DOI:
10.1039/D5RA10041K
(Review Article)
RSC Adv., 2026,
16, 11478-11495
Recent developments in the organocatalytic asymmetric cycloaddition/annulation reactions involving indolylmethanols
Received
27th December 2025
, Accepted 19th February 2026
First published on 27th February 2026
Abstract
Enantioenriched indole frameworks constitute the structural core of a wide array of biologically active natural products and therapeutic agents; as such, their synthesis has emerged as a central and enduring objective in organic synthesis. In this regard, suitably substituted indolylmethanols have emerged as versatile precursors to a series of catalytic asymmetric transformations to construct complex indole-based architectures. This article provides a concise overview of the fundamental chemistry of n-indolylmethanols, with a specific emphasis on their role as key substrates in enantioselective cycloadditions catalyzed by chiral phosphoric acids.
 Arun Patel | Arun Patel was born in Rewa, Madhya Pradesh, India. He received his B.Sc. and M.Sc. (Chemistry) from Maharishi Dayanand University (Rohtak), Haryana, India. Currently, he is working as a UGC-JRF for his doctoral degree under the guidance of Dr. Indresh Kumar at BITS Pilani. His main research interests are asymmetric catalysis and the development of new synthetic methods for bridged polycyclic heterocycles. |
 Harshit | Harshit, originally from Charkhi Dadri, Haryana, India, completed his B.Sc. at Maharishi Dayanand University (Rohtak) and his M.Sc. in Chemistry at CUH, Haryana. He is currently pursuing a doctoral degree as a research scholar under the supervision of Dr. Indresh Kumar at BITS Pilani. His research focuses on organocatalysis and the development of novel synthetic methodologies for bridged polycyclic heterocycles. |
 Eldhose Iype | Dr. Eldhose Iype was born in Kerala, India. After earning his master’s degree in chemical engineering in 2008, he completed his PhD in reactive molecular dynamics in 2014. Following two years of postdoctoral research, he transitioned to an academic career. He is now an assistant professor at the American University of the Middle East in Kuwait and has made notable contributions to integrating machine learning with molecular simulations. |
 Indresh Kumar | Dr. Indresh Kumar is a Professor of Chemistry at the Birla Institute of Technology and Science, Pilani, Pilani campus (Rajasthan). He completed his PhD at the National Chemical Laboratory (CSIR), Pune, followed by a postdoctoral fellowship at Tokyo University of Science. Prof. Kumar receives the CRSI Bronze Medal, the Professor D.K. Banerjee Memorial Lecture Award, and the ISCB Young Scientist Award. His primary research interest is the development of novel synthetic methods to access bioactive nitrogen-heterocyclic compounds under metal-free conditions. |
1. Introduction
The indole scaffold belongs to a class of electron-rich heteroaromatic rings and is a perennially hot topic in both academic and industrial chemistry.1,2 Indole-containing frameworks occupy a privileged position in organic chemistry, serving as a critical structural element in natural products, pharmaceuticals, agrochemicals, and functional materials (Fig. 1).3 The enantioselective construction of indole-based polycyclic architectures remains a central objective, driven by the profound influence of stereochemistry on biological activity and molecular recognition. Among the diverse strategies, cycloaddition reactions provide a robust, convergent approach to accessing structurally complex indole-fused polycycles, such as cyclopenta[b]indoles, spirooxindoles, and tetrahydro-β-carbolines. These scaffolds are the core of numerous alkaloids, anticancer agents, and CNS drugs, underpinning their diverse pharmacological profiles,4a which include antitumor,4b antimalarial,4c antiimplantation,4d and antioxidants.4e
 |
| | Fig. 1 Representative natural and bioactive molecules containing an indole unit. | |
A paradigm shift occurred with the recognition that strategically functionalizing the indole nucleus enables the creation of versatile “platform molecules” with diversified and controllable reactivity, thereby unlocking new pathways for asymmetric synthesis.5 The most efficient way to access enantioenriched compounds is through catalytic asymmetric synthesis, a field honoured with the 2001 Nobel Prize in Chemistry.6
In this context, the field has developed rapidly, yet the catalytic asymmetric synthesis of indole-based chiral heterocycles constitutes a significantly underexplored area.7 Moreover, one of the most straightforward and efficient ways to make indole derivatives is to start from existing indole-containing building blocks.8 In recent years, among the indole derivatives, indolylmethanols have proven to be versatile programmable platform molecules for asymmetric synthesis of indole derivatives.9 Under Brønsted acid catalysis, indolylmethanols readily undergo dehydration to generate vinyliminium or carbocationic intermediates, which can be intercepted in a highly stereocontrolled manner.10 The exceptional stability of these intermediates is resonance-stabilized, best described as a blend of a carbocation and a vinyliminium ion. Depending on substitution patterns and reaction partners, these intermediates enable indolylmethanols to function as three-carbon or four-carbon synthons, unlocking a remarkable diversity of cycloaddition manifolds. The electrophilicity at the C-3 position in the cationic intermediates generated from 3-indolylmethanols provides a versatile platform for interception by a diverse range of nucleophiles. Consequently, these species serve as pivotal substrates for well-documented catalytic enantioselective nucleophilic substitution reactions. Although the C-2 position of the indole nucleus is typically nucleophilic and unreactive in electrophilic pathways, 3-indolylmethanol derivatives can be employed as three-carbon synthons in the ring-construction reactions (Scheme 1).
 |
| | Scheme 1 Profile of catalytic asymmetric reactions of 3-indolylmethanols. | |
Similarly, 2-indolylmethanols have been established as powerful reactants for constructing enantioenriched indole architectures. Their synthetic potential is often greater than that of 3-indolylmethanols, a consequence of the more complex electrophilic profile of their reaction intermediates, which feature multiple, distinct reactive centres amenable to catalysis. The synthetic versatility of 2-indolylmethanols is further exemplified by their suitability as electrophiles at either the C-2 position (normal) or the C-3 position (abnormal). Moreover, these compounds also behave as three-atom building blocks in enantioselective [3 + n] cyclizations, providing direct access to diverse indole-fused chiral cyclic architectures (Scheme 2). Due to the multiple reactive sites inherent in their electrophilic intermediates, 2- or 3-indolylmethanols provide a versatile platform for generating a vast spectrum of structurally and functionally diverse products.11–14 Overall, the key advantage of these transformations is their inherent atom-economy, with water as the sole byproduct, which aligns with the principles of green chemistry.
 |
| | Scheme 2 Profile of catalytic asymmetric reactions of 2-indolylmethanols. | |
Catalytic asymmetric cycloaddition/annulation reactions have played a prominent role as fundamental synthetic methodologies15 for the enantioselective construction of various cyclic heterocycles.16 These are pivotal disconnection strategies, offering a direct and logical path to introduce intricate stereochemistry with inherent modularity.17 Nowadays, there is an exponential increase in focus on creating versatile and efficient ways to build cyclic molecules with specific spatial orientations to address diverse needs in chemistry and medicine.18 In this context, the development of catalytic asymmetric cycloadditions using n-indolylmethanols offers a promising route to enantiopure indole-based heterocycles.19
Over the past decade, chiral phosphoric acid (CPA) catalysis has emerged as a particularly effective platform for controlling the reactivity and stereochemical outcome of indolylmethanol-derived intermediates. Through finely balanced ion-pairing interactions, hydrogen-bonding networks, and confinement effects, CPAs enable precise differentiation of enantiotopic faces in highly reactive cationic species. A large number of such chiral CPAs have been utilized in asymmetric [m + n] cycloaddition and annulation reactions (Fig. 2). Progress has advanced over the past decade, with several comprehensive reviews documenting the broader reactivity of indolylmethanols in asymmetric catalysis.20–25 During the preparation of this manuscript, Shi and coworkers summarized indolylmethanols as versatile indole-based platform molecules for the catalytic asymmetric construction of chiral indole scaffolds.26 Nevertheless, a focused and mechanistically oriented analysis of catalytic asymmetric cycloaddition/annulation reactions involving indolylmethanols remains timely and necessary. In particular, a unified discussion that compares activation modes, stereocontrol strategies, and transition-state models across different [m + n] cycloaddditon is required. chiral indole frameworks. In this review, we provide a critical and systematic overview of recent developments in organocatalytic asymmetric cycloaddition and annulation reactions involving indolylmethanols. Emphasis is placed on mechanistic principles and stereochemical models governing reactivity in [3 + n] and [4 + n] cycloaddition pathways, and it is anticipated that the review will enable and inspire the construction of more complex chiral indole architectures.
 |
| | Fig. 2 List of various chiral acid catalysts utilized for developing asymmetric cycloadditions. | |
2. Organocatalytic [3 + n] cycloaddition involving indolylmethanol
Recent studies have highlighted the significant potential of n-indolylmethanols as versatile intermediates in organic synthesis. These compounds have proven to be highly reactive partners, enabling a range of catalytic enantioselective nucleophilic substitution reactions. Indolylmethanol, serving as a three-carbon (3C) synthon in catalytic asymmetric cycloadditions, particularly in enantioselective [3 + n] cycloadditions, has recently attracted attention (Scheme 3).
 |
| | Scheme 3 Utilization of n-indolylmethanol in enantioselective transformations. | |
2.1 [3 + 2] Cycloaddition involving indolylmethanol
Developing a catalytic asymmetric [3 + 2] cycloaddition of n-indolylmethanols is an attractive yet demanding goal in synthetic organic chemistry, as it offers a direct route to chiral cyclopenta[b]indole scaffolds (Scheme 4), which are significant structural motifs in bioactive molecules. The C-2 position of indole exhibits relatively low nucleophilicity, and the resulting cycloadduct contains three contiguous stereocenters within a rigid framework, demanding precise synthetic design.
 |
| | Scheme 4 Profile of n-indolylmethanol involved in [3 + 2] cycloadditions. | |
In this direction, in pursuit of the synthetic objective, Shi and coworkers demonstrated two similar chiral phosphoric acid (CPA)-catalyzed formal [3 + 2] cyclizations employing isatin-derived 3-indolylmethanols to assemble cyclopenta[b]indole frameworks (Scheme 5). In both studies, CPA-mediated dehydration of the 3-indolylmethanol 1/2 generates a vinyliminium intermediate, which undergoes [3 + 2] cycloaddition with high efficiency and excellent stereocontrol. In the first case, the reaction between 3-indolylmethanol 1 (R = Bn)and 3-methyl-2-vinylindoles 3 proceeds via a dual-activation mode using catalyst (CPA-X) enabling a vinylogous Michael addition and subsequent intramolecular Friedel–Crafts reaction to form spiro cyclopenta[b]indole-1,3′-oxindoles 4.27 In another case, N-protected 2-vinylindoles 5 react with vinyliminium intermediate as shown in TS-I, in situ generated from 3-indolylmethanol 2 (R = Me) using catalyst (CPA-IX) to a stereoselective vinylogous addition followed by intramolecular Michael cyclization to cyclopenta[b]indole framework 6.28 Collectively, these studies highlight the mechanistic versatility of CPA catalysis and the synthetic value of 3-indolylmethanols in accessing enantioenriched cyclopenta[b]indole scaffolds.
 |
| | Scheme 5 Organocatalytic asymmetric formal [3 + 2] cycloadditions to construct a spiro cyclopenta[b]indole framework. | |
Masson and coworkers reported asymmetric [3 + 2] cycloaddition between enecarbamates 7 and 3-indolylmethanols 8, which affords trisubstituted 3-aminocyclopenta[b]indoles 9. The mechanism involves the phosphoric acid-promoted dehydration of 3-indolylmethanols to a vinyliminium ion, followed by a vinylogous Mannich addition, and then a Pictet–Spengler reaction through TS-II. This one-pot process yields structurally diverse chiral cyclopenta[b]indoles with excellent stereocontrol (Scheme 6).29
 |
| | Scheme 6 Organocatalytic asymmetric [3 + 2] cycloaddition to access trisubstituted 3-aminocyclopenta[b]indoles. | |
Soon after, Shi and coworkers introduced the first catalytic asymmetric [3 + 2] cycloaddition of C-3 unsubstituted 2-indolylmethanols 10 with 3-vinylindoles 11, using a chiral phosphoric acid (CPA-VIII) as the catalyst. The reaction efficiently constructs biologically significant cyclopenta[b]indole 12 frameworks with excellent regio-, diastereo-, and enantioselectivity. Key innovations include overcoming challenges in 2-indolylmethanol transformations and employing a stepwise cascade mechanism via dual activation (TS-III), which involves both hydrogen bonding and ion pair interactions between the substrates and the chiral phosphoric acid catalyst, supported by control experiments. A series of complex indole-based architectures was accessed with excellent stereocontrol along with scalability, and derivatisation to interesting indole derivatives (Scheme 7).30
 |
| | Scheme 7 Organocatalytic asymmetric [3 + 2] cycloadditions using 2-indolylmethanols and 3-vinyl-indoles as building blocks. | |
In the same year, by altering the nucleophilicity of indolylmethanol, Schneider and coworkers reported a highly enantioselective one-step [3 + 2] cycloaddition method for the synthesis of pyrrolo[1,2-a]indoles 14, key scaffolds in natural products and drugs. The in situ generated 2-methide-2H-indoles from 3-substituted 2-indolylmethanols 13, catalyzed by a BINOL-derived Brønsted acid catalyst (CPA-III), seem crucial for the [3 + 2] cycloaddition with 2-vinylindoles 3. The transition state (TS-IV) was proposed to be highly organized through double hydrogen bonding involving the catalyst, which is essential for controlling stereoselectivity, as confirmed by control experiments. This method affords products with three contiguous stereocenters in excellent yields and enantioselectivities, typically as single diastereomers, and has been successfully applied on a large scale to access biologically relevant heterocycles (Scheme 8).31
 |
| | Scheme 8 Enantioselective[3 + 2] cycloaddition between 2-indolylmethanols and 2-vinyl-indoles to access pyrrolo[1,2-a]indoles. | |
At the same time, asymmetric synthesis of indolo[1,2-a]indoles 16 was disclosed by Bera and coworkers through an enantioselective [3 + 2] cycloaddition of 3-substituted 2-indolylmethanols 13 with cyclic enamides 15 using chiral phosphoric acid catalysis. Under optimized conditions, the chiral catalyst (CPA-IV) generated 2-methide-2H-indoles in situ from 13, which underwent [3 + 2] cycloaddition with enamides 15, yielding products 16 with three contiguous stereocenters in high yields and enantioselectivities (up to >99:1 er) under mild conditions. Mechanistic studies suggest dual activation by CPA via TS-V proceeds through a stepwise mechanism involving conjugate addition and aminalization to access indole-fused scaffolds relevant for drug discovery (Scheme 9).32
 |
| | Scheme 9 Brønsted acid-catalysed asymmetric formal [3 + 2] cycloaddition of cyclic enamides with 2-methide-2H-indoles. | |
A year later, Meng and coworkers used p-hydroxystyrenes 17 as suitable precursors to synthesise cyclopenta[b]indole 18 scaffolds. This method involves a chiral phosphoramide catalysed (CPA-XVI) enantioselective [3 + 2] cycloaddition between 2-indolylmethanols 10 and p-hydroxystyrenes 17. The proposed mechanistic pathway consists of the phosphoramide anion simultaneously forming both an ion pair and a hydrogen-bonding interaction with the carbocation intermediate via TS-VI, while concurrently activating the p-hydroxystyrene reactant via a second hydrogen bond to its free hydroxyl group. Control experiments rigorously confirmed the necessity of both the free –NH and –OH groups for successful cycloaddition, verifying that this dual activation is key to the mechanism for delivering products in outstanding yields and excellent enantioselectivities (Scheme 10).33
 |
| | Scheme 10 Catalytic enantioselective [3 + 2] cycloaddition to access cyclopenta[b]indole scaffolds. | |
Focusing on cyclopent[b]indole, Deng and coworkers demonstrated a synergistic catalytic system by combining the enantioselective [3 + 2] cycloaddition of 2-indolylmethanols 10 with α,β-unsaturated aldehydes 19. This important synthetic strategy utilizes a synergistic catalytic system comprising a Brønsted acid, a palladium complex, and a chiral secondary amine catalyst (cat-C1). The proposed mechanistic pathway involves dual activation, shown in TS-VII, which drives the reaction of dienamine-intermediate with Pd-complex, followed by intramolecular cyclization to construct the synthetically important cyclopenta[b]indole 20 scaffolds with high enantioselectivities (Scheme 11).34
 |
| | Scheme 11 Synergistic catalysis for asymmetric [3 + 2] cycloadditions of 2-indolylmethanols with α,β-unsaturated aldehydes. | |
In 2020, Fabian and coworkers reported a palladium-catalyzed, enantioselective [3 + 2] cycloannulation between alkylidene 2-H-indoles 13 and β-keto esters 21, enabling access to complex N-fused polycyclic indoles as a challenging unit. A single chiral Pd-catalyst (cat-C2) achieves dual activation to generate in situ both reactive chiral metal enolates and 2-methide-2H-indoles as vinylogous iminium ions. The method affords products 22 with high yields (up to 99%), excellent diastereo- and enantioselectivity (up to >99% ee), and broader substrate scope. Mechanistic insights from ESI-MS support a cooperative catalytic cycle. Additionally, the products undergo diverse functionalizations, showcasing their synthetic utility in constructing bioactive fused heterocycles (Scheme 12).35
 |
| | Scheme 12 Palladium-catalyzed, enantioselective [3 + 2] cycloannulation between β-keto esters and alkylidene 2-H-indoles. | |
More recently, the involvement of two various indolylmethanols as suitable substrates to access chiral pyrrolo[1,2-a]indoles was shown by Shi and coworkers. They reported the first catalytic asymmetric formal [3 + 2] cycloaddition of methyl-substituted 2-indolylmethanols 23 with 3-substituted-2-indolylmethanols 13 using chiral phosphoric acids (CPA-V). The reaction delivers chiral pyrrolo[1,2-a]indoles 24 in high yields, excellent diastereoselectivities (>95:5 dr), and good enantioselectivities (up to 94% ee). Mechanistic insights were supported by DFT calculations and control experiments, revealing dual hydrogen bonding by (CPA-V) and ion-pairing activation between the substrates through TS-VIII. The method demonstrates broad substrate scope, functional group tolerance, and scalability to access enantioenriched indole-based scaffolds (Scheme 13).36
 |
| | Scheme 13 Catalytic asymmetric formal [3 + 2] cycloaddition of methyl-substituted 2-indolylmethanols with 3-substituted-2-indolylmethanols. | |
Very recently, the construction of spiro-bis-N-heterocyclic lactam backbones 26 was achieved by Wang and coworkers through a highly efficient chiral phosphoric acid-catalyzed asymmetric formal [3 + 2] cycloaddition between 2-indolylmethanol 13 and 3-methylene isoindolinone 25. Mechanistically, the chiral catalyst CPA-XII plays an essential role in the simultaneous activation of both 2-indolylmethanol 13 for generating a vinyliminium intermediate and 3-methylene isoindolinone 25, due to the crucial necessity of the NH bond for reactivity, as shown in TS-IX. The utility of this approach is demonstrated to a diverse scope of substrates and corresponding products 26 were obtained with excellent enantioselectivity (up to 99% ee) (Scheme 14).37
 |
| | Scheme 14 Catalytic asymmetric [3 + 2] cycloaddition to access chiral spirobis-N-heterocyclic lactams. | |
2.2 [3 + 3] Cycloaddition involving indolylmethanol
Pursuing a catalytic asymmetric [3 + 3] cycloaddition of n-indolylmethanols presents a compelling yet challenging objective in synthetic organic chemistry, offering a streamlined pathway to chiral indole-fused scaffolds. Such a cycloaddition reaction requires a 1,4-donor–acceptor precursor, which can participate in the cycloaddition reaction with n-indolylmethanols under mild conditions (Scheme 15). In this direction, initial breakthroughs were reported by Shi and coworkers for an enantioselective three-component [3 + 3] cycloaddition using chiral phosphoric acid (Scheme 16). Initially, isatin-derived 3-indolylmethanols 1 underwent a [3 + 3] cycloaddition with in situ generated azomethine ylides from aromatic aldehydes 27 and amino-ester 28 through chiral catalyst (CPA-II), affording spiroindoline-3,4′-pyridoindoles 29 in high yields and enantioselectivities (up to 99% ee) (Scheme 16).38 Later, the same group utilized isatin 30 as a reacting partner with amino-ester 28 for in situ generation of azomethine ylides, which subsequently participate in a [3 + 3] cycloaddition with 3-indolylmethanols 1 under chiral catalyst (CPA-X). A series of bispirooxindoles 31 with quaternary stereocenters was accessed with high stereoselectivity (>95:5 dr, 98:2 er) (Scheme 16).39 In a typical reaction mechanism, the reaction proceeds through the TS-X, in which a chiral Brønsted acid catalyst activates both the reacting partners, such as azomethine ylides and the in situ generated vinyliminium ion intermediate, for a Michael addition followed by intramolecular cyclization. Variations with diverse aldehydes and isatins were tested to assess a large number of structurally complex spirooxindoles.
 |
| | Scheme 15 Profile of 3-indolylmethanol involved [3 + 3] cycloaddition. | |
 |
| | Scheme 16 Three-component cascade reaction for enantioselective synthesis of bispirooxindoles. | |
Building on the initial work, the same group in 2016 advanced a catalytic enantioselective [3 + 3] cycloaddition involving C3-unsubstituted 2-indolylmethanols 10 as 3-carbon synthons with in situ generated azomethine ylides from aldehydes 27 and amino esters 28, catalyzed by chiral phosphoric acid (CPA-V). A series of chiral tetrahydro-γ-carbolines 32 was accessed in good yield and with excellent enantioselectivities (up to 99% ee) using catalyst CPA-V (Scheme 17).40
 |
| | Scheme 17 Catalytic asymmetric [3 + 3] cycloaddition of azomethine ylides with C3-substituted 2-indolylmethanols. | |
Whereas, this idea was extended to C3-substituted 2-indolylmethanols 13 for CPA-X catalyzed [3 + 3] cycloaddition with in situ generated azomethine ylides derived from a similar set of starting materials to access tetrahydropyrimido[1,6-a]indoles 33 with good yields and high enantioselectivity (up to 96% ee) (Scheme 17).41 The high stereochemical outcome was explained by the fact that CPA drives dehydration to vinyliminium ions, and simultaneous activation of azomethine ylides occurs through a compact TS-XI to access diverse indole frameworks.
Continuing their efforts in this area, the same research group reported a chiral phosphoric acid CPA-V catalyzed asymmetric [3 + 3] cycloaddition between C3-substituted 2-indolylmethanols 13 and isatin 30 derived azomethine ylides to construct chiral spiro-oxindoles 34. Mechanistic studies suggest a CPA-V-facilitated tandem cyclization pathway involving an azomethine ylide and a vinyl iminium intermediate, via a transition state (TS-XII), to access chiral spiro-oxindole scaffolds 34 with high yields and enantioselectivity (Scheme 18).42 The methodology showed broad substrate scope, tolerating diverse substitutions on both indolylmethanols 13 and isatins 30.
 |
| | Scheme 18 Enantioselective construction of spirooxindole scaffolds through a catalytic asymmetric [3 + 3] cycloaddition. | |
Shi and co-workers developed a highly regio- and enantioselective [3 + 3] cycloaddition of nitrones with 2-indolylmethanols, providing a robust synthetic route to indole-fused six-membered heterocycles. This methodology marks the first organocatalytic asymmetric [3 + 3] cycloaddition of nitrones 35 and is facilitated by a cooperative catalytic system consisting of a chiral phosphoric acid (CPA-X) and hexafluoroisopropanol (HFIP). The reaction proceeds with excellent yields (up to 98%) and enantioselectivities (up to 96% ee), utilizing the C3-nucleophilicity of the 2-indolylmethanols 10. Mechanistic insights and DFT calculations revealed that HFIP plays a critical role as a co-catalyst, enhancing both reactivity and stereocontrol by stabilizing the transition states through hydrogen-bonding networks (TS-XIII), thereby expanding the utility of cooperative organocatalysis in the construction of complex, bioactive indole derivatives 36 (Scheme 19).43
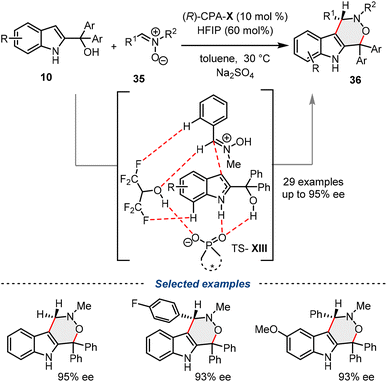 |
| | Scheme 19 Catalytic asymmetric [3 + 3] cycloaddition of nitrones with 2-indolylmethanols. | |
In 2021, Christopher and coworkers reported a novel, highly stereoselective [3 + 3] cycloannulation strategy that enables the one-step synthesis of oxa-bridged azepino[1,2-a]indoles 38. This process involves phosphoric acid-catalyzed activation of the electrophilic partner through hydrogen bonding and protonation, generating a stabilized vinyliminium intermediate. The key innovation lies in the use of cooperative catalysis: a rhodium catalyst generates carbonyl ylides from α-diazo esters 37, while a chiral phosphoric acid (CPA-XI) catalyst activates indolyl-2-methides through a transition state (TS-XIV). High enantioselectivities (up to 98% ee) and good yields are achieved across a broad substrate scope (Scheme 20).44
 |
| | Scheme 20 [3 + 3] Cycloannulation to access oxa-bridged azepino[1,2-a]indoles. | |
In 2024, Shi and coworkers reported the catalytic asymmetric [3 + 3] cycloaddition between two different types of 2-indolylmethanols, simple 2-indolylmethanols 10 and 3-alkyl(aryl)-substituted ones 13 under chiral acid catalysis. The reaction proceeds under chiral phosphoric acid (CPA-X) catalysis, with both coupling partners cooperatively activated. Initially, the acid protonates and dehydrates the indolylmethanol (or related alcohol precursor) to generate a resonance-stabilized vinyliminium intermediate through TS-XV. The reaction delivers a wide variety of enantioenriched indole-fused six-membered heterocycles 39 with high yields and enantioselectivities (Scheme 21).45 This work offers a novel strategy for constructing biologically significant indole-fused frameworks and broadens the utility of indolylmethanol derivatives in asymmetric cycloaddition chemistry.
 |
| | Scheme 21 Catalytic asymmetric [3 + 3] cycloaddition between two different 2-indolylmethanols. | |
In 2024, Zou and coworkers developed an efficient asymmetric [3 + 3] cycloaddition reaction between 2-indolylmethanols 10 and cinnamaldehyde-derived N-aryl nitrones 35, catalyzed by a chiral phosphoric acid (CPA-III). This methodology provides a direct route to various indole-fused 1,2-oxazines 40, with high yields and exceptional enantioselectivities (up to 99% ee). Mechanistic analysis revealed that the reaction proceeds through dual hydrogen-bonding activation through TS-XVI. The substrate scope included a range of indolylmethanols 10 and nitrones 35 under metal-free conditions to access high-value scaffolds in medicinal chemistry (Scheme 22).46
 |
| | Scheme 22 CPA-catalyzed [3 + 3] cycloaddition of cinnamaldehyde-derived N-aryl nitrones with 2-indolylmethanols. | |
In a follow-up study, the same research group reports a novel chiral phosphoric acid and HFIP co-catalyzed asymmetric [3 + 3] cycloaddition of 2-indolylmethanols 10 with N-vinyl oxindole nitrones 41, enabling the efficient synthesis of spirooxindole[1,2]oxazines 42 after cleavage of N-vinyl moiety of nitrone, bearing a tetrasubstituted chiral center. The mechanism involves CPA activating the nitrone, followed by HFIP-assisted dehydration, cyclization, and hydrolysis as shown in TS-XVII. The enantioselectivity is controlled by the chiral catalyst (CPA-XIII) through hydrogen-bonding interactions to access biologically critical six-membered spiroheterocycles with high enantioselectivity (Scheme 23).47
 |
| | Scheme 23 Catalytic asymmetric [3 + 3] cycloaddition of N-vinyloxindole nitrones with 2-indolylmethanols. | |
In a recent contribution, Studer and coworkers reported the first efficient, enantioselective synthesis of tetrahydro-1H-1,3-methanocarbazoles 44 using chiral phosphoric acid derivatives to catalyze the formal [3 + 3] cycloaddition between 1H-indol-3-yl(hetero)aryl methanol 8 and bicyclo[1.1.0]butanes 43. The bicyclo[1.1.0]butanes (BCBs) 43 were used as three-carbon components, leveraging their inherent strain in the cycloaddition process to form a more rigid, three-dimensional bridged architecture. The chiral Brønsted acid catalyst (CPA-VI) facilitates the nucleophilic ring-opening of the BCB by a transient carbocation with a chiral iminium ion intermediate through TS-XVIII, leading to ring expansion and the formation of chiral tetrahydro-1H-1,3-methanocarbazoles 44 with a broad substrate scope and high enantioselectivity (up to 98:2 er) (Scheme 24).48
 |
| | Scheme 24 Enantioselective organocatalytic formal [3 + 3] cycloaddition of bicyclo[1.1.0] butanes with 1H-indol-3-yl((hetero)aryl)methanol. | |
2.3 [3 + 4] Cycloaddition involving indolylmethanol
The development of a catalytic asymmetric [3 + 4] cycloaddition of n-indolylmethanols represents a highly appealing approach for a direct and efficient route to chiral cyclohepta[b]indole derivatives (Scheme 25). In this direction, Masson and coworkers reported a highly enantio- and diastereoselective formal [3 + 4] cycloaddition in 2018 that employs a chiral phosphoric acid catalyst to facilitate the reaction between 3-indolylmethanols 8 and 1,3-diene-1-carbamates 45 for the construction of cyclohepta[b]indole 46 with high diastereo- and enantioselectivity (up to 99% ee). The reaction demonstrates a broad substrate scope, tolerating various substituents on both the diene and the indolylmethanol precursor, including electron-rich, electron-deficient, and sterically demanding aryl groups under mild reaction conditions. A mechanistic study suggests that cycloaddition occurs stepwise, in which a favored ion pair consisting of the chiral phosphate anion and the newly formed indolium cation is held together with the dienecarbamate 45, activated and positioned through a hydrogen bond between its NH group and the Lewis basic phosphoryl oxygen of the chiral catalyst (CPA-III). This dual activation creates a highly organized arrangement that dictates the enantioselectivity of the subsequent C–C bond formation. The final ring-closing step is hypothesized to proceed via a bent-boat-like conformation (TS-XIX) to yield the thermodynamically favoured all-cis diastereomer by addressing the challenges associated with the conversion (Scheme 26).49
 |
| | Scheme 25 Profile of 3-indolylmethanol involved in [3 + 4] cycloaddition. | |
 |
| | Scheme 26 Catalytic enantioselective formal [3 + 4] cycloaddition of 3-indolylmethanols with 1,3-diene-1-carbamates. | |
In 2019, Shi and coworkers reported the first catalytic asymmetric [3 + 4] cyclization of 2-indolylmethanols 10 with in situ generated ortho-quinone methides (o-QMs), yielding seven-membered heterocycles in high yields and with excellent enantioselectivity. This method addresses the significant challenges associated with constructing seven-membered rings, providing access to chiral oxepino[2,3-b]indoles (48/50) in high yields and with excellent enantioselectivities (up to 98% ee). Two distinct compounds, such as ortho-hydroxybenzyl alcohols 47 and para-quinone methide derivatives 49, were tested as suitable precursors for the in situ generation of ortho-quinone methides (o-QMs) under acid catalysis. In mechanistic terms, the catalysts (CPA-X and CPA-XIV) simultaneously activate both the 2-indolylmethanol 10 and the o-QM via hydrogen-bonding interactions, facilitating the enantioselective nucleophilic addition to the intermediate, followed by an intramolecular cyclization through TS-XX and TS-XXI, simultaneously. This method offers a versatile route to a series of chiral oxepino[2,3-b]indole derivatives (Scheme 27).50
 |
| | Scheme 27 Catalytic asymmetric [3 + 4] cyclizations of 2-indolylmethanols with in situ generated ortho-quinone methides. | |
The same research group designed a novel class of axially chiral aryl-alkene-indole frameworks and achieved their first catalytic asymmetric construction via organocatalytic [3 + 4] cyclization of 3-alkynyl-2-indolylmethanols 51 with 2-naphthols or phenols 52. The process initiates with the 3-alkynyl-2-indolylmethanol 51 transforming into an allene-iminium intermediate. Subsequently, the CPA catalyst (CPA-XV) activates both substrates through hydrogen-bonding and ion-pairing interactions through TS-XXII. The mechanism involves, firstly, the nucleophilic addition creating axial chirality, followed by rearomatization, dehydration, and an intramolecular nucleophilic addition, resulting in the chiral oxepino[2,3-b]indoles 53 with an excellent efficiency >95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 (E/Z) ratio), and enantioselectivity (up to 97% ee) (Scheme 28).51 The method expands the atropisomer family to alkene-heteroaryl atropisomers and enables access to bioactive scaffolds.
:5 (E/Z) ratio), and enantioselectivity (up to 97% ee) (Scheme 28).51 The method expands the atropisomer family to alkene-heteroaryl atropisomers and enables access to bioactive scaffolds.
 |
| | Scheme 28 Catalytic asymmetric [3 + 4] cyclization to construct axially chiral aryl-alkene-indole frameworks. | |
A reliable synthetic route for an asymmetric [3 + 4] cycloaddition between dialkyl-substituted 2-indolylmethanols 10 and dienolsilanes 54 was introduced by List and coworkers in 2022. By employing a strongly acidic and confined imidodiphosphorimidate (IDPi-C3) catalyst, they achieved the enantioselective synthesis of bicyclo[3.2.2]cyclohepta[b]indoles 55 with high yield and excellent enantioselectivity (up to 98:2 er) by overcoming the earlier limitations in this direction (Scheme 29).52 This powerful catalytic system, which generates a reactive silylium species in situ which is essential for the concerted but asynchronous [3 + 4] cycloaddition via TS-XXIII. The straightforward access to such valuable molecular frameworks is expected to facilitate the synthesis of potentially bioactive natural products.
 |
| | Scheme 29 Organocatalytic asymmetric [3 + 4] cycloaddition of 2-indolylalcohols with dienolsilanes. | |
Further expanding the scope of the accessible framework, Lin and coworkers in 2024 reported a chiral acid (CPA-VII)-catalyzed enantioselective [3 + 4] cyclization of 2-indolylmethanols 10 with α-(3-isoindolinonyl) propargylic alcohols 56, yielding spiro-fused isoindolinone-oxepino-indoles 57 with high yields and excellent enantioselectivity (up to 95% ee) (Scheme 30).53 Control experiments and HRMS studies suggested a mechanism involving propargylic N-acyl imine intermediates and 2-indolylmethanols 10, which act as 1,4-donor-donor precursors. At the same time, the CPA catalyst plays a dual role by activating both the electrophilic species and the nucleophilic 2-indolylmethanol via a precise hydrogen-bonding network, as shown in transition state (TS-XXIV), guiding the formal [3 + 4] cycloaddition. Furthermore, the researchers demonstrated the practical utility of the method through gram-scale synthesis and the discovery of bright fluorescence in the resulting products, suggesting that this scaffold holds promise not only for medicinal chemistry but also for the development of novel optoelectronic materials.
 |
| | Scheme 30 Organocatalytic enantioselective [3 + 4] cyclization to access spiro-fused heterocycles. | |
Very recently, Chen and coworkers employed a completely different catalytic strategy, a dual photoredox chiral phosphoric acid relay, to access seven-membered heterocycles via a three-component cyclization of 2-vinylphenol 47, methylbromodifluoroacetate 58, and 2-indolylmethanols 10. Under visible light, an excited-state iridium(III) catalyst (PC-I) initiates a single electron transfer (SET) reduction of methyl bromodifluoroacetate 58 to generate a reactive difluoroalkyl radical, which undergoes regioselective addition to 2-vinylphenol 47, and subsequently, deprotonation results in a transient ortho-quinone methide intermediate. This ortho-quinone methide intermediate reacts with 2-indolylmethanols 10, involving chiral acid (CPA-II) activation, facilitated by hydrogen bonding through TS-XXV, enabling highly enantioselective conjugate addition and subsequent cyclization as an overall redox-neutral [3 + 4] process (Scheme 31).54 This cyclization provides de novo access to valuable, diversely substituted seven-membered oxygen-containing heterocycles.
 |
| | Scheme 31 Dual photoredox/chiral phosphoric [3 + 4] cyclization to access seven-membered heterocycles. | |
2.4 [4 + n] Cycloaddition involving indolylmethanol
Specially designed indolylmethanols can efficiently function as four-atom synthons in stereoselective [4 + n] cycloadditions, exploiting the nucleophilicity of the C-3 position of the indole and suitably substituted hydroxyl groups at the C4-position of indole (Scheme 32). This methodology enables the assembly of diverse chiral indole architectures, including those relevant to medicinal chemistry, such as spirocyclic and axially chiral frameworks. Notably, [4 + 3] annulations provide efficient routes to complex azepinoindole systems, while [4 + 4] or [4 + 5] variants grant access to eight or nine-membered heterocyclic scaffolds. This strategy thus facilitates concise access to privileged chiral structures from simple precursors. In this direction, Deng and coworkers in 2021 reported the first iridium-catalyzed asymmetric [4 + 3] cycloaddition that unites 4-indolyl allylic alcohols 60 with azomethine ylides 61 to access seven-membered azepino[3,4,5-cd]indole 62 with high yields and enantioselectivity (up to 99% ee). The key to the high stereoselectivity for [4 + 3] is the nucleophilic attack of the indole C-3 position onto the π-allyl-Ir species (allylation), followed by an intramolecular condensation between the C4-tethered iminium species as ylide frameworks activated by Zn(OTf)2 in the presence of ligand (L1) through TS-XXVI which promotes the subsequent Pictet–Spengler cyclization (Scheme 33).55 Furthermore, the broad substrate scope, including tolerance for sensitive functional groups and heteroaromatic systems, underscores its utility for medicinal chemistry.
 |
| | Scheme 32 Profile of well-designed 3-indolylmethanol involved [4 + n] cycloaddition. | |
 |
| | Scheme 33 Organocatalytic enantioselective [4 + 3] cycloaddition to access azepino[3,4,5-cd] indoles. | |
Very recently, Shi and coworkers developed the first catalytic enantioselective [4 + 4] annulation of o-hydroxybenzyl alcohols 47 with 4-indolylmethanols 63 in the presence of chiral phosphoric acid (CPA-V) catalyst to access a variety of chiral indole-based eight-membered heterocycle scaffolds. In the mechanistic study, CPA-V first dehydrates o-hydroxybenzyl alcohols 47 to form the o-quinone methide intermediate, which undergoes an enantioselective 1,4-addition utilizing C3-nucleophilicity of 4-indolylmethanols 63, followed by CPA-catalyzed dehydration, and intramolecular cyclization to complete the eight-membered ring formation through TS-XXVII. The precise spatial confinement imposed by the CPA anion governs both regioselectivity and enantioinduction, leading to excellent asymmetric control across a broad substrate scope. This innovative protocol offers a novel, high-yield approach for the construction of enantioenriched medium-sized complex indole motifs (Scheme 34).56
 |
| | Scheme 34 Organocatalytic enantioselective [4 + 4] cycloaddition to access cycloocta[b]indole. | |
3 Miscellaneous examples
A novel enantioselective and diastereodivergent synthesis of spirocyclization 66 was achieved by Masson and coworkers in 2021, involving a [4 + 2] cycloaddition between 2-substituted 3-indolylmethanols 8 and 1,3-dienecarbamates 65, using a chiral phosphoric acid (CPA-V). The mechanistic investigation, including experimental studies and density functional theory (DFT), suggested a stepwise process in which a transient carbocation–phosphate ion pair forms, with the chiral catalyst dictating the facial selectivity of the initial nucleophilic attack and subsequent ring closure via TS-XXVIII, furnishes polysubstituted spirocyclohexyl-indolenines 66 having up to four contiguous stereocontrolled chiral centers, including a quaternary one with excellent enantio- and diastereoselectivity (Scheme 35).57 This methodology not only advances the structural diversity available from indole-based scaffolds but also provides a profound mechanistic framework for understanding how non-covalent interactions and reaction conditions can be leveraged to override intrinsic substrate bias in complex cycloadditions.
 |
| | Scheme 35 Enantioselective and diastereodivergent synthesis of spiroindolenines. | |
Indolylmethanols serve as versatile indole-based platform molecules essential for the catalytic asymmetric construction of chiral indole scaffolds. In this context, to broaden the synthetic utility, innovative strategies have been established in which indolylmethanol serves as a 4C synthon. In this direction, the Shi group has designed a novel class of 2,3-indolyldimethanols, in which the hydroxymethyl groups are present at both the C2- and C3-positions of the indole ring. Such indolylmethanols can serve as 1,4-dielectrophiles to catalyze asymmetric (4 + n) annulations with dinucleophiles, thereby accessing indole-based fused scaffolds. The first catalytic asymmetric [4 + n] cycloadditions of 2,3-indolyldimethanols 68 with indole 67 derivatives and 2-naphthols 52 using chiral phosphoric acids as catalysts to construct enantioenriched indole-fused six and seven-membered rings with high yields and excellent enantioselectivities (Scheme 36).58 The work addresses challenges in regioselectivity control and dinucleophile compatibility, leveraging indoles and 2-naphthols 52 as reaction partners. Mechanistically, the reaction proceeds through the CPA-mediated dehydration of indolyldimethanols to generate highly reactive vinyliminium or carbocationic intermediates, which serve as versatile multi-atom synthons. These intermediates undergo formal [4 + 2] or [4 + 3] annulations with indoles or 2-naphthols, where the CPA not only controls the facial selectivity but also dictates the chemoselectivity of the cyclization through TS-XXIX and TS-XXX. DFT calculations further elucidate the reaction pathways and dual activation modes by chiral phosphoric acid, highlighting the role of hydrogen bonding in stereocontrol. This strategy advances indole chemistry by providing a versatile platform for the synthesis of chiral heterocycles.
 |
| | Scheme 36 Catalytic enantioselective dinucleophile-compatible [4 + n] cycloadditions of 2,3-indolyldimethanols. | |
In a significant advancement for indole-fused architectures, Lin and coworkers in 2024 developed an organocatalytic asymmetric [4 + 3] cycloaddition that yields optically active cyclohepta-fused diindoles with exceptional precision. Interestingly, the unprecedented use of 3-methyl-2-indolylmethanol 71 as a four-carbon (4C) synthon has been explored for [4 + 3] cycloaddition with distinctly substituted 2-indolylmethanols 1/8. The mechanism involves chiral phosphoric acids (CPA-IX and CPA-XVI) promoting the dehydration of the substrates to generate indole-2,3-quinodimethane (IQDM), and vinyliminium intermediates via the respective transition states TS-XXXI and TS-XXXII, which bridge the two indole units into a seven-membered ring system (Scheme 37).59 Overall, the work presents a novel approach to the synthesis of a series of structurally complex molecules 72/73, with high yields and excellent enantioselectivities (up to >99% ee).
 |
| | Scheme 37 Catalytic asymmetric [4 + 3] cyclizations of 2-indolylmethanols with in situ generated ortho-quinone methides. | |
Further to showcase the utility of indolylmethanol as containing 4C synthons, Shi and coworkers developed the catalytic asymmetric synthesis of furan-indole compounds 75 with both axial and central chirality through an organocatalytic [4 + 2] annulation of 2,3-indolyldimethanols 68 with achiral furan-indoles 74, using chiral phosphoric acid (CPA-IX). A mechanistic picture emerging from DFT studies features a catalyst-organized transition state, TS-XXXIII, that imposes uncommon regioselectivity in the annulation pathway, rationalizing how rotational barriers are raised to lock axial chirality while the prochiral center is set enantioselectively. Methodologically, the work is notable for (i) using metal-free organocatalysis, aligning with sustainability goals, (ii) establishing general access to five-membered furan-based frameworks that are difficult to assemble by conventional routes, and (iii) revealing a rare regio-control paradigm in furan chemistry that should be extensible to related heteroaryl systems. This developed protocol demonstrates a new strategy for the axially chiral furan-based compounds with high enantioselectivity (Scheme 38).60
 |
| | Scheme 38 Organocatalytic enantioselective [4 + 2] cycloaddition to access furan-indoles. | |
Very recently, Shi and coworkers reported a challenging, atroposelective, catalytic construction of axially chiral arylalkene-fused nine-membered rings via first organocatalytic asymmetric [4 + 5] cycloaddition. This strategy utilizes 3-alkynyl-2-indolylmethanols 51 and 2-indolylethanols 76 as versatile building blocks, facilitated by a chiral phosphoric acid (CPA) catalyst. Mechanistically, the CPA-XVII operates through a dual activation mode: it promotes the generation of a reactive vinylogous iminium or carbocationic intermediate while precisely orienting the nucleophilic 2-indolylethanol via transition state TS-XXXIV, stabilized by a network of hydrogen-bonding and ion-pairing interactions. The high level of enantioselectivity (up to 98% ee) is achieved by establishing crucial hydrogen-bonding interactions with the substrates' NH groups throughout the reaction pathway, thereby enabling the creation of structurally diverse alkenylindole-fused scaffolds (Scheme 39).61 Furthermore, the authors utilize theoretical calculations to provide deep insights into the reaction pathway and demonstrate the practical utility of the products in asymmetric catalysis and medicinal chemistry (e.g., antitumor activity). By expanding the toolkit for accessing configurationally stable nine-membered atropisomers, this work sets a new benchmark for complexity in cycloaddition chemistry.
 |
| | Scheme 39 Atroposelective construction of axially chiral alkenylindole-fused nine-membered rings [4 + 5] organocatalytic enantioselective cycloaddition. | |
4. Conclusion and remarks
In this review, we present recent developments in organocatalytic asymmetric [m + n] cycloaddition/annulation reactions involving indolylmethanols and provide insights into catalyst design, stereocontrol during substrate activation, and scope. The unique reactivity of indolylmethanols enables access to structurally complex, medium-sized indole-fused frameworks that are often difficult to construct due to inherent thermodynamic and stereochemical challenges. These indolylmethanols are readily accessible and can be used in a wide range of transformations. Despite remarkable progress in this direction, there is still potential to develop novel transformations in an asymmetric fashion by addressing the limitations associated with substrate generality, especially with unsubstituted and alkyl-substituted indolylmethanols. On the other hand, various substrates having distinct reactive sites have yet to be tested with indolylmethanols. Moreover, developments are expected to arise from expanding substituted indolylmethanol platforms, improving catalytic efficiency, and integrating with emerging paradigms such as photoredox, electrochemical catalysis, and other synergistic catalytic strategies. Overall, indolylmethanol-based asymmetric cycloaddition chemistry represents a powerful and evolving tool for the synthesis of biologically relevant chiral indole frameworks and will continue to inspire innovation in asymmetric catalysis.
Conflicts of interest
There are no conflicts to declare.
Data availability
This review article does not contain any experimental data. All the schemes were drawn using ChemDraw and have not been copied or taken from elsewhere.
Acknowledgements
Arun Patel, thank UGC-New Delhi, and Harshit thanks BITS Pilani for Research Fellowships.
References
- G. Bartoli, G. Bencivennia and R. Dalpozzob, Chem. Soc. Rev., 2010, 39, 4449 RSC.
- G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875 CrossRef CAS PubMed.
- A. J. Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489 CrossRef PubMed.
-
(a) P. A. Yakkala, V. Erram, S. A. Begum and A. Kamal, RSC Med. Chem., 2025, 16, 2483 Search PubMed;
(b) E. O. M. Orlemans, W. Verboom, M. W. Scheltinga, D. N. Reinhoudt, P. Lelieveld, H. H. Fiebig, B. R. Winterhalter, J. A. Double and M. C. Bibby, J. Med. Chem., 1989, 32, 1612 CrossRef CAS PubMed;
(c) L. S. Fernandez, M. L. Sykes, K. T. Andrews and V. M. Avery, Int. J. Antimicrob. Agents., 2010, 36, 275 CrossRef CAS PubMed;
(d) Y. C. Kong, K. H. Ng, K. H. Wat, A. Wong, I. F. Saxena, K. F. Cheng, P. P. H. But and H. T. Chang, Planta Med., 1985, 51, 304 Search PubMed;
(e) O. Talaz, I. Gülçin, S. Göksu and N. Saracoglu, Bioorg. Med. Chem., 2009, 17, 6583 CrossRef CAS PubMed.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538 RSC.
- J. M. Brown, Chem. Soc. Rev., 1993, 22, 25 RSC.
- J.-B. Chen and Y.-X. Jia, Org. Biomol. Chem., 2017, 15, 3550 RSC.
- H. F. Zheng, X. H. Liu, C. R. Xu, Y. Xia, L. L. Lin and X. M. Feng, Angew. Chem., Int. Ed., 2015, 54, 10958 CrossRef CAS PubMed.
- A. Palmieri, M. Petrini and R. R. Shaikh, Org. Biomol. Chem., 2010, 8, 1259 RSC.
- M. M. Xu, H. Q. Wang, Y. Wan, S. L. Wang and F. Shi, J. Org. Chem., 2017, 82, 10226 CrossRef CAS PubMed.
- D. R. Fandrick, C. A Hart, I. S. Okafor, M. A. Mercadante, S. Sanyal, J. T. Masters, M. Sarvestani, K. R. Fandrick, J. L. Stockdill, N. Grinberg, N. Gonnella, H. Lee and C. H. Senanayake, Org. Lett., 2016, 18, 6192 CrossRef CAS PubMed.
- M. Bandini, Org. Biomol. Chem., 2013, 11, 5206 RSC.
- X. Zhong, Y. Li and F.-S. Han, Chem.–Eur. J., 2012, 18, 9784 CrossRef CAS PubMed.
- S. J. Liu, T. Z. Li, N. Y. Wang, Q. Cheng, Y. Jiao, Y. C. Zhang and F. Shi, Org. Chem. Front., 2024, 11, 4812 RSC.
-
(a) K. Takao, R. Munakata and K. Tadano, Chem. Rev., 2005, 105, 4779 CrossRef CAS PubMed;
(b) C.-L. J. Wang, Chem. Rev., 2103, 113, 7545 Search PubMed;
(c) H. Gong, L. Zong, J. Ma and Z.-Q. Liu, Org. Chem. Front., 2023, 10, 2616 RSC.
-
(a) M. Trost and C. Jiang, Synthesis, 2006, 3, 369 CrossRef;
(b) H. Huang, Z. Liu, X. Huang, C. Zhang and G. Li, RSC Adv., 2024, 14, 4786 Search PubMed.
- L. Klier, F. Tur, P. H. Poulsen and K. A. Jørgensen, Chem. Soc. Rev., 2017, 46, 1080 RSC.
- X. Liu, H. Zheng, Y. Xia, L. Lin and X. Feng, Acc. Chem. Res., 2017, 50, 2621 CrossRef CAS PubMed.
- A. Fanourakis, P. J. Docherty, P. Chuentragool and R. J. Phipps, ACS Catal., 2020, 10, 10672 CrossRef CAS PubMed.
- L. Wang, Y. Chen and J. Xiao, Asian J. Org. Chem., 2014, 3, 1036 CrossRef CAS.
- H. Wu, Y. P. He and F. Shi, Synthesis, 2015, 47, 1990 CrossRef CAS.
- G.-J. Mei and F. Shi, J. Org. Chem., 2017, 82, 7695 CrossRef CAS PubMed.
- Y.-C. Zhang, F. Jiang and F. Shi, Acc. Chem. Res., 2020, 53, 425 CrossRef CAS PubMed.
- M. Petrini, Adv. Synth. Catal., 2020, 362, 1214 CrossRef CAS.
- Z. Honghao and F. Shi, Chin. J. Org. Chem., 2022, 42, 3351 CrossRef.
- Y. D. Wang, W. Y. Luan, C. Ma and F. Shi, Chem. Commun., 2026, 62, 1109. RSC.
- W. Tan, X. Li, Y.-X. Gong, M.-D. Ge and F. Shi, Chem. Commun., 2014, 50, 15901 RSC.
- T. Fan, H. H. Zhang, C. Li, Y. Shen and F. Shi, Adv. Synth. Catal., 2016, 358, 2017 CrossRef CAS.
- C. Lebée, A. O. Kataja, F. Blanchard and G. Masson, Chem.–Eur. J., 2015, 21, 8399 CrossRef PubMed.
- Z. Q. Zhu, Y. Shen, X. X. Sun, J. Y. Tao, J. X. Liu and F. Shi, Adv. Synth. Catal., 2016, 358, 3797 CrossRef CAS.
- K. Bera and C. Schneider, Chem.–Eur. J., 2016, 22, 7074 CrossRef CAS PubMed.
- K. Bera and C. Schneider, Org. Lett., 2016, 18, 5660 CrossRef CAS PubMed.
- M. M. Xu, H. Q. Wang, Y. Wan, S. L. Wang and F. Shi, J. Org. Chem., 2017, 82, 10226 CrossRef CAS PubMed.
- J. Mao, H. Zhang, X.-F. Ding, X. Luo and W.-P. Deng, J. Org. Chem., 2019, 84, 11186 CrossRef CAS PubMed.
- F. Göricke and C. Schneider, Org. Lett., 2020, 22, 6101 CrossRef PubMed.
- S.-J. Liu, T.-Z. Li, N.-Y. Wang, Q. Cheng, Y. Jiao, Y.-C. Zhang and F. Shi, Org. Chem. Front., 2024, 11, 4812 RSC.
- Q. Li, P. Han, T. Wang and Y.-Q. Wang, Org. Lett., 2025, 27, 13318 CrossRef CAS PubMed.
- F. Shi, R. Y. Zhu, W. Dai, C. S. Wang and S. J. Tu, Chem.–Eur. J., 2014, 20, 2597 CrossRef CAS PubMed.
- W. Dai, H. Lu, X. Li, F. Shi and S. J. Tu, Chem.–Eur. J., 2014, 20, 11382 CrossRef CAS PubMed.
- X. X. Sun, H. H. Zhang, G. H. Li, Y. Y. He and F. Shi, Chem.–Eur. J., 2016, 22, 17526 CrossRef CAS PubMed.
- X. X. Sun, C. Li, Y. Y. He, Z. Q. Zhu, G. J. Mei and F. Shi, Adv. Synth. Catal., 2017, 359, 2660 CrossRef CAS.
- C. Li, H. Lu, X.-X. Sun, G.-J. Mei and F. Shi, Org. Biomol.
Chem., 2017, 15, 4794 RSC.
- T. Z. Li, S. J. Liu, Y. W. Sun, S. Deng, W. Tan, Y. Jiao, Y. C. Zhang and F. Shi, Angew. Chem., Int. Ed., 2021, 60, 2355 CrossRef CAS PubMed.
- H. J. Loui, A. Suneja and C. Schneider, Org. Lett., 2021, 23, 2578 CrossRef CAS PubMed.
- T. Li, S. Liu, S. Wu, Q. Cheng, Q. Chen, Y. Jiao, Y. Zhang and F. Shi, Sci. China Chem., 2024, 67, 2629 CrossRef CAS.
- N. Zou, Y.-Z. Wu, Z.-W. Shang, Y.-W. Cao, L.-M. Liao, C. Wei, D.-L. Mo and W.-J. Zhou, Org. Biomol. Chem., 2024, 22, 9047 RSC.
- N. Zou, Y. Z. Wu, X. Y. Zhong, C. Wei, L. M. Liao, D. L. Mo and W. J. Zhou, Adv. Synth. Catal., 2024, 366, 4960 CrossRef CAS.
- S. Dutta, C. G. Daniliuc, C. M. Lichtenfeld and A. Studer, J. Am. Chem. Soc., 2025, 147, 4249 CrossRef CAS PubMed.
- C. Gelis, G. Levitre, J. Merad, P. Retailleau, L. Neuville and G. Masson, Angew. Chem., Int. Ed., 2018, 57, 12121 CrossRef CAS PubMed.
- S. Meng, C. Ma, S. J. Zhou, S. F. Lou, J. Xiao, Y. Jiao and F. Shi, Angew. Chem., Int. Ed., 2019, 131, 8795 CrossRef.
- W. C. Shuai, T. Z. Li, S. J. Liu, Y. C. Zhang, S. Deng, Y. Jiao and F. Shi, Chin. J. Chem., 2020, 38, 543 CrossRef.
- R. Maji, M. Leutzsch, B. Mitschke and B. List, J. Am. Chem. Soc., 2022, 144, 8460 CrossRef PubMed.
- K. Rui, S. Huang, Y. Wu, H. Shen and X. Lin, Chem. Commun., 2024, 60, 9400 Search PubMed.
- D. Liang, P. Gao, Z. Zhang, W. Xiao and J. Chen, Green Synth. Catal., 2025, 6, 282 CAS.
- W. L. Yang, T. Ni and W. P. Deng, Org. Lett., 2021, 23, 588 CrossRef CAS PubMed.
- W.-Y. Luan, T.-Y. Fang, Y.-F. Wu, X.-Y. Xiang, C. Ma and F. Shi, Org. Lett., 2025, 27, 7406 CrossRef CAS PubMed.
- T. Varlet, M. Matisic, E. Elslande, L. Neuville, V. Gandon and G. Masson, J. Am. Chem. Soc., 2021, 143, 11611 CrossRef CAS PubMed.
- J. Y. Zhang, J. Y. Chen, C. H. Gao, L. Yu, S. F. Ni, W. Tan and F. Shi, Angew. Chem., Int. Ed., 2023, 62, e202305450 CrossRef CAS PubMed.
- K. Rui, H. Shen and X. Lin, J. Org. Chem., 2024, 89, 14348 CrossRef CAS PubMed.
- J. Wang, C. Gao, C. Ma, Y. Wu, S. Ni, W. Tan and F. Shi, Angew. Chem., Int. Ed., 2024, 63, e202316454 CrossRef CAS PubMed.
- S.-J. Liu, X. Wang, J.-X. Yang, X.-S. Ao, S.-F. Ni, Y.-C. Zhang and F. Shi, Nat. Commun., 2025, 16, 6605 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b and
Indresh Kumar
b and
Indresh Kumar












































