DOI:
10.1039/D5RA09654E
(Paper)
RSC Adv., 2026,
16, 26273-26291
New phenylcyclopropane-carbohydrazide furan derivatives with potent anticancer activity and EGFR inhibitory potential
Received
13th December 2025
, Accepted 13th April 2026
First published on 18th May 2026
Abstract
Fifteen new phenylcyclopropane-carbohydrazide-containing furan derivatives are synthesised and thoroughly characterized using FT-IR, 1H-NMR, 13C-NMR, and HRMS spectroscopy techniques in the search for new anticancer drugs. Using the MTT assay, this work examines the antiproliferative capability of these newly synthesized phenylcyclopropane hybrid compounds against cancer cell lines of the liver (HepG-2), lung (A549), and breast (MCF-7 and MDA-MB-231). Results indicated that with IC50 values of 2.08 ± 0.45, 2.15 ± 0.29, 2.17 ± 0.13, and 3.10 ± 0.32 µM for the MCF-7 cell line, compounds 8e, 8i, 8m, and 8n with p-dimethylamino, p-methoxy, p-hydroxy, and p-chloro-o-methoxy groups, respectively, showed the strongest antiproliferative activity, surpassing the reference drug doxorubicin in some cases. Among the synthesized compounds, 8e, 8j, 8m and 8n were the most active compounds with the EGFRWT inhibitory effect, exhibiting IC50 values of 1.79 ± 0.10, 1.59 ± 0.03, 0.87 ± 0.08, and 0.90 ± 0.60 µM, respectively. Molecular docking studies against the human breast cancer therapy compound and epidermal growth factor receptor (EGFR) (PDB codes: 3HB5 & 1M17) revealed that the furan derivatives exhibited excellent binding affinity (−10.70 and −8.54 kcal mol−1) through favorable van der Waals, electrostatic, HB-bonding and CH-bonding interactions within the active site. Furthermore, the ADME-T (absorption, distribution, metabolism, excretion, and toxicity) profiling of the synthesized compounds revealed balanced pharmacokinetic profiles and favorable drug-like features, confirming their potential as viable candidates for additional pharmaceutical development. The established procedure is a promising solution for a variety of applications since it uses low-toxic cyclopropane-1-carbohydrazide, furan with a broad substrate scope, and readily available substrates and requires only a short reaction time. Molecule 8n is the most stable conformer among all the compounds optimized at the B3LYP/6-31G level, with significant variations observed in the dipole moments and HOMO–LUMO gaps affecting their stability and reactivity.
Introduction
Cancer is still a major fatal and public health problem in the 21st century. With an anticipated 21.4 million new cases and 11.7 million deaths in 2025, cancer is the second largest cause of death globally according to the most recent Global Cancer Statistics 2025 report. According to this global survey, liver cancer (8.9%) and lung cancer (18.7% of all cancer deaths) rank first and fourth in terms of cancer mortality, respectively.1 In 2022, there were an estimated 20 million new instances of cancer, including 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 296840 cases of breast cancer, which led to 666103 fatalities.2 Numerous individuals in India have advanced stages of different types of cancer, according to the National Cancer Registry Programme (NCRP) reports: 57.6% for the breast, 60.0% for the cervix, 66.6% for the head and neck, and 50.8% for the stomach. Additionally, it was found that the incidence of distant metastases in lung cancer was gender-based, with 47.6% of cases occurring in females and 44.0% in males.3 Malignancies are likely to develop in both sexes equally. Prostate and colon cancers are frequent in men, while breast, cervix, and lung cancers are common in women.4 It is crucial to develop innovative anticancer treatments that can specifically target malignant cells and successfully inhibit their growth in light of the rising incidence of cancer.
296840 cases of breast cancer, which led to 666103 fatalities.2 Numerous individuals in India have advanced stages of different types of cancer, according to the National Cancer Registry Programme (NCRP) reports: 57.6% for the breast, 60.0% for the cervix, 66.6% for the head and neck, and 50.8% for the stomach. Additionally, it was found that the incidence of distant metastases in lung cancer was gender-based, with 47.6% of cases occurring in females and 44.0% in males.3 Malignancies are likely to develop in both sexes equally. Prostate and colon cancers are frequent in men, while breast, cervix, and lung cancers are common in women.4 It is crucial to develop innovative anticancer treatments that can specifically target malignant cells and successfully inhibit their growth in light of the rising incidence of cancer.
Because of its rising mortality rate, breast cancer is becoming one of the deadliest cancers in the world. The discovery of novel therapeutic targets and the subsequent development of numerous potent chemotherapeutic medications have been made possible by a thorough understanding of the molecular biology of breast cancer. However, the significant side effects of current chemotherapies, multidrug resistance, and metastatic breast cancer (MBC) remain important obstacles to disease treatment. As a result, research on new medications is still in progress.5 For many anticancer drugs on the market today, tyrosine kinases are the most researched therapeutic targets.6 The epidermal growth factor receptor (EGFR), a kind of tyrosine kinase membrane receptor, triggers a signaling cascade that controls cellular processes, such as angiogenesis, migration, differentiation, and proliferation, through ligand-induced dimerization. EGFR overexpression and oncogenic mutations in its catalytic domain have a major impact on the development of cancer and multidrug resistance. Most solid tumors, including breast, colorectal, and non-small cell lung cancer (NSCLC), overexpress the EGFR. Inhibiting EGFR expression and function is therefore a key focus in anticancer research according to studies.7
Cyclopropane is a common minor chemical compound in medications and natural goods.8 One structurally stable bio isostere that can be used to swap out carbon–carbon double bonds is cyclopropane. Among its effects are increased medication efficacy, improved metabolic stability, decreased off-target effects, increased degree of drug dissociation, and increased receptor affinity.9,10 As a result, drug design makes extensive use of it. Cyclopropane-containing compounds have biological and pharmacological effects, such as anticancer,11 antiviral,12 antibacterial,13 antifungal,14 antidepressant15 and antioxidant.16 Because of their high biological activity, heterocyclic molecules are essential in organic and medical chemistry. Due to their presence in a variety of contemporary medicinal agents, furan (a five-membered ring containing an oxygen heteroatom) and its derivatives are pharmacologically significant and serve as essential building blocks for the development of novel drugs.17 Numerous biological activities, such as anticancer, antibacterial, antiviral, analgesic, anti-inflammatory, antioxidant, antihyperglycemic, anticonvulsant, antihypertensive, antitubercular, antidepressant, and antifungal properties, are possessed by compounds with a furan ring.18–20 Consequently, because of their biological activity and low toxicity, molecules with phenylcyclopropane carbohydrazide and furan structures have drawn a lot of attention. Compared to medications containing biphenyl amide analogues, those containing aliphatic cyclic amide analogues have shown greater anticancer efficacy in colon, breast, and melanoma cell lines.21
A variety of phenylcyclopropane carbohydrazides and furan derivatives were designed, synthesised, and tested against breast cancer cell lines, including MCF-7, MDA-MB-231 (breast cancer), A549 (lung cancer), and HepG-2 (liver cancer), to build on the strong activity observed in phenylcyclopropane derivatives. To optimize these medications, computational methods, including pharmacokinetic predictions (ADMET), docking simulations, and molecular modelling, are crucial. The hybridization approach has several disadvantages, such as complicated synthesis, pharmacokinetic problems, higher molecular weight, possible toxicity, and doubts regarding efficacy.22 Additionally, molecular docking studies have shown that these substances can attach to protein targets linked to cancer, including kinases and cell cycle-related enzymes. This research not only aims to contribute to the development of anticancer drugs but also seeks to provide valuable insights into the design of hybrid compounds with enhanced pharmacological properties (Fig. 1).
 |
| | Fig. 1 Structures of commercial drugs of cyclopropane and furan. | |
Results and discussion
Design and synthesis
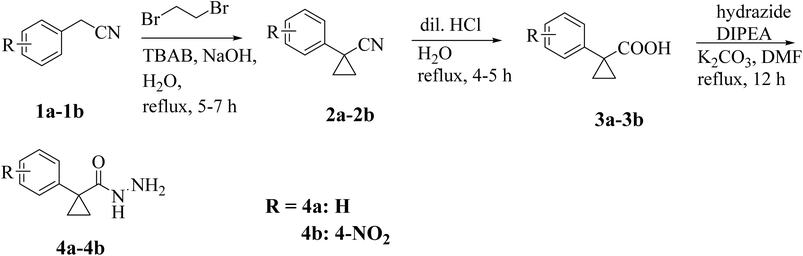
A conventional synthetic approach was employed to synthesize 1-phenylcyclopropane-1-carbohydrazide derivatives (4a, 4b).23
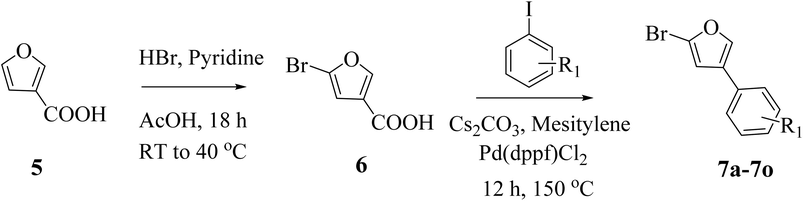
The synthesis of 1-phenylcyclopropane-1-carbohydrazide was carried out in three steps. In order to create phenylcyclopropane-1-carbonitriles (2a, 2b), 2-phenyl acetonitrile (1a, 1b) was first refluxed with 1,2-dibromoethane in water (20 mL) containing sodium hydroxide and TBAB (tetrabutylammonium bromide) for 5–7 hours. The second phase involved refluxing substituted carbonitriles (2) with dil. HCl in 10 mL of water for 4–5 hours at 90 °C. This was followed by neutralization with sodium carbonate, filtration, and cold water washing. Flash column chromatography was used to purify the final products (3a and 3b), which were then crystallized in methanol. Furthermore, a condensation reaction between hydrazide and 1-phenylcyclopropane-1-carboxylic acids is aided by a base catalyst. DIPEA (N,N-diisopropylethylamine) and K2CO3 (potassium carbonate) are two alkaline reagents used to aid in this condensation. Key intermediates (4a and 4b) are produced by the reaction, which takes place in a DMF solvent at 70–80 °C for 12 hours (Scheme 1). The synthesis of furan-based compounds 7a–o is shown in Scheme 2. Furan-3-carboxylic acid (5) was used to synthesize the primary starting intermediate 2-bromo-4-phenylfuran (7a–o), followed by 5-bromofuran-3-carboxylic acid (6). 5-Bromofuran-3-carboxylic acid (6) was produced by the substituted reaction of furan-3-carboxylic acid (5) with HBr in the presence of acetic acid in pyridine. Additionally, 2-bromo-4-phenylfurans (7a–o) were generated via the C–C bond synthesis of 6 with matching iodobenzene in the presence of Cs2CO3 (cesium carbonate) and Pd(dppf)Cl2([1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II)) catalyst in mesitylene at 150 °C for 12 hours.
 |
| | Scheme 1 Synthesis of 1-phenylcyclopropane-1-carbohydrazide derivatives. | |
 |
| | Scheme 2 Synthesis of furan-substituted intermediates. | |
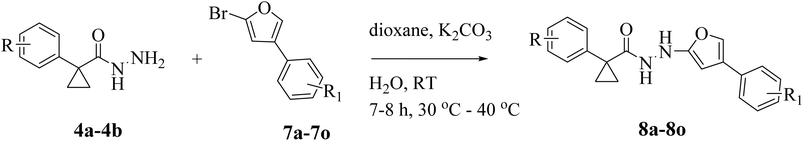
Different substituted 2-bromo-4-phenylfurane compounds (7a–o) are coupled with two distinct substituted 1-phenylcyclopropanecarbohydrazide derivatives (4a, 4b), which were previously prepared (Scheme 3). The synthetic methods used to create different targets are shown in Fig. 2. Several bases and solvents were used in our C–N coupling reaction experiment. We initially attempted a coupling reaction between 4a and 7a to obtain 8a. A nucleophilic aromatic substitution mechanism drives the process. With the help of the various substituents, the nitrogen atom of 1-phenylcyclopropanecarbohydrazide targets the aromatic ring's electron-deficient carbon in 2-bromo-4-phenylfurane, forming the desired molecules. Using various solvents, bases, temperatures, and reaction periods, the optimization of the reaction conditions for the synthesis of compounds 8a was methodically investigated (Table 1). The findings showed that temperature, base, and solvent selection had a significant impact on the reaction yield and product formation. Initial attempts to synthesise compound 8a using water and NaHCO3 in DMF at room temperature (RT) (entry 1) produced yields of 38% even after 7 hours. However, employing NaHCO3 as the base in DMF (entry 2) and conducting the reaction in a pressure reactor at 50 °C resulted in a notable improvement, producing compound 8a in a low yield of 46% in 7 hours. Similarly, a low yield of 40% was obtained when the base was changed to NaH in DMF and water was utilized at RT (entry 3), demonstrating weak reactivity under these conditions. Additionally, preliminary experiments employing NaH in DMSO at 40–50 °C for the production of compound 8a produced modest to good yields (entries 4 and 5). When Na2CO3 was utilized as the base at RT for seven hours (entry 6), the yield increased, emphasizing the significance of strong base Na2CO3 conditions and a moderate temperature for effective product production. When Na2CO3 was added to DMSO at 40 °C for seven hours (entry 7), a high yield product was formed; however, a tarry substance was occasionally formed. The reaction results emphasize that under these conditions, Na2CO3 works better than NaH. In the synthesis of compound 8a, the reaction proved challenging under various conditions. Interestingly, a breakthrough was achieved when the reaction was conducted at 40 °C with K2CO3 as the base in 1,4-dioxane (entry 8), which afforded an 84% yield after 7.5 h. However, the reaction with K2CO3 under similar conditions produced the product in an excellent yield (entry 9, 88%).
 |
| | Scheme 3 Synthesis of phenylcyclopropane-carbohydrazide-bearing furan scaffolds. | |
 |
| | Fig. 2 Structures of the synthesised molecules (8a–o). | |
Table 1 Optimization of reaction conditions for the synthesis of compound 8aa
| Entry |
Reactants |
Solvent |
Baseb |
Time (h) |
Temp (°C) |
Yields (%) |
| Reactions on a 2.8 mmol scale of reactants 4a and 7a. 4.2 mmol. Isolated yield of pure products. |
| 1 |
4a |
7a |
DMF/H2O |
NaHCO3 |
7 |
27 |
38 |
| 2 |
4a |
7a |
DMF/H2O |
NaHCO3 |
7 |
50 |
46 |
| 3 |
4a |
7a |
DMF/H2O |
NaH |
8 |
27 |
40 |
| 4 |
4a |
7a |
DMSO/H2O |
NaH |
8 |
40 |
50 |
| 5 |
4a |
7a |
DMSO/H2O |
NaH |
8 |
50 |
58 |
| 6 |
4a |
7a |
DMSO/H2O |
Na2CO3 |
7 |
27 |
63 |
| 7 |
4a |
7a |
Dioxane/H2O |
Na2CO3 |
7 |
40 |
70 |
| 8 |
4a |
7a |
Dioxane/H2O |
K2CO3 |
7.5 |
40 |
84 |
| 9 |
4a |
7a |
Dioxane/H2O |
K2CO3 |
8 |
40 |
88c |
The synthetic phenylcyclopropane-1-carbohydrazides (8a–o) were characterized by 1H-NMR, 13C-NMR, IR and HRMS spectroscopic methods. Compound 8a's 1H-NMR spectra showed two distinct NH proton signals as singlets at δ 10.40 and 8.41 ppm. In the 1H-NMR spectrum, the furan protons were detected at δ 7.84 and 6.88 ppm, and two doublets were ascribed to p-methyl phenyl protons at δ 7.72 and 7.60. The 1H-NMR spectra of compound 8a revealed cyclopropane protons at δ 1.70–1.88, while aromatic signals ranged from δ 7.32 to 7.37. Aromatic carbon signals between δ 144.05 and 108.21 were observed in compound 8a. The ketone carbonyl carbon is responsible for the signal at δ 182.51. δ 32.55 and 16.39 ppm were the measured values for this cyclopropyl group. In the 13C-NMR spectra, the phenyl ring connected to the methyl moiety showed a resonance signal at 21.44 ppm. For sp2 C–H bonds, the synthesised cyclopropane derivative (8a) shows a significant stretching frequency of approximately 3150 cm−1, while for sp3C–H bonds, the stretching frequency was 2923 cm−1. The stretching frequencies of the ether (COC) and carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) groups are 1164 cm−1 and 1682 cm−1, respectively. Between 1510 and 1610 cm−1, more noteworthy stretching frequencies for the CC bonds are discovered. Doublet signals at δ 6.89 and 7.37 for dimethyl amino phenyl and 7.74 and 8.18 ppm for nitrophenyl protons in the 1H-NMR spectrum indicate that compound 8l has p-substituted phenyl rings. Although the other two singlets appeared for the furan ring at δ 7.88 and 6.58 ppm, respectively, the proton signals corresponding to NH were between δ 10.48 and 8.35 ppm. However, compound 8l, which has a cyclopropyl ring, displayed dimethyl amino protons as a singlet at δ 2.93 and methylene protons at δ 1.52–1.71. Aromatic carbon signals were detected in the δ 154.07–107.09 ppm range of the 13C-NMR spectra. The ketone carbonyl, coupled with a saturated system, has been reported to show a 13C-NMR signal at about δ 181.20 ppm. The ([M + H]+) ion peak, which corresponds to the chemical formula C22H22N4O4, is observed at m/z = 407.1385 according to the mass spectrum characterization of molecule 8l.
O) groups are 1164 cm−1 and 1682 cm−1, respectively. Between 1510 and 1610 cm−1, more noteworthy stretching frequencies for the CC bonds are discovered. Doublet signals at δ 6.89 and 7.37 for dimethyl amino phenyl and 7.74 and 8.18 ppm for nitrophenyl protons in the 1H-NMR spectrum indicate that compound 8l has p-substituted phenyl rings. Although the other two singlets appeared for the furan ring at δ 7.88 and 6.58 ppm, respectively, the proton signals corresponding to NH were between δ 10.48 and 8.35 ppm. However, compound 8l, which has a cyclopropyl ring, displayed dimethyl amino protons as a singlet at δ 2.93 and methylene protons at δ 1.52–1.71. Aromatic carbon signals were detected in the δ 154.07–107.09 ppm range of the 13C-NMR spectra. The ketone carbonyl, coupled with a saturated system, has been reported to show a 13C-NMR signal at about δ 181.20 ppm. The ([M + H]+) ion peak, which corresponds to the chemical formula C22H22N4O4, is observed at m/z = 407.1385 according to the mass spectrum characterization of molecule 8l.
Anticancer activity
The cytotoxic activity of the synthesized compounds 8a–o was evaluated against breast (MCF-7, MDA-MB-231), lung (A549) and liver (HepG2) cancer cell lines using the MTT assay;24,25 the cell lines were procured from the National Centre for Cell Science (NCCS), Pune, India. The IC50 values are summarized in Table 2. In this case, doxorubicin (DXN) was used as a reference drug molecule. With IC50 values ranging from 2.08 to 47.90 µM, the majority of the assessed drugs demonstrated moderate to good cytotoxicity against all cancer cell lines. With an IC50 value ranging from 2.10 to 3.59 µM, DXN, a well-known anticancer medication, exhibited a notably significant cytotoxic effect, demonstrating its strong anticancer efficacy. Compound 8c demonstrated moderate to good cytotoxicity against HepG2 (IC50 = 6.70 ± 0.39 µM), but good activity against MCF-7 and MDA-MB-231 breast cancer cells (IC50 = 4.75 ± 0.09, 3.90 ± 1.12 µM). On all cancer cell lines, however, the methyl (8a) and methoxy (8b) groups of the phenyl moiety to the furan ring caused reduced activity (IC50 = 13.77 ± 1.02 to 40.21 ± 1.02 µM), while 8d showed moderate to good anticancer activity towards breast cancer cell lines MCF-7 and MDA-MB-231 (IC50 = 19.72 ± 0.08 and 16.90 ± 1.10 µM). p-Substituted-cyclopropane-1-carbohydrazide hybrids were also found to be potent against breast and lung cancer, and compound 8e demonstrated good activity against MCF-7, MDA-MB-231, A549, and HepG2 with IC50 values of 2.08 ± 0.45, 3.17 ± 0.55, 4.58 ± 0.07 and 6.70 ± 0.39 µM, respectively, when compared to DXN (2.10–3.59 µM) (Fig. 3).
Table 2 In vitro MTT-based cytotoxicity activity of selected compounds against human breast, lung and liver cancer cell lines (IC50 in µM)
| Entry |
Cytotoxicity IC50 (µM)a |
EGFRWT (IC50, µM)b |
| MCF-7 |
MDA-MB-231 |
A549 |
HepG-2 |
| IC50 values of the in vitro anti-proliferative activities of the tested compounds against MCF-7, MDA-MB-231, A549 and HepG2 cell lines from three independent experiments. 50% Inhibition of EGFR activity; NT: compounds not tested. NC, noncytotoxic. IC50 > 50 µM denoted as noncytotoxic; these results were expressed as mean value ± standard deviation (SD). |
| 8a |
20.78 ± 0.68 |
NC |
34.58 ± 0.90 |
40.21 ± 1.02 |
NT |
| 8b |
13.77 ± 1.02 |
33.28 ± 1.13 |
NC |
24.57 ± 0.83 |
NT |
| 8c |
4.75 ± 0.09 |
3.90 ± 1.12 |
8.90 ± 1.11 |
10.68 ± 0.40 |
1.79 ± 0.10 |
| 8d |
19.72 ± 0.08 |
16.90 ± 1.10 |
18.90 ± 0.09 |
NC |
NT |
| 8e |
2.08 ± 0.45 |
3.17 ± 0.55 |
4.58 ± 0.07 |
6.70 ± 0.39 |
2.55 ± 0.16 |
| 8f |
41.40 ± 1.13 |
32.17 ± 0.08 |
NC |
25.68 ± 1.10 |
NT |
| 8g |
NC |
21.54 ± 1.13 |
17.48 ± 0.08 |
15.92 ± 0.09 |
NT |
| 8h |
19.43 ± 1.13 |
20.78 ± 0.06 |
21.04 ± 1.32 |
NC |
NT |
| 8i |
2.15 ± 0.29 |
3.71 ± 0.03 |
2.58 ± 0.27 |
3.10 ± 1.54 |
2.70 ± 0.07 |
| 8j |
3.88 ± 1.15 |
2.85 ± 0.37 |
NC |
4.68 ± 0.10 |
1.59 ± 0.03 |
| 8k |
29.03 ± 0.69 |
23.57 ± 1.10 |
32.56 ± 1.19 |
47.90 ± 1.15 |
NT |
| 8l |
25.83 ± 0.75 |
NC |
31.20 ± 1.03 |
NC |
NT |
| 8m |
2.17 ± 0.13 |
3.15 ± 0.84 |
2.67 ± 0.42 |
3.27 ± 0.34 |
0.87 ± 0.08 |
| 8n |
3.10 ± 0.32 |
2.89 ± 0.37 |
5.79 ± 0.28 |
7.80 ± 0.50 |
0.90 ± 0.60 |
| 8o |
NC |
15.47 ± 0.42 |
NC |
20.98 ± 1.01 |
NT |
| DXN |
2.30 ± 0.15 |
3.50 ± 0.22 |
2.10 ± 0.10 |
3.59 ± 0.08 |
NT |
| Erlotinib |
NT |
NT |
NT |
NT |
0.94 ± 0.09 |
 |
| | Fig. 3 (A) % of Cell growth inhibition by 8e and 8i in the MCF-7 and HepG-2 cell lines, respectively. (B) Log dose–response curve of 8e and 8i at different concentrations (0.5, 1.0, 5.0, 10.0, 25.0, and 50.0 µM). | |
Compounds 8g and 8h from this series demonstrated moderate inhibition against MDA-MB-231 and A549 cell lines and robustly regulated the progression of cancer (IC50 = 17.48 to 21.54 µM). With an IC50 value of 25.68 ± 1.10–41.40 ± 1.13 µM, molecule 8f from this series exhibited the lowest cancer potential against all cell lines. In contrast, compounds 8i and 8j demonstrated exceptional cytotoxicity potential against MDA-MB-231 (3.71 ± 0.03, 2.85 ± 0.37 µM), A549 (2.58 ± 0.27 µM), HepG2 (3.10 ± 1.54, 4.68 ± 0.10 µM), and MCF-7 (2.15 ± 0.29, 3.88 ± 1.15 µM) (Fig. 3). With IC50 values of 2.17 ± 1.03, 3.15 ± 1.16, 2.67 ± 0.52, and 3.27 ± 1.04 µM against breast, lung, and liver cancer cell lines, the p-hydroxyphenyl substituted molecule (8m) demonstrated strong anticancer activity. The halogenated derivative (8n) showed the strongest antitumor activity. The most promising inhibition was shown by 8n (4-Cl, & 2-OMe), with IC50 values of 2.89 ± 1.07 µM (MDAMB-231) and 3.10 ± 1.32 µM (MCF-7). Moreover, 8o(4-Cl) had a mild anticancer effect. According to these data, all the produced compounds had moderate activity against liver and lung cancer and superior activity against breast cancer.
Aberrant EGFR trafficking, often resulting from oncogenic mutations, disrupts regulatory processes, leading to hyperactivation of downstream signaling and contributing to cancer development. Despite extensive research, many questions remain regarding the mechanisms and interaction networks governing EGFR endocytosis and degradation. The EGFR is widely expressed in non-small cell lung cancer (NSCLC) and is an important target in NSCLC. However, the majority of NSCLC tumors express EGFR wild type (EGFRwt) and do not respond to EGFR inhibition. Nevertheless, EGFR ligands are commonly expressed in lung cancer. Furthermore, constitutive overexpression-induced EGFRwt signaling has also been reported. Thus, it is possible that EGFRwt could play an oncogenic role in lung cancer.26 Compounds 8c, 8e, 8i, 8j, 8m, and 8n showed potential cytotoxicity; additional testing was conducted to determine whether they had any inhibitory effects on EGFRWT. Since erlotinib is a potent EGFR inhibitor, it was employed as a reference drug.27 Table 2 provides a summary of the findings. The EGFRWT cytotoxicity of the most potent chemicals was also contrasted with that of erlotinib, a popular anticancer medication that demonstrated an IC50 value of more than 0.94 ± 0.09 µM. In comparison to erlotinib, all assessed drugs showed moderate to increased cytotoxicity. Compounds with comparatively stronger EGFRWT inhibitory effects among the tested compounds included 8c (IC50 = 1.79 ± 0.10 µM), 8j (IC50 = 1.59 ± 0.03 µM), 8m (IC50 = 0.87 ± 0.08 µM), and 8n (IC50 = 0.90 ± 0.60 µM). The investigated drugs exhibit moderate cytotoxicity against the EGFRWT enzyme; according to the results of the MTT experiment, their cytotoxic effects are noticeably greater than those of erlotinib. To balance the cytotoxicity and therapeutic efficacy of these compounds and maximize their therapeutic potential as anticancer medicines, more research is required.
Compounds 8e, 8i, and 8m showed no significant cytotoxicity on HCC827 cells across the tested concentration range (2–32 µM), with cell viability remaining above ∼70% even at the highest concentration (Table 3). This demonstrates their low toxicity and ability to sustain cellular metabolic activity within this range. Among the three, 8e exhibited the highest cell viability, indicating that it was the least cytotoxic compared to 8m and 8i (Fig. 4). These findings suggest that all three compounds possess a favorable safety profile and potential for further study.
Table 3 Assessment of cytotoxicity against EGFR-dependent normal cell line HCC827
| Concentration (µM) |
% Cell viability |
| 8e |
8i |
8m |
| Control |
100 |
100 |
100 |
| 2 |
98.50 ± 1.07 |
94.90 ± 1.17 |
95.52 ± 0.20 |
| 4 |
91.30 ± 1.34 |
89.10 ± 1.02 |
86.34 ± 1.02 |
| 8 |
87.72 ± 1.03 |
82.02 ± 1.03 |
80.12 ± 1.03 |
| 16 |
82.01 ± 1.01 |
79.05 ± 0.91 |
76.08 ± 0.91 |
| 32 |
76.60 ± 0.98 |
71.50 ± 1.18 |
69.50 ± 1.18 |
 |
| | Fig. 4 Graph representing the % cell viability of different concentrations of 8e, 8i, and 8m on the HCC827 cell line. | |
SAR studies
It was found that the aryl group (Ar) had a big effect on the anti-proliferative activities of the target imidazole derivatives 8a–o when tested against cancer cells in a lab setting. It was observed that the introduction of a para dimethyl amino group at the phenyl ring (compound 8e) led to a significant increase in anticancer activity against all the cancer cell lines, and the para hydroxyphenyl group (compound 8f) appears to be the least favourable functional group for this series of compounds. The better effect of compound 8j can be attributed to its structural similarity to 8k, which allows these compounds with the m, p-electron donating groups to reach their anticancer activity more easily than o, p-EDG. Unexpectedly, compound 8i with p-methoxyphenyl substitution showed strong anticancer activity compared to compound 8h, which is probably due to the p-methylphenyl orientation of this compound in the EGFR enzyme active site, consistent with the good molecular docking results. The introduction of a hydroxyl group at the para-position of the phenyl ring (compound 8m) and para-chloro, ortho-methoxy phenyl ring significantly increased the anti-proliferative activity and EGFR inhibitory effect (Fig. 5). Additionally, the placement of an electron-withdrawing group like –Cl at the para position of the phenyl ring (compound 8o) is not suitable for anticancer activity and EGFR inhibitory activity compared to the electron-donating groups (compounds 8i and 8m). Overall, the biological results revealed that the nature and position of the phenyl-ring substitutions had a significant effect on the EGFR inhibitory activity of the synthesized compounds.
 |
| | Fig. 5 Structure activity relationship (SAR) of the final derived furan compounds. | |
Docking analysis
In breast tumour tissue, 17β-hydroxysteroid dehydrogenase type 1 (17β-HSD1) catalyzes the final stage of oestradiol and androstenediol production. When aromatase in the tumour is inhibited and oestrogen receptors are blocked simultaneously, the enzyme's high expression and activity indicate that its inhibition is a prerequisite for breast cancer treatment. Protein PDB-3HB5 is used for the analysis of the interaction of the active compounds.28
In solid tumours, members of the EGFR family are usually hyperactive. Several treatment strategies have been investigated that disrupt abnormal EGFR family signalling. Small compounds competing with ATP are a relatively novel therapeutic strategy for kinase inhibition. Numerous studies have demonstrated the effectiveness and selectivity of specific 4-anilino quinazoline derivatives as EGFR kinase inhibitors. There is structural information available for compounds of this general family that bind to the mitogen-activated protein kinase p38 (P38) and the distantly related intracellular kinase CDK2. Protein PDB-1M17 is used to analyze the interaction of the active compounds.29
PyRx software with Autodock Vina was used to conduct molecular docking investigations.30 ChemDraw was used to illustrate chemical compounds 8a–o. They were converted to pdbqt files, and the structures were minimized using a universal force field. Proteins with co-crystal ligands were used as binding sites in order to analyze the binding affinity. The coordinates were center_x = 10.11, center_y = 8.45, center_z = −11.15, size_x = 32.92, size_y = 36.64 and size_z = 33.44 for protein 3HB5 and center_x = 30.03, center_y = 5.85, center_z = 59.51, size_x = 37.98, size_y = 45.90 and size_z = 37.33 for protein 1M17. The interactions of the compounds with the proteins with good activity are tabulated in Tables 3 and 4. Compounds 8e, 8i, 8m and 8n showed good activity against breast cancer-related targeted proteins with docking scores of −10.5, 10.5, 10.7 and −10.8 kcal mol−1, respectively, and compounds 8e, 8j, 8m, and 8n showed good activity against EFRG with docking scores of −10.5, −8.1, −8.3, −8.4 and −8.2 kcal mol−1, respectively (Table 5) (Fig. 6).
Table 4 Docking pose interactions with protein EGFR (PDB-1M17)
| S. no. |
Compound |
Docking interactions (protein-1M17) |
Docking score (kcal mol−1) |
| 1 |
8e |
Hydrogen bond interactions: LYS-721, pi–pi-interactions-PHE-699, pi-anion-ASP-831, van der Waals interactions-GLN-767, MET-769, LEU-694, GLY-772, THR-830, pi-sigma interaction-LEU-820 |
−8.1 |
| 2 |
8j |
Pi-sigma interactions-LYS-721, pi-anion and cation-PHE-699, ASP-831, van der Waals interactions-GLU-738, ILE-765, LEU-764, MET-742, THR-766, ALA-719, THR-830 |
−8.3 |
| Alkyl-pi alkyl-LEU-723, ILE-735, ALA-731, VAL-702, LEU-820 |
| 3 |
8m |
Hydrogen bond-MET-769 |
−8.4 |
| Alkyl-pi-alkyl ALA-719, LEU-694, VAL-702, LEU-820 van der Waals-ASP-776, GLU-780, LEU-764, THR-766, GLN-767, LEU-768, PRO-770, CYS-773, TYR-777, THR-830, MET-742 |
| 4 |
8n |
van der Waals interactions-MET-742, THR-766, CYS-751, GLN-767, MET-769, GLY-772, LEU-768 |
−8.2 |
| Pi–pi sigma interactions-PHE-699 |
| Alkyl-pi-alkyl interactions-ALA-719, LEU-694, VAL-702, LYS-721 |
| Carbon hydrogen interactions-ASP-831 |
| 5 |
Co-crystal |
ASN-152, MET-193, van der Waals interactions: TYR-218, SER-222, MET-279, HIS-221, GLU-282, VAL-283, CYS-185, THR-140, VAL-143, SER-142, GLY-186, PHE-192, ILE-14, hydrogen bond interactions: GLY-141, LYS-159, pi-sigma-LEU-149, alkyl interactions: VAL-225, PHE-226, PRO-187, TYR-155, VAL-188, PHE-259 |
−6.9 |
Table 5 Docking pose interactions with protein MCF-17 (PDB-3HB5)
| S. no. |
Compound |
Docking interactions (protein-3HB5) |
Docking score (kcal mol−1) |
| 1 |
8e |
Hydrogen bond interactions: TYR-155, GLY-144, VAL-143, SER-142, GLY-186, CYS-185 van der Waals interactions: THR-190, PHE-259, SER-222 |
−10.5 |
| Pi-sigma-ILE-14, LEU-149 |
| Alkyl and pi-alkyl interactions: VAL-188, PHE-226, PRO-187, TYR-218, VAL-225 |
| Pi–pi stacked interaction: PHE-192 |
| 2 |
8i |
Hydrogen bond interactions: ILE-14, TYR-155, CYS-185, VAL-143, SER-142, GLY-144, GLY-186 van der Waals interactions: THR-140, ASN-90, GLY-13, GLY-15, SER-122 |
−10.2 |
| Pi-sigma-LEU-149 |
| Alkyl and pi-alkyl: VAL-225, HIS-221, TYR-218, VAL-188, PHE-226 |
| Pi–pi stacked: PHE-192 |
| 3 |
8m |
Hydrogen bond interactions: TYR-155, GLY-144, VAL-143, SER-142, GLU-282, CYS-185 van der Waals interaction: ASN-152, LEU-96, VAL-196, LEU-262, MET-279, HIS-221, PHE-259, GLY-186 |
−10.7 |
| Pi-sigma interaction: LEU-149 |
| Alkyl and pi-alkyl interactions: VAL-188, VAL-225, PRO-187, MET-193, PHE-192, PRO-187, VAL-225 |
| 4 |
8n |
Hydrogen bond interactions: ILE-14, TYR-155, GLY-186, GLY-144, SER-142, CYS-185 van der Waals interactions: GLY-15, THR-140, THR-190, ASN-90, GLY-13, SER-222 |
−10.8 |
| Pi–pi-stacked: PHE-192 |
| Alkyl and pi-alkyl interactions: HIS-228, TYR-218, MET-147, VAL-225, SER-142, CYS-185, GLY-144 |
| 5 |
Co-crystal |
Hydrogen bond interactions: GLY-141, LYS-152, van der Waals interactions: ASN-152, MET-193, TYR-218, SER-222, MET-279, HIS-221, GLU-282, VAL-283, CYS-185, THR-140, VAL-143, SER-142, GLY-186, PHE-192, pi-sigma interactions: LEU-149, alkyl interactions: VAL-225, PHE-226, PRO-187, TYR-155, VAL-188, PHE-259 |
−11.4 |
 |
| | Fig. 6 Docking images. (A) Re-docking of the co-crystal structure in the protein (1M17) pocket. (B) Interaction image of ligand 8j with protein-1M17. (C) Interaction image of ligand 8m with protein-1M17. (D) Interaction image of ligand 8n with protein-1M17. | |
Protein 1M17 forms important interactions with the synthesized active compound 8m. The hydrogen bond is formed with the oxygen of the 2-methoxyphenyl furan and the hydrogen of amino acid MET-769. Amino acids ALA-719 and LEU-820 formed alkyl and pi-alkyl interactions with the hydrogen of the 2-methoxyphenyl furan. The oxygen of the 4-hydroxy phenyl formed a hydrogen bond with amino acid LYS-721. It also formed alkyl and pi-alkyl interactions with VAL-702 and LEU-820. The cyclopropane formed an alkyl interaction with amino acid LEU-694. The 4-nitrophenyl form pi-sigma bonds with amino acid GLY-772 (Fig. 7).
 |
| | Fig. 7 Docking images. (A) Interaction image of ligand 8e with protein-3HB5. (B) Interaction image of ligand 8i with protein-3HB5. (C) Interaction image of ligand 8m with protein-3HB5. (D) Interaction image of ligand 8n with protein-3HB5. | |
Protein 3HB5 forms important interactions with the synthesized compound 8e. The hydrogen of the carbohydrazide forms a hydrogen bond with GLY-186, CYS-185 and SER-142. The oxygen of the carbohydrazide forms a hydrogen bond with the hydrogen of amino acid TYR-155. The oxygen of the furan-5-yl ring forms a hydrogen bond with amino acids VAL-143 and GLY-144. The pi-sigma bond is formed between ILE-14 and LEU-149 with the hydrogens of the phenyl and phenyl cyclopropane. The hydrogen of the dimethyl phenyl group forms alkyl bonds with amino acids TYR-218, VAL-225, and HIS-221. The pi–pi stacked interactions are formed between phenyl and PHE-192. The phenyl also formed pi alkyl interactions with the amino acids VAL-188 and PHE-226.
ADME-Tox prediction
A molecule must have both a favorable ADME/T profile and a significant level of biochemical activity in order to be considered a promising candidate for drug selection. To predict the ADME-Tox features of possible anticancer medication candidates, a wide range of tools and online platforms have surfaced in recent years. Using Protox3 (https://tox.charite.de/protox3/index.php?site=compound_input) and the SwissADME (https://www.swiss.adme.ch/) online program, the physicochemical, pharmacokinetic, and ADME parameters were estimated from computational investigations.31–34 Novel medications and formulations were chosen based on their physicochemical characteristics, such as solubility, molecular weight, TPSA, molar refractivity, and hydrogen-bonding capabilities. Very good bioavailability is demonstrated by the TPSA values, which range from 54.27 to 117.64 Å, and the drug-likeness scores, which may be anticipated between 0.01 and −0.37 (Fig. 8). According to the results, we found that indomethacin and all amides meet the drug-likeness criteria (Lipinski, Egan, Veber, Mugge, and Golden rules) and are authorized as distinct drug candidates. Caco-2 cell permeability has thus been a crucial marker for novel therapeutics. The target chemical is thought to have appropriate Caco-2 permeability when its predicted value is more than 5.15log cm s−1. Based on this criterion, we found that all the compounds 8a–o have suitable Caco-2 permeability ranging from −4.65 to −4.96 log cm s−1 when compared to the unsuitable value of −6.77log cm s−1 for DXN (Table 6). The pink area in the radar figure represents the appropriate range of physicochemical space for optimal oral bioavailability (Fig. 9).
 |
| | Fig. 8 Drug-likeness scores of 8a, 8f and 8l. | |
Table 6 Physicochemical properties and drug likeness parameters of the target compounds and DXN
| S. no. |
TPSA |
logP |
nHet |
HBA |
HBD |
nROTB |
Lipinski |
Veber |
Egan |
Mugge |
Golden |
Drug-likeness |
| 8a |
54.27 |
4.42 |
4.0 |
4.0 |
2.0 |
6.0 |
Yes |
Yes |
Yes |
Yes |
Yes |
−0.17 |
| 8b |
63.50 |
3.98 |
5.0 |
5.0 |
2.0 |
7.0 |
Yes |
Yes |
Yes |
Yes |
Yes |
−0.23 |
| 8c |
72.73 |
3.54 |
6.0 |
6.0 |
2.0 |
8.0 |
Yes |
Yes |
Yes |
Yes |
Yes |
0.06 |
| 8d |
72.73 |
3.69 |
6.0 |
6.0 |
2.0 |
8.0 |
Yes |
Yes |
Yes |
Yes |
Yes |
0.01 |
| 8e |
57.51 |
4.02 |
5.0 |
5.0 |
2.0 |
7.0 |
Yes |
Yes |
Yes |
Yes |
Yes |
−0.14 |
| 8f |
74.50 |
3.59 |
5.0 |
5.0 |
3.0 |
6.0 |
Yes |
Yes |
Yes |
Yes |
Yes |
0.08 |
| 8g |
63.50 |
4.32 |
6.0 |
5.0 |
2.0 |
7.0 |
Yes |
Yes |
Yes |
Yes |
Yes |
0.06 |
| 8h |
97.41 |
4.28 |
7.0 |
7.0 |
2.0 |
7.0 |
Yes |
Yes |
Yes |
Yes |
Yes |
−0.37 |
| 8i |
106.64 |
3.47 |
8.0 |
8.0 |
2.0 |
8.0 |
Yes |
Yes |
Yes |
Yes |
Yes |
−0.19 |
| 8j |
115.87 |
2.90 |
9.0 |
9.0 |
2.0 |
9.0 |
Yes |
Yes |
Yes |
Yes |
Yes |
−0.22 |
| 8k |
115.87 |
3.33 |
9.0 |
9.0 |
2.0 |
9.0 |
Yes |
Yes |
Yes |
Yes |
Yes |
−0.21 |
| 8l |
100.65 |
3.49 |
8.0 |
8.0 |
2.0 |
8.0 |
Yes |
Yes |
Yes |
Yes |
Yes |
−0.23 |
| 8m |
117.64 |
3.28 |
8.0 |
8.0 |
3.0 |
7.0 |
Yes |
Yes |
Yes |
Yes |
Yes |
−0.17 |
| 8n |
106.64 |
4.23 |
9.0 |
8.0 |
2.0 |
8.0 |
Yes |
Yes |
Yes |
Yes |
Yes |
−0.16 |
| 8o |
97.41 |
4.52 |
8.0 |
7.0 |
2.0 |
7.0 |
Yes |
Yes |
Yes |
Yes |
Yes |
−0.20 |
| DXN |
206.07 |
1.20 |
12.0 |
12.0 |
7.0 |
5.0 |
Yes |
Yes |
Yes |
Yes |
Yes |
0.16 |
 |
| | Fig. 9 Red circles represent the suggested medications. | |
It was also calculated how the tested cyclopropane carbohydrazide may interact with cytochrome P450. The data obtained indicate that although none of the tested drugs are CYP2E1 inhibitors, all of them are CYP3A4 inhibitors, almost all of them are CYP2C19 inhibitors (with the exception of substances 8e, 8f, and 8l), and all of them are moderate CYP1A2 and CYP2C9 inhibitors (Table 7). Drug–drug interactions must be considered while developing new compounds. Although a modest inhibitor, CYP2D6 accounts for around 65% of drug metabolism. The intestinal or brain-assessed permeability technique (BOILED-Egg), a successful prediction model based on small-molecule lipophilicity and polarity calculations, was established in order to accomplish this goal. The BOILED-Egg model offers a rapid, simple, dependable, and statistically unmatched method for predicting the better brain permeability and GI absorption of small compounds for drug research and discovery (Fig. 10). The physicochemical zone of chemicals most likely to be absorbed by the GI tract seems to be the yolk or white portion of the egg. A region of chemical and physical defense against chemicals that can potentially enter the brain is the blood–brain barrier (BBB), which is shown in yellow. P-glycoprotein, a protein that pumps foreign chemicals out of cells, was unable to transport indomethacin or any of the most active drugs out of the cells. This suggests that the compounds can stay longer in the cells and have a greater effect on the receptors.
Table 7 Pharmacokinetic properties of the target compounds (8a–o)
| S. no. |
Cyp1 A2 |
Cyp2 C19 |
Cyp2 C9 |
Cyp2 D6 |
Cyp3 A4 |
Cyp2 E1 |
MDCK |
Caco-2 |
Aquatic tox |
Skin sensi |
hERG |
AMES |
| 8a |
0.85 |
0.57 |
0.66 |
0.54 |
0.50 |
0.99 |
−4.65 |
−4.68 |
1 |
2 |
0.50 |
0.71 |
| 8b |
0.78 |
0.51 |
0.65 |
0.62 |
0.50 |
0.99 |
−4.51 |
−4.84 |
1 |
2 |
0.25 |
0.78 |
| 8c |
0.79 |
0.50 |
0.69 |
0.68 |
0.55 |
1.00 |
−4.61 |
−4.79 |
1 |
4 |
0.19 |
0.71 |
| 8d |
0.79 |
0.51 |
0.68 |
0.67 |
0.55 |
1.00 |
−4.53 |
−4.75 |
1 |
2 |
0.27 |
0.81 |
| 8e |
0.87 |
0.66 |
0.55 |
0.53 |
0.50 |
0.99 |
−4.59 |
−4.95 |
1 |
2 |
0.17 |
0.85 |
| 8f |
0.80 |
0.66 |
0.54 |
0.67 |
0.68 |
0.99 |
−4.71 |
−4.85 |
1 |
2 |
0.17 |
0.70 |
| 8g |
0.73 |
0.51 |
0.75 |
0.55 |
0.52 |
0.99 |
−4.68 |
−4.68 |
2 |
2 |
0.30 |
0.65 |
| 8h |
0.74 |
0.63 |
0.57 |
0.70 |
0.53 |
0.99 |
−4.52 |
−4.65 |
1 |
2 |
0.20 |
0.94 |
| 8i |
0.73 |
0.58 |
0.61 |
0.67 |
0.51 |
1.00 |
−4.58 |
−4.76 |
1 |
4 |
0.19 |
0.94 |
| 8j |
0.73 |
0.58 |
0.60 |
0.67 |
0.51 |
1.00 |
−4.41 |
−4.67 |
1 |
2 |
0.26 |
0.96 |
| 8k |
0.74 |
0.65 |
0.52 |
0.67 |
0.52 |
0.99 |
−4.23 |
−4.50 |
1 |
2 |
0.23 |
0.90 |
| 8l |
0.79 |
0.71 |
0.53 |
0.78 |
0.59 |
0.99 |
−4.41 |
−4.67 |
1 |
2 |
0.26 |
0.96 |
| 8m |
0.71 |
0.57 |
0.65 |
0.66 |
0.52 |
0.99 |
−4.70 |
−4.96 |
1 |
2 |
0.17 |
0.94 |
| 8n |
0.70 |
0.64 |
0.57 |
0.64 |
0.53 |
0.99 |
−4.43 |
−4.68 |
2 |
2 |
0.22 |
0.91 |
| 8o |
0.71 |
0.61 |
0.56 |
0.68 |
0.55 |
0.99 |
−4.49 |
−4.71 |
2 |
2 |
0.29 |
0.93 |
| DXN |
0.99 |
0.97 |
0.73 |
0.92 |
0.98 |
0.99 |
−5.53 |
−6.77 |
2 |
8 |
0.16 |
0.99 |
 |
| | Fig. 10 Boiled egg representation of the intestinal absorption and permeation through the blood–brain barrier for diquinothiazines 8a–o and DXN. | |
To use them as a model for permeability screening, Madin–Darby canine kidney (MDCK) cells were selected. Although DXN displayed a value of −5.53 cm s−1, Table 8 illustrates that the most active cyclopropane carbohydrazide had a high passive MDCK permeability of more than 20 × 10−6 cm s−1 in the range of −4.23 to −4.70 cm s−1. The findings of the computational analysis of all the synthesised cyclopropane carbohydrazides showed good pharmacokinetic properties for oral bioavailability. The predicted input compound's results provide details on the dataset's prepared fifteen structures, the distribution of the input compounds' physicochemical properties, and the acute toxicity class (predicted median lethal dose in weight, toxicity class, and prediction accuracy) (Table 7 and Fig. 11). A table with the predicted class and confidence score for each of the following is presented: hepatotoxicity, neurotoxicity, nephrotoxicity, cardiotoxicity (organ toxicity), carcinogenicity, mutagenicity, BBB-barrier, clinical toxicity (toxicity end points), oestrogen receptor alpha, mitochondrial membrane potential, GABA receptor, molecular initiating events, and metabolism. A toxicity radar graphic for active class prediction is another way to display prediction results. For an input chemical, the results of the active and inactive classes are also shown as a network plot (Fig. 12). Molecular docking predicted a high binding affinity of −10.8 kcal mol−1 with the target 8n, driven by hydrogen bondings with ILE-14, TYR-155, and CYS-185. The calculated parameters (logP 4.23 and TPSA 106.64 Å2) fall within the recommended ranges for oral bioavailability, predicting high gastrointestinal absorption and low probability of toxicity (Table 8).
Table 8 Toxicity properties of the final prepared cyclopropane-1-carbohydrazide derivatives
| S. no. |
Hep. |
Neur. |
Neph. |
Cardio. |
Carcino. |
Muta. |
BBB |
Clin. tox. |
ER |
MMP |
GABA |
| 8a |
Active |
Active |
Inactive |
Inactive |
Active |
Active |
Active |
Active |
Inactive |
Active |
Inactive |
| 8b |
Active |
Active |
Inactive |
Inactive |
Active |
Active |
Active |
Active |
Inactive |
Inactive |
Inactive |
| 8c |
Active |
Inactive |
Inactive |
Inactive |
Active |
Inactive |
Active |
Active |
Inactive |
Inactive |
Inactive |
| 8d |
Active |
Inactive |
Inactive |
Inactive |
Active |
Inactive |
Active |
Active |
Inactive |
Active |
Inactive |
| 8e |
Active |
Active |
Inactive |
Inactive |
Active |
Inactive |
Active |
Active |
Inactive |
Active |
Inactive |
| 8f |
Active |
Inactive |
Inactive |
Inactive |
Active |
Active |
Active |
Active |
Inactive |
Inactive |
Inactive |
| 8g |
Active |
Active |
Inactive |
Inactive |
Active |
Inactive |
Active |
Active |
Inactive |
Active |
Inactive |
| 8h |
Active |
Inactive |
Inactive |
Inactive |
Active |
Active |
Active |
Inactive |
Inactive |
Active |
Inactive |
| 8i |
Active |
Inactive |
Inactive |
Inactive |
Active |
Active |
Active |
Inactive |
Inactive |
Active |
Inactive |
| 8j |
Active |
Inactive |
Inactive |
Inactive |
Active |
Active |
Active |
Inactive |
Inactive |
Active |
Inactive |
| 8k |
Active |
Inactive |
Inactive |
Inactive |
Active |
Active |
Active |
Inactive |
Inactive |
Active |
Inactive |
| 8l |
Active |
Inactive |
Inactive |
Inactive |
Inactive |
Active |
Active |
Inactive |
Inactive |
Active |
Inactive |
| 8m |
Active |
Inactive |
Inactive |
Inactive |
Inactive |
Active |
Active |
Inactive |
Inactive |
Active |
Inactive |
| 8n |
Active |
Inactive |
Active |
Inactive |
Active |
Active |
Active |
Inactive |
Inactive |
Active |
Inactive |
| 8o |
Active |
Inactive |
Active |
Inactive |
Active |
Active |
Active |
Inactive |
Inactive |
Active |
Inactive |
| DXN |
In active |
Active |
Active |
Active |
Inactive |
Active |
Inactive |
Active |
Inactive |
Inactive |
Inactive |
 |
| | Fig. 11 Comparison of the input compound (8a) with dataset compounds. | |
 |
| | Fig. 12 Toxicity radar charts for compound 8a and DXN (blue: probability for activity; orange: average for activity). | |
Computational calculations
A total of 15 molecules (8a–o) were optimized using the B3LYP/6-31G level of theory. The optimized energy, relative energy (with respect to the most stable structure), dipole moment, and HOMO–LUMO gap for each molecule are summarized (Table 8). All optimized structures correspond to true minima on the potential energy surface, as confirmed by the absence of imaginary frequencies. Notably, molecule 8n is the most stable conformer and is thus used as the reference for calculating relative energies. Molecules 8g, 8o, 8k and 8j exhibit moderate stability with relative energies (ΔE) less than 220 kcal mol−1, while molecules 8a–f are significantly less stable with ΔE values exceeding 400 kcal mol−1, indicating poor thermodynamic viability. The computed dipole moments span from 1.39 D to 6.05 D, with molecules 8j to 8o showing higher polarity (µ > 4 D), which may enhance their infrared activity and solvent interactions. The HOMO–LUMO gaps range from 2.012 eV to 5.184 eV. Molecules 8a, 8f and 8o display relatively large gaps, suggesting higher electronic stability and lower chemical reactivity, whereas molecules 8h to 8l show smaller gaps, indicating higher reactivity and greater electronic delocalization.
A narrow gap suggests that a molecule can easily donate electrons (from HOMO) or accept them (into LUMO). This often correlates with higher biological activity (e.g., 8e, 8j, 8m and 8n in anticancer agents) because the drug can readily engage in charge-transfer interactions with proteins. A smaller HOMO–LUMO gap can indicate a better anticancer capacity. Generally, high biological activity often correlates with a certain range of dipole moments (often lower than 10–13 debye for many oral drugs), which aids in solubility and membrane permeability. From the results of DFT analysis, ligands 8e, 8m and 8n showed acceptable dipole moment values of 1.876039, 4.811753 and 4.388619 debye, respectively. From this analysis, a narrow HOMO–LUMO gap usually suggests higher reactivity, a specific dipole moment suggests better receptor binding, and structural stability indicates that the molecule will survive to reach the active site (Table 9).
Table 9 Toxicity properties of the final prepared cyclopropane-1-carbohydrazide derivatives
| Molecule |
Point group |
E (Hartree) |
Relative energy (kcal mol−1) |
Dipole moment (D) |
HOMO–LUMO gap (eV) |
Imag. freq. |
| 8a |
C1 |
−1072.124136 |
462.26 |
2.1099 |
5.184 |
0 |
| 8b |
C1 |
−1147.322762 |
415.99 |
1.395499 |
4.973 |
0 |
| 8c |
C1 |
−1261.830961 |
345.39 |
1.810336 |
4.956 |
0 |
| 8d |
C1 |
−1261.836048 |
345.07 |
1.924019 |
4.889 |
0 |
| 8e |
C1 |
−1088.162927 |
453.84 |
1.876039 |
4.517 |
0 |
| 8f |
C1 |
−1108.022932 |
442.55 |
2.817164 |
5.059 |
0 |
| 8g |
C1 |
−1606.926201 |
128.17 |
3.777786 |
4.965 |
0 |
| 8h |
C1 |
−1276.604444 |
335.55 |
3.938784 |
2.706 |
0 |
| 8i |
C1 |
−1351.803027 |
288.39 |
3.374133 |
2.493 |
0 |
| 8j |
C1 |
−1466.311188 |
216.27 |
6.047712 |
2.472 |
0 |
| 8k |
C1 |
−1466.316805 |
215.91 |
5.017866 |
2.398 |
0 |
| 8l |
C1 |
−1292.643537 |
325.96 |
5.661041 |
2.012 |
0 |
| 8m |
C1 |
−1312.503226 |
313.42 |
4.811753 |
2.587 |
0 |
| 8n |
C1 |
−1811.406551 |
0 |
4.388619 |
2.781 |
0 |
| 8o |
C1 |
−1531.729782 |
175.33 |
5.516639 |
5.127 |
0 |
Experimental section
Materials and methods
Several necessary reagents for furan scaffolds carrying cyclopropane-1-carbohydrazide were purchased from Sigma-Aldrich. TLC plates coated with 254 were used to verify the progress of the reactions (Merck, Germany). Under UV light with wavelengths of 366 and 254 nm, TLC dots were visible. With dimethyl sulfoxide (DMSO-d6) used as the reference solvent, the structures of these recently synthesised compounds were examined using a frequency of 500 MHz and 1H NMR at a 13C NMR frequency of 125 MHz. FT-IR spectra were acquired using spectra manager software on a Jasco instrument (model FT/IR-4100 type A). 3–5 mg of the chemical was combined with 250–300 mg of KBr salt to create the sample. The high-resolution electron impact mass spectra were interpreted using mass spectrometry.
Synthesis of 4a–b
The 1-phenylcyclopropane-1-carbohydrazide derivatives (2, 3)23 were synthesised using a traditional synthetic method, as detailed in our previous study. K2CO3 (32.36 mmol, 4.46 g) was added to a solution containing compound 3a (21.57 mmol, 3.5 g), hydrazide (43.15 mmol, 1.4 g), and DIPEA (25.89 mmol, 3.34 g) in 35 mL DMF for 30 minutes at below 10 °C. For 12 hours, the reaction mixture was swirled at 80 °C. Following TLC, the reaction was diluted with 5 mL of EtOAc, filtered through a tiny silica plug, and then rinsed with 10 mL of EtOAc. After vacuuming out, the solvent was dried. A pure white solid 4a was obtained by purifying the crude component using flash column chromatography with ethyl acetate/hexene (8:2) (62%). Mp: 98–100 °C.
Synthesis of 6
Furan-3-carboxylic acid (5) (28.57 mmol, 3.2 g), HBr (31.42 mmol, 2.54 g), pyridine (42.85 mmol, 3.38 g), and 30 mL of acetic acid were added to a 250 mL round-bottom flask submerged in an ice-water bath. The reaction mixture was then allowed to sit at 0 °C for 1 hour. For 18 hours, the reaction mixture was agitated at 40 °C. Once the reaction was finished, it was allowed to cool to ambient temperature before being filtered through a Celite pad. After being vacuum-evaporated, the filtrate was diluted with 100 mL of water and extracted using 150 mL of EtOAc. After cleaning with a saturated HCl solution, the CH2Cl2 layer was dried over anhydrous sodium sulphate. The crude product was purified by flash column chromatography using hexane:ethyl acetate (8:2) as an eluent to afford pure compound 6 as a brownish solid (93%). Mp 170–172 °C.
General procedure for the synthesis of furan derivatives (7a–o)
Pd(dppf)Cl2 (7.89 mmol, 5.76 g), the corresponding iodobenzene (15.78 mmol, 3.44 g), a mild base Cs2CO3 (18.94 mmol, 6.15 g), and a suitable organic solvent (mesitylene) were added to a solution of compound 6 (15.78 mmol, 3.0 g). At 150 °C, the reaction mixture was agitated for 12 hours. The ice bath was left to thaw and heated to room temperature during the process. After that, the solvent was dried by evaporation. EtOAc was used to extract the resultant mixture after it was diluted with water. A saturated aqueous NaCl solution was used to wash the mixed organic layers, which were then dried on MgSO4, filtered, and evaporated until completely dry. Mp 81–83 °C.
General procedure for the synthesis of cyclopropane-1-carbohydrazide bearing furans (8a–o)
K2CO3 (21.29 mmol, 2.93 g) and 1-phenylcyclopropane-1-carbohydrazide (4a) (14.19 mmol, 2.5 g) were combined in 25 mL of 1,4-dioxane and agitated at room temperature for 30 minutes. Next, water (12.5 mL) was added to 2-bromo-4-phenylfurane (14.19 mmol, 3.36 g), and the reaction was agitated for 7.5 h at 40 °C. Following TLC, the reaction was diluted with 100 mL of EtOAc, filtered through a tiny silica plug, and rinsed with 25 mL of diluted HCl. After vacuuming out, the solvent was dried. Using ethyl acetate/hexene (2:8) and flash column chromatography, the crude chemical was refined to produce a pure white solid 8a (88%) (Mp 157–159 °C).
Spectroscopic data
1-Phenyl-N′-(4-(p-tolyl)furan-2-yl)cyclopropane-1-carbohydrazide (8a). White solid, yields 88%, Mp 157–159 °C. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.40 (s, 1H, CONH), 8.41 (s, 1H, NH), 7.84 (s, 1H, ArH), 7.72 (d, 2H, J = 9.8 Hz, ArH), 7.60 (d, 2H, J = 9.8 Hz, ArH), 7.37 (m, 3H, ArH), 7.32 (m, 2H, ArH), 6.88 (s, 1H, ArH), 2.20 (s, 3H, CH3), 1.88 (m, 2H, CH2), 1.70 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 182.51, 148.27, 144.05, 138.21, 137.42, 134.83, 134.12, 129.85, 128.01, 127.37, 124.46, 117.04, 108.21, 32.55, 21.44, 16.39. IR (KBr, cm−1): ν 3419 (NH), 3150 (CH), 2923, 2835 (CH), 1682 (CO), 1548 (CC), 1342 (NN), 1164 (COC). HRMS: m/z = 333.1029 [M + H]+.
N′-(4-(4-Methoxyphenyl)furan-2-yl)-1-phenylcyclopropane-1-carbohydrazide (8b). White solid, yields 84%, Mp 150–151 °C. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.12 (s, 1H, CONH), 8.30 (s, 1H, NH), 7.90 (s, 1H, ArH), 7.64 (d, 2H, J = 9.8 Hz, ArH), 7.45 (m, 2H, ArH), 7.25 (m, 3H, ArH), 7.02 (d, 2H, J = 9.8 Hz, ArH), 6.62 (s, 1H, ArH), 3.70 (s, 3H, OCH3), 2.04 (m, 2H, CH2), 1.80 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 181.50, 161.66, 145.74, 142.11, 141.29, 137.35, 131.69, 130.68, 128.17, 128.04, 127.15, 122.03, 116.89, 108.15, 55.69, 31.67, 16.60. IR (KBr, cm−1): ν 3424 (NH), 2922, 2853 (CH), 1693 (CO), 1584, 1510 (CC), 1340 (NN), 1152 (COC). HRMS: m/z = 349.0694 [M + H]+.
N′-(4-(3,4-Dimethoxyphenyl)furan-2-yl)-1-phenylcyclopropane-1-carbohydrazide (8c). White solid, yields 80%, Mp 148–150 °C. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.46 (s, 1H, CONH), 8.52 (s, 1H, NH), 7.86 (s, 1H, ArH), 7.62 (d, 1H, J = 10.0 Hz, ArH), 7.40 (m, 2H, ArH), 7.18 (m, 3H, ArH), 7.10 (s, 1H, ArH), 6.79 (d, 1H, J = 10.0 Hz, ArH), 6.65 (s, 1H, ArH), 3.88 (s, 3H, OCH3), 3.82 (s, 3H, OCH3), 1.64 (m, 2H, CH2), 1.37 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 182.02, 150.75, 149.13, 145.36, 138.02, 137.59, 134.57, 132.78, 127.58, 127.10, 123.32, 122.06, 114.40, 108.14, 55.61, 55.48, 32.09, 16.41. IR (KBr, cm−1): ν 3425 (NH), 3071 (CH), 2922, 2850 (CH), 1725 (CO), 1597, 1515 (CC), 1330 (NN), 1206, 1150 (COC). HRMS: m/z = 379.1701 [M + H]+.
N′-(4-(2,4-Dimethoxyphenyl)furan-2-yl)-1-phenylcyclopropane-1-carbohydrazide (8d). White solid, yields 78%, Mp 137–139 °C. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.49 (s, 1H, CONH), 8.33 (s, 1H, NH), 7.80 (s, 1H, ArH), 7.63 (d, 1H, J = 10.0 Hz, ArH), 7.45 (m, 2H, ArH), 7.39 (m, 3H, ArH), 7.08 (s, 1H, ArH), 7.02 (d, 1H, J = 10.0 Hz, ArH), 6.82 (s, 1H, ArH), 3.69 (s, 3H, OCH3), 3.55 (s, 3H, OCH3), 1.70 (m, 2H, CH2), 1.43 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 182.78, 163.57, 161.60, 143.91, 138.26, 137.25, 131.62, 131.63, 128.08, 128.01, 127.04, 124.48, 117.01, 106.83, 104.20, 98.26, 61.37, 58.78, 31.26, 16.64. IR (KBr, cm−1): ν 3393 (NH), 2925, 2852 (CH), 1678 (CO), 1589, 1510 (CC), 1326 (NN), 1154 (COC). HRMS: m/z = 379.0821 [M + H]+.
N′-(4-(4-(Dimethylamino)phenyl)furan-2-yl)-1-phenylcyclopropane-1-carbohydrazide (8e). Pale yellow solid, yields 82%, Mp 167–169 °C. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.32 (s, 1H, CONH), 8.32 (s, 1H, NH), 7.78 (s, 1H, ArH), 7.62 (d, 2H, J = 9.8 Hz, ArH), 7.47 (m, 2H, ArH), 7.33 (m, 3H, ArH), 7.22 (d, 2H, J = 9.8 Hz, ArH), 6.83 (s, 1H, ArH), 3.12 (s, 6H, N(CH3)2), 1.95 (m, 2H, CH2), 1.79 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 180.57, 152.81, 147.97, 144.39, 140.10, 134.50, 132.22, 128.03, 126.51, 125.18, 124.42, 117.23, 114.45, 108.34, 45.89, 31.39, 17.23. IR (KBr, cm−1): ν 3414, 3308 (NH), 2928, 2851 (CH), 1716 (CO), 1604 (CC), 1344 (NN), 1180 (COC). HRMS: m/z = 362.1281 [M + H]+.
N′-(4-(4-Hydroxyphenyl)furan-2-yl)-1-phenylcyclopropane-1-carbohydrazide (8f). White solid, yields 88%, Mp 199–200 °C. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.35 (s, 1H, CONH), 9.50 (s, 1H, OH), 8.18 (s, 1H, NH), 7.87 (s, 1H, ArH), 7.64 (d, 2H, J = 9.8 Hz, ArH), 7.42 (m, 2H, ArH), 7.34 (m, 3H, ArH), 6.89 (d, 2H, J = 9.8 Hz, ArH), 6.68 (s, 1H, ArH), 1.87 (m, 2H, CH2), 1.66 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 180.30, 157.29, 146.24, 145.07, 139.23, 138.21, 137.46, 134.55, 132.79, 129.90, 127.55, 123.31, 116.39, 108.30, 32.79, 16.41. IR (KBr, cm−1): ν 3422 (OH), 3361 (NH), 2923 (CH), 1679 (CO), 1524 (CC), 1351 (NN), 1152 (COC). HRMS: m/z = 335.0977 [M + H]+.
N′-(4-(4-Chloro-2-methoxyphenyl)furan-2-yl)-1-phenylcyclopropane-1-carbohydrazide (8g). White solid, yields 89%, Mp 137–139 °C. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.40 (s, 1H, CONH), 8.35 (s, 1H, NH), 7.86 (s, 1H, ArH), 7.69 (d, 1H, J = 10.0 Hz, ArH), 7.48 (m, 2H, ArH), 7.25 (s, 1H, ArH), 7.18 (m, 3H, ArH), 6.98 (d, 1H, J = 10.0 Hz, ArH), 6.45 (s, 1H, ArH), 3.86 (s, 3H, OCH3), 1.85 (m, 2H, CH2), 1.67 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 182.79, 157.24, 148.25, 144.06, 138.26, 137.45, 134.53, 132.78, 128.04, 127.50, 124.45, 123.33, 117.04, 114.20, 108.05, 55.81, 31.37, 17.43. IR (KBr, cm−1): ν 3405 (NH), 3074 (CH), 2920, 2852 (CH), 1713 (CO), 1562, 1509 (CC), 1353 (NN), 1201 (COC). HRMS: m/z = 383.1540 [M + H]+, 384.1274 [M + 2H]+.
1-(4-Nitrophenyl)-N′-(4-(p-tolyl)furan-2-yl)cyclopropane-1-carbohydrazide (8h). Light yellow solid, yields 85%, Mp 171–173 °C. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.36 (s, 1H, CONH), 8.38 (s, 1H, NH), 8.20 (d, 2H, J = 9.8 Hz, ArH),7.89 (s, 1H, ArH), 7.75 (d, 2H, J = 9.8 Hz, ArH), 7.24 (d, 2H, J = 9.8 Hz, ArH), 7.15 (d, 2H, J = 9.8 Hz, ArH), 6.46 (s, 1H, ArH), 2.27 (s, 3H, CH3), 1.64 (m, 2H, CH2), 1.48 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 180.17, 157.27, 148.19, 144.42, 141.27, 137.43, 134.56, 134.42, 132.79, 130.14, 128.91, 127.52, 127.16, 123.38, 107.56, 32.79, 23.38, 17.16. IR (KBr, cm−1): ν 3415 (NH), 3063 (CH), 2927, 2858 (CH), 1721 (CO), 1610, 1556 (CC), 1339 (NN), 1156 (COC). HRMS: m/z = 378.0494 [M + H]+.
N′-(4-(4-Methoxyphenyl)furan-2-yl)-1-(4-nitrophenyl)cyclopropane-1-carbohydrazide (8i). Light yellow solid, yields 81%, Mp 170–172 °C. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.30 (s, 1H, CONH), 8.40 (s, 1H, NH), 8.28 (d, 2H, J = 9.8 Hz, ArH), 7.86 (s, 1H, ArH), 7.72 (m, 4H, ArH), 7.10 (d, 2H, J = 9.8 Hz, ArH), 6.64 (s, 1H, ArH), 3.88 (s, 3H, OCH3), 1.70 (m, 2H, CH2), 1.61 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 180.38, 160.34, 153.30, 145.39, 144.16, 138.35, 137.69, 134.80, 134.15, 130.02, 129.93, 127.41, 114.35, 107.27, 56.04, 30.12, 17.27. IR (KBr, cm−1): ν 3419 (NH), 3057 (CH), 2928 (CH), 1712 (CO), 1585 (CC), 1330 (NN), 1208 (COC). HRMS: m/z = 394.1288 [M + H]+.
N′-(4-(3,4-Dimethoxyphenyl)furan-2-yl)-1-(4-nitrophenyl)cyclopropane-1-carbohydrazide (8j). Light yellow solid, yields 72%, Mp 174–176 °C. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.19 (s, 1H, CONH), 8.38 (s, 1H, NH), 8.24 (d, 2H, J = 9.8 Hz, ArH), 7.85 (s, 1H, ArH), 7.68 (d, 2H, J = 9.8 Hz, ArH), 7.30 (d, 1H, J = 10.0 Hz, ArH), 7.20 (d, 1H, J = 10.0 Hz, ArH), 7.12 (s, 1H, ArH), 6.62 (s, 1H, ArH), 3.86 (s, 3H, OCH3), 3.77 (s, 3H, OCH3), 1.87 (m, 2H, CH2), 1.69 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 181.23, 151.46, 148.72, 144.37, 140.18, 131.52, 130.29, 129.13, 127.58, 125.55, 124.80, 123.02, 116.27, 115.48, 108.14, 56.73, 31.45, 17.33. IR (KBr, cm−1): ν 3411 (NH), 3082 (CH), 2929, 2855 (CH), 1729 (CO), 1574 (CC), 1330 (NN), 1132 (COC). HRMS: m/z = 424.1436 [M + H]+.
N′-(4-(2,4-Dimethoxyphenyl)furan-2-yl)-1-(4-nitrophenyl)cyclopropane-1-carbohydrazide (8k). Light yellow solid, yields 69%, Mp 163–165 °C. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.59 (s, 1H, CONH), 8.40 (s, 1H, NH), 8.26 (d, 2H, J = 9.8 Hz, ArH), 7.86 (d, 1H, J = 10.0 Hz, ArH), 7.75 (d, 2H, J = 9.8 Hz, ArH), 7.67 (s, 1H, ArH), 6.92 (s, 1H, ArH), 6.85 (d, 1H, J = 10.0 Hz, ArH), 6.70 (s, 1H, ArH), 3.84 (s, 3H, OCH3), 3.71 (s, 3H, OCH3), 1.77 (m, 2H, CH2), 1.63 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 182.18, 163.36, 162.38, 149.72, 148.66, 143.93, 132.47, 131.62, 130.36, 130.06, 129.37, 127.69, 126.75, 123.93, 115.98, 108.76, 99.34, 55.20, 54.77, 31.52, 18.76. IR (KBr, cm−1): ν 3331 (NH), 3022 (CH), 2844 (CH), 1708 (CO), 1603 (CC), 1344 (NN), 1173 (COC). HRMS: m/z = 424.1288 [M + H]+.
N′-(4-(4-(Dimethylamino)phenyl)furan-2-yl)-1-(4-nitrophenyl)cyclopropane-1-carbohydrazide (8l). Yellow solid, yields 78%, Mp 190–191 °C. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.48 (s, 1H, CONH), 8.35 (s, 1H, NH), 8.18 (d, 2H, J = 9.8 Hz, ArH), 7.88 (s, 1H, ArH), 7.74 (d, 2H, J = 9.8 Hz, ArH), 7.37 (d, 2H, J = 9.8 Hz, ArH), 6.89 (d, 2H, J = 9.8 Hz, ArH), 6.58 (s, 1H, ArH), 2.93 (s, 6H, N(CH3)2), 1.71 (m, 2H, CH2), 1.52 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 181.20, 154.07, 149.22, 143.77, 141.89, 134.35, 131.91, 131.56, 130.12, 127.38, 127.25, 124.48, 124.36, 123.74, 115.51, 107.09, 43.57, 31.07, 17.09. IR (KBr, cm−1): ν 3415 (NH), 3001 (CH), 2813 (CH), 1726 (CO), 1600, 1514 (CC), 1342 (NN), 1176 (COC). HRMS: m/z = 407.1385 [M + H]+.
N′-(4-(4-Hydroxyphenyl)furan-2-yl)-1-(4-nitrophenyl)cyclopropane-1-carbohydrazide (8m). Light yellow solid, yields 74%, Mp 211–213 °C. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.45 (s, 1H, CONH), 9.80 (s, 1H, OH), 8.38 (s, 1H, NH), 8.19 (d, 2H, J = 9.8 Hz, ArH), 7.89 (s, 1H, ArH), 7.69 (d, 2H, J = 9.8 Hz, ArH), 7.59 (d, 2H, J = 9.8 Hz, ArH), 6.92 (d, 2H, J = 9.8 Hz, ArH), 6.64 (s, 1H, ArH), 1.74 (m, 2H, CH2), 1.53 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 182.20, 157.89, 148.66, 141.70, 132.45, 131.69, 130.17, 129.85, 129.13, 127.07, 123.51, 122.46, 119.75, 116.87, 115.39, 107.18, 31.09, 17.37. IR (KBr, cm−1): ν 3452 (OH), 3089 (CH), 2813 (CH), 1666 (CO), 1600, 1519 (CC), 1350 (NN), 1176 (COC). HRMS: m/z = 380.1586 [M + H]+.
N′-(4-(4-Chloro-2-methoxyphenyl)furan-2-yl)-1-(4-nitrophenyl)cyclopropane-1-carbohydrazide (8n). Light yellow solid, yields 86%, Mp 141–143 °C. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.14 (s, 1H, CONH), 8.41 (s, 1H, NH), 8.24 (d, 2H, J = 9.8 Hz, ArH), 7.87 (s, 1H, ArH), 7.75 (d, 2H, J = 9.8 Hz, ArH), 7.62 (d, 1H, J = 10.0 Hz, ArH), 7.33 (s, 1H, ArH), 7.25 (d, 1H, J = 10.0 Hz, ArH), 6.80 (s, 1H, ArH), 3.80 (s, 3H, OCH3), 1.58 (m, 2H, CH2), 1.32 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 182.46, 160.38, 151.19, 149.38, 144.67, 142.45, 134.47, 132.11, 131.68, 130.19, 127.22, 127.09, 124.44, 123.74, 116.01, 107.42, 56.80, 31.86, 17.42. IR (KBr, cm−1): ν 3271 (NH), 3014 (CH), 2854 (CH), 1685 (CO), 1585, 1517 (CC), 1342 (NN), 1184, 1114 (COC), 854 (CCl). HRMS: m/z = 428.0627 [M + H]+, 429.0852 [M + 2H]+.
N′-(4-(4-Chlorophenyl)furan-2-yl)-1-(4-nitrophenyl)cyclopropane-1-carbohydrazide (8o). Light yellow solid, yields 91%, Mp 163–165 °C. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 10.17 (s, 1H, CONH), 8.37 (s, 1H, NH), 8.19 (d, 2H, J = 9.8 Hz, ArH), 7.77 (s, 1H, ArH), 7.68 (d, 2H, J = 9.8 Hz, ArH), 7.54 (d, 2H, J = 9.8 Hz, ArH), 7.41 (d, 2H, J = 9.8 Hz, ArH), 6.86 (s, 1H, ArH), 1.64 (m, 2H, CH2), 1.38 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 181.39, 152.44, 148.22, 146.27, 134.28, 131.32, 130.39, 129.23, 127.98, 125.51, 124.82, 124.10, 123.03, 116.25, 115.45, 106.73, 31.32, 17.39. IR (KBr, cm−1): ν 3274 (NH), 3016 (CH), 2843 (CH), 1684 (CO), 1580, 1510 (CC), 1340 (NN), 1186 (COC), 856 (CCl). HRMS: m/z = 398.0703 [M + H]+, 399.1243 [M + 2H]+.The experimental procedure for biological evaluation, docking protocol, and spectroscopic copies of the 1H/13C NMR, HRMS, and IR spectra (Fig. S1–S60) are included in the SI documentation; the data supporting this study's findings are included in its SI.
Conclusion
In conclusion, a number of furan-containing cyclopropane-1-carbohydrazide derivatives were developed, and their anticancer properties were assessed in vitro. High-resolution mass spectrometry (HRMS), infrared spectroscopy, and nuclear magnetic resonance spectroscopy (1H/13C) were used to analyze the resultant molecules. All the substances described exhibited good activity against liver, lung, and breast cancers. The strongest antiproliferative action was demonstrated by compounds 8e, 8i, and 8m, which had IC50 values of 4.58 ± 0.07, 2.58 ± 0.27, and 2.67 ± 0.52 µM, respectively. In comparison to erlotinib (0.94 µM), the investigated drugs exhibited moderate to good EGFR inhibition, with IC50 inhibition ranging from 0.87 to 2.70 µM. Molecular docking studies revealed strong binding affinities of these compounds to the epidermal growth factor receptor (EGFR), particularly through HB-interactions, π–π stacking, and hydrophobic interactions, with 8m demonstrating the lowest binding energy (−8.4 kcal mol−1). Additionally, the pharmacokinetic parameters of the synthesised hybrids were assigned using the SwissADME, ADMETlab3.0, and Protox-II online tools. To support the experimental portion of the investigation, computational studies were conducted to create precursor chemicals. Therefore, it is hypothesized that chemical scaffolds have produced novel single molecules with anticancer properties, at least in part, by targeting the EGFR enzyme. The analogues revealed in this study should support international efforts to find potential lead compounds for the subsequent development of novel entities with anticancer inhibitory characteristics.
Ethical statement
This study involved only established human cell lines and did not include experiments on human participants or animals. All procedures were conducted in accordance with institutional biosafety and laboratory practice guidelines.
Author contributions
Suresh Patagani: methodology, investigation, visualization, and writing – original draft. Kolli Balakrishna: supervision, and writing – review & editing. Mahadevappa Naganathappa: formal analysis. Dhilli Rao Gorja: resources, and funding acquisition. Naresh Kumar Katari: validation, funding acquisition, and writing – review & editing. Vani Madhuri Velavalapalli: software, and data curation. Rambabu Gundla: conceptualization, and project administration.
Conflicts of interest
The authors declare that no competing interests.
Data availability
All data supporting the findings of this study are available in the manuscript or supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5ra09654e.
Acknowledgements
RG acknowledges GITAM for providing financial assistance and facilities.
References
- R. L. Siegel, T. B. Kratzer, A. N. Giaquinto, H. Sung and A. Jemal, Cancer statistics, 2025, Ca-Cancer J. Clin., 2025, 75(1), 10–45 CrossRef PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, Cancer statistics, 2015, Chin. J. Cancer, 2015, 65(1), 5–29 CrossRef PubMed.
- P. Mathur, K. Sathishkumar, M. Chaturvedi, P. Das, K. L. Sudarshan and S. Santhappan, et al., Cancer statistics, 2020: report from national cancer registry programme, India JCO Glob. Oncol., 2020, 6, 1063–1075 CrossRef PubMed.
- M. Danihelová, M. Veverka, E. Šturdík and S. Jantová, Antioxidant action and cytotoxicity on HeLa and NIH-3T3 cells of new quercetin derivatives, Interdiscip. Toxicol., 2013, 6(4), 209–216 Search PubMed.
- A. M. El-Naggar, A. Zidan, E. B. Elkaeed, M. S. Taghour and W. A. Badawi, Design, synthesis and docking studies of new hydrazinyl-thiazole derivatives as anticancer and antimicrobial agents, J. Saudi Chem. Soc., 2022, 26(4), 101488 CrossRef CAS.
- A. Ayati, S. Emami, S. Moghimi and A. Foroumadi, Thiazole in the targeted anticancer drug discovery, Future Med. Chem., 2019, 11(16), 1929–1952 CrossRef CAS PubMed.
- N. S. Habib, R. Soliman, A. A. El-Tombary, S. A. El-Hawash and O. G. Shaaban, Synthesis and biological evaluation of novel series of thieno [2, 3-d] pyrimidine derivatives as anticancer and antimicrobial agents, Med. Chem. Res., 2013, 22, 3289–3308 CrossRef CAS.
- C. J. Thibodeaux, W. Chang and H. Liu, Enzymatic chemistry of cyclopropane, epoxide, and aziridine biosynthesis, Chem. Rev., 2012, 112, 1681–1709 CrossRef CAS PubMed.
- T. T. Talele, The “cyclopropyl fragment” is a versatile player that frequently appears in preclinical/clinical drug molecules, J. Med. Chem., 2016, 59, 8712–8756 CrossRef CAS PubMed.
- H. Abe, S. Kikuchi, K. Hayakawa, T. Iida, N. Nagahashi, K. Maeda, J. Sakamoto, N. Matsumoto, T. Miura and K. Matsumura, et al., Discovery of a highly potent and selective MEK inhibitor: GSK1120212 (JTP-74057 DMSO solvate), ACS Med. Chem. Lett., 2011, 2, 320–324 CrossRef CAS PubMed.
- A. Oubella, M. Fawzi, A. Auhmani, A. Riahi, H. Morjani, A. Robert and M. Y. Ait Itto, Synthesis and antitumor activity of novel heterocyclic systems with monoterpenic skeleton combining dichlorocyclopropane and 1,3,4-thiadiazole nucleus, ChemistrySelect, 2020, 5, 6403–6406 CrossRef CAS.
- C. Köllmann, S. M. Wiechert, P. G. Jones, T. Pietschmann and D. B. Werz, Synthesis of 4′/5′-spirocyclopropanated uridine and d-xylouridine derivatives and their activity against the human respiratory syncytial virus, Org. Lett., 2019, 21, 6966–6971 CrossRef PubMed.
- T. V. Kharlamova, A. V. Gabdrakipov, P. B. Seidakhmetova and K. D. Praliyev, Antibacterial activity of synthesized derivatives of purpurin containing cyclopropane and cyclobutane fragment, Eurasian Chem.-Technol. J., 2020, 22, 213–218 CrossRef CAS.
- D. Du, C. Ji, S. Zheng, Y. Chen, H. Cai, Z. Li, D. Yan and H. Teng, Synthesis of difluorocyclopropanes via visible-light-mediated [1+2] cycloaddition of diazo esters with gem-difluoroalkenes and evaluation of their antifungal activity, Adv. Synth. Catal., 2024, 366, 1738–1743 CrossRef CAS.
- S. Ulrich, R. Ricken, P. Buspavanich, P. Schlattmann and M. Adli, Efficacy and adverse effects of tranylcypromine and tricyclic antidepressants in the treatment of depression: A systematic review and comprehensive meta-analysis, J. Clin. Psychopharmacol., 2020, 40, 63–74 CrossRef PubMed.
- M. Pourshab, S. Asghari, M. Tajbakhsh and A. Khalilpour, Diastereoselective sonochemical synthesis of spirocyclopropaneoxindoles and evaluation of their antioxidant and cytotoxic activities, Chem. Biodivers., 2019, 16, e1900087 CrossRef PubMed.
- P. Patel, R. Shakya, Vishakha, V. Asati, B. Das Kurmi, S. K. Verma, G. D. Gupta and H. Rajak, Furan and benzofuran derivatives as privileged scaffolds as anticancer agents: SAR and docking studies (2010 to till date), J. Mol. Struct., 2024, 1299, 137098 CrossRef CAS.
- S. Shukla, A. Srivastava, P. Tandon, J. Jamalis and R. B. Singh, Structural properties of a novel heterocyclic chalcones derivative, (E)-3-(5-methyl furan-2-yl)-1-phenyl prop-2-en-1-one: A spectroscopic and DFT perception, J. Mol. Struct., 2021, 1244, 130973 CrossRef CAS.
- H. Kwiecień, M. Perużyńska, K. Stachowicz, K. Piotrowska, J. Bujak, P. Kopytko and M. Droździk, Synthesis and biological evaluation of 3-functionalized 2-phenyl- and 2-alkylbenzo[b]furans as antiproliferative agents against human melanoma cell line, Bioorg. Chem., 2019, 88, 102930 CrossRef PubMed.
- Z.-Y. Qi, H. Shu-Yi, H.-Z. Tian, H.-L. Bian, L. Hui and S.-W. Chen, Synthesis and biological evaluation of 1-(benzofuran-3-yl)-4-(3,4,5-trimethoxyphenyl)-1H-1,2,3-triazole derivatives as tubulin polymerization inhibitors, Bioorg. Chem., 2020, 94, 103392 CrossRef CAS PubMed.
- R. R. Ruddarraju, A. C. Murugulla, R. Kotla, M. C. B. Tirumalasetty, R. Wudayagiri, S. Donthabakthuni and R. Maroju, Medchemcomm, 2017, 8, 176–183 RSC.
- A. K. Singh, A. Kumar, H. Singh, P. Sonawane, H. Paliwal and S. Thareja, et al., Concept of hybrid drugs and recent advancements in anticancer hybrids, Pharmaceuticals, 2022, 15(9), 1071 CrossRef CAS PubMed.
- S. Patagani, K. Balakrishna, N. Kumar Katari, M. Panasa and R. Gundla, ChemistrySelect, 2024, 9, e202402656 CrossRef CAS.
- S. T. Al-Rashood, A. R. Hamed, G. S. Hassan, H. M. Alkahtani, A. A. Almehizia, A. Alharbi, M. M. Al-Sanea and W. M. Eldehna, Antitumor properties of certain spiro oxindoles towards hepatocellular carcinoma endowed with antioxidant activity, J. Enzyme Inhib. Med. Chem., 2020, 35(1), 831–839 CrossRef CAS PubMed.
- M. Thabrew, R. D. Hughes and I. G. Mcfarlane, Screening of hepatoprotective plant components using a HepG2 cell cytotoxicity assay, J. Pharm. Pharmacol., 1997, 49(11), 1132–1135 CrossRef CAS PubMed.
- Q. Liu, EGFR-TKIs resistance via EGFR-independent signaling pathways, Mol. Cancer, 2018, 17, 53 CrossRef PubMed.
- J. Stamos, M. X. Sliwkowski and C. Eigenbrot, Structure of the Epidermal Growth Factor Receptor Kinase Domain Alone and in Complex with a 4-Anilinoquinazoline Inhibitor, J. Biol. Chem., 2002, 277(48), 46265–46272 CrossRef CAS PubMed.
- M. Mazumdar, et al.. “Binary and Ternary Crystal Structure Analyses of a Novel Inhibitor with 17β-HSD Type 1: A Lead Compound for Breast Cancer Therapy, Biochem. J., 2009, 424(3), 357–366 CrossRef CAS PubMed.
- K. R. Cousins, J. Am. Chem. Soc., 2011, 133(21), 8388 CrossRef CAS PubMed.
- S. Dallakyan and A. J. Olson, Small-Molecule Library Screening by Docking with PyRx, Methods Mol. Biol., 2015, 1263, 243–250 CrossRef CAS PubMed.
- B. K. Park, A. Boobis, S. Clarke, C. E. Goldring, D. Jones, J. G. Kenna, C. Lambert, H. G. Laverty, D. J. Naisbitt and S. Nelson, Managing the challenge of chemically reactive metabolites in drug development, Nat. Rev. Drug Discov., 2011, 10(4), 292–306 CrossRef CAS PubMed.
- S. M. Huang, J. M. Strong, L. Zhang, K. S. Reynolds, S. Nallani, R. Temple, S. Abraham, S. A. Habet, R. K. Baweja and G. J. Burckart, New era in drug interaction evaluation: US Food and Drug Administration update on CYP enzymes, transporters, and the guidance process, J. Clin. Pharmacol., 2008, 48(6), 662–670 CrossRef CAS PubMed.
- A. B. Reddy, T. R. Allaka, V. S. R. Avuthu, K. Chepuri, M. Z. Ahmed and H. Nagarajaiah, New Quinazolinone-1,2,4-Triazole Analogues: Synthesis, Anticancer Evaluation, Molecular Docking, and In Silico ADMET Prediction, J. Mol. Struct., 2025, 1334, 141850 CrossRef CAS.
- P. Banerjee, E. Kemmler, M. Dunkel and R. Preissner, ProTox 3.0: a webserver for the prediction of toxicity of chemicals, Nucleic Acids Res., 2024, 52(W1), W513–W520 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *df,
Vani Madhuri Velavalapalli
*df,
Vani Madhuri Velavalapalli













