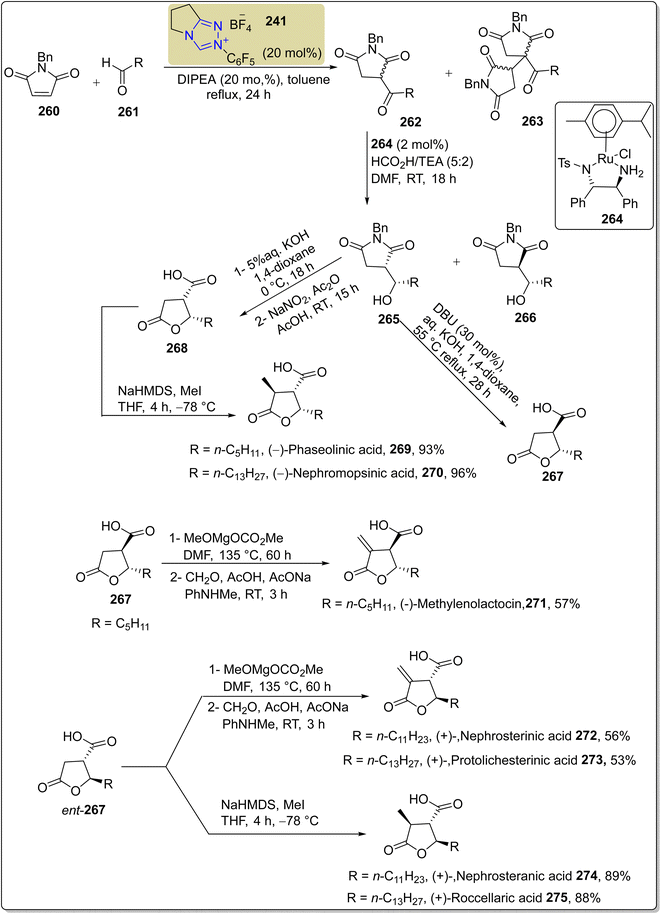 |
| | Scheme 28 Asymmetric synthesis of (−)-phaseolinic acid 269, (−)-nephromopsinic acid 270, (−)-methylenolactocin 271, (+)-nephrosterinic acid 272, (+)-protolichesterinic acid 273, (+)-nephrosteranic acid 274 and (+)-roccellaric acid 275. | |
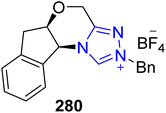
Fredericamycin A 282 is a naturally occurring aromatic pentadecaketide that was isolated from the Streptomyces griseus by Pandey and co-workers in 1981.182,183 Fredericamycin A 282 was observed to exhibit cytotoxic activity against a number of cell lines and moderate anticancer bioactivity.183 Owing to its medicinal importance, Ren et al., in 2025, reported an efficient asymmetric approach to synthesize fredericamycin A 282 by utilizing NHC 280 as a catalyst. Initially, phthalide 277 was prepared from compound 276 in a few steps. Parallel to this, dienophile 279 was furnished from compound 278 in a few steps. Next, phthalide 277 and dienophile 279 were allowed to undergo NHC 280 catalyzed asymmetric reaction in the presence of N-methylynetoluenesulfonamide as coupling reagent and DABCO as base to generate the intermediate 281,182 which was reported earlier by Kita et al., in 1999, and utilized for the synthesis of fredericamycin A 282 (Scheme 29).184
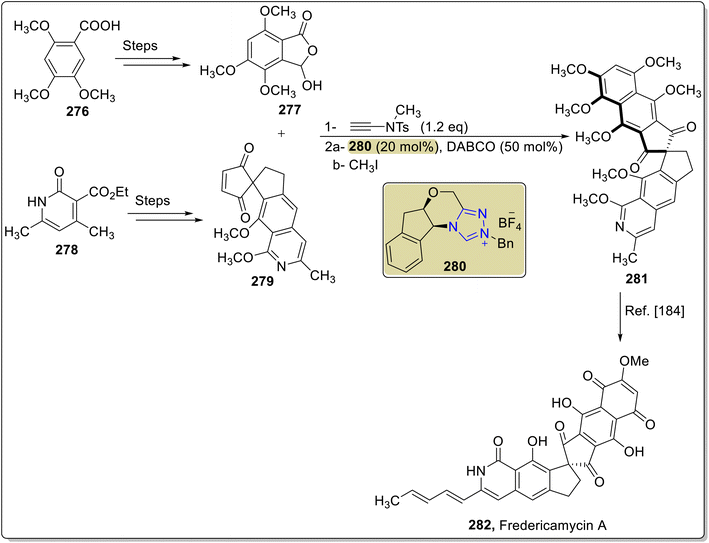 |
| | Scheme 29 Asymmetric synthesis of fredericamycin A 282. | |
2.2 Pharmaceutical drugs
2.2.1 Synthesis of antidepressant drugs. (R)-(−)-Thiazesim 284 is an antidepressant drug that is available as its hydrochloride salt, i.e., Altinil and diltiazem. (R)-(−)-Thiazesim 284 is utilized for the amelioration of hypertension and angina.185 Owing to its pharmaceutical importance, Li et al., in 2016, reported an asymmetric hydrogenation approach to furnish (R)-(−)-thiazesim 284 by utilizing ruthenium NHC (S,S)-30 as a catalyst in a single step. The synthesis began with the asymmetric hydrogenation of unsaturated 1,5-benzothiazepinone 283 in the presence of NHC (S,S)-30 as ligand with ruthenium catalyst by employing KOtBu in n-hexane under 100 bar H2 at room temperature to synthesize (R)-(−)-thiazesim 284 in 78% yield with 93% enantiomeric excess (Scheme 30).186
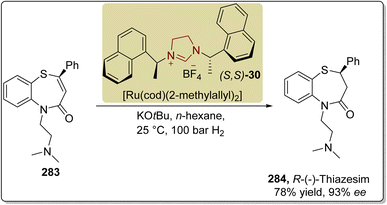 |
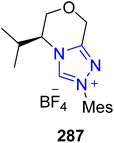
| | Scheme 30 Asymmetric synthesis of (R)-(−)-thiazesim 284. | |
In 2017, Fang et al. also reported a novel enantioselective strategy to synthesize (R)-thiazesim 284 by utilizing the triazolium-based NHC 287 as a catalyst. The synthesis commenced with the [3 + 4] annulation of α-bromoenal 285 with 2-aminobenzenethiol 286 in the presence of NHC 287 (catalyst) and NaOAc (base) in PhMe (solvent) at room temperature to furnish 1,5-benzothiazepine 288 in 53% yield with 90.7% ee. In the next step, 1,5-benzothiazepine 288 was refluxed with 2-chloro-N,N-dimethylethan-1-amine and K2CO3 in AcOEt/H2O for 12 hours to generate (R)-thiazesim 284 in 91% yield with 93% ee (Scheme 31).187
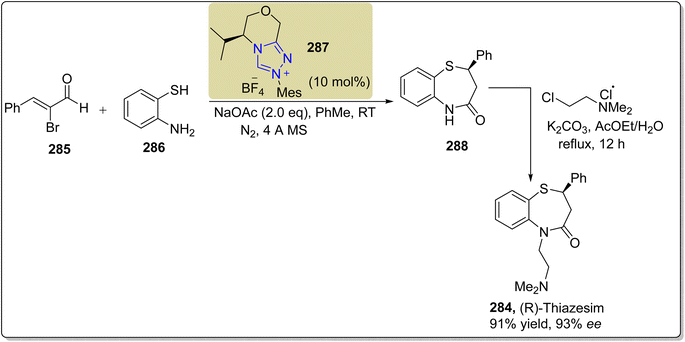 |
| | Scheme 31 Asymmetric synthesis of (R)-(−)-thiazesim 284. | |
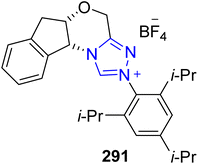
The pharmaceutical product paroxetine 296 has been used to treat a number of anxiety disorders, namely generalized anxiety disorder, major depressive disorder, obsessive-compulsive disorder, premenstrual dysphoric disorder, panic disorder, post-traumatic disorder, and social anxiety disorder.188 Owing to its high medicinal importance, Porey et al., in 2019, demonstrated an enantioselective strategy towards the formal synthesis of paroxetine 295 by utilizing oxidative NHC 291 catalysis. The synthesis commenced with the [3 + 3] annulation of enal 289 and malonamide 290 by utilizing NHC 291 (catalyst) and DBU in THF to furnish compound 292 in 72% yield, 97% enantioselectivity and 4:1 diastereoselectivity. In the next step, compound 292 was allowed to undergo transamidation to install the PMP-group, followed by imide reduction by utilizing LiAlH4 in THF to synthesize compound 293. Next, Boc-protection of compound 293 was performed, and subsequent DIBAL reduction generated the important intermediate alcohol 294.189 The synthesized intermediate 294 had been reported earlier by Hughes et al. in the synthesis of paroxetine 295 (Scheme 32).190
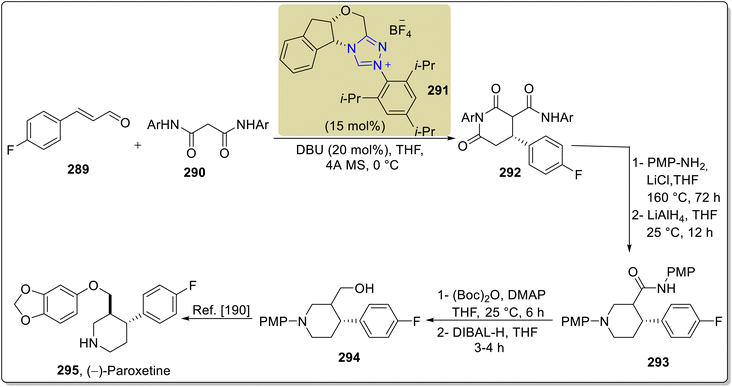 |
| | Scheme 32 Asymmetric synthesis of paroxetine 295. | |
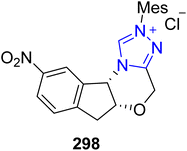
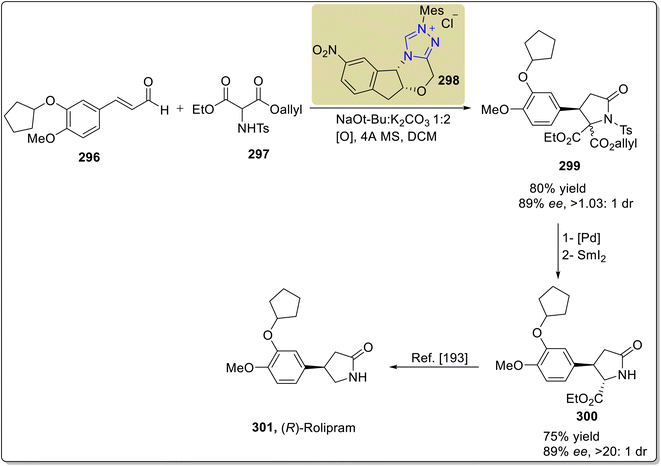
Rolipram 301 is a phosphodiesterase inhibitor that was initially formulated to treat depression, but has also been used to treat asthma, Huntington's disease, arthritis, multiple sclerosis, human immunodeficiency virus (HIV) infections, traumatic brain injury (TBI) and Alzheimer's disease.191 In this regard, Zhang et al., in 2021, reported an asymmetric strategy towards the formal synthesis of (R)-rolipram 301 by employing oxidative NHC 298 catalysis. In this synthetic protocol, enal 296 and N-Ts allyl-ethyl aminomalonate 297 were allowed to undergo [3 + 2] annulation by utilizing NHC 298 (catalyst) and NaOtBu:K2CO3 (base) in DCM to furnish the γ-lactam 299 in 80% yield, 90% enantiomeric excess and 1.03:1 diastereoselectivity ratio. Next, the γ-lactam 299 was treated with a palladium catalyst to remove the allyl ester, followed by detosylation using SmI2 to synthesize an intermediate γ-lactam 300,192 which was reported earlier for the synthesis of rolipram 301 (Scheme 33).193
 |
| | Scheme 33 Asymmetric synthesis of (R)-rolipram 301. | |
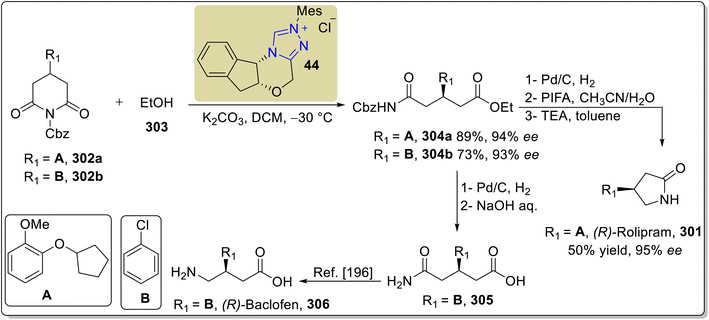
A year later, in 2022, Hu et al. reported an asymmetric approach towards the total synthesis of (R)-rolipram 301 by utilizing NHC 44 as a catalyst. The synthesis began with the imide C–N bond cleavage for the desymmetrization of N-Cbz cyclic imide 302a with alcohol 303 in the presence of triazolium-based NHC 44 (as a catalyst) with K2CO3 (base) in DCM at −30 °C to furnish the amido ester 304a in 89% yield with 94% enantiomeric excess. In the next step, amido ester 304a was allowed to undergo decarbobenzyloxylation followed by decarbonylation in conjunction with intramolecular lactamization to furnish (R)-rolipram 301 in 50% yield with 95% enantioselectivity.194 Along with (R)-rolipram 301, Hu et al. also reported the formal synthesis of (R)-baclofen 306, which is used to treat spasticity linked to brain and spinal cord injuries, alcoholism and drug addiction, overactive bladder and cancer pain.195 (R)-Baclofen 306 synthesis commenced with the desymmetrization of N-Cbz cyclic imide 302b with alcohol 303, under similar conditions to those used for 304a, to furnish the amido ester 304b. Next, amido ester 304b was subjected to decarbobenzyloxylation in conjunction with hydrolysis to synthesize the key intermediate 305,194 which was reported earlier by Ji et al. for the synthesis of (R)-baclofen 306 (Scheme 34).196
 |
| | Scheme 34 Asymmetric synthesis of (R)-rolipram 301 and (R)-baclofen 306. | |
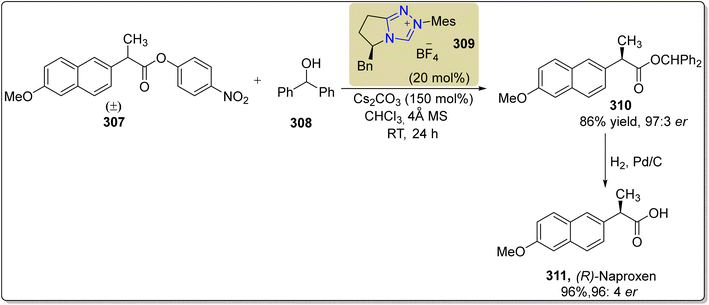
2.2.2 Synthesis of NSAIDs. Naproxen 311, containing free carboxylic acid functionality has been known to demonstrate analgesic, anticancer, antipyretic and antiinflammatory activity.197 Owing to its high pharmaceutical importance, Chen et al., in 2016, reported a synthetic protocol to synthesize asymmetric (R)-naproxen 311 by utilizing NHC 309 as a catalyst. The synthesis commenced with dynamic kinetic resolution (DKR) of racemic α,α-disubstituted carboxylic ester 307 with alcohol 308 in the presence of triazolium-based NHC 309 (as a catalyst) with Cs2CO3 (as base) in CHCl3 at room temperature to deliver the transesterified product 310 in 86% yield with 97:3 enantioselectivity ratio. Next, compound 310 was allowed to undergo hydrogenolysis to furnish (R)-naproxen 311 in 96% yield and 96:4% enantioselectivity ratio (Scheme 35).198
 |
| | Scheme 35 Asymmetric synthesis of naproxen 311. | |
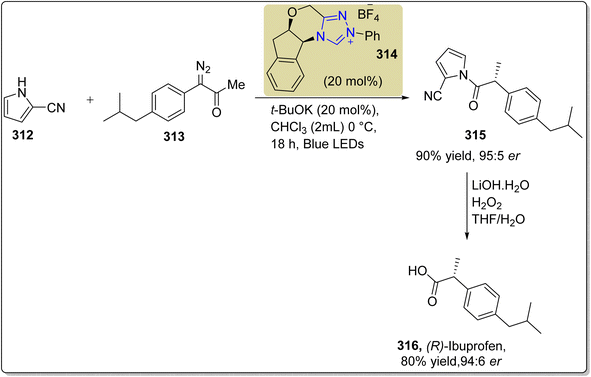
Ibuprofen 316 is an important non-steroidal anti-inflammatory drug (NSAID) used to treat rheumatoid arthritis, musculoskeletal pain and osteoarthritis.199 Owing to its medicinal importance, Wang et al., in 2024, reported an asymmetric approach to furnish (R)-ibuprofen 316 by utilizing visible-light-induced NHC 314 cooperative catalysis. In this synthetic protocol, pyrole-2-carbonitrile 312 and α-diazoketones 313 were allowed to undergo α-amination by utilizing NHC 314 (as a catalyst), potassium t-butoxide (as base), in CHCl3 (as solvent) under blue LED irradiation to synthesize the chiral product 315 in 90% yield with 95:5 enantioselectivity ratio. Next, compound 315 was treated with LiOH·H2O and H2O2 in THF/H2O to deliver (R)-ibuprofen 316 in 80% yield with 94:6 enantioselectivity ratio (Scheme 36).200
 |
| | Scheme 36 Asymmetric synthesis of ibuprofen 316. | |
2.2.3 Synthesis of analgesics and antibacterial drugs. In 2019, Liu et al. reported an asymmetric approach to synthesize talmetacin 323 (analgesic), talniflumate 324 (analgesic) and talampicillin 325 (antibacterial) drugs by utilizing triazolium-based NHC 319 as a catalyst. In this synthetic protocol, carboxylic acid 317 was allowed to undergo acetalization with o-phthalaldehyde 318 in the presence of NHC 319 (catalyst), quinone 184 (oxidant), K2CO3 (base) and LiCl (additive) in CHCl3 to synthesize talmetacin 323 in 80% yield and 98:2 enantioselectivity ratio, talniflumate 324 in 96% yield and 95:5 enantioselectivity ratio, and talampicillin 325 in 58% yield with 6:1 diastereoselectivity ratio (Scheme 37).201
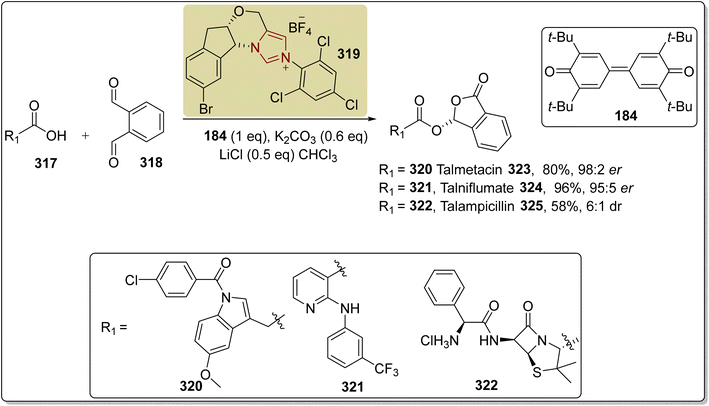 |
| | Scheme 37 Asymmetric synthesis of talmetacin 323, talniflumate 324 and talampicillin 325. | |
A muscle relaxant exhibiting anticholinergic pharmacology called tolterodine tartrate 331 is used to treat discomfort, urgency and frequency of urination in patients with unstable bladders.202 In this regard, Guduguntla et al. (2017) demonstrated an asymmetric strategy to synthesize tolterodine 331 by utilizing NHC 328 as a ligand, with a copper catalyst. The synthesis commenced with the enantioselective allylic arylation of aryl bromide 326 with aryllithium reagent 327 in the presence of Cu/NHC 328 (catalyst/ligand) in CH2Cl2 at −80 °C to furnish compound 329, which was further allowed to undergo hydroboration–oxidation to synthesize a key intermediate 330,203 which was reported earlier by Sun et al. in 2014 and utilized for the synthesis of tolterodine 331 (Scheme 38).204
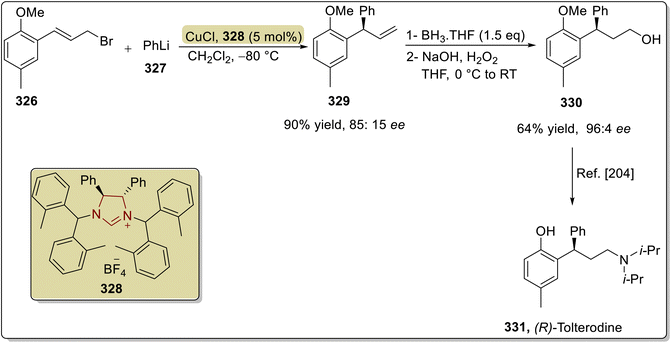 |
| | Scheme 38 Asymmetric synthesis of tolterodine 331. | |
3 Conclusion
This review provides a comprehensive perspective on NHC-catalyzed asymmetric formal and total synthesis of natural products, along with pharmaceutical drugs, reported since 2016. The synthesis of different classes of natural products, including alkaloids, terpenoids, lignans, polyketides, pentadecaketides, and flavonoids, alongwith pharmaceutical drugs, such as NSAIDs, antidepressants, antibacterial and analgesics, employing asymmetric NHC catalysis, has been summarized. During the thorough assessment, it was observed that asymmetric NHC catalysis delivered products with excellent stereochemical selectivity and substantial yields. Despite various synthetic applications of NHCs, the enhanced synthetic use of NHC catalysis in the synthesis of a wide range of other natural products and pharmaceutical drugs is yet to be explored. Further studies are needed on asymmetric NHC catalysis, which might open new doors to access more important bioactive molecular scaffolds, drugs and important natural products. Moreover, greener, more economical and sustainable synthetic protocols (utilizing asymmetric NHC catalysis) targeting diverse stereoenriched organic compounds need to be developed.
Conflicts of interest
The authors declare no conflicts of interest.
Data availability
All data are contained in the manuscript.
Acknowledgements
Authors are grateful for the facilities provided by the Government College University Faisalabad, Pakistan. A. Irfan extends his appreciation to the Deanship of Research and Graduate Studies at King Khalid University for funding this work through the Large Groups Research Project under grant number (RGP2/74/46). The authors are thankful to the Deanship of Graduate Studies and Scientific Research at the University of Bisha for supporting this work through the Fast-Track Research Support Program.
References
- X. Chen, H. Wang, Z. Jin and Y. R. Chi, N-heterocyclic carbene organocatalysis: activation modes and typical reactive intermediates, Chin. J. Chem., 2020, 38, 1167–1202 CrossRef CAS.
- J.-B. Dumas and E. M. Peligot, Mémoire sur l'esprit de bois et sur les divers composés éthérés qui en proviennent, 1835 Search PubMed.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, An overview of N-heterocyclic carbenes, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- W. A. Herrmann and C. Köcher, N-Heterocyclic carbenes, Angew Chem. Int. Ed. Engl., 1997, 36, 2162–2187 CrossRef CAS.
- D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Organocatalytic reactions enabled by N-heterocyclic carbenes, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed.
- P. Chauhan and D. Enders, N-heterocyclic carbene catalyzed activation of esters: a new option for asymmetric domino reactions, Angew. Chem., Int. Ed., 2014, 53, 1485–1487 CrossRef CAS PubMed.
- T. K. Das and A. T. Biju, Imines as acceptors and donors in N-heterocyclic carbene (NHC) organocatalysis, Chem. Commun., 2020, 56, 8537–8552 RSC.
- C. De Risi, O. Bortolini, G. Di Carmine, D. Ragno and A. Massi, Kinetic resolution, dynamic kinetic resolution and asymmetric desymmetrization by N-heterocyclic carbene catalysis, Synthesis, 2019, 51, 1871–1891 CrossRef CAS.
- A. Grossmann and D. Enders, N-heterocyclic carbene catalyzed domino reactions, Angew. Chem., Int. Ed., 2012, 51, 314–325 CrossRef CAS PubMed.
- H. Ohmiya, N-Heterocyclic carbene-based catalysis enabling cross-coupling reactions, ACS Catal., 2020, 10, 6862–6869 CrossRef CAS.
- R. S. Menon, A. T. Biju and V. Nair, Recent advances in employing homoenolates generated by N-heterocyclic carbene (NHC) catalysis in carbon–carbon bond-forming reactions, Chem. Soc. Rev., 2015, 44, 5040–5052 RSC.
- D. Enders, O. Niemeier and A. Henseler, Organocatalysis by N-heterocyclic carbenes, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed.
- V. Nair, S. Vellalath and B. P. Babu, Recent advances in carbon–carbon bond-forming reactions involving homoenolates generated by NHC catalysis, Chem. Soc. Rev., 2008, 37, 2691–2698 RSC.
- E. M. Phillips, A. Chan and K. A. Scheidt, Discovering new reactions with N-heterocyclic carbene catalysis, Aldrichimica Acta, 2009, 42, 55 CAS.
- S. J. Ryan, L. Candish and D. W. Lupton, Acyl anion free N-heterocyclic carbene organocatalysis, Chem. Soc. Rev., 2013, 42, 4906–4917 RSC.
- G. Bertrand, Carbene Chemistry: from Fleeting Intermediates to Powerful Reagents, CRC Press, 2002 Search PubMed.
- L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz and V. Cesar, Synthetic routes to N-heterocyclic carbene precursors, Chem. Rev., 2011, 111, 2705–2733 CrossRef CAS PubMed.
- R. Bharti, M. Verma, A. Thakur and R. Sharma, N-Heterocyclic Carbenes (NHCs): An Introduction, in Carbene, ed. S. Saha and A. Manna, IntechOpen, London, 2022, pp. 43–62 Search PubMed.
- X. Bugaut and F. Glorius, Organocatalytic umpolung: N-heterocyclic carbenes and beyond, Chem. Soc. Rev., 2012, 41, 3511–3522 RSC.
- X.-Y. Chen, Z.-H. Gao and S. Ye, Bifunctional N-heterocyclic carbenes derived from l-pyroglutamic acid and their applications in enantioselective organocatalysis, Acc. Chem. Res., 2020, 53, 690–702 CrossRef CAS PubMed.
- F. Lu, F. Su, S. Pan, X. Wu, X. Wu and Y. R. Chi, N-Heterocyclic Carbene Enabled Functionalization of Inert C (Sp3)− H Bonds via Hydrogen Atom Transfer (HAT) Processes, Chem.–Eur. J., 2024, 30, e202401811 CrossRef CAS PubMed.
- Q. Tang, D. Du and J. Gao, Radical Acylation of Alkenes by NHC-Organocatalysis, Eur. J. Org Chem., 2023, 26, e202300832 CrossRef CAS.
- G. Pisanò and C. S. J. Cazin, General mechanochemical synthetic protocol to late transition metal–NHC (N-Heterocyclic Carbene) complexes, ACS Sustain. Chem. Eng., 2021, 9, 9625–9631 CrossRef.
- A. D. Obi, N. C. Frey, D. A. Dickie, C. E. Webster and R. J. Gilliard Jr, N-Heterocyclic Carbene-Assisted Reversible Migratory Coupling of Aminoborane at Magnesium, Angew. Chem., 2022, 134, e202211496 CrossRef.
- P. Gao and M. Szostak, Hydration reactions catalyzed by transition metal–NHC (NHC= N-heterocyclic carbene) complexes, Coord. Chem. Rev., 2023, 485, 215110 CrossRef CAS PubMed.
- B. C. Lee, C.-F. Liu, L. Q. H. Lin, K. Z. Yap, N. Song, C. H. M. Ko, P. H. Chan and M. J. Koh, N-Heterocyclic carbenes as privileged ligands for nickel-catalysed alkene functionalisation, Chem. Soc. Rev., 2023, 52, 2946–2991 RSC.
- R. Ye, A. V. Zhukhovitskiy, R. V. Kazantsev, S. C. Fakra, B. B. Wickemeyer, F. D. Toste and G. A. Somorjai, Supported Au nanoparticles with N-heterocyclic carbene ligands as active and stable heterogeneous catalysts for lactonization, J. Am. Chem. Soc., 2018, 140, 4144–4149 CrossRef CAS PubMed.
- D. O. Prima, N. S. Kulikovskaya, R. A. Novikov, A. Y. Kostyukovich, J. V. Burykina, V. M. Chernyshev and V. P. Ananikov, Revealing the mechanism of combining best properties of homogeneous and heterogeneous catalysis in hybrid Pd/NHC systems, Angew. Chem., Int. Ed., 2024, 63, e202317468 CrossRef CAS PubMed.
- M. Koy, P. Bellotti, M. Das and F. Glorius, N-Heterocyclic carbenes as tunable ligands for catalytic metal surfaces, Nat. Catal., 2021, 4, 352–363 CrossRef CAS.
- C. Eisen, J. M. Chin and M. R. Reithofer, Catalytically Active Gold Nanomaterials Stabilized by N-heterocyclic Carbenes, Chem.–Asian J., 2021, 16, 3026–3037 CrossRef CAS PubMed.
- T. Scattolin, A. A. Logvinov, N. V. Tzouras, C. S. J. Cazin and S. P. Nolan, Advances in the synthesis and applications of N-heterocyclic carbene metal complexes with a focus on the weak base route, Organometallics, 2023, 42, 2692–2730 CrossRef CAS.
- B. Qie, Z. Wang, J. Jiang, Z. Zhang, P. H. Jacobse, J. Lu, X. Li, F. Liu, A. N. Alexandrova and S. G. Louie, Synthesis and characterization of low-dimensional N-heterocyclic carbene lattices, Science, 2024, 384, 895–901 CrossRef CAS PubMed.
- L. Tschugajeff, M. Skanawy-Grigorjewa, A. Posnjak and M. Skanawy-Grigorjewa, Über Die Hydrazin-Carbylamin-Komplexe des Platins, Z. fur Anorg. Allg. Chem., 1925, 148, 37–42 CrossRef.
- E. O. Fischer and A. Maasböl, On the existence of a tungsten carbonyl carbene complex, Angew Chem. Int. Ed. Engl., 1964, 3, 580–581 CrossRef.
- M. C. Jahnke and F. E. Hahn, Introduction to N-Heterocyclic Carbenes: Synthesis and Stereoelectronic Parameters, 2016 Search PubMed.
- A. J. Arduengo Iii, R. L. Harlow and M. Kline, A stable crystalline carbene, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef.
- M. Pareek, Y. Reddi and R. B. Sunoj, Tale of the Breslow intermediate, a central player in N-heterocyclic carbene organocatalysis: then and now, Chem. Sci., 2021, 12, 7973–7992 RSC.
- S. Mondal, A. Ghosh and A. T. Biju, N-Heterocyclic Carbene (NHC)-Catalyzed Transformations Involving Azolium Enolates, Chem. Rec., 2022, 22, e202200054 CrossRef CAS PubMed.
- J. Gao, J. Feng and D. Du, Generation of azolium dienolates as versatile nucleophilic synthons via N-heterocyclic carbene catalysis, Org. Chem. Front., 2021, 8, 6138–6166 RSC.
- K.-Q. Chen, H. Sheng, Q. Liu, P.-L. Shao and X.-Y. Chen, N-heterocyclic carbene-catalyzed radical reactions, Sci. China:Chem., 2021, 64, 7–16 CrossRef CAS.
- T. Jousseaume, N. E. Wurz and F. Glorius, Highly enantioselective synthesis of α-amino acid derivatives by an NHC-catalyzed intermolecular Stetter reaction, Angew. Chem., Int. Ed., 2011, 50, 1410–1414 CrossRef CAS PubMed.
- D. A. DiRocco and T. Rovis, Catalytic asymmetric intermolecular Stetter reaction of enals with nitroalkenes: enhancement of catalytic efficiency through bifunctional additives, J. Am. Chem. Soc., 2011, 133, 10402–10405 CrossRef CAS PubMed.
- S. J. Ryan, L. Candish and D. W. Lupton, N-heterocyclic carbene-catalyzed (4+ 2) cycloaddition/decarboxylation of silyl dienol ethers with α, β-unsaturated acid fluorides, J. Am. Chem. Soc., 2011, 133, 4694–4697 CrossRef CAS PubMed.
- Z. Fu, J. Xu, T. Zhu, W. W. Y. Leong and Y. R. Chi, β-Carbon activation of saturated carboxylic esters through N-heterocyclic carbene organocatalysis, Nat. Chem., 2013, 5, 835–839 CrossRef CAS PubMed.
- Z. Jin, S. Chen, Y. Wang, P. Zheng, S. Yang and Y. R. Chi, β-Functionalization of Carboxylic Anhydrides with β-Alkyl Substituents through Carbene Organocatalysis, Angew. Chem., Int. Ed., 2014, 53, 13506–13509 CrossRef CAS PubMed.
- C. Guo, M. Fleige, D. Janssen-Müller, C. G. Daniliuc and F. Glorius, Switchable selectivity in an NHC-catalysed dearomatizing annulation reaction, Nat. Chem., 2015, 7, 842–847 CrossRef CAS PubMed.
- Y. Nakano and D. W. Lupton, Enantioselective N-Heterocyclic Carbene Catalysis by the Umpolung of α, β-Unsaturated Ketones, Angew. Chem., Int. Ed., 2016, 55, 3135–3139 CrossRef CAS PubMed.
- X. Y. Chen, J. W. Xiong, Q. Liu, S. Li, H. Sheng, C. von Essen, K. Rissanen and D. Enders, Control of N-Heterocyclic Carbene Catalyzed Reactions of Enals: Asymmetric Synthesis of Oxindole-γ-Amino Acid Derivatives, Angew. Chem., Int. Ed., 2018, 57, 300–304 CrossRef CAS PubMed.
- F. Ritter, D. Mukherjee, T. P. Spaniol, A. Hoffmann and J. Okuda, A masked cuprous hydride as a catalyst for carbonyl hydrosilylation in aqueous solutions, Angew. Chem., Int. Ed., 2019, 58, 1818–1822 CrossRef CAS PubMed.
- G. Yang, D. Guo, D. Meng and J. Wang, NHC-catalyzed atropoenantioselective synthesis of axially chiral biaryl amino alcohols via a cooperative strategy, Nat. Commun., 2019, 10, 3062 CrossRef PubMed.
- Y.-T. Wu, R. Zhang, X.-Y. Duan, H.-F. Yu, B.-Y. Sun and J. Qi, Access to dihydropyrano [3, 2-b] pyrrol-5-ones skeletons by N-heterocyclic carbene-catalyzed [3+ 3] annulations, Chem. Commun., 2020, 56, 9854–9857 RSC.
- I. Kim, H. Im, H. Lee and S. Hong, N-Heterocyclic carbene-catalyzed deaminative cross-coupling of aldehydes with Katritzky pyridinium salts, Chem. Sci., 2020, 11, 3192–3197 RSC.
- A. V. Bay, E. J. Farnam and K. A. Scheidt, Synthesis of cyclohexanones by a tandem photocatalyzed annulation, J. Am. Chem. Soc., 2022, 144, 7030–7037 CrossRef CAS PubMed.
- D. Sarkar, S. Barik, S. Shee, R. G. Gonnade and A. T. Biju, NHC-Catalyzed Enantioselective Synthesis of Tetracyclic δ-Lactones by (4+ 2) Annulation of ortho-Quinodimethanes with Activated Ketones, Org. Lett., 2023, 25, 7852–7857 CrossRef CAS PubMed.
- X. Y. Chen, Q. Liu, P. Chauhan, S. Li, A. Peuronen, K. Rissanen, E. Jafari and D. Enders, N-Heterocyclic Carbene Catalyzed [4+ 2] Annulation of Enals via a Double Vinylogous Michael Addition: Asymmetric Synthesis of 3, 5-Diaryl Cyclohexenones, Angew. Chem., 2017, 129, 6337–6341 CrossRef.
- R. N. Mitra, K. Show, D. Barman, S. Sarkar and D. K. Maiti, NHC-catalyzed dual Stetter reaction: a mild cascade annulation for the syntheses of naphthoquinones, isoflavanones, and sugar-based chiral analogues, J. Org. Chem., 2018, 84, 42–52 CrossRef PubMed.
- D. Barman, T. Ghosh, K. Show, S. Debnath, T. Ghosh and D. K. Maiti, NHC-Mediated Stetter-Aldol and Imino-Stetter-Aldol Domino Cyclization to Naphthalen-1 (2 H)-ones and Isoquinolines, Org. Lett., 2021, 23, 2178–2182 CrossRef CAS PubMed.
- K. Parmar, P. Haghshenas and M. Gravel, Total Synthesis of (+)-Hyacinthacine A1 using a chemoselective cross-benzoin reaction and a furan photooxygenation–amine cyclization strategy, Org. Lett., 2021, 23, 1416–1421 CrossRef CAS PubMed.
- I. Baranska, B. Ośmiałowski, K. Rafinska and Z. Rafinski, Construction of highly functionalized 2-styrylfurans by N-heterocyclic carbene/Brønsted acid catalysis, Org. Lett., 2024, 26, 3514–3518 CrossRef CAS PubMed.
- K. Liu, M. Schwenzer and A. Studer, Radical NHC catalysis, ACS Catal., 2022, 12, 11984–11999 CrossRef CAS.
- N. Zhou, F. Zhao, L. Wang, X. Gao, X. Zhao and M. Zhang, NHC-Catalyzed Regioselective Intramolecular Radical Cyclization Reaction for the Synthesis of Benzazepine Derivatives, Org. Lett., 2023, 25, 6072–6076 CrossRef CAS PubMed.
- A. Mushtaq, A. F. Zahoor, M. N. Ahmad, S. G. Khan, N. Akhter, U. Nazeer, A. Mansha, H. Ahmad, A. R. Chaudhry and A. Irfan, Accessing the synthesis of natural products and their analogues enabled by the Barbier reaction: a review, RSC Adv., 2024, 14, 33536–33567 RSC.
- L. Katz and R. H. Baltz, Natural product discovery: past, present, and future, J. Ind. Microbiol. Biotechnol., 2016, 43, 155–176 CrossRef CAS PubMed.
- S. Munawar, A. F. Zahoor, S. M. Hussain, S. Ahmad, A. Mansha, B. Parveen, K. G. Ali and A. Irfan, Steglich esterification: A versatile synthetic approach toward the synthesis of natural products, their analogues/derivatives, Heliyon, 2024, 10, 1–37 CrossRef PubMed.
- J. Ceramella, D. Iacopetta, A. Franchini, M. De Luca, C. Saturnino, I. Andreu, M. S. Sinicropi and A. Catalano, A look at the importance of chirality in drug activity: Some significative examples, Appl. Sci., 2022, 12, 10909 CrossRef CAS.
- N. Marion, S. Díez-González and S. P. Nolan, N-heterocyclic carbenes as organocatalysts, Angew. Chem., Int. Ed., 2007, 46, 2988–3000 CrossRef CAS PubMed.
- J. Izquierdo, G. E. Hutson, D. T. Cohen and K. A. Scheidt, A Continuum of Progress: Applications of N-Hetereocyclic Carbene Catalysis in Total Synthesis, Angew. Chem., Int. Ed., 2012, 51, 11686–11698 CrossRef CAS PubMed.
- H. U. Vora, P. Wheeler and T. Rovis, Exploiting Acyl and Enol Azolium Intermediates via N-Hetero-cyclic Carbene-Catalyzed Reactions of α-Reducible Aldehydes, Adv. Synth. Catal., 2012, 354, 1617–1639 CrossRef CAS PubMed.
- S. R. Yetra, A. Patra and A. T. Biju, Recent advances in the N-heterocyclic carbene (NHC)-organocatalyzed Stetter reaction and related chemistry, Synthesis, 2015, 47, 1357–1378 CrossRef CAS.
- M. H. Wang and K. A. Scheidt, Cooperative catalysis and activation with N-heterocyclic carbenes, Angew. Chem., Int. Ed., 2016, 55, 14912–14922 CrossRef CAS PubMed.
- S. Mondal, S. R. Yetra, S. Mukherjee and A. T. Biju, NHC-catalyzed generation of α, β-unsaturated acylazoliums for the enantioselective synthesis of heterocycles and carbocycles, Acc. Chem. Res., 2019, 52, 425–436 CrossRef CAS PubMed.
- A. Takada, Y. Hashimoto, H. Takikawa, K. Hikita and K. Suzuki, Total synthesis and absolute stereochemistry of seragakinone A, Angew. Chem., Int. Ed., 2011, 50, 2297–2301 CrossRef CAS PubMed.
- M. He, M. Rommel and J. W. Bode, Formal Synthesis of (±)-clausenamide by NHC-Catalyzed γ-lactam formation, Heterocycles, 2012, 86, 1689–1696 CrossRef CAS PubMed.
- Y. Koyama, R. Yamaguchi and K. Suzuki, Total synthesis and structure assignment of the anthrone C-glycoside cassialoin, Angew. Chem., Int. Ed. Engl., 2008, 47, 1084 CrossRef CAS PubMed.
- M. Yang, F. Yin, H. Fujino and S. A. Snyder, The total synthesis of chalcitrin, J. Am. Chem. Soc., 2019, 141, 4515–4520 CrossRef CAS PubMed.
- H. Mao, H. Jeong, J. Yang, H. J. Ha and J. W. Yang, Preparation of Chiral Contiguous Epoxyaziridines and Their Regioselective Ring-Opening for Drug Syntheses, Chem.–Eur. J., 2018, 24, 2370–2374 CrossRef CAS PubMed.
- X. Chen, R. Song, Y. Liu, C. Y. Ooi, Z. Jin, T. Zhu, H. Wang, L. Hao and Y. R. Chi, Carbene and acid cooperative catalytic reactions of aldehydes and o-hydroxybenzhydryl amines for highly enantioselective access to dihydrocoumarins, Org. Lett., 2017, 19, 5892–5895 CrossRef CAS.
- G.-T. Li, Z.-K. Li, Q. Gu and S.-L. You, Asymmetric synthesis of 4-Aryl-3, 4-dihydrocoumarins by N-heterocyclic carbene catalyzed annulation of phenols with enals, Org. Lett., 2017, 19, 1318–1321 CrossRef CAS PubMed.
- S. Dong, M. Frings, H. Cheng, J. Wen, D. Zhang, G. Raabe and C. Bolm, Organocatalytic kinetic resolution of sulfoximines, J. Am. Chem. Soc., 2016, 138, 2166–2169 CrossRef CAS PubMed.
- J. C. Sheehan and D. H. Hunneman, Homogeneous asymmetric catalysis, J. Am. Chem. Soc., 1966, 88, 3666–3667 CrossRef CAS.
- D. Enders, K. Breuer, J. Runsink and J. H. Teles, The first asymmetric intramolecular Stetter reaction. Preliminary communication, Helv. Chim. Acta, 1996, 79, 1899–1902 CrossRef CAS.
- K. Nagao and H. Ohmiya, N-Heterocyclic Carbene (NHC)/Metal Cooperative Catalysis, Top. Curr. Chem., 2019, 377, 35 CrossRef PubMed.
- N. Mukherjee, B. Mondal, T. N. Saha and R. Maity, Palladium, iridium, and rhodium complexes bearing chiral N-heterocyclic carbene ligands applied in asymmetric catalysis, Appl. Organomet. Chem., 2024, 38, e6794 CrossRef CAS.
- M. Zhang, X. Yang, X. Peng, X. Li and Z. Jin, Asymmetric construction of axial and planar chirality with N-heterocyclic carbene (NHC) organocatalysis, Sci. China:Chem., 2025, 68, 815–825 CrossRef CAS.
- S. Li, D. Ling, Y. Deng, M. Zhang, L. Chen and Z. Jin, Enantioselective electrophilic activation of aldehydes, esters and imines via N-heterocyclic carbene catalysis, Chem. Commun., 2025, 61, 5540–5555 RSC.
- H. Cai, X. Yang, S.-C. Ren and Y. R. Chi, Radical Reactions with N-Heterocyclic Carbene (NHC)-Derived Acyl Azoliums for Access to Multifunctionalized Ketones, ACS Catal., 2024, 14, 8270–8293 CrossRef CAS.
- Y. Que and H. He, Advances in N-Heterocyclic Carbene Catalysis for Natural Product Synthesis, Eur. J. Org. Chem., 2020, 2020, 5917–5925 CrossRef CAS.
- L.-Z. Lin, G. A. Cordell, C.-Z. Ni and J. Clardy, Oxindole Alkaloids from Gelsemium Elegans, 1991 Search PubMed.
- M. Kitajima, N. Kogure, K. Yamaguchi, H. Takayama and N. Aimi, Structure reinvestigation of gelsemoxonine, a constituent of Gelsemium elegans, reveals a novel, azetidine-containing indole alkaloid, Org. Lett., 2003, 5, 2075–2078 CrossRef CAS PubMed.
- M. Kitajima, Y. Arai, H. Takayama and N. Aimi, A chemical study on “Yakatsu (_?? _?? _)” stored in Shosoin repository Isolation and characterization of four indole alkaloids from a 1250 year-old sample of the Chinese toxic medicine, Proc. Jpn. Acad., Ser. B, 1998, 74, 159–163 CrossRef.
- M. Kitajima, T. Nakamura, N. Kogure, M. Ogawa, Y. Mitsuno, K. Ono, S. Yano, N. Aimi and H. Takayama, Isolation of Gelsedine-Type Indole Alkaloids from Gelsemium e legans and Evaluation of the Cytotoxic Activity of Gelsemium Alkaloids for A431 Epidermoid Carcinoma Cells, J. Nat. Prod., 2006, 69, 715–718 CrossRef CAS PubMed.
- M. Kitajima, T. Nakamura, N. Kogure, M. Ogawa, Y. Mitsuno, K. Ono, S. Yano, N. Aimi and H. Takayama, Erratum: Isolation of gelsedine-type indole alkaloids from Gelsemium elegans and evaluation of the cytotoxic activity of Gelsemium alkaloids for A431 epidermoid carcinoma cells (Journal of Natural Products (2006) 69), J. Nat. Prod., 2007, 70, 142 CrossRef CAS.
- Q.-C. Zhao, W. Hua, L. Zhang, T. Guo, M.-H. Zhao, M. Yan, G.-B. Shi and L.-J. Wu, Antitumor activity of two gelsemine metabolites in rat liver microsomes, J. Asian Nat. Prod. Res., 2010, 12, 731–739 CrossRef CAS PubMed.
- C. Zhao, F. Li and J. Wang, N-Heterocyclic Carbene Catalyzed Dynamic Kinetic Resolution of Pyranones, Angew. Chem., Int. Ed., 2016, 55, 1820–1824 CrossRef CAS PubMed.
- J. Shimokawa, T. Harada, S. Yokoshima and T. Fukuyama, Total synthesis of gelsemoxonine, J. Am. Chem. Soc., 2011, 133, 17634–17637 CrossRef CAS PubMed.
- H. Shao, W. Bao, Z.-R. Jing, Y.-P. Wang, F.-M. Zhang, S.-H. Wang and Y.-Q. Tu, Construction of the [6, 5, 7, 5] tetracyclic core of Calyciphylline A type alkaloids via a tandem semipinacol rearrangement/Nicholas reaction, Org. Lett., 2017, 19, 4648–4651 CrossRef CAS PubMed.
- G. Coussanes and J. Bonjoch, Synthesis of the tetracyclic ABCD ring domain of calyciphylline A-type alkaloids via reductive radical cyclizations, Org. Lett., 2017, 19, 878–881 CrossRef CAS PubMed.
- H. Zhang, S. L. Shyaula, J.-Y. Li, J. Li and J.-M. Yue, Himalensines A and B, alkaloids from Daphniphyllum himalense, Org. Lett., 2016, 18, 1202–1205 CrossRef CAS PubMed.
- H. Shi, I. N. Michaelides, B. Darses, P. Jakubec, Q. N. N. Nguyen, R. S. Paton and D. J. Dixon, Total synthesis of (−)-himalensine A, J. Am. Chem. Soc., 2017, 139, 17755–17758 CrossRef CAS PubMed.
- A. G. Kozlovskiĭ, T. F. Solov'eva, V. G. Sakharovskiĭ and V. M. Adanin, Biosynthesis of “unusual” ergot alkaloids by the fungus Penicillium aurantio-virens, Dokl. Akad. Nauk SSSR, 1981, 260, 230–233 Search PubMed.
- H.-C. Lin, T. C. McMahon, A. Patel, M. Corsello, A. Simon, W. Xu, M. Zhao, K. N. Houk, N. K. Garg and Y. Tang, P450-mediated coupling of indole fragments to forge communesin and unnatural isomers, J. Am. Chem. Soc., 2016, 138, 4002–4005 CrossRef CAS PubMed.
- J. A. May and B. Stoltz, The structural and synthetic implications of the biosynthesis of the calycanthaceous alkaloids, the communesins, and nomofungin, Tetrahedron, 2006, 62, 5262–5271 CrossRef CAS.
- A. Numata, C. Takahashi, Y. Ito, T. Takada, K. Kawai, Y. Usami, E. Matsumura, M. Imachi, T. Ito and T. Hasegawa, Communesins, cytotoxic metabolites of a fungus isolated from a marine alga, Tetrahedron Lett., 1993, 34, 2355–2358 CrossRef CAS.
- W. Li, M. Wollenburg and F. Glorius, Enantioselective synthesis of 2-oxazolidinones by ruthenium (II)–NHC-catalysed asymmetric hydrogenation of 2-oxazolones, Chem. Sci., 2018, 9, 6260–6263 RSC.
- J. Park, D.-H. Kim, T. Das and C.-G. Cho, Intramolecular Fischer indole synthesis for the direct synthesis of 3, 4-fused tricyclic indole and application to the total synthesis of (−)-aurantioclavine, Org. Lett., 2016, 18, 5098–5101 CrossRef CAS PubMed.
- M. Chen, L. Gan, S. Lin, X. Wang, L. Li, Y. Li, C. Zhu, Y. Wang, B. Jiang and J. Jiang, Alkaloids from the root of Isatis indigotica, J. Nat. Prod., 2012, 75, 1167–1176 CrossRef CAS.
- Y.-L. Ho and Y.-S. Chang, Studies on the antinociceptive, anti-inflammatory and antipyretic effects of Isatis indigotica root, Phytomedicine, 2002, 9, 419–424 CrossRef PubMed.
- P. Zou, Y. Hong and H. L. Koh, Chemical fingerprinting of Isatis indigotica root by RP-HPLC and hierarchical clustering analysis, J. Pharm. Biomed. Anal., 2005, 38, 514–520 CrossRef CAS PubMed.
- J.-F. Liu, Z.-Y. Jiang, R.-R. Wang, Y.-T. Zheng, J.-J. Chen, X.-M. Zhang and Y.-B. Ma, Isatisine A, a novel alkaloid with an unprecedented skeleton from leaves of Isatis indigotica, Org. Lett., 2007, 9, 4127–4129 CrossRef CAS PubMed.
- J. B. Bremner, H. F. Russell, B. W. Skelton, A. H. White and N. Indole-fusod, Mediumsized Ring Heterocycles via Chloroacetamide Photochemistry, Heterocycles, 2000, 53, 277–290 CrossRef CAS.
- M. M. Ahire, M. D. Pol, D. S. Kavale, R. G. Gonnade and S. B. Mhaske, Stereoselective construction of deoxy-cruciferane alkaloids by NHC-catalyzed intramolecular annulation of homoenolate with quinazolinone, Org. Biomol. Chem., 2019, 17, 7135–7139 RSC.
- Y. Ming-He, C. Yan-Yong and H. Liang, Three novel cyclic amides from Clausena lansium, Phytochemistry, 1988, 27, 445–450 CrossRef.
- M. Stadler, J. Bitzer, A. Mayer-Bartschmid, H. Müller, J. Benet-Buchholz, F. Gantner, H.-V. Tichy, P. Reinemer and K. B. Bacon, Cinnabaramides A− G: analogues of lactacystin and salinosporamide from a terrestrial streptomycete, J. Nat. Prod., 2007, 70, 246–252 CrossRef CAS PubMed.
- L. Yang, Q.-Y. Zheng, D.-X. Wang, Z.-T. Huang and M.-X. Wang, Reversal of nucleophilicity of enamides in water: Control of cyclization pathways by reaction media for the orthogonal synthesis of dihydropyridinone and pyrrolidinone Clausena alkaloids, Org. Lett., 2008, 10, 2461–2464 CrossRef CAS PubMed.
- Z. Feng, X. Li, G. Zheng and L. Huang, Synthesis and activity in enhancing long-term potentiation (LTP) of clausenamide stereoisomers, Bioorg. Med. Chem. Lett., 2009, 19, 2112–2115 CrossRef CAS PubMed.
- X. Li, C. Zhu, C. Li, K. Wu, D. Huang and L. Huang, Synthesis of N-substituted Clausenamide analogues, Eur. J. Med. Chem., 2010, 45, 5531–5538 CrossRef CAS PubMed.
- S. Chu, S. Liu, W. Duan, Y. Cheng, X. Jiang, C. Zhu, K. Tang, R. Wang, L. Xu and X. Wang, The anti-dementia drug candidate,(−)-clausenamide, improves memory impairment through its multi-target effect, Pharmacol. Ther., 2016, 162, 179–187 CrossRef CAS PubMed.
- Z. Hu, Y. Zhu, Z. Fu and W. Huang, Asymmetric synthesis of enantioenriched 6-hydroxyl butyrolactams promoted by N-heterocyclic carbene, J. Org. Chem., 2019, 84, 10328–10337 CrossRef CAS PubMed.
- Y. M. L. Pan and H. S. Wang, Novel Method for Synthesizing Clausenamide, Chinese Pat., CN105017124A, 2015 Search PubMed.
- L. E. Overman, K. L. Bell and F. Ito, Enantioselective total syntheses of pumiliotoxin B and pumiliotoxin 251D. A general entry to the pumiliotoxin A alkaloids via stereospecific iminium ion-vinylsilane cyclizations, J. Am. Chem. Soc., 1984, 106, 4192–4201 CrossRef CAS.
- H. Sorek, A. Rudi, S. Gueta, F. Reyes, M. J. Martin, M. Aknin, E. Gaydou, J. Vacelet and Y. Kashman, Netamines A–G: seven new tricyclic guanidine alkaloids from the marine sponge Biemna laboutei, Tetrahedron, 2006, 62, 8838–8843 CrossRef CAS.
- Y. Sun, Y. Zhou, Y. Shi, J. Del Pozo, S. Torker and A. H. Hoveyda, Copper–Hydride-Catalyzed Enantioselective Processes with Allenyl Boronates. Mechanistic Nuances, Scope, and Utility in Target-Oriented Synthesis, J. Am. Chem. Soc., 2019, 141, 12087–12099 CrossRef CAS PubMed.
- N.-H. Lin, L. E. Overman, M. H. Rabinowitz, L. A. Robinson, M. J. Sharp and J. Zablocki, Efficient Total Syntheses of Pumiliotoxins A and B. Applications of Iodide-Promoted Iminium Ion−Alkyne Cyclizations in Alkaloid Construction, J. Am. Chem. Soc., 1996, 118, 9062–9072 CrossRef CAS.
- N. R. Jabir, C. K. Firoz, T. A. Zughaibi, M. A. Alsaadi, A. M. Abuzenadah, A. I. Al-Asmari, A. Alsaieedi, B. A. Ahmed, A. K. Ramu and S. Tabrez, A literature perspective on the pharmacological applications of yohimbine, Ann. Med., 2022, 54, 2849–2863 CrossRef PubMed.
- E. R. Miller, M. T. Hovey, K. A. Scheidt and A. Concise, Enantioselective Approach for the Synthesis of Yohimbine Alkaloids, J. Am. Chem. Soc., 2020, 142, 2187–2192 CrossRef CAS PubMed.
- B. M. Trost, M. Osipov, S. Krüger and Y. Zhang, A catalytic asymmetric total synthesis of (−)-perophoramidine, Chem. Sci., 2015, 6, 349–353 RSC.
- J. H. Seo, G. D. Artman and S. M. Weinreb, Synthetic Studies on Perophoramidine and the Communesins: Construction of the Vicinal Quaternary Stereocenters, J. Org. Chem., 2006, 71, 8891–8900 CrossRef CAS PubMed.
- Y.-H. Wen, Z.-J. Zhang, S. Li, J. Song and L.-Z. Gong, Stereodivergent propargylic alkylation of enals via cooperative NHC and copper catalysis, Nat. Commun., 2022, 13, 1344 CrossRef CAS PubMed.
- T. Ozawa, M. Kanematsu, H. Yokoe, M. Yoshida and K. Shishido, Total Synthesis of Debromoflustramines B and E Based on the Intramolecular Carbamoylketene–Alkene [2 + 2] Cycloaddition, J. Org. Chem., 2012, 77, 9240–9249 CrossRef CAS PubMed.
- T. Fan, J. Song and L. Z. Gong, Asymmetric Redox Allylic Alkylation to Access 3, 3′-Disubstituted Oxindoles Enabled by Ni/NHC Cooperative Catalysis, Angew. Chem., 2022, 134, e202201678 CrossRef.
- H. Miyamoto, T. Hirano, Y. Okawa, A. Nakazaki and S. Kobayashi, Stereoselective synthesis of spirocyclic oxindoles based on a one-pot Ullmann coupling/Claisen rearrangement and its application to the synthesis of a hexahydropyrrolo[2,3-b]indole alkaloid, Tetrahedron, 2013, 69, 9481–9493 CrossRef CAS.
- L. Peng, M. Wang, J. Huang, C. Guo, L.-Z. Gong and J. Song, Enantio- and Diastereodivergent N-Heterocyclic Carbene/Nickel Dual-Catalyzed Umpolung Propargylic Substitutions of Enals, J. Am. Chem. Soc., 2023, 145, 28085–28095 CrossRef CAS PubMed.
- T. Ozawa, M. Kanematsu, H. Yokoe, M. Yoshida and K. Shishido, Synthesis of (±)-Esermethole via an Intramolecular Carbamoylketene-Alkene [2+ 2] Cycloaddition, Heterocycles, 2012, 85, 2927–2932 CrossRef CAS.
- S. Dutta, A. Porey and J. Guin, N-Heterocyclic carbene catalyzed desymmetrization of diols: access to enantioenriched oxindoles having a C3-quaternary stereocenter, Chem. Commun., 2023, 59, 5771–5774 RSC.
- S. Akai, T. Tsujino, E. Akiyama, K. Tanimoto, T. Naka and Y. Kita, Enantiodivergent Preparation of Optically Active Oxindoles Having a Stereogenic Quaternary Carbon Center at the C3 Position via the Lipase-Catalyzed Desymmetrization Protocol: Effective Use of 2-Furoates for Either Enzymatic Esterification or Hydrolysis, J. Org. Chem., 2004, 69, 2478–2486 CrossRef CAS PubMed.
- A. Ashimori, B. Bachand, M. A. Calter, S. P. Govek, L. E. Overman and D. J. Poon, Catalytic Asymmetric Synthesis of Quaternary Carbon Centers. Exploratory Studies of Intramolecular Heck Reactions of (Z)-α,β-Unsaturated Anilides and Mechanistic Investigations of Asymmetric Heck Reactions Proceeding via Neutral Intermediates, J. Am. Chem. Soc., 1998, 120, 6488–6499 CrossRef CAS.
- A. Cave, M. Leboeuf, H. Moskowitz, A. Ranaivo, I. Bick, W. Sinchai, M. Nieto, T. Sevenet and P. Cabalion, Alkaloids of Cryptocarya phyllostemon, Aust. J. Chem., 1989, 42, 2243–2263 CrossRef CAS.
- M. Lebœuf, A. Cavé, A. Ranaivo and H. Moskowitz, Cryptowolinol et cryptowolidine, nouveaux alcaloïdes de type dibenzopyrrocoline, Can. J. Chem., 1989, 67, 947–952 CrossRef.
- T. Miyakoshi, D. Kronenberg, S. Tamaki, R. Lombardi and O. Baudoin, Studies towards the Enantioselective Synthesis of Cryptowolinol via Pd0-Catalyzed C(sp3)–H Arylation/Parallel Kinetic Resolution, Org. Lett., 2024, 26, 2923–2927 CrossRef CAS PubMed.
- X. Li, Q. Zheng, J. Yin, W. Liu and S. Gao, Chemo-enzymatic synthesis of equisetin, Chem. Commun., 2017, 53, 4695–4697 RSC.
- F. Meng, X. Li, S. Torker, Y. Shi, X. Shen and A. H. Hoveyda, Catalytic enantioselective 1,6-conjugate additions of propargyl and allyl groups, Nature, 2016, 537, 387–393 CrossRef CAS PubMed.
- K. Yuki, M. Shindo and K. Shishido, Enantioselective total synthesis of (−)-equisetin using a Me3Al-mediated intramolecular Diels–Alder reaction, Tetrahedron Lett., 2001, 42, 2517–2519 CrossRef CAS.
- H. Tang, W. Wei, J. Wu, X. Cui, W. Wang, T. Qian, J. Wo and B.-C. Ye, Engineering Streptomyces albus B4 for Enhanced Production of (R)-Mellein: A High-Titer Heterologous Biosynthesis Approach, J. Agric. Food Chem., 2024, 72, 17499–17509 CrossRef CAS PubMed.
- P. Reveglia, M. Masi and A. Evidente, Melleins—Intriguing natural compounds, Biomolecules, 2020, 10, 772 CrossRef CAS PubMed.
- C. Donner and M. Gill, Pigments of Fungi. LXIX. Total Synthesis of (R)-Ochratoxin α and the Formal Total Synthesis of Ochratoxin, Aust. J. Chem., 2002, 55, 213–217 CrossRef CAS.
- W. Li, M. P. Wiesenfeldt and F. Glorius, Ruthenium–NHC–Diamine Catalyzed Enantioselective Hydrogenation of Isocoumarins, J. Am. Chem. Soc., 2017, 139, 2585–2588 CrossRef CAS PubMed.
- I. P. Singh, K. E. Milligan and W. H. Gerwick, Tanikolide, a Toxic and Antifungal Lactone from the Marine Cyanobacterium Lyngbya majuscula, J. Nat. Prod., 1999, 62, 1333–1335 CrossRef CAS PubMed.
- J. Krauss, Total Synthesis of (±) Tanikolide, Nat. Prod. Lett., 2001, 15, 393–399 CrossRef CAS PubMed.
- H. Jinnouchi, H. Nambu, T. Fujiwara and T. Yakura, Divergent synthesis of (+)-tanikolide and its analogues employing stereoselective rhodium(II)-catalyzed reaction, Tetrahedron, 2018, 74, 1059–1070 CrossRef CAS.
- H. Nambu, H. Jinnouchi, T. Fujiwara and T. Yakura, Total Synthesis of (+)-Tanikolide by a Traceless Stereoinduction Method Using Rhodium (II)-Catalyzed Oxonium Ylide Formation–[2, 3]-Sigmatropic Rearrangement and NHC-Catalyzed Ring-Expansion Lactonization, Synlett, 2016, 27, 1106–1109 CrossRef CAS.
- W.-G. Wang, A. Li, B.-C. Yan, S.-B. Niu, J.-W. Tang, X.-N. Li, X. Du, G. L. Challis, Y. Che, H.-D. Sun and J.-X. Pu, LC-MS-Guided Isolation of Penicilfuranone A: A New Antifibrotic Furancarboxylic Acid from the Plant Endophytic Fungus Penicillium sp. sh18, J. Nat. Prod., 2016, 79, 149–155 CrossRef CAS PubMed.
- Y. Ding, X. Long, J. Zhang, C. Qu, P. Wang, X. Yang, P.-T. Puno and J. Deng, Asymmetric total synthesis of penicilfuranone A through an NHC-catalyzed umpolung strategy, Chem. Sci., 2025, 16, 8302–8308 RSC.
- M. T. Hovey, D. T. Cohen, D. M. Walden, P. H. Y. Cheong and K. A. Scheidt, A carbene catalysis strategy for the synthesis of protoilludane natural products, Angew. Chem., 2017, 129, 9996–9999 CrossRef.
- B. M. Trost and K. Dogra, Synthesis of (-)-Δ9-trans-Tetrahydrocannabinol: Stereocontrol via Mo-Catalyzed Asymmetric Allylic Alkylation Reaction, Org. Lett., 2007, 9, 861–863 CrossRef CAS PubMed.
- M. M. Radwan, S. Chandra, S. Gul and M. A. ElSohly, Cannabinoids, Phenolics, Terpenes and Alkaloids of Cannabis, Molecules, 2021, 26, 2774 CrossRef CAS PubMed.
- A. Ametovski and D. W. Lupton, Enantioselective Total Synthesis of (−)-Δ9-Tetrahydrocannabinol via N-Heterocyclic
Carbene Catalysis, Org. Lett., 2019, 21, 1212–1215 CrossRef CAS PubMed.
- J. A. Ciaccio, R. Alam, C. D. D'Agrosa, A. E. Deal and D. Marcelin, Isolation and Identification of Nepetalactone Diastereomers from Commercial Samples of Nepeta cataria L. (Catnip): An Introductory Organic Laboratory Experiment, J. Chem. Educ., 2013, 90, 646–650 CrossRef CAS.
- W. Harnying, J.-M. Neudörfl and A. Berkessel, Enantiospecific Synthesis of Nepetalactones by One-Step Oxidative NHC Catalysis, Org. Lett., 2020, 22, 386–390 CrossRef CAS PubMed.
- T. Huang, S.-H. Ying, J.-Y. Li, H.-W. Chen, Y. Zang, W.-X. Wang, J. Li, J. Xiong and J.-F. Hu, Phytochemical and biological studies on rare and endangered plants endemic to China. Part XV. Structurally diverse diterpenoids and sesquiterpenoids from the vulnerable conifer Pseudotsuga sinensis, Phytochemistry, 2020, 169, 112184 CrossRef CAS PubMed.
- S. B. Solabannavar, P. P. Wadgaonkar, U. V. Desai and R. B. Mane, A SHORT SYNTHESIS OF ar-TODOMATUIC ACID AND DIHYDRO-ar-TURMERONE BY THE HECK REACTION, Org. Prep. Proced. Int., 2003, 35, 418–420 CrossRef CAS.
- A. Khatua, S. Pal, M. K. Das and V. Bisai, Asymmetric total syntheses of (–)-ar-turmerone, (–)-dihydro-ar-turmerone, (–)-ar-dehydrocurcumene, and (–)-ar-himachalene via a key allylic oxidative rearrangement, Tetrahedron Lett., 2021, 73, 153105 CrossRef CAS.
- L.-L. Zhang, Y.-Z. Gao, S.-H. Cai, H. Yu, S.-J. Shen, Q. Ping and Z.-P. Yang, Ni-catalyzed enantioconvergent deoxygenative reductive cross-coupling of unactivated alkyl alcohols and aryl bromides, Nat. Commun., 2024, 15, 2733 CrossRef CAS PubMed.
- Y. M. Xia, Q. R. Liang, X. L. Wang, X. P. Cao and X. F. Pan, Total Synthesis of (-)-Kaerophyllin,(-)-Hinokinin and (±)-Isohinokinin, Chin. J. Chem., 2003, 21, 1540–1542 CrossRef CAS.
- M. C. Marcotullio, A. Pelosi and M. Curini, Hinokinin, an emerging bioactive lignan, Molecules, 2014, 19, 14862–14878 CrossRef PubMed.
- J. M. V. Timple, L. G. Magalhães, K. C. Souza Rezende, A. C. Pereira, W. R. Cunha, M. L. Andrade e Silva, O. V. Mortensen and A. C. K. Fontana, The Lignan (−)-Hinokinin Displays Modulatory Effects on Human Monoamine and GABA Transporter Activities, J. Nat. Prod., 2013, 76, 1889–1895 CrossRef CAS PubMed.
- S. Singha, E. Serrano, S. Mondal, C. G. Daniliuc and F. Glorius, Diastereodivergent synthesis of enantioenriched α,β-disubstituted γ-butyrolactones via cooperative N-heterocyclic carbene and Ir catalysis, Nat. Catal., 2020, 3, 48–54 CrossRef CAS.
- Y.-P. Jiang, Q.-L. Guo, Y.-F. Liu and J.-G. Shi, Codonopiloneolignanin A, a polycyclic neolignan with a new carbon skeleton from the roots of Codonopsis pilosula, Chin. Chem. Lett., 2016, 27, 55–58 CrossRef CAS.
- X. Li, H. Yong, X. Fan, Y. Zheng, Z. Wang and Z. Xie, Scalable Total Synthesis of (+)- and (−)-Codonopiloneolignanin A via Ti(IV)/NHC Cooperative Control Highly Enantioselective Dimerization of Multisubstituted Cinnamaldehyde, Org. Lett., 2021, 23, 6573–6577 CrossRef CAS PubMed.
- S. Perveen, S. Yang, M. Meng, W. Xu, G. Zhang and X. Fang, Asymmetric total synthesis of rotenoids via organocatalyzed dynamic kinetic resolution, Commun. Chem., 2019, 2, 8 CrossRef.
- W. X. Wu Xin, L. F. Li FeiLong, L. H. Liao HongBo, Q. S. Qiu ShuangLi, W. H. Wu Hua and Z. X. Zhu XiaoHui, 12a-hydroxymunduserone induces apoptosis of human hepatocarcinoma cells through Wnt/β-catenin pathway, Genomics Appl. Biol., 2016, 35, 1881–1886 Search PubMed.
- K. Umehara, K. Nemoto, A. Matsushita, E. Terada, O. Monthakantirat, W. De-Eknamkul, T. Miyase, T. Warashina, M. Degawa and H. Noguchi, Flavonoids from the Heartwood of the Thai Medicinal Plant Dalbergia parviflora and Their Effects on Estrogenic-Responsive Human Breast Cancer Cells, J. Nat. Prod., 2009, 72, 2163–2168 CrossRef CAS PubMed.
- K. Iwai, M. Ono, Y. Nanjo and T. Ema, Minimization of Amounts of Catalyst and Solvent in NHC-Catalyzed Benzoin Reactions of Solid Aldehydes: Mechanistic Consideration of Solid-to-Solid Conversion and Total Synthesis of Isodarparvinol B, ACS Omega, 2020, 5, 10207–10216 CrossRef CAS PubMed.
- Y. Mu, T. Jin, G. W. Kim, J. S. Kim, S. S. Kim, Y. S. Tian, C. Y. Oh and W. H. Ham, Total Syntheses of d-xylo-and d-arabino-Phytosphingosine Based on the Syntheses of Chiral 1, 3-Oxazines, Eur. J. Org Chem., 2012, 2012, 2614–2620 CrossRef CAS.
- P. Haghshenas and M. Gravel, Chemo- and Diastereoselective N-Heterocyclic Carbene-Catalyzed Cross-Benzoin Reactions Using N-Boc-α-amino Aldehydes, Org. Lett., 2016, 18, 4518–4521 CrossRef CAS PubMed.
- B. Kang, Y. Wang, S. Kuwano, Y. Yamaoka, K. Takasu and K.-i. Yamada, Site-selective benzoin-type cyclization of unsymmetrical dialdoses catalyzed by N-heterocyclic carbenes for divergent cyclitol synthesis, Chem. Commun., 2017, 53, 4469–4472 RSC.
- M. P. Thomas, S. J. Mills and B. V. L. Potter, The “other” inositols and their phosphates: synthesis, biology, and medicine (with recent advances in myo-inositol chemistry), Angew. Chem., Int. Ed., 2016, 55, 1614–1650 CrossRef CAS PubMed.
- B. K. Park, M. Nakagawa, A. Hirota and M. Nakayama, Methylenolactocin, a novel antitumor antibiotic from Penicillium sp, J. Antibiot., 1988, 41, 751–758 CrossRef CAS PubMed.
- T. Pengsuparp, L. Cai, H. Constant, H. H. S. Fong, L.-Z. Lin, A. D. Kinghorn, J. M. Pezzuto, G. A. Cordell, K. Ingolfsdöttir, H. Wagner and S. H. Hughes, Mechanistic Evaluation of New Plant-Derived Compounds That Inhibit HIV-1 Reverse Transcriptase, J. Nat. Prod., 1995, 58, 1024–1031 CrossRef CAS PubMed.
- Y.-P. Liu, W.-H. Zhao, X.-Y. Feng, Z.-J. Zhang, K. Zong, Z.-G. Sun, Y.-T. Zheng and Y.-H. Fu, Novel tetrahydrofuran derivatives from Trigonostemon howii with their potential anti-HIV-1 activities, Bioorg. Chem., 2018, 79, 111–114 CrossRef CAS PubMed.
- A. K. Perepogu, D. Raman, U. S. N. Murty and V. J. Rao, Stereoselective Synthesis of (+)-Nephrosteranic Acid by Ring-Closing Metathesis and Its Biological Evaluation, Synth. Commun., 2010, 40, 686–696 CrossRef CAS.
- A. M. Sarkale, V. Maurya, S. Giri and C. Appayee, Stereodivergent Synthesis of Chiral Paraconic Acids via Dynamic Kinetic Resolution of 3-Acylsuccinimides, Org. Lett., 2019, 21, 4266–4270 CrossRef CAS PubMed.
- P. Ren, Q. Zhao, Y. R. Chi and T. Zhu, Carbene-catalyzed enantioselective construction of a quasi-symmetrical spirocyclic hydroquinone with a minor chiral distinction, Chem. Sci., 2025, 16, 8940–8945 RSC.
- Y. Chen, E. Wendt-Pienkowski and B. Shen, Identification and Utility of FdmR1 as a Streptomyces Antibiotic Regulatory Protein Activator for Fredericamycin Production in Streptomyces griseus ATCC 49344 and Heterologous Hosts, J. Bacteriol., 2008, 190, 5587–5596 CrossRef CAS PubMed.
- Y. Kita, K. Higuchi, Y. Yoshida, K. Iio, S. Kitagaki, S. Akai and H. Fujioka, Asymmetric total synthesis of fredericamycin A, Angew. Chem., Int. Ed., 1999, 38, 683–686 CrossRef CAS PubMed.
- S. Meninno, C. Volpe and A. Lattanzi, Catalytic Enantioselective Synthesis of Protecting-Group-Free 1, 5-Benzothiazepines, Chem.–Eur. J., 2017, 23, 4547–4550 CrossRef CAS PubMed.
- W. Li, C. Schlepphorst, C. Daniliuc and F. Glorius, Asymmetric Hydrogenation of Vinylthioethers: Access to Optically Active 1, 5-Benzothiazepine Derivatives, Angew. Chem., Int. Ed., 2016, 55, 3300–3303 CrossRef CAS PubMed.
- C. Fang, T. Lu, J. Zhu, K. Sun and D. Du, Formal [3 + 4] Annulation of α,β-Unsaturated Acyl Azoliums: Access to Enantioenriched N–H-Free 1,5-Benzothiazepines, Org. Lett., 2017, 19, 3470–3473 CrossRef CAS PubMed.
- M. Kowalska, J. Nowaczyk, Ł. Fijałkowski and A. Nowaczyk, Paroxetine—overview of the molecular mechanisms of action, Int. J. Mol. Sci., 2021, 22, 1662 CrossRef CAS PubMed.
- A. Porey, S. Santra and J. Guin, Highly Enantioselective Synthesis of Functionalized Glutarimide Using Oxidative N-Heterocyclic Carbene Catalysis: A Formal Synthesis of (−)-Paroxetine, J. Org. Chem., 2019, 84, 5313–5327 CrossRef CAS PubMed.
- G. Hughes, M. Kimura and S. L. Buchwald, Catalytic Enantioselective Conjugate Reduction of Lactones and Lactams, J. Am. Chem. Soc., 2003, 125, 11253–11258 CrossRef CAS PubMed.
- S. M. Schaal, M. S. Garg, M. Ghosh, L. Lovera, M. Lopez, M. Patel, J. Louro, S. Patel, L. Tuesta and W.-M. Chan, The therapeutic profile of rolipram, PDE target and mechanism of action as a neuroprotectant following spinal cord injury, PLoS One, 2012, 7, 1–22 Search PubMed.
- Y. Zhang, Y. Zhang, J. Guo, J. Han, X. Zhou and Z. Fu, Asymmetric synthesis of γ-lactams under low-loading N-heterocyclic carbene catalysis, Org. Chem. Front., 2021, 8, 5087–5091 RSC.
- J. Caruano, G. G. Muccioli and R. Robiette, Biologically active γ-lactams: synthesis and natural sources, Org. Biomol. Chem., 2016, 14, 10134–10156 RSC.
- Z. Hu, C. Wei, Q. Shi, X. Hong, J. Liu, X. Zhou, J. Han, W. Cao, A. K. Gupta, X. Zhang, D. Wei, Z. Fu and W. Huang, Desymmetrization of N-Cbz glutarimides through N-heterocyclic carbene organocatalysis, Nat. Commun., 2022, 13, 4042 CrossRef CAS PubMed.
- M. I. Attia, C. Herdeis and H. Bräuner-Osborne, GABAB-agonistic activity of certain baclofen homologues, Molecules, 2013, 18, 10266–10284 CrossRef CAS PubMed.
- L. Ji, Y. Ma, J. Li, L. Zhang and L. Zhang, An efficient synthesis of (R)- and (S)-baclofen via desymmetrization, Tetrahedron Lett., 2009, 50, 6166–6168 CrossRef CAS.
- Y. Li, Z. You, V. K. W. Wong, M. Chen, K. Zhang, W. Liu and S. Zhao, Synthesis and Biological Evaluation of Naproxen Derivatives as Novel NLRP3 Inhibitors, Chem. Pharm. Bull., 2024, 72, 979–988 CrossRef CAS PubMed.
- X. Chen, J. Z. M. Fong, J. Xu, C. Mou, Y. Lu, S. Yang, B.-A. Song and Y. R. Chi, Carbene-Catalyzed Dynamic Kinetic Resolution of Carboxylic Esters, J. Am. Chem. Soc., 2016, 138, 7212–7215 CrossRef CAS PubMed.
- F. Seraj, Kanwal, K. M. Khan, A. Khan, M. Ali, R. Khalil, Z. Ul-Haq, S. Hameed, M. Taha and U. Salar, Biology-oriented drug synthesis (BIODS), in vitro urease inhibitory activity, and in silico studies on ibuprofen derivatives, Mol. Diversity, 2021, 25, 143–157 CrossRef CAS PubMed.
- J. Wang, Y. Chen, S. Miao, C. Yao and K. Zhang, Synthesis of α-chiral amides via synergistic visible-light-induced Wolff rearrangement and asymmetric NHC catalysis, New J. Chem., 2024, 48, 15287–15291 RSC.
- Y. Liu, Q. Chen, C. Mou, L. Pan, X. Duan, X. Chen, H. Chen, Y. Zhao, Y. Lu, Z. Jin and Y. R. Chi, Catalytic asymmetric acetalization of carboxylic acids for access to chiral phthalidyl ester prodrugs, Nat. Commun., 2019, 10, 1675 CrossRef PubMed.
- K. Srinivas, N. Srinivasan, K. S. Reddy, M. Ramakrishna, C. R. Reddy, M. Arunagiri, R. L. Kumari, S. Venkataraman and V. T. Mathad, An Improved, Scalable, and Impurity-Free Process for Tolterodine Tartrate, Org. Process Res. Dev., 2005, 9, 314–318 CrossRef CAS.
- S. Guduguntla, V. Hornillos, R. Tessier, M. Fañanás-Mastral and B. L. Feringa, Chiral Diarylmethanes via Copper-Catalyzed Asymmetric Allylic Arylation with Organolithium Compounds, Org. Lett., 2016, 18, 252–255 CrossRef CAS PubMed.
- C. Sun, B. Potter and J. P. Morken, A Catalytic Enantiotopic-Group-Selective Suzuki Reaction for the Construction of Chiral Organoboronates, J. Am. Chem. Soc., 2014, 136, 6534–6537 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Ameer Fawad Zahoor
b,
Ameer Fawad Zahoor








































































