DOI:
10.1039/D5RA09733A
(Paper)
RSC Adv., 2026,
16, 20822-20839
Deprotonation-controlled copper-free Pd-catalyzed Sonogashira coupling versus the Kumada–Tamao–Corriu reaction: a DFT investigation toward anticancer carborane alkynes
Received
16th December 2025
, Accepted 13th April 2026
First published on 21st April 2026
Abstract
Carborane derivatives have emerged as valuable hydrophobic pharmacophores in medicinal chemistry owing to their unique electronic characteristics and rigid three-dimensional architectures. Here, density functional theory (DFT) calculations are used to dissect the copper-free Pd-catalyzed Sonogashira coupling that leads to 3-quinolylethynyl carborane alkynes and to benchmark this transformation against the Pd-catalyzed Kumada–Tamao–Corriu reaction for B–C bond formation. The Sonogashira manifold is analyzed in terms of four limiting scenarios: a carbopalladation route, cationic and anionic deprotonation pathways and an ionic pathway involving base-assisted chloride substitution at palladium. The carbopalladation route, although overall exergonic, is rendered kinetically inaccessible by a prohibitively high barrier for vinylic C–H deprotonation, whereas the cationic and ionic deprotonation mechanisms display substantially lower, but still moderate, activation free energies. In contrast, the anionic deprotonation pathway—initiated by base-promoted deprotonation of the terminal alkyne and electronic stabilization of the resulting acetylide by the 3-quinolyl group—features the lowest overall Gibbs free energy barrier and therefore emerges as the dominant mechanism under copper-free conditions. Comparison with the Kumada cycle shows that, while the latter is thermodynamically feasible, the key B–C bond-forming reductive elimination step is associated with a significantly higher barrier and delivers a less stable product than the corresponding Sonogashira outcome. Taken together with available experimental data, these results indicate that copper-free Sonogashira coupling with Pd(PPh3)2Cl2 is both kinetically and thermodynamically preferred for accessing 3-quinolylethynyl carborane alkynes, highlight these motifs as promising anticancer pharmacophores and provide mechanistic guidelines for the rational design of B–C bond-forming reactions in carborane chemistry.
1. Introduction
Building larger, more complex molecules from simple fragments is one of the central goals of synthetic chemistry, and metal-catalyzed cross-coupling reactions have become the method of choice for achieving this efficiently.1–7 Such transformations rely on adaptable catalysts and reagents that can operate under mild conditions and tolerate a broad range of functional groups. In this context, palladium-catalyzed cross-coupling protocols have proved particularly powerful, enabling the preparation of targets that span from active pharmaceutical ingredients to advanced functional materials.8–16 The emergence of modern theoretical and computational tools has greatly deepened our understanding of how these Pd-catalyzed reactions proceed at the molecular level. In particular, density functional theory (DFT) now allows realistic modelling of entire catalytic cycles, providing quantitative information on energy barriers, relative stabilities of intermediates and the influence of ligands and solvents on catalytic turnover. These insights are not only useful for rationalizing experimental observations, but also for anticipating the behaviour of new catalytic systems before they are explored in the laboratory.17–23 Palladium catalyzes efficient carbon–carbon and carbon–heteroatom bond formation in cross-coupling reactions, typically offering mild reaction conditions, high yields and excellent selectivity.24–29
In many cases, only low catalyst loadings are required, which reduces both cost and waste generation and makes these methods attractive from a green chemistry perspective. Recent work has focused on developing well-defined molecular precatalysts and on extending Pd-catalyzed cross-coupling chemistry beyond classical organic substrates to more unconventional frameworks, most notably boron-rich clusters such as carboranes.30–49 A prominent polyhedral boron cluster of medicinal significance is the icosahedral closo-C2B10H12, which exists in three isomeric forms: ortho- (1,2-C2B10H12), meta- (1,7-C2B10H12) and para-carborane (1,12-C2B10H12) (Fig. 1).50–53 The combination of an electron-deficient framework with three-dimensional aromaticity makes these clusters particularly appealing building blocks for functionalization by transition-metal-catalyzed cross-coupling, allowing the attachment of organic fragments while tuning the overall electronic profile. Pd-catalyzed aryl halide cross-couplings and related metal-mediated transformations have, in this way, opened up straightforward synthetic access to a wide range of functionalized carboranes that are now being explored in both materials science and medicinal chemistry.54–59 Carboranes are especially attractive for biomedical applications because they combine several advantageous features: high boron content (ideal for boron neutron capture therapy, BNCT), exceptional thermal and chemical stability, marked hydrophobicity and finely tunable electronic properties.60–81 Their ability to form robust metallacarboranes and water-soluble derivatives further supports their use as pharmacophores in anticancer drug candidates, targeted delivery systems and molecular imaging agents. This study examines the Pd-catalyzed Sonogashira and Kumada cross-coupling reactions for forming B–C bonds, thereby enabling the incorporation of organic fragments into carborane frameworks. The Sonogashira reaction, in particular, is recognized as one of the most versatile tools for constructing C(sp)–C(sp2) bonds in organic synthesis (Scheme 1).82–87 In copper-free Sonogashira reactions (HCS protocol), several mechanistic pathways have been proposed (Fig. 2), including carbopalladation, deprotonation and ionic routes.88,89 More recently, an alternative bimetallic Pd/Pd pathway has been suggested, in which two palladium centres cooperate through an intermetallic, transmetallation-like step that effectively replaces the role of copper in the traditional mechanism and helps to account for efficient catalytic turnover under copper-free conditions. In all of these scenarios, the catalytic cycle begins with oxidative addition of the organohalide (carborane–X) to a Pd(0)L2 complex, followed by coordination of the alkyne to give a π-complex (Fig. 2). From this point, the mechanisms diverge. In the carbopalladation pathway, the alkyne inserts directly into the Pd–C bond, whereas in the deprotonation pathway a base first abstracts the proton from the terminal alkyne to generate a nucleophilic alkynyl species that subsequently binds to Pd. It is important to emphasize that, in copper-free systems, this step is not a genuine transmetalation event, because no second metal is present; rather, it is more accurately described as an inner-sphere ligand exchange followed by intramolecular acetylide transfer at palladium. Distinguishing between these different mechanistic pictures is essential for understanding reactivity, regioselectivity and kinetic control. Significant contributions to clarifying these mechanisms have been made by Jutand et al.90,91 and Mårtensson et al.92 The latter provided experimental evidence that argues against a purely carbopalladation-based mechanism and instead supports two competing deprotonation pathways—cationic and anionic—operating in the palladium-catalyzed, copper-free Sonogashira reaction (Fig. 3). Despite these advances, truly side-by-side computational comparisons of the Sonogashira and Kumada–Tamao–Corriu (KTC) mechanisms within a single, internally consistent framework are still relatively rare. The present work therefore aims to provide a systematic theoretical assessment of the relative favourability of copper-free Sonogashira versus Kumada cross-couplings, using both thermodynamic and kinetic criteria, for the synthesis of 3-quinolylethynyl carborane, a compound with potential anticancer activity. To this end, DFT calculations including explicit solvation and entropy corrections are used to construct Gibbs free energy profiles for several candidate pathways, namely carbopalladation, cationic and anionic deprotonation and an ionic route. The resulting picture not only clarifies the energetics of the competing mechanisms, but also offers deeper mechanistic insight into Pd-mediated B–C coupling in boron cluster chemistry, which can in turn guide future experimental exploration. The Kumada reaction, which couples organohalides with Grignard reagents (RMgX), represents a complementary and robust strategy for forging C–C bonds and accessing key intermediates of biologically active molecules (Scheme 2).93–97 The Schlenk equilibrium (2 RMgCl ⇌ R2Mg + MgCl2) represents a potential source of complexity in Grignard-mediated couplings. Our computational protocol employs R–MgCl reagents in 1,4-dioxane – a strong donor solvent that disrupts Mg–X–Mg bridges through competitive coordination. This approach is supported by experimental precedent: Sinha et al. report 91% isolated yield and >95% Br-selectivity using 4-cyanophenyl magnesium chloride (a related Grignard reagent) under similar conditions.98 By comparing its reaction energetics and transition states with those of the copper-free Sonogashira coupling, valuable information is obtained on how nucleophile strength, potential metal–metal cooperation and solvent effects together dictate the overall catalytic efficiency of Pd-mediated systems.
 |
| | Fig. 1 The three isomeric closo-carboranes with formula C2B10H12, depicted in their icosahedral structures. | |
 |
| | Scheme 1 Overall representation of the Pd-catalyzed Sonogashira reaction. | |
 |
| | Fig. 2 Proposed reaction pathways for the copper-free Sonogashira reaction: carbopalladation (left) and deprotonation-based mechanisms (right). | |
 |
| | Fig. 3 Copper-free Sonogashira manifold relying on direct alkyne coordination and proceeding via cationic, anionic and ionic pathways. | |
 |
| | Scheme 2 General framework for the Pd-catalyzed Kumada–Tamao–Corriu (Kumada) reaction. | |
2. Computational details
All quantum mechanical calculations in this research were conducted at the DFT level using the dispersion-corrected B3LYP (B3LYP-D3) functional,99 as implemented in the Gaussian 09 (Revision D) software package,100 with 1,4-dioxane as the solvent. Implicit solvation models such as PCM, COSMO and SMD were used to describe the solvent effects of 1,4-dioxane, which is a polar aprotic medium with suitable polarity, chemical stability and solubility for organic substrates. For geometry optimizations, the 6–31G*(d,p) basis set101 was employed for H, B, C, N, P and Cl atoms, whereas the LANL2DZ effective core potential102,103 was used for Pd and Mg atoms. This mixed basis set is referred to as BSL. The chosen computational level has been extensively applied in theoretical studies of related Pd-catalyzed cross-coupling reactions and affords reliable structural and energetic data.104–108 Prior to selecting the final computational protocol, several alternative functionals were tested in order to evaluate their suitability for the present catalytic system. In particular, CAM-B3LYP and M06, which are often recommended for systems with significant charge-transfer character and dispersion interactions, were explored during the initial stages of this work. However, for the large catalytic models considered here (exceeding 110 atoms and containing flexible Pd–phosphine coordination environments), these functionals exhibited significant convergence difficulties. In particular:
• CAM-B3LYP failed to converge for several key transition states associated with the Pd–phosphine catalytic manifold.
• M06 calculations encountered repeated SCF and geometry convergence failures for multiple stationary points along the catalytic pathway.
To assess the potential influence of long-range exchange effects, single-point energy calculations were performed using the CAM-B3LYP functional on the B3LYP-D3 optimized geometries of representative intermediates and transition states along the Kumada pathway. Although the absolute energies differ from those obtained at the B3LYP-D3 level, the relative ordering of intermediates and transition states remains unchanged. In particular, the same transition state is identified as the highest-energy point along the pathway. These results indicate that the proposed mechanistic conclusions are qualitatively robust with respect to the choice of exchange–correlation functional. A detailed comparison of the two computational approaches is provided in the SI (Table S2).
All reactants, intermediates and products were fully optimized at the B3LYP-D3/BSL level in 1,4-dioxane without imposing symmetry constraints, and no spurious interactions involving the hydrogen atoms of the triphenylphosphine (PPh3) ligands were detected during optimization. Harmonic vibrational frequency calculations at the same level were used to confirm that minima exhibit no imaginary frequencies, whereas each transition state has a single imaginary frequency. To further validate the transition states, intrinsic reaction coordinate (IRC) calculations were carried out at the B3LYP-D3/BSL level, ensuring that each saddle point connects the appropriate reactant and product minima along the reaction pathway (Fig. S7).109
Single-point electronic energies were subsequently refined at the B3LYP-D3/def2-TZVPP level on the B3LYP-D3/BSL-optimized geometries. Gibbs free energies in solution were obtained by combining these B3LYP-D3/def2-TZVPP electronic energies with thermal and entropic corrections evaluated at the B3LYP-D3/BSL level. To gain insight into electronic structure and bonding changes along the reaction coordinates, natural bond orbital (NBO) analyses were performed at the B3LYP-D3/def2-TZVPP//B3LYP-D3/BSL level, and Wiberg bond indices (WBIs) were extracted as quantitative descriptors of bond formation and cleavage in the key transition states.110
3. Results and discussion
3.1. Copper-free Pd-catalyzed B–C bond formation in carborane-based Sonogashira coupling
The Sonogashira reaction mechanism consists of four elementary steps: oxidative addition (OA), cis–trans isomerization, transmetalation (TM) and reductive elimination (RE) (Fig. 2). In this study, theoretical investigations were conducted on carborane substrates utilizing the Pd(PPh3)2Cl2 complex as the catalyst for the Sonogashira cross-coupling reaction. The geometry of the Pd(PPh3)2Cl2 complex is approximately square planar around the palladium center, a characteristic feature commonly observed in many palladium(II) complexes.111–115 The Sonogashira reaction mechanism was further elucidated using 3-ethynylquinoline and 2-Cl-p-carborane as the coupling partners, with pyrrolidine serving as the base and 1,4-dioxane as the solvent (Scheme 3).116 The analysis of the oxidative addition step is crucial for understanding the initial interactions between the reactants and the palladium catalyst. As depicted in Fig. 4, the first step of the reaction involves the oxidative addition of 2-Cl-p-carborane to the palladium complex [Pd(PPh3)2]. The starting reagents, [Pd(PPh3)2] and 2-Cl-p-carborane, are assigned a relative energy of 0.0 kcal mol−1, providing a reference point for evaluating the energy changes along the reaction pathway (The optimized structures of Int1, Int2, TS1 and TS2 are shown in Fig. 5). Upon interaction with the palladium center, an intermediate Int1 is formed, stabilized at −81.5 kcal mol−1. This substantial drop in energy indicates that formation of Int1 is highly favorable, reflecting strong interactions between the palladium center and the carborane moiety. The subsequent step involves oxidative addition of the B(carborane)–Cl bond of 2-Cl-p-carborane to the palladium center. During this process, the B(carborane)–Cl bond is cleaved and new Pd–B(carborane) and Pd–Cl bonds are formed, resulting in an increase in the oxidation state of palladium from 0 to +2. The transition state for this step (TS1) has a relative energy of −0.13 kcal mol−1, indicating that it is slightly more stable than the separated reactants but still represents a high-energy configuration along the reaction coordinate. Following TS1, another intermediate Int2 is generated with a relative energy of −48.4 kcal mol−1, further stabilizing the system as it progresses toward product formation. Importantly, in this transition state TS1, the Pd–B(carborane), Pd–Cl and B(carborane)–Cl bond lengths are 2.0, 3.1 and 3.0 Å, respectively. These structural parameters are consistent with partial cleavage of the B–Cl bond and concurrent formation of the Pd–B and Pd–Cl bonds. It is worth noting that the oxidative addition step is a common feature in all Sonogashira reaction pathways considered in this work. The next step, a cis–trans isomerization (Fig. 4), results in a product with a trans configuration. The isomerization between square-planar cis and trans isomers of metal complexes, particularly in the context of oxidative addition and transmetalation processes, is a well-recognized feature of cross-coupling catalysis. The cis isomer is often formed as an intermediate during oxidative addition, yet it is rarely isolated. This suggests that, while it may be a transient species, the trans isomer plays a more pivotal role in subsequent catalytic steps, such as transmetalation. The conversion from the square-planar cis isomer to the trans isomer occurs via a four-coordinate mechanism. This pathway enables a direct transformation without altering the coordination number of the metal, which is crucial for maintaining the stability of the complex throughout the reaction117 (Fig. 6). This step proceeds through transition state TS2 (−42.1 kcal mol−1), which requires an activation barrier of 6.23 kcal mol−1 (Fig. 4). As shown in Fig. S1, this contrasts with intermediate Int2 (cis-[Pd(p-carborane)(Cl)(PPh3)2]), which adopts a cis configuration. Int2 exhibits Pd–B(carborane), Pd–Cl, Pd–P(1) and Pd–P(2) bond lengths of 2.1, 2.4, 2.7 and 2.3 Å, respectively, along with P(1)–Pd–P(2) and Cl–Pd–B bond angles of 103.19° and 91.42°, respectively.
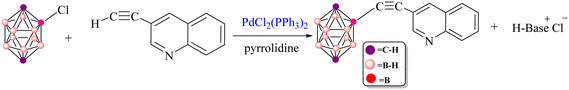 |
| | Scheme 3 Copper-free Pd-catalyzed Sonogashira coupling between 2-Cl-p-carborane and 3-ethynylquinoline. | |
 |
| | Fig. 4 Gibbs free-energy profile for the Pd-catalyzed copper-free Sonogashira reaction, including oxidative addition and cis–trans isomerization, computed at the B3LYP-D3/def2-TZVPP//B3LYP-D3/BSL level in 1,4-dioxane. | |
In comparison, TS2 shows Pd–B(carborane), Pd–Cl, Pd–P(1) and Pd–P(2) bond lengths of 2.0, 2.6, 2.4 and 2.4 Å, respectively, and P(1)–Pd–P(2) and Cl–Pd–B bond angles of 146.80° and 132.23°, respectively. The optimized TS2 thus shows moderate changes in bond lengths and angles compared to Int2, reflecting the structural rearrangements associated with cis–trans isomerization. This process ultimately leads to the formation of the trans isomer Int3 (trans-[Pd(p-carborane)(Cl)(PPh3)2]), which has an energy of −46.7 kcal mol−1. Int3 exhibits Pd–B(carborane), Pd–Cl, Pd–P(1) and Pd–P(2) bond lengths of 2.1, 2.5, 2.4 and 2.4 Å, respectively, along with P(1)–Pd–P(2) and Cl–Pd–B bond angles of 154.79° and 153.09°, respectively. The trans isomer is therefore more stable than both the cis isomer and TS2 (−46.7 vs. −48.4 and −42.1 kcal mol−1), suggesting that it is thermodynamically favored as a resting state for subsequent reactions. Notably, such isomerization is a consistent feature in all Sonogashira reaction pathways. As previously discussed, oxidative addition followed by cis-to-trans isomerization constitutes a common initial sequence in all proposed pathways for the Sonogashira reaction in the absence of copper. Therefore, we proceed to analyze the subsequent steps of the reaction and compare the proposed pathways for the Sonogashira reaction, starting from complex trans-[Pd(p-carborane)(Cl)(PPh3)2].
 |
| | Fig. 5 Optimized geometries of key intermediates (Int2, Int3) and transition states along the copper-free Sonogashira pathway to 3-quinolylethynyl carborane, obtained at the B3LYP-D3/BSL level in 1,4-dioxane. | |
 |
| | Fig. 6 Cis–trans isomerization of [Pd(p-carborane)(Cl)(PPh3)2] at palladium. | |
3.2. Carbopalladation-based copper-free Sonogashira pathway to 3-quinolylethynyl carborane
Fig. 7 summarizes the carbopalladation-type pathway explored for the copper-free Sonogashira reaction of 3-ethynylquinoline, catalyzed by the Pd(PPh3)2Cl2 complex. This mechanism starts with substitution of one phosphine ligand by 3-ethynylquinoline to form intermediate Int4. This associative substitution is endergonic, with a relative free energy of −43.6 kcal mol−1 (referenced to the separated reactants), a trend similar to that observed for the corresponding deprotonation-based pathways. The transition state for this step, TS3, lies at 39.1 kcal mol−1 and is characterized by Pd–P(1), Pd–C(1) and Pd–C(2) distances of 5.6, 2.2 and 2.5 Å, respectively. Following formation of the Pd–alkyne complex, the carbopalladation step proceeds via transition state TS1-C, which also has a relative energy of −43.6 kcal mol−1. In TS1-C, the Pd–B(carborane) bond length is 2.2 Å, whereas the Pd–C(2) and B–C(1) distances are both 2.1 Å, consistent with simultaneous formation of Pd–C and B–C bonds along the alkyne. The reaction then evolves to a highly stabilized intermediate, RE-Int1-C, located at −63.6 kcal mol−1 relative to the reactants, with an overall barrier of only 8.5 kcal mol−1 for this transformation. Subsequent coordination of a phosphine ligand occurs through TS2-C (−25.6 kcal mol−1), featuring Pd–B and Pd–P(1) distances of 3.7 and 2.4 Å, respectively. In the final step of this pathway, the alkenyl fragment in RE-Int1-C undergoes deprotonation by an external base via transition state RE-TS1-C, yielding the coupled product 3-quinolylethynyl carborane and regenerating the active catalyst. However, this deprotonation step carries a very high activation barrier of 52.87 kcal mol−1, reflecting both the substantial stabilization of RE-Int1-C and the intrinsic difficulty of deprotonating a vinylic C–H bond. Overall, although the carbopalladation pathway is globally exergonic (ΔG = −80.6 kcal mol−1), the large barrier associated with the final deprotonation renders this mechanism kinetically inaccessible under typical copper-free Sonogashira conditions. This theoretical conclusion is consistent with the experimental observations of Mårtensson et al.,92 who reported that a complex analogous to RE-Int1-C failed to deliver the cross-coupled product under standard reaction conditions.
 |
| | Fig. 7 Gibbs free-energy diagram for the carbopalladation-based copper-free Sonogashira pathway, catalyzed by Pd with pyrrolidine as base, computed at the B3LYP-D3/def2-TZVPP//B3LYP-D3/BSL level in 1,4-dioxane. | |
3.3. Deprotonation-driven copper-free Sonogashira manifold under Pd catalysis
The copper-free Sonogashira reaction was also examined through a mechanism in which C–H deprotonation plays the key activating role. As outlined in the introduction, this manifold involves ligand exchange at the palladium centre and can proceed through two distinct pathways, cationic and anionic, depending on the sequence of ligand substitution and deprotonation events (Fig. 3).92,118,119 The order of these steps determines which deprotonation pathway is energetically preferred. In the cationic pathway, ligand substitution at complex 3 first generates a cationic palladium species, cis-[Pd(carborane)(alkyne)(L)2]+. The coordinated alkyne is then deprotonated by an external base, and the resulting acetylide complex undergoes reductive elimination to furnish the cross-coupled product. In contrast, the anionic pathway begins with deprotonation of the free alkyne, giving an anionic complex cis-[Pd(carborane)(acetylide)(X)(L)]− (Fig. 3). After this initial deprotonation, ligand substitution at palladium takes place, followed by reductive elimination to deliver the desired product.
3.3.1. Cationic deprotonation sequence in the Pd-catalyzed copper-free Sonogashira manifold. The cationic pathway of the copper-free Sonogashira reaction between 3-ethynylquinoline and 2-Cl-p-carborane proceeds through a characteristic sequence of ligand substitution and deprotonation steps. The computed Gibbs free energy profile (Fig. 8) reveals the key intermediates and transition states that define this mechanism. The reaction begins with formation of a cationic palladium complex, followed by substitution of the chloride ligand by a phosphine ligand to give the ion-paired intermediate Int1-DC at −13.3 kcal mol−1. The associated transition state TS1-DC has a barrier of 36.9 kcal mol−1 and features Pd–P(1) and Pd–Cl distances of 2.6 and 3.7 Å, respectively. Subsequent deprotonation of the coordinated alkyne occurs via TS2-DC, located at −1.9 kcal mol−1. This transition state is characterized by Pd–Cl, C(1)–H, N–H and Cl–N distances of 4.1, 2.8, 2.2 and 3.5 Å, respectively, and leads to intermediate RE-Int1, in which the two organic fragments adopt a cis arrangement around palladium. The final step involves reductive elimination from RE-Int1, with a Gibbs energy of −38.4 kcal mol−1. This process proceeds through TS-RE1 (−31.7 kcal mol−1), where the Pd–B, Pd–C(1) and B–C(1) distances are 2.2, 1.9 and 2.0 Å, respectively, and furnishes the cross-coupled product together with regeneration of the palladium catalyst. The overall Gibbs free energy change for the cationic pathway is −80.6 kcal mol−1. Fig. 5 depicts the optimized geometries of TS1-DC, TS2-DC and TS-RE1, highlighting the structural features that govern the reaction dynamics. Comparison of this energy profile with that of the anionic pathway (Section 3.3.2) underscores the differences in deprotonation sequences and ligand-substitution events, thereby refining the mechanistic picture of copper-free Sonogashira reactions.
 |
| | Fig. 8 Gibbs free-energy diagram for the cationic copper-free Sonogashira pathway, catalyzed by Pd with pyrrolidine as base, computed at the B3LYP-D3/def2-TZVPP//B3LYP-D3/BSL level in 1,4-dioxane. | |
3.3.2. Anionic deprotonation sequence in the Pd-catalyzed copper-free Sonogashira manifold. The anionic pathway of the Sonogashira reaction features a deprotonation sequence that differs fundamentally from the cationic mechanism. In this case, deprotonation of the alkyne in complex 3 by an external base is the initial step, setting off a series of transformations that ultimately yield the cross-coupled product. As depicted in Fig. 9, the alkyne is deprotonated via transition state TS1-DA, located at −26.6 kcal mol−1. This step affords the ion-pair intermediate Int1-DA at −50.6 kcal mol−1, comprising an anionic palladium acetylide complex and the protonated base. In TS1-DA, the Pd–C(1), C(2)–H and H–N distances are 1.9, 1.1 and 1.8 Å, respectively. Following formation of Int1-DA, a chloride-for-phosphine substitution takes place, proceeding through TS2-DA at −29.9 kcal mol−1 and leading to intermediate RE-Int1 with a Gibbs energy of −39.7 kcal mol−1. TS2-DA is characterized by Pd–P(1), Pd–Cl and Cl–H distances of 2.6, 6.3 and 1.9 Å, respectively. In the final step, reductive elimination from RE-Int1 generates the cross-coupled product and regenerates the palladium catalyst; this step is thermodynamically highly favorable, with an overall Gibbs free energy of −80.6 kcal mol−1. The optimized geometries of TS1-DA and TS2-DA are shown in Fig. 5. Taken together, the anionic pathway underscores how the sequence of alkyne deprotonation and ligand substitution modulates the energetics and efficiency of the copper-free Sonogashira reaction. The maximum activation free energy along the anionic Sonogashira pathway corresponds to the TS2-DA transition state, which lies 20.7 kcal mol−1 above the reference intermediate Int1-DA. The free energies of these species are −29.9 kcal mol−1 for TS2-DA and −50.6 kcal mol−1 for Int1-DA, giving ΔG_max = 20.7 kcal mol−1. This value therefore represents the highest barrier along the productive anionic catalytic pathway relative to the resting-state intermediate Int1-DA.
 |
| | Fig. 9 Gibbs free-energy diagram for the anionic copper-free Sonogashira pathway, catalyzed by Pd with pyrrolidine as base, computed at the B3LYP-D3/def2-TZVPP//B3LYP-D3/BSL level in 1,4-dioxane. | |
3.3.3. Comparison of the cationic and anionic deprotonation pathways. Computational results show that chloride–phosphine exchange at palladium is associated with sizeable energy barriers in both the cationic and anionic manifolds. In the cationic sequence, ligand substitution precedes alkyne deprotonation: in complex 3, this step proceeds via TS1-DC, and the corresponding Gibbs free energy barrier is within the range expected for a kinetically viable process under typical Sonogashira conditions. In contrast, in the anionic sequence, deprotonation of the alkyne occurs first, and the chloride-for-phosphine substitution takes place later, from Int1-DA via TS2-DA. The calculated overall Gibbs free energy barriers indicate that both pathways are, in principle, kinetically accessible. However, the free-energy profiles point to a clear preference for product formation along the anionic pathway, owing to the additional stabilization of the anionic intermediate by the 3-quinolyl–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C fragment. This conjugated group effectively delocalizes negative charge, lowering the energy of the anionic species and thereby rendering the anionic deprotonation sequence more favorable than the cationic alternative.
C fragment. This conjugated group effectively delocalizes negative charge, lowering the energy of the anionic species and thereby rendering the anionic deprotonation sequence more favorable than the cationic alternative.
3.4. Ionic mechanism as an alternative pathway
The transmetalation step in Pd-catalyzed cross-couplings is a key elementary process that involves ligand exchange at the metal centre. In many cases, transmetalation proceeds through an ionic pathway in which an external ligand or base replaces a halide (such as chloride) at palladium, thereby facilitating cross-coupling reactivity.120–122 The nature of the external ligand can markedly influence both the kinetics and thermodynamics of this step, often enhancing overall catalytic efficiency. In the present system, transmetalation is described by an ionic mechanism in which ligand substitution occurs directly at the palladium centre. Unlike the cationic and anionic deprotonation pathways, where chloride dissociation occurs only after alkyne coordination, the ionic route features base-assisted chloride substitution at an early stage (Fig. 3). The resulting changes in the coordination environment around palladium play an important role in controlling reaction efficiency and selectivity. As shown in Fig. 10, the reaction of the cationic palladium complex Int1-I proceeds via TS1-I to give an intermediate with a Gibbs free energy of −31.0 kcal mol−1. TS1-I is characterized by Pd–N and Pd–Cl distances of 2.3 and 4.3 Å, respectively, indicating substantial reorganization at the metal centre. The corresponding activation barrier for this ionic transmetalation step is 22.1 kcal mol−1, notably lower than the barrier associated with alkyne coordination to Int3 via TS3, which suggests that transmetalation is relatively facile compared with subsequent steps in the mechanism. Computational analysis further shows that 3-ethynylquinoline can displace one of the phosphine ligands in Int1-I without encountering a significant energy barrier, affording an isoenergetic species denoted Int2-RE2. This essentially barrierless ligand exchange simplifies the catalytic manifold by removing additional energetic penalties for phosphine dissociation/association. From Int2-RE2, the system follows a common reductive-elimination pathway via TS1-RE2, which has an activation barrier of 13.2 kcal mol−1. TS1-RE2 exhibits B–C(1), Pd–B and Pd–C(1) distances of 2.0, 2.2 and 1.96 Å, respectively, and a C(1)–Pd–B bond angle of 67.1°. (The optimized structures of TS1-I and TS1-RE2 are depicted in Fig. 5). In summary, and as illustrated in Fig. 11, the carbopalladation pathway is disfavoured under standard conditions due to its prohibitively high activation barrier, whereas the cationic, anionic and ionic deprotonation manifolds all display substantially lower Gibbs free-energy barriers and are therefore mechanistically viable. The computed profiles indicate that these three pathways can, in principle, compete, and that subtle changes in solvent, ligands, substrates or base may shift the mechanistic preference toward one route or another. In the present case, the anionic pathway emerges as both thermodynamically and kinetically most favourable.
 |
| | Fig. 10 Gibbs free-energy diagram for the ionic copper-free Sonogashira pathway, catalyzed by Pd with pyrrolidine as base, computed at the B3LYP-D3/def2-TZVPP//B3LYP-D3/BSL level in 1,4-dioxane. | |
3.5. Pd-catalyzed Kumada B–C bond-forming reaction
The Kumada cross-coupling reaction proceeds through three fundamental steps: oxidative addition, transmetalation and reductive elimination (Fig. 12). In the present study, the Pd(PPh3)2Cl2 complex is employed as the catalyst for B–C bond formation in carborane substrates, and the mechanism is analysed using 3-quinolylethynyl magnesium chloride and 2-Cl-p-carborane as coupling partners in 1,4-dioxane (Scheme 4). This section assesses the computational feasibility of the Kumada versus Sonogashira cross-coupling pathways for the synthesis of product 5. As shown in Fig. 13, the oxidative addition step is common to both mechanisms, after which the pathways diverge. The computed Kumada manifold highlights an overall efficient route to 3-quinolylethynyl carborane. The sequence begins with formation of intermediate Int2 from the separated reactants via a three-centre transition state TS1 at −0.13 kcal mol−1, affording Int2 at −51.6 kcal mol−1. We performed additional calculations to evaluate the effect of magnesium speciation on the key intermediate Int3-k. In particular, Int3 was modeled using both monomeric MgCl2 and a representative dimeric MgCl2 species, which serves as a simple model for aggregated magnesium structures commonly present in Grignard equilibria. The results show (Table S1) that the monomeric Int3 structure is 9.34 kcal mol−1 more stable than the corresponding dimeric model relative to Int2. Importantly, although the absolute stabilization differs slightly depending on the aggregation state, the qualitative energetic picture of the Kumada pathway remains unchanged. This transformation involves migration of MgCl2 from Int3-K to the borate moiety, promoting cleavage of the Pd–Cl bond and formation of a Pd–C bond; at this stage, intermediate Int3-K is located at −64.4 kcal mol−1, with the aryl fragment coordinated to both palladium and magnesium. In the final phase of the catalytic cycle, B–C bond formation occurs via reductive elimination from Int5, which lies at −72.2 kcal mol−1. This step proceeds through transition state RE1-TS at −28.0 kcal mol−1 and requires an activation barrier of approximately 30.6 kcal mol−1 relative to Int4. The value of 30.6 kcal mol−1 corresponds to the intrinsic reductive-elimination (RE) barrier in the Kumada pathway, defined as the free-energy difference between the post-transmetalation intermediate (Int4-K) and the corresponding transition state (TS-RE1). The catalytic cycle is completed by release of the cross-coupled product and regeneration of Pd(PPh3)2 at −68.8 kcal mol−1. Comparison of this profile with the copper-free Sonogashira pathways (Sections 3.2–3.4) provides a quantitative basis for evaluating the relative kinetic and thermodynamic viability of Kumada versus Sonogashira B–C bond-forming strategies in carborane chemistry. Overall, the key B–C bond-forming reductive elimination thus proceeds with a substantially higher barrier in the Kumada manifold (30.6 kcal mol−1 from Int4) than in the anionic copper-free Sonogashira pathway, and delivers a less stable product (ΔG = −68.8 vs. −80.7 kcal mol−1), underscoring the kinetic and thermodynamic preference for the Sonogashira route.
 |
| | Fig. 11 Gibbs free‑energy profiles for all proposed mechanisms of the copper‑free Sonogashira reaction, computed at the B3LYP‑D3/def2‑TZVPP//B3LYP‑D3/BSL level in 1,4‑dioxane. | |
 |
| | Scheme 4 Pd-catalyzed Kumada coupling between 2-Cl-p-carborane and 3-quinolylethynyl magnesium chloride. | |
 |
| | Fig. 12 Pd‑catalyzed Kumada–Tamao–Corriu (Kumada) cross‑coupling of a representative 2‑Cl‑p‑carborane. OA = oxidative addition, TM = transmetalation, RE = reductive elimination. | |
 |
| | Fig. 13 Gibbs free‑energy diagram for the Pd‑catalyzed Kumada mechanism, computed at the B3LYP‑D3/def2‑TZVPP//B3LYP‑D3/BSL level in 1,4‑dioxane. | |
3.6. Which route is more favorable for 3-quinolylethynyl carborane: Sonogashira or Kumada?
In 2004, Irina P. Beletskaya and co-workers reported the synthesis of 3-quinolylethynyl carborane via both Sonogashira and Kumada cross-coupling reactions (Scheme 5).116 They employed 3-quinolyl–CC–X (X = H or MgCl) as the coupling partner. Under standard Sonogashira conditions using Pd(PPh3)2Cl2 as the catalyst, pyrrolidine as the base and 1,4-dioxane as the solvent, the 3-quinolylalkyne product was obtained in 80% yield, underscoring the synthetic efficiency of this route. Theoretical calculations carried out in this work further support the superiority of the Sonogashira pathway over the Kumada alternative for constructing the B–C bond between 3-quinolyl–CC–X and 2-Cl-p-carborane. In particular, the activation barrier for the key reductive-elimination transition state TS-RE1 in the Kumada mechanism is significantly higher (22.7 kcal mol−1) than that of the corresponding anionic Sonogashira pathway, indicating a kinetic preference for the latter. In particular, the relative free-energy difference between the key reductive-elimination transition state (TS-RE1) of the Kumada mechanism and the corresponding transition state in the anionic Sonogashira pathway is calculated to be 22.7 kcal mol−1. This value reflects the energy separation between the two competing pathways when referenced to a common starting point, as illustrated in the comparative energy profiles. Accordingly, it serves as a measure for cross-pathway comparison and should not be interpreted as an intrinsic activation barrier for any individual elementary step. The lower transition-state energy associated with the anionic pathway is consistent with its preferred reactivity under the studied conditions. In addition, comparison of product stabilities shows that the Sonogashira product (−80.7 kcal mol−1) is more stable than the Kumada product (−68.8 kcal mol−1), demonstrating that thermodynamic factors also favour the Sonogashira route. Taken together, these kinetic and thermodynamic data indicate that, when Pd(PPh3)2 is used as the catalyst, the anionic copper-free Sonogashira pathway is the most advantageous strategy for synthesizing 3-quinolylethynyl carborane. The combination of experimental observations and DFT results thus highlights the efficiency and reliability of this synthetic approach. Fig. S1–S6 and S8 provide a comprehensive overview of the optimized structures, bond lengths (Å) and Wiberg bond indices (WBIs) for the transition states involved in the various Sonogashira and Kumada manifolds considered. Specifically, Fig. S1 summarizes the common structural motifs for the standard Sonogashira reaction, Fig. S2 addresses the copper-free carbopalladation variant, Fig. S3 details the cationic copper-free Sonogashira pathway, Fig. S4 and S5 depict key intermediates for the anionic and ionic copper-free mechanisms, and Fig. S6 presents the optimized structures for the Kumada cross-coupling reaction. All calculations were performed at the B3LYP-D3/BSL level with 1,4-dioxane as the solvent.
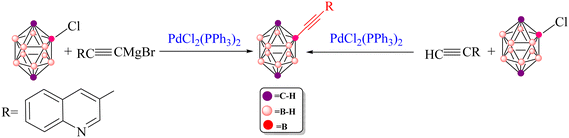 |
| | Scheme 5 Preparation of 3-quinolylethynyl carborane via copper-free Sonogashira and Kumada cross coupling reactions. | |
4. Conclusion
The growing body of work on carborane chemistry underscores the importance of these boron-rich clusters in both fundamental studies and applied research, particularly in medicinal chemistry. Their rigid three-dimensional architectures and distinctive reactivity profiles make them attractive building blocks for the design of new therapeutic agents and functional materials. Metal-catalyzed cross-coupling reactions are central to the functionalization of carboranes, and this study has focused on Pd-catalyzed B–C bond formation through copper-free Sonogashira and Kumada manifolds. DFT calculations at the B3LYP-D3 level were used to analyse the copper-free Sonogashira coupling between 3-ethynylquinoline and 2-Cl-p-carborane via three mechanistic scenarios: carbopalladation, deprotonation (cationic and anionic) and an ionic pathway. The carbopalladation route exhibits a prohibitively high activation barrier and can therefore be ruled out under standard conditions. By contrast, the cationic and anionic deprotonation pathways, as well as the ionic variant in which the base initially replaces the halide at palladium, all display substantially lower Gibbs energy barriers and are mechanistically viable. Among these, the anionic pathway emerges as both thermodynamically and kinetically most favourable for the present system. The comparison between copper-free Sonogashira and Pd-catalyzed Kumada cross-coupling is particularly informative in the context of 3-quinolylethynyl carborane synthesis. The calculations show that the key reductive-elimination barrier in the Kumada manifold is higher and that the resulting product is less stable than in the corresponding anionic Sonogashira pathway, indicating that both kinetic and thermodynamic factors favour the latter. In line with the experimental study by Beletskaya and co-workers, the combined computational and experimental evidence supports copper-free Sonogashira coupling with Pd(PPh3)2 as the most advantageous strategy for accessing 3-quinolylethynyl carborane under the conditions examined. Overall, this work illustrates how detailed mechanistic analysis can guide the selection and optimization of cross-coupling conditions for carborane-based targets. The results highlight that relatively subtle changes in base, ligands, substrates or solvent can shift the balance between competing carbopalladation, deprotonation and ionic pathways, and that, for the system studied here, the anionic copper-free Sonogashira route provides the most favourable B–C bond-forming platform for 3-quinolylethynyl carborane.
Conflicts of interest
The authors have declared no conflict of interest.
Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5ra09733a.
Acknowledgements
The authors acknowledge Iran National Science Foundation (INSF) for financial support of this work (Grant number: 4021695).
References
- Z. Ahmadvand and M. Bayat, Competition between the Hiyama and Suzuki–Miyaura Pd-catalyzed cross-coupling reaction mechanisms for the formation of some regioselective derivatives of quinoxaline and benzofuran: which reaction mechanism is more favorable?, J. Mol. Liq., 2021, 325, 115174 CrossRef CAS.
- A. Biffis, P. Centomo, A. Del Zotto and M. Zecca, Pd metal catalysts for cross-couplings and related reactions in the 21st century: a critical review, Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS.
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, The merger of transition metal and photocatalysis, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- P. Ruiz-Castillo and S. L. Buchwald, Applications of palladium-catalyzed C–N cross-coupling reactions, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Palladium-catalyzed cross-coupling: a historical contextual perspective to the 2010 Nobel Prize, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- I. P. Beletskaya and V. P. Ananikov, Transition-metal-catalyzed C–S, C–Se and C–Te bond formation via cross-coupling and atom-economic addition reactions, Chem. Rev., 2011, 111, 1596–1636 CrossRef CAS.
- J. F. Hartwig, Evolution of a fourth-generation catalyst for the amination and thioetherification of aryl halides, Acc. Chem. Res., 2008, 41, 1534–1544 CrossRef CAS.
- L. Li, C. Y. Wang, R. Huang and M. R. Biscoe, Stereoretentive Pd-catalysed Stille cross-coupling reactions of secondary alkyl azastannatranes and aryl halides, Nat. Chem., 2013, 5, 607–612 CrossRef CAS.
- G. J. Perry, J. M. Quibell, A. Panigrahi and I. Larrosa, Transition-metal-free decarboxylative iodination: new routes for decarboxylative oxidative cross-couplings, J. Am. Chem. Soc., 2017, 139, 11527–11536 CrossRef CAS PubMed.
- G. Tan, L. Zhu, X. Liao, Y. Lan and J. You, Rhodium/copper cocatalyzed highly trans-selective 1,2-diheteroarylation of alkynes with azoles via C–H addition/oxidative cross-coupling: a combined experimental and theoretical study, J. Am. Chem. Soc., 2017, 139, 15724–15737 CrossRef CAS PubMed.
- Y. Xia and J. Wang, Transition-metal-catalyzed cross-coupling with ketones or aldehydes via N-tosylhydrazones, ACS Catal., 2020, 10, 10592–10605 CrossRef.
- Q. Dai, W. Li, Z. Li and J. Zhang, P-chiral phosphines enabled by palladium/Xiao-Phos-catalyzed asymmetric P–C cross-coupling of secondary phosphine oxides and aryl bromides, J. Am. Chem. Soc., 2019, 141, 20556–20564 CrossRef CAS.
- C. H. Lim, M. Kudisch, B. Liu and G. M. Miyake, C–N cross-coupling via photoexcitation of nickel–amine complexes, J. Am. Chem. Soc., 2018, 140, 7667–7673 CrossRef CAS PubMed.
- G. Li, Y. Nieves-Quinones, H. Zhang, Q. Liang, S. Su, Q. Liu, M. C. Kozlowski and T. Jia, Transition-metal-free formal cross-coupling of aryl methyl sulfoxides and alcohols via nucleophilic activation of C–S bonds, Nat. Commun., 2020, 11, 2890 CrossRef CAS PubMed.
- M. Cortes-Clerget, N. Akporji, J. Zhou, F. Gao, P. Guo, M. Parmentier, F. Gallou and J. Y. Berthon, Bridging the gap between transition metal- and biocatalysis via aqueous micellar catalysis, Nat. Commun., 2019, 10, 2169 CrossRef PubMed.
- G. Li and M. Szostak, Highly selective transition-metal-free transamidation of amides and amidation of esters at room temperature, Nat. Commun., 2018, 9, 4165 CrossRef.
- S. Kumar, J. Jyoti, D. Gupta, G. Singh and A. Kumar, A decade of exploration of transition-metal-catalyzed cross-coupling reactions: an overview, SynOpen, 2023, 7, 580–614 CrossRef CAS.
- J. Li, Q. Ren, X. Cheng, K. Karaghiosoff and P. Knochel, Chromium(II)-catalyzed diastereoselective and chemoselective C(sp2)–C(sp3) cross-couplings using organomagnesium reagents, J. Am. Chem. Soc., 2019, 141, 18127–18135 CrossRef CAS.
- L. C. Campeau and N. Hazari, Cross-coupling and related reactions: connecting past success to the development of new reactions for the future, Organometallics, 2018, 38, 3–35 CrossRef.
- M. Garcia-Melchor, A. A. C. Braga, A. Lledós, G. Ujaque and F. Maseras, Computational perspective on Pd-catalyzed C–C cross-coupling reaction mechanisms, Acc. Chem. Res., 2013, 46, 2626–2634 CrossRef CAS PubMed.
- G. Nagendrappa and Y. C. Sunil Kumar, The 2010 chemistry Nobel Prize: Pd(0)-catalyzed organic synthesis, Resonance, 2011, 16, 152–164 CrossRef CAS.
- T. Ljungdahl, T. Bennur, A. Dallas, H. Emtenäs and J. Mårtensson, Two competing mechanisms for the Cu-free Sonogashira cross-coupling reaction, Organometallics, 2008, 27, 2490–2498 CrossRef CAS.
- E. Negishi, A genealogy of Pd-catalyzed cross-coupling, J. Organomet. Chem., 2002, 653, 34–40 CrossRef CAS.
- M. Balasubramanian, Industrial scale palladium chemistry, Tetrahedron Org. Chem. Ser., 2007, 26, 587–620 CAS.
- C. Torborg and M. Beller, Recent applications of palladium-catalyzed coupling reactions in the pharmaceutical, agrochemical, and fine chemical industries, Adv. Synth. Catal., 2009, 351, 3027–3043 CrossRef CAS.
- L. Xue and Z. Lin, Theoretical aspects of palladium-catalysed carbon–carbon cross-coupling reactions, Chem. Soc. Rev., 2010, 39, 1692–1705 RSC.
- F. Schoenebeck and K. N. Houk, Ligand-controlled regioselectivity in palladium-catalyzed cross-coupling reactions, J. Am. Chem. Soc., 2010, 132, 2496–2497 CrossRef CAS PubMed.
- G. Nagendrappa and Y. C. Sunil Kumar, The 2010 chemistry Nobel Prize: Pd(0)-catalyzed organic synthesis, Org. Magn. Reson., 2011, 16, 152–164 CAS.
- M. Garcia-Melchor, M. C. Pacheco, C. Nájera, A. Lledós and G. Ujaque, Mechanistic exploration of the Pd-catalyzed Cu-free Sonogashira reaction, ACS Catal., 2012, 2, 135–144 CrossRef CAS.
- S. Kozuch, C. Amatore, A. Jutand and S. Shaik, What makes for a good catalytic cycle? A theoretical study of the role of an anionic palladium(0) complex in the cross-coupling of an aryl halide with an anionic nucleophile, Organometallics, 2005, 24, 2319–2330 CrossRef CAS.
- L. J. Goossen, D. Koley, H. L. Hermann and W. Thiel, Mechanistic pathways for oxidative addition of aryl halides to palladium(0) complexes: a DFT study, Organometallics, 2005, 24, 2398–2410 CrossRef CAS.
- N. Hazari, P. R. Melvin and M. M. Beromi, Well-defined nickel and palladium precatalysts for cross-coupling, Nat. Rev. Chem., 2017, 1, 0025 CrossRef CAS.
- Z. Qiu, Recent advances in transition metal-mediated functionalization of o-carboranes, Tetrahedron Lett., 2015, 56, 963–971 CrossRef CAS.
- R. M. Dziedzic and A. M. Spokoyny, Metal-catalyzed cross-coupling chemistry with polyhedral boranes, Chem. Commun., 2019, 55, 430–442 RSC.
- K. Aizawa, K. Ohta and Y. Endo, Synthesis of 3-aryl-1,2-dicarba-closo-dodecaboranes by Suzuki–Miyaura coupling reaction, Heterocycles, 2010, 80, 369–378 CrossRef CAS.
- S. Alassadi, M. J. Pisani and N. J. Wheate, Luminescent metal–carborane complexes: an overview, Dalton Trans., 2022, 51, 10823–10833 RSC.
- T. D. Moseev, T. A. Idrisov, M. V. Varaksin, A. N. Tsmokaluk, V. N. Charushin and O. N. Chupakhin, Copper-catalyzed Sonogashira-type coupling reaction of vinylacetylene ortho-carborane with boronic acid in the synthesis of luminophores with phosphorescent emission, React. Chem. Eng., 2024, 5, 868–882 CAS.
- Z. Kelemen, A. Pepiol, M. Lupu, R. Sillanpää, M. M. Hänninen, F. Teixidor and C. Viñas, Icosahedral carboranes as scaffolds for congested regioselective polyaryl compounds: the distinct distance tuning of C–C and its antipodal B–B, Chem. Commun., 2019, 55, 8927–8930 RSC.
- K. P. Anderson, H. A. Mills, C. Mao, K. O. Kirlikovali and J. C. Axtell, Improved synthesis of icosahedral carboranes containing exopolyhedral B–C and C–C bonds, Tetrahedron, 2019, 75, 187–191 CrossRef CAS PubMed.
- R. M. Dziedzic, L. M. A. Saleh, J. C. Axtell, J. L. Martin, S. L. Stevens, A. T. Royappa and A. L. Rheingold, B–N, B–O and B–C–N bond formation via palladium-catalyzed cross-coupling of B-bromo-carboranes, J. Am. Chem. Soc., 2016, 138, 9081–9084 CrossRef CAS PubMed.
- L. Eriksson, K. J. Winberg, R. T. Claro and S. Sjöberg, Palladium-catalyzed Heck reactions of styrene derivatives and 2-iodo-p-carborane, J. Org. Chem., 2003, 68, 3569–3573 CrossRef CAS PubMed.
- B. Ringstrand, H. Monobe and P. Kaszynski, Anion-driven mesogenicity: ionic liquid crystals based on the [closo-1-CB9H10]− cluster, J. Mater. Chem., 2009, 19, 4805–4812 RSC.
- I. P. Beletskaya, V. I. Bregadze, K. Z. Kabytaev, G. G. Zhigareva, P. V. Petrovskii and I. V. Glukhov, Palladium-catalyzed amination of 2-iodo-para-carborane, Organometallics, 2007, 26, 2340–2347 CrossRef CAS.
- Y. Sevryugina, R. L. Julius and M. F. Hawthorne, Novel approach to aminocarboranes by mild amidation of selected iodo-carboranes, Inorg. Chem., 2010, 49, 10627–10634 CrossRef CAS.
- R. M. Dziedzic, J. L. Martin, J. C. Axtell, L. M. A. Saleh, T. C. Ong, Y. F. Yang, M. S. Messina and A. L. Rheingold, Cage-walking: vertex differentiation by palladium-catalyzed isomerization of B(9)-bromo-meta-carborane, J. Am. Chem. Soc., 2017, 139, 7729–7732 CrossRef CAS.
- K. Z. Kabytaev, S. N. Mukhin, I. V. Glukhov, Z. A. Starikova, V. I. Bregadze and I. P. Beletskaya, Boron–oxygen bond formation by palladium-catalyzed etheration of 2-iodo-para-carborane, Organometallics, 2009, 28, 4758–4763 CrossRef CAS.
- S. A. Jasper Jr, R. B. Jones, J. Mattern, J. C. Huffman and L. J. Todd, Palladium-mediated substitution reactions of polyhedral borane anions, Inorg. Chem., 1994, 33, 5620–5624 CrossRef.
- A. M. Spokoyny, C. D. Lewis, G. Teverovskiy and S. L. Buchwald, Extremely electron-rich, boron-functionalized, icosahedral carborane-based phosphinoboranes, Organometallics, 2012, 31, 8478–8481 CrossRef CAS PubMed.
- K. Z. Kabytaev, T. A. Everett, A. V. Safronov, Y. V. Sevryugina, S. S. Jalisatgi and M. F. Hawthorne, B-mercaptocarboranes: a new synthetic route, Inorg. Chem., 2013, 52, 2488–2491 Search PubMed.
- M. Brighi, F. Murgia, Z. Łodziana and R. Cerny, Structural phase transitions in closo-dicarbadodecaboranes C2B10H12, Inorg. Chem., 2022, 61, 5813–5823 CrossRef CAS PubMed.
- R. J. Grams, W. L. Santos, I. R. Scorei, A. Abad-García, C. A. Rosenblum, A. Bita and H. Cerecetto, The rise of boron-containing compounds: advancements in synthesis, medicinal chemistry, and emerging pharmacology, Chem. Rev., 2024, 124, 2441–2511 CrossRef CAS PubMed.
- R. K. Bohn and M. D. Bohn, Molecular structures of 1,2-, 1,7-, and 1,12-dicarba-closo-dodecaborane(12), B10C2H12, Inorg. Chem., 1971, 10, 350–355 CrossRef CAS.
- A. R. Turner, H. E. Robertson, K. B. Borisenko, D. W. H. Rankin and M. A. Fox, Gas-phase electron diffraction studies of the icosahedral carbaboranes, ortho-, meta- and para-C2B10H12, Dalton Trans., 2005, 7, 1310–1318 RSC.
- R. M. Dziedzic and A. M. Spokoyny, Metal-catalyzed cross-coupling chemistry with polyhedral boranes, Chem. Commun., 2019, 55, 430–442 RSC.
- M. Scholz and E. Hey-Hawkins, Carbaboranes as pharmacophores: properties, synthesis, and application strategies, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS PubMed.
- J. Poater, M. Solà, C. Viñas and F. Teixidor, Hückel’s rule of aromaticity categorizes aromatic closo boron hydride clusters, Chem.–Eur. J., 2016, 22, 7437–7443 CrossRef CAS PubMed.
- J. Poater, C. Viñas, I. Bennour and S. Escayola, Too persistent to give up: aromaticity in boron clusters survives radical structural changes, J. Am. Chem. Soc., 2020, 142, 9396–9407 CrossRef CAS PubMed.
- G. M. A. Junqueira, Remarkable aromaticity of cobalt bis(dicarbollide) derivatives: a NICS study, Theor, Chem. Acc., 2018, 137, 92 CrossRef.
- C. Viñas, R. Núñez, I. Bennour and F. Teixidor, Periphery-decorated and core-initiated neutral and polyanionic borane large molecules: forthcoming and promising properties for medicinal applications, Curr. Med. Chem., 2019, 26, 5036–5076 CrossRef PubMed.
- A. F. Armstrong and J. F. Valliant, The bioinorganic and medicinal chemistry of carboranes: from new drug discovery to molecular imaging and therapy, Dalton Trans., 2007, 38, 4240–4251 RSC.
- R. F. Barth, J. A. Coderre, M. G. H. Vicente and T. E. Blue, Boron neutron capture therapy of cancer: current status and future prospects, Clin. Cancer Res., 2005, 11, 3987–4002 CrossRef CAS PubMed.
- V. I. Bregadze and S. A. Glazun, Metal-containing carboranes with antitumor activity, Russ. Chem. Bull., 2007, 56, 643–659 CrossRef CAS.
- V. I. Bregadze, I. B. Sivaev, D. Gabel and D. Wöhrle, Polyhedral boron derivatives of porphyrins and phthalocyanines, J. Porphyrins Phthalocyanines, 2001, 5, 767–781 CrossRef CAS.
- E. L. Crossley, E. J. Ziolkowski, J. A. Coderre and L. M. Rendina, Boronated DNA-binding compounds as potential agents for boron neutron capture therapy, Mini-Rev. Med. Chem., 2007, 7, 303–313 CrossRef CAS PubMed.
- Y. Endo, K. Ohta and T. Yoshimi, A new application of inorganic clusters, carboranes, for medicinal drug design and molecular construction, Phosphorus, Sulfur Silicon Relat. Elem., 2004, 179, 799–802 CrossRef CAS.
- Y. Endo, T. Yoshimi, K. Ohta, T. Suzuki and S. Ohta, Potent estrogen receptor ligands based on bisphenols with a globular hydrophobic core, J. Med. Chem., 2005, 48, 3941–3944 CrossRef CAS PubMed.
- M. F. Hawthorne and M. W. Lee, A critical assessment of boron target compounds for boron neutron capture therapy, J. Neuro-Oncol., 2003, 62, 33–45 CrossRef PubMed.
- M. F. Hawthorne and A. Maderna, Applications of radiolabeled boron clusters to the diagnosis and treatment of cancer, Chem. Rev., 1999, 99, 3421–3434 CrossRef CAS PubMed.
- Z. J. Lesnikowski, Boron units as pharmacophores: new applications and opportunities of boron cluster chemistry, Collect. Czech. Chem. Commun., 2007, 72, 1646–1658 CrossRef CAS.
- Z. J. Lesnikowski, Boron clusters: a new entity for DNA-oligonucleotide modification, Eur. J. Org Chem., 2003, 23, 4489–4500 CrossRef.
- Z. J. Lesnikowski, J. Shi and R. F. Schinazi, Nucleic acids and nucleosides containing carboranes, J. Organomet. Chem., 1999, 581, 156–169 CrossRef CAS.
- A. F. Mironov and M. A. Grin, Synthesis of chlorin and bacteriochlorin conjugates for photodynamic and boron neutron capture therapy, J. Porphyrins Phthalocyanines, 2008, 12, 1163–1172 CrossRef CAS.
-
(a) V. I. Bregadze, Anticancer Agents Med. Chem., 2006, 6, 75 CrossRef CAS PubMed;
(b) I. B. Sivaev and V. I. Bregadze, Eur. J. Inorg. Chem., 2009, 2009, 1433–1450 Search PubMed.
- I. B. Sivaev, V. I. Bregadze and N. T. Kuznetsov, Derivatives of the closo-dodecaborate anion and their application in medicine, Russ. Chem. Bull., 2002, 51, 1362–1374 CrossRef CAS.
- W. Tjarks, The use of boron clusters in the rational design of boronated nucleosides for neutron capture therapy of cancer, J. Organomet. Chem., 2000, 614, 37–47 CrossRef.
- W. Tjarks, R. Tiwari, Y. Byun, S. Narayanasamy and R. F. Barth, Carboranyl thymidine analogues for neutron capture therapy, Chem. Commun., 2007, 47, 4978–4980 RSC.
- V. Tolmachev and S. Sjöberg, Polyhedral boron compounds as potential linkers for attachment of radiohalogens to targeting proteins and peptides: a review, Collect. Czech. Chem. Commun., 2002, 67, 913–935 CrossRef CAS.
- J. F. Valliant, K. J. Guenther, A. S. King, P. Morel, P. Schaffer, O. O. Sogbein and K. A. Stephenson, The medicinal chemistry of carboranes, Coord. Chem. Rev., 2002, 232, 173–230 CrossRef CAS.
- R. Satapathy, B. P. Dash and J. A. Maguire, New developments in the medicinal chemistry of carboranes, Collect. Czech. Chem. Commun., 2010, 75, 995–1022 CrossRef CAS.
- Z. J. Lesnikowski, New opportunities in boron chemistry for medical applications, in Boron Science: New Technologies and Applications, 2011, pp. 3–19 Search PubMed.
- V. M. Ahrens, R. Frank, S. Stadlbauer, A. G. Beck-Sickinger and E. Hey-Hawkins, Incorporation of ortho-carbaboranyl-Nε-modified L-lysine into neuropeptide Y receptor Y1- and Y2-selective analogues, J. Med. Chem., 2011, 54, 2368–2377 CrossRef CAS PubMed.
- K. Sonogashira, Development of the Sonogashira reaction, J. Organomet. Chem., 2002, 653, 46–49 CrossRef CAS.
- E. Negishi and L. Anastasia, Palladium-catalyzed alkynylation, Chem. Rev., 2003, 103, 1979–2018 CrossRef CAS PubMed.
- R. R. Tykwinski, Evolution in the palladium-catalyzed cross-coupling of sp- and sp2-hybridized carbon atoms, Angew. Chem., Int. Ed., 2003, 42, 1566–1568 CrossRef CAS PubMed.
- R. Chinchilla and C. Nájera, The Sonogashira reaction: a booming methodology in synthetic organic chemistry, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed.
- R. Chinchilla and C. Nájera, Recent advances in Sonogashira reactions, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC.
- U. H. F. Bunz, Poly(aryleneethynylene)s: syntheses, properties, structures, and applications, Chem. Rev., 2000, 100, 1605–1644 CrossRef CAS PubMed.
- A. H. Dieck and F. R. Heck, Palladium-catalyzed synthesis of aryl, heterocyclic and vinylic acetylene derivatives, J. Organomet. Chem., 1975, 93, 259–263 CrossRef.
- A. Soheili, J. Albaneze-Walker, J. A. Murry, P. G. Dormer and D. L. Hughes, Efficient and general protocol for the Cu-free Sonogashira coupling of aryl bromides at room temperature, Org. Lett., 2003, 5, 4191–4194 CrossRef CAS PubMed.
- C. Amatore, S. Bensalem, S. Ghalem, A. Jutand and Y. Medjour, Mechanistic aspects of copper-free Sonogashira reactions, Eur. J. Org Chem., 2004, 366–375 CrossRef CAS.
- A. Tougerti, S. Negri and A. Jutand, Mechanism of the copper-free palladium-catalyzed Sonogashira reactions: multiple role of amines, Chem.–Eur. J., 2007, 13, 666–676 CrossRef CAS PubMed.
- T. Ljungdahl, T. Bennur, A. Dallas, H. Emtenäs and J. Mårtensson, Two competing mechanisms for the Cu-free Sonogashira cross-coupling reaction, Organometallics, 2008, 27, 2490–2498 CrossRef CAS.
- M. M. Heravi and P. Hajiabbasi, Recent advances in Kumada–Tamao–Corriu cross-coupling reaction catalyzed by different ligands, Monatsh. Chem., 2012, 143, 1575–1592 CrossRef CAS.
- M. M. Heravi, V. Zadsirjan, P. Hajiabbasi and H. Hamidi, Advances in Kumada–Tamao–Corriu cross-coupling reaction: an update, Monatsh. Chem., 2019, 150, 535–591 CrossRef CAS.
- A. Joshi-Pangu, C. Y. Wang and M. R. Biscoe, Nickel-catalyzed Kumada cross-coupling reactions of tertiary alkylmagnesium halides and aryl bromides/triflates, J. Am. Chem. Soc., 2011, 133, 8478–8481 CrossRef CAS PubMed.
- J. Mao, F. Liu, M. Wang, L. Wu, B. Zheng, S. Liu and P. J. Walsh, Cobalt–bisoxazoline-catalyzed asymmetric Kumada cross-coupling of racemic α-bromo esters with aryl Grignard reagents, J. Am. Chem. Soc., 2014, 136, 17662–17668 CrossRef CAS PubMed.
- S. Lou and G. C. Fu, Nickel/bis(oxazoline)-catalyzed asymmetric Kumada reactions of alkyl electrophiles:
cross-couplings of racemic α-bromoketones, J. Am. Chem. Soc., 2010, 132, 1264–1266 CrossRef CAS PubMed.
- A. Sinha, C. M. Simon and M. G. Organ, Fast Room Temperature Cross-Coupling Reactions with Grignard Reagents, Chem.–Eur. J., 2019, 25, 5178–5182 Search PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel et al., Gaussian 09, Revision D.01, Gaussian Inc., Wallingford, CT, 2013 Search PubMed.
- W. J. Hehre, Ab initio molecular orbital theory, Acc. Chem. Res., 1976, 9, 399–406 CrossRef CAS.
- P. J. Hay and W. R. Wadt, Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- L. J. Goossen, D. Koley, H. L. Hermann and W. Thiel, The palladium-catalyzed cross-coupling reaction of carboxylic anhydrides with arylboronic acids: a DFT study, J. Am. Chem. Soc., 2005, 127, 11102–11114 CrossRef CAS PubMed.
- M. García-Melchor, The Cu-free Sonogashira reaction mechanism. A theoretical study of Pd-catalyzed C–C cross-coupling reactions, in Springer Science Reviews, Springer, 2013, vol. 1, pp. 89–111 Search PubMed.
- R. Álvarez, M. Perez, O. N. Faza and A. de Lera, Associative transmetalation in the Stille cross-coupling reaction to form dienes: theoretical insights into the open pathway, Organometallics, 2008, 27, 3378–3389 CrossRef.
- A. Ariafard and B. F. Yates, Subtle balance of ligand steric effects in Stille transmetalation, J. Am. Chem. Soc., 2009, 131, 13981–13991 CrossRef CAS PubMed.
- Z. Kelemen, A. Pepiol, M. Lupu, R. Sillanpää, M. M. Hänninen, F. Teixidor and C. Viñas, Icosahedral carboranes as scaffolds for congested regioselective polyaryl compounds: the distinct distance tuning of C–C and its antipodal B–B, Chem. Commun., 2019, 55, 8927–8930 RSC.
- M. Finze, Carba-closo-dodecaborates with one or two alkynyl substituents bonded to boron, Inorg. Chem., 2008, 47, 11857–11867 CrossRef CAS PubMed.
- A. P. Chan, J. A. Parkinson, G. M. Rosair and A. J. Welch, Bis(phosphine) hydridorhodacarborane derivatives of 1,1′-bis(ortho-carborane) and their catalysis of alkene isomerization and the hydrosilylation of acetophenone, Inorg. Chem., 2020, 59, 2011–2023 CrossRef CAS PubMed.
- L. J. Goossen, D. Koley, H. L. Hermann and W. Thiel, Palladium monophosphine intermediates in catalytic cross-coupling reactions: a DFT study, Organometallics, 2006, 25, 54–67 CrossRef CAS.
- L. N. Cui, Z. F. Li, J. Wen, Y. H. Jiang, Q. H. Jin and H. L. Gong, Crystal structure of bis(triphenylphosphine-κP)dichloropalladium(II) acetonitrile(1:1), [PdCl2(C18H15P)2]·CH3CN, Z. Kristallogr. NCS, 2011, 226, 591–592 CAS.
- F. E. Hahn, T. Lügger and M. Beinhoff, Palladium(II) complexes with benzoxazol-2-ylidene ligands: crystal structures of trans-chloro(benzoxazol-2-ylidene)bis(triphenylphosphine)palladium(II) chloride and cis-diiodo(benzoxazol-2-ylidene)(triphenylphosphine)palladium(II), Z. Naturforsch., B, J. Chem. Sci., 2004, 59, 196–201 CAS.
- V. V. Grushin and H. Alper, Alkali-induced disproportionation of palladium(II) tertiary phosphine complexes, [L2PdCl2], to L4Pd and palladium(0): key intermediates in the biphasic carbonylation of ArX catalyzed by [L2PdCl2], Organometallics, 1993, 12, 1890–1901 CrossRef CAS.
- C. S. Horbaczewskyj and I. J. S. Fairlamb, Pd-catalyzed cross-couplings: on the importance of the catalyst quantity descriptors, mol% and ppm, Org. Process Res. Dev., 2022, 26, 2240–2269 CrossRef CAS PubMed.
- M. S. Inkpen, A. J. P. White, T. Albrecht and N. J. Long, Rapid Sonogashira cross-coupling of iodoferrocenes and the unexpected cyclo-oligomerization of 4-ethynylphenylthioacetate, Chem. Commun., 2013, 49, 5663–5665 RSC.
- I. P. Beletskaya, V. I. Bregadze, V. A. Ivushkin, P. V. Petrovskii, I. B. Sivaev and S. Sjöberg, New B-substituted derivatives of m-carborane, p-carborane and cobalt bis(1,2-dicarbollide) anion, J. Organomet. Chem., 2004, 689, 2920–2929 CrossRef CAS.
- A. A. C. Braga, G. Ujaque and F. Maseras, A DFT study of the full catalytic cycle of the Suzuki–Miyaura cross-coupling on a model system, Organometallics, 2006, 25, 3647–3658 CrossRef CAS.
- M. Garcia-Melchor, M. C. Pacheco, C. Nájera, A. Lledós and G. Ujaque, Mechanistic exploration of the Pd-catalyzed Cu-free Sonogashira reaction, ACS Catal., 2012, 2, 135–144 CrossRef CAS.
- P. Veerakumar, P. Thanasekaran, K. L. Lu, K. C. Lin and S. Rajagopal, Computational studies of versatile heterogeneous palladium-catalyzed Suzuki, Heck and Sonogashira coupling reactions, ACS Sustainable Chem. Eng., 2017, 5, 8475–8490 CrossRef CAS.
- M. Garcia-Melchor, B. Fuentes, A. Lledós, J. A. Casares, G. Ujaque and P. Espinet, Cationic intermediates in the Pd-catalyzed Negishi coupling: kinetic and density functional theory study of alternative transmetalation pathways in the Me–Me coupling of ZnMe2 and trans-[PdMeCl(PMePh2)2], J. Am. Chem. Soc., 2011, 133, 13519–13526 CrossRef CAS PubMed.
- M. Beaupérin, E. Fayad, R. Amardeil, H. Cattey, P. Richard, S. Brandès and J.-C. Hierso, First copper(I) ferrocenyltetraphosphine complexes: possible involvement in Sonogashira cross-coupling reaction?, Organometallics, 2008, 27, 1506–1513 CrossRef.
- M. Beaupérin, A. Job, H. Cattey, S. Royer, P. Meunier and J.-C. Hierso, Copper(I) iodide polyphosphine adducts at low loading for Sonogashira alkynylation of demanding halide substrates: ligand exchange study between copper and palladium, Organometallics, 2010, 29, 2815–2822 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *b
*b

















