DOI:
10.1039/D5RA09444E
(Paper)
RSC Adv., 2026,
16, 18187-18198
Porous hydroxyapatite microparticles loaded with 169Er for treatment of chronic inflammatory disease of small joints: harnessing mechanochemistry for synthesis of an advanced biomaterial
Received
6th December 2025
, Accepted 20th March 2026
First published on 7th April 2026
Abstract
In the present study, nanoporous hydroxyapatite microparticles (HA MPs) were synthesized using a D(+)-glucose template mediated solid state mechanochemical approach. The synthesized nanoporous HA MPs were extensively characterized and found to have a particle size distribution in the range of 1–10 µm. The nanoporous HA MPs demonstrated high sorption capacity for 169Er (285 ± 8 mg g−1), which in turn facilitated loading of clinically relevant doses of low specific activity 169Er produced in a medium neutron flux research reactor. The sorption of 169Er in nanoporous HA followed the Langmuir–Freundlich adsorption isotherm and pseudo second order kinetics. The details of the sorption mechanism were investigated using density functional theory (DFT)-based calculations. The 169Er-loaded nanoporous HA ([169Er]Er-HA) MPs retained their radiochemical integrity (>98%) over a period of 14 days in physiological media. As a proof of concept, [169Er]Er-HA MPs were administered intra-articularly in one of the ankle joints of healthy Wistar rats. The homogeneous diffusion of the MPs formulation in the joint cavity was ascertained by SPECT/CT imaging and ex vivo biodistribution studies. Overall, the suitability of nanoporous HA MPs synthesized by a solid state route for loading low specific activity therapeutic radionuclides was established, which makes them an ideal agent for treatment of inflammatory small joint disorders and other ailments.
1. Introduction
Synthesis of porous biomaterials has gained huge momentum over the last few years because of the excellent capability of this class of materials to interact with atoms, ions and molecules not only at their surfaces but throughout the bulk leading to their suitability for a wide range of biomedical applications.1–4 Among various biomaterials studied, hydroxyapatite (HA) has gained remarkable attention due to its excellent physical, chemical, and biological properties.5–9 The most interesting aspects favoring use of HA are its unique properties such as biocompatibility, osteo-reconstruction conductivity and bone integration. Owing to these attributes, HA has been extensively employed in a substantive role as a drug delivery vehicle, coating material, dental and bone implant, scaffold material in tissue engineering and antimicrobial membrane.10–16 The increasing clinical demand of HA in various biomedical applications has sparked rapid interest towards synthesis of engineered materials with porous structures, which exhibit higher bone forming ability and greater drug loading capacity.5,8
The porosity in a material can be introduced by using various methods such as phase separation, salt leaching, freeze-drying, gas foaming, sintering and the use of sacrificial templates.17–21 One of the most viable approaches to introduce porosity in HA is the use of sacrificial templates in which HA is synthesized in presence of organic molecules which can subsequently be removed by burning off the synthesized material.22–24 This process controls the surface area, customize pore size and volume of the HA matrix, and generate stable, cohesive framework with active pore surface.25 This in turn would facilitate the conversion of surface-active sites or functionalization, thereby fulfilling the criteria of an ‘advanced biomaterial’ for diverse applications. The commonly used strategies using sacrificial templates for synthesis of porous biomaterials are usually time and energy consuming and sometimes lead to generation of large volume of liquid waste. Additionally, scalability of many such processes might be an issue which negatively impacts their translational prospects. An alternate solution is the use of environment friendly mechanochemical synthesis, which is suitable for large-scale production of functional materials.26 Mechanochemistry is not a new concept and has been applied in various areas such as metallurgy, mineral processing and synthesis of organic compounds for the last several years.27–29 The advantages of mechanochemical synthesis include its solvent-free nature, which significantly reduces the environmental impact and precludes the need of hazardous chemicals. Moreover, mechanochemical process consumes minimal energy and time compared to other conventional methods, making it more cost-effective. Additionally, mechanochemistry allows precise control over reaction conditions, resulting in the formation of high-quality, reproducible materials. These features make it an attractive option for large-scale, sustainable production in various industrial sectors. Of late, this field is again gaining momentum and is experiencing a renaissance because of its successful implementation in synthesis of new generation of biomaterials thereby demonstrating its tremendous potential for chemical and pharmaceutical industries.
Size controlled inorganic/organic MPs loaded with suitable therapeutic radionuclides are used in nuclear medicine procedures as radiation synovectomy (RSV) agents for treatment of chronic inflammatory joint disorders and have been successfully translated for routine clinical use in few countries.8,30–35 In this procedure, suitable β− emitting radionuclides are loaded in MPs of the size range 1–10 µm and the formulation is administered intra-articularly into the inflamed joint under fluoroscopic or ultrasonic guidance. Despite excellent attributes in relieving pain, a major problem associated with RSV is the unwanted radiation dose to non-targeted organs primarily, the liver, lymph nodes and spleen. This is a consequence of leakage of smaller sized particles via capillary fenestration or leakage of radioactivity from the MPs leading to uptake in healthy organs.36,37 Use of HA MPs of required size range (1–10 µm) as the carrier matrix of RSV agent has the potential to circumvent most of these issues because it is radiation resistant and several therapeutic radionuclides, especially radiolanthanides can be irreversibly loaded in the HA matrix.8,38
Various radionuclides have been proposed for RSV of different types of inflammatory joint disorders.39,40 Among them, 169Er [T½ = 9.4 days, Eβ-(max) = 342 keV (45%) and 351 keV (55%), Eγ = 110.5 keV (0.0014%)] is the most preferred one for treatment of digital joints of fingers.41 This is attributed to the suitable half-life of 169Er and emission of two low energy β− particles having maximum soft tissue penetration of 1 mm, with a mean range between 0.2 and 0.3 mm. Erbium-169 can be produced in a nuclear research reactor by thermal neutron capture via 168Er (n, γ) 169Er reaction.42 Because of the low thermal cross section of 168Er (σ = 1.95 b), 169Er will be produced in low specific activity even in reactors with moderate to high thermal neutron flux. Therefore, commercially available bulk HA becomes inappropriate for loading of low specific activity 169Er due to its low sorption capacity. This mandates the need for synthesis of porous HA having high sorption capacity so that it can be used to prepare RSV agents using 169Er produced in medium flux nuclear reactors (ϕ ∼ 1014 n cm−2 s−1). According to the IAEA database, such reactors are widely available with good geographical distribution potentially favoring widescale use of 169Er in nuclear medicine practices worldwide.43
In the present study, we report the synthesis of nanoporous HA by solid state mechanochemical approach using glucose as a template. The conditions for loading of 169Er in nanoporous HA matrix was optimized and the in vitro and in vivo stabilities of the radioactive MPs were established. To understand the sorption mechanism of 169Er on surface of nanoporous HA, a DFT based calculation was performed and subsequently sorption isotherms and kinetics were determined to corroborate the theoretical findings. Finally, as a proof of concept, SPECT/CT images were acquired post administration of [169Er]Er-HA MPs in the ankle joints of healthy Wistar rats to visualize the retention of the radioactive formulation in the synovial cavity. The overall schematic illustration of the synthesis procedure of nanoporous HA MPs, loading of 169Er in nanoporous HA MPs and their potential application for RSV is depicted in Fig. 1A.
 |
| | Fig. 1 (A) Schematic illustration of the solid state mechanochemical synthesis of nanoporous HA MPs, loading of 169Er in the HA matrix and their potential application in RSV of small joint arthritis. (B) (i) XRD pattern of nanoporous HA MPs (ii) FT-IR spectra and (iii) Raman spectra of nanoporous HA MPs. | |
2. Experimental
2.1 Chemicals
Calcium nitrate tetrahydrate (≥99%), diammonium hydrogen phosphate (≥99%), sodium hydroxide (≥99%) and D(+)-glucose were purchased from Sigma-Aldrich. Isotopically enriched Er2O3 (98.2% in 168Er, >99.99%) used as the target material for production of 169Er were procured from Isoflex, Russia. Erbium oxide (Er2O3) of natural isotopic composition (>99.99%) was purchased from Sigma-Aldrich. For comparative studies, bulk HA MPs (5.8 ± 1.3 µm) was purchased from Sigma-Aldrich.
2.2 Synthesis of nanoporous HA MPs
A solid-state mechanochemical approach was employed for the synthesis of nanoporous HA MPs. In this approach, a mixture of 0.01 mol of calcium nitrate and 0.006 mol of diammonium hydrogen phosphate was prepared. Subsequently 0.036 mol of D-(+)-glucose and 0.01 mol of sodium hydroxide were added and the resultant mixture was grinded thoroughly in an agate mortar for 2 h at room temperature (25 °C). Subsequently, the resulting mixture was transferred in a porcelain crucible and heated in a furnace at 700 °C for 2 h. The material was removed from the furnace after allowing it to attain room temperature. Then, the material was crushed mechanically and sieved with an automated sieved shaker (Lab India Analytical, India) to obtain particles in the range of 1–10 µm. Finally, the crushed and sieved material was taken for characterization using various analytical tools.
2.3 Characterization of nanoporous HA MPs
The formation of HA MPs was ascertained by powder X-ray diffraction (XRD) using monochromatized Cu-Kα radiation on a PANalytical X-ray diffractometer (X'pert PRO). For further confirmation, Raman spectra and Fourier transform infrared spectra (FT-IR) were recorded using STR-300, SEKI Technotron (Japan) micro-Raman spectrometer and Bruker ALPHA II ECO-ATR spectrophotometer, respectively. The morphology of nanoporous HA MPs was imaged using field emission scanning electron microscopy (FESEM) (Carl Zeiss, model: GeminiSEM 300) which was operated at a 2 kV accelerating voltage. High-resolution transmission electron microscopy (HRTEM) image was recorded using a Philips CM 200 TEM system to determine the lattice parameter as well as to demonstrate the polycrystalline nature of the nanoporous HA MPs. In order to determine the pore size on the surface of the synthesize HA, small-angle neutron scattering (SANS) experiments were carried out at MSANS facility of Dhruva research reactor at Bhabha Atomic Research Centre. The double crystal based MSANS facility cover the low Q (4π![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ/λ, 2θ is the scattering angle, λ is the wavelength of neutron). In order to cover the higher Q, a lab-based small-angle X-ray scattering (SAXS) facility was utilized. Thermal stability of the synthesized nanoporous HA MPs was investigated by thermogravimetric analysis (TGA) using Mettler Toledo TG/DSC STARe system. The particle size distribution of synthesized nanoporous HA MPs was determined using laser diffraction particle size analyzer (Cilas Particle Size, USA). The photoluminescence (PL) property of the Er-loaded nanoporous HA MPs was investigated by recording PL spectra using FLS1000 fluorescence spectrometer (Edinburgh Instruments, UK). The SPECT/CT images were recorded using GE Discovery NM/CT 670 scanner.
sinθ/λ, 2θ is the scattering angle, λ is the wavelength of neutron). In order to cover the higher Q, a lab-based small-angle X-ray scattering (SAXS) facility was utilized. Thermal stability of the synthesized nanoporous HA MPs was investigated by thermogravimetric analysis (TGA) using Mettler Toledo TG/DSC STARe system. The particle size distribution of synthesized nanoporous HA MPs was determined using laser diffraction particle size analyzer (Cilas Particle Size, USA). The photoluminescence (PL) property of the Er-loaded nanoporous HA MPs was investigated by recording PL spectra using FLS1000 fluorescence spectrometer (Edinburgh Instruments, UK). The SPECT/CT images were recorded using GE Discovery NM/CT 670 scanner.
2.4 Production of 169Er
Erbium-169 was produced by irradiation of enriched (98.2% in 168Er) Er2O3 target in the Dhruva reactor at Bhabha Atomic Research Centre at a thermal neutron flux of 1 × 1014 n cm−2 s−1 for 21 days. The radiochemical processing of irradiated target was carried out by dissolving it in 0.1 M HCl with gentle heating inside a lead shielded glove box fitted with mechanical tongs. The final product obtained was [169Er]ErCl3 solution in 0.01 M HCl, which was used in further studies. In order to determine the radionuclidic purity of [169Er]ErCl3, an aliquot of known volume of this solution was withdrawn in a glass vial and its volume was adjusted up to 1 mL using deionized water. Subsequently, the γ-spectrum of this solution was acquired using a pre-calibrated HPGe detector (Eurysis, Canberra), coupled with multichannel analyzer (MCA). The dead time of the detector was ensured to be <2%. The radiochemical purity of 169Er in the form of Er3+ ions was determined by radio-thin layer chromatography (radio-TLC) developed in ammonia:ethanol:water (1:10:20 v/v).42
As, γ-photon abundance of 169Er was too low to acquire any SPECT image, ∼5 mg of Er2O3 of natural isotopic composition was also irradiated at the same flux for the same time period so that appreciable amount of 171Er [T½ = 7.5 h, Eγ = 111.5 keV (21%), 124 keV (0.93%)] is co-produced with 169Er, which was used as imaging surrogate of 169Er during in vivo evaluation of radioactive nanoporous HA MPs in Wistar rats.
2.5 Optimization of conditions for loading of 169Er in nanoporous HA MPs
In order to load 169Er in nanoporous HA MPs, systematic optimization of involved parameters was carried out. For the optimization studies, 169Er produced from the irradiation of enriched (98.2% of 168Er) Er2O3 target was used. Yield of [169Er]Er-HA was determined at different concentrations of nanoporous HA (0.5–20 mg mL−1), while keeping the pH of the solution fixed (pH ∼6). For this, suspension of nanoporous HA MPs of various concentration (0.5–20 mg mL−1) were taken in vials where ∼185 MBq (5 mCi) activity of 169Er was added and mixed thoroughly. The resultant suspensions were incubated at room temperature for 1 h and centrifuged subsequently. The supernatant was carefully separated from the pellet of 169Er-loaded HA MPs and discarded. The radiolabeled microparticles left were washed and re-dispersed in 1 mL of physiological saline. The activity associated with [169Er]Er-HA MPs was measured using a dose calibrator and compared with the radioactivity of [169Er]ErCl3 solution added at radiolabeling step. The loading yield was determined using following formula:| |
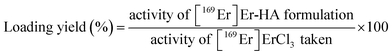 | (1) |
Similarly, loading yield was obtained at different pH of the incubation medium (3–7), while keeping the concentration of nanoporous HA fixed (5 mg mL−1).
2.6 Investigation of mechanism of Er-loading onto nanoporous HA MPs using DFT study
Vienna ab initio simulations package (VASP) has been used to perform all the spin polarized density functional theory (DFT) calculations.44–46 Generalized Gradient Approximation (GGA) of Perdew–Burke–Ernzerhof (PBE) has been used to treat the exchange–correlation energy density functional.47 Kohn–Sham equations of the valence electrons were expanded through using the plane-wave basis sets with 550 eV kinetic energy cutoff. Interaction between the valence and core electrons were treated through projector augmented wave (PAW) method.48 Dispersion interactions were incorporated into the total energies using the Grimme's D3 semi empirical method (PBE-D3) with Becke–Jonson damping.49 A vacuum layer of around 15 Å is considered along the non-periodic direction to minimize the image–image interactions. For charged systems, mono-pole corrections were applied to the total energies calculated. All the structural optimizations were carried out at constant volume with a force cut-off of 0.01 eV Å−1. Graphical software VESTA has been used to generate the reported figures of the optimized structures.50
2.7 Experimental determination of sorption isotherms and kinetics of loading of 169Er onto nanoporous HA MPs
The sorption capacity (qe, mg g−1) of nanoporous HA MPs for 169Er was determined by batch equilibrium method as reported earlier.8,13 For this, ∼5 mg of nanoporous HA was taken in a glass vial and equilibrated with 5 mL of ErCl3 solutions of various concentrations (0.125–1 mg mL−1) spiked with ∼370 kBq (10 µCi) of 169Er radioactivity in each. Then, the pH of these suspensions was adjusted to ∼6 and incubated at room temperature for 2 h. Subsequently, the suspensions were centrifuged at 5000 rpm for 20 min. Then the supernatants were withdrawn, the radioactivity associated with the supernatant was measured using a well type NaI(Tl) detector and compared with that of the standard solution taken from the reaction mixture before equilibration with nanoporous HA MPs. All the measurements were carried out in triplicate. The sorption capacity was calculated using the following expression:| |
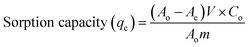 | (2) |
where Ao and Ae are the radioactivity of 169Er in 1 mL of supernatant solution before and after sorption, respectively, Co was the total Er content in 1 mL of solution before sorption, V was the volume of solution and m was the mass (g) of nanoporous HA MPs. Subsequently, equilibrium concentration of Er3+ (Ce) was determined for each initial concentration of Er3+. Then, qe and Ce values were converted into required algebraic form and fitted linearly in to the various isotherm. In order to determine the kinetics of sorption of Er3+ on the surface of nanoporous HA, sorption capacity was determined at various time intervals for a fixed concentration of Er3+ (0.25 mg mL−1) spiked with ∼370 kBq (10 µCi) of 169Er, as described above.
2.8 In vitro stability [169Er]Er-HA MPs
Under the optimized conditions, [169Er]Er-HA MPs formulation was prepared. At the same time a ‘blank’ solution of equal volume was prepared with same amount of activity of [169Er]ErCl3 without adding nanoporous HA MPs. Then [169Er]Er-HA MPs formulation was centrifuged at 5000 rpm for 20 min and a measured aliquot (100 µL) was withdrawn from the supernatant and measured the activity (R) using same NaI(Tl) detector using earlier. Subsequently, an aliquot of equal volume (100 µL) was taken out from ‘blank’ solution and measured the activity (B) using same NaI(Tl) detector. The radiochemical purity was determined using the following expression:| |
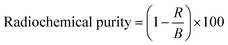 | (3) |
The in vitro stability of [169Er]Er-HA MPs was then investigated in PBS and human serum media. For this, the supernatant of [169Er]Er-HA MPs formulation after centrifugation was discarded and the left [169Er]Er-HA MPs were incubated with 1 mL of PBS solution and 1 mL of human serum medium at 37 °C separately. Subsequently, the radiochemical purity was determined over periods of 14 days using eqn (3).
2.9 SPECT/CT imaging and biodistribution studies after administration of [169Er]Er-HA MPs in the ankle joints of healthy Wistar rats
Biological studies in animals were carried out with prior approval of Institutional Animal Ethics Committee of Bhabha Atomic Research Centre (Reference: BAEC/12/2024). All procedures involving animals were performed in strict adherence to the relevant ethical guidelines and regulations approved by the Institutional Animal Ethics Committee. The in vivo stability and pharmacokinetics behaviour of [169+171Er]Er-HA MPs formulation was demonstrated by acquiring SPECT/CT images of healthy Wistar rats. For this study, radiolabeling of HA MPs was performed using [169+171Er]ErCl3 which was produced by the irradiation of natural Er2O3 target. The [169+171Er] Er-HA MPs formulation was prepared following optimized protocol and an aliquot of 0.1 mL (15–20 MBq) was injected intra-articularly into one of the ankle joints of a healthy Wistar rat (weight ∼300 g). Subsequently the rat was anesthetized and whole-body SPECT/CT images were acquired at different time points post injection (p.i.). For quantitative assessment the retention of [169Er]Er-HA MPs inside the synovial ankle joint cavity and estimation of the uptake of radioactivity in non-targeted organs/tissues, if any, ex vivo biodistribution study was performed after administration of [169+171Er]Er-HA MPs in healthy Wistar rats. The ex vivo biodistribution study was carried out as per the protocol discussed in the SI.
2.10 Statistics
All experimental data regarding determination sorption capacity, radiolabeling yield and ex vivo biodistribution of radiolabeled formulation are presented as mean ± SD (standard deviation) of the measured observations from 3 independent experiments by performing one-way analysis of variance (ANOVA). A p-value < 0.05 was considered statistically significant.
3. Results and discussion
3.1 Synthesis and characterization of nanoporous HA MPs
In the present study, nanoporous HA MPs was synthesized using mechanical action offered by the grinding of the chemical precursors in the solid state. Porosity was introduced in HA MPs using glucose as a sacrificial template which could easily be removed by calcination at an elevated temperature. To optimize the calcination temperature, experiments were carried out at different temperatures (300 °C to 900 °C). During the heating process, glucose would be decomposed to release CO2 gas which arrested collapsing of pores and resulted in formation of nanoporous HA.25 The optimum heating temperature for removing the glucose template was found to be 700 °C because below this temperature sorption capacity of HA was low and beyond this temperature there was no significant improvement in the sorption capacity of HA for 169Er (Table S1). Also, at lower temperature some black particles were visible in nanoporous HA MPs due to incomplete conversion of carbonaceous material to gaseous form, which were absent when heating was performed at higher temperatures (Fig. S1).
The X-ray diffraction pattern of the synthesized powder was shown in Fig. 1B(i). The diffracted X-ray peaks matched well with standard crystalline hydroxyapatite (JCPDS data No. 9-432). Thus, the formation of hydroxyapatite was confirmed from XRD study. The formation of hydroxyapatite was again ascertained by FT-IR and Raman spectroscopy. In the FT-IR spectrum (Fig. 1B(ii)), peak at ∼3310 cm−1 corresponded to the stretching vibration of OH− ion.13 The peaks at 1090 cm−1,1036 cm−1 and 962 cm−1 were attributed to starching bands of P–O of PO43− ion and peaks at 603 cm−1 and 565 cm−1 corresponded to the bending modes of P–O–P group.13,51 In the Raman spectra (Fig. 1B(iii)), the peak at 383 cm−1 was attributed to bending vibrations in calcium hydroxide. The Raman peaks at 410 cm−1, 520 cm−1 and 585 cm−1 could be assigned to the bending modes of HPO4− and PO43−.8 The vibrational modes of HPO42− appeared at 874 cm−1. The intense peaks at 963 cm−1 and 1035 cm−1 could be attributed to the symmetric stretching of phosphate P–O.8 Clearly, the appearance of characteristic peaks in FT-IR and Raman spectra indicated the formation of hydroxyapatite. The complete removal of glucose template was ascertained by TGA study (Fig. S2), where no weight loss in the sample was found beyond 100 °C temperature. The weight loss in the temperature range 0–100 °C might be attributed to physically adsorbed water molecules on the surface of the nanoporous HA MPs. Had there been any carbonaceous moiety attached to nanoporous HA MPs, then weight loss of the sample beyond 200 °C would be observed in the TGA plot. This study clearly indicated the complete removal of carbonaceous material from nanoporous HA MPs.
The morphological analysis of the synthesized hydroxyapatite was done using FESEM and TEM studies. The FESEM image showed agglomerated porous structures of HA MPs (Fig. 2A). The particle size distribution of the nanoporous hydroxyapatite MPs is shown in Fig. 2B which demonstrated that >95% of the particles falls in the range of 1–10 µm with mean particle size 6.2 ± 0.6 µm. Therefore, these particles meet the size criteria for application in RSV.36 The HRTEM image (Fig. 2C) showed that the MPs are composed of hydroxyapatite nanorods. The lattice fringe of the hydroxyapatite could be seen in the higher magnified scale of HRTEM (Fig. 2D) and from the lattice fringe d-spacing the hydroxyapatite crystal was determined to be 0.267 nm which is very close to d-spacing value of (211) plane of hydroxyapatite.52 Also selected area electron diffraction (SAED) pattern of HRTEM (inset of Fig. 2D) revealed polycrystalline nature of synthesized nanoporous HA MPs.
 |
| | Fig. 2 (A) Field emission scanning electron microscopy image of nanoporous HA MPs (B) particle size distribution of nanoporous HA MPs (C and D) high-resolution transmission electron microscopy (HRTEM) image of nanoporous HA MPs. The inset of image (D) shows the selected area electron diffraction (SAED) dotted pattern of nanoporous HA MPs signifying their crystalline nature. | |
The SAXS and SANS experiments were carried out to determine the porosity of the synthesized HA MPs. The scattering intensity from the HA aggregates arises due to intergranular (pore radius < 50 nm) and intragranular pores (pore radius > 50 nm). The scattering intensity can be expressed the sum of scattering contributions from these pores.
| | |
I(Q) = Ipore1(Q) + Ipore2(Q)
| (4) |
Generally, shape and size of both the intragranular and intergranular pores are not very well defined. Spherical shape of pore with lognormal size distribution is assumed for fitting the of the experimental profile. The scattering intensity can be expressed as,
| | |
Ipore(q) = C∫D(r)P(Q, r)r6dr
| (5) |
Here,
p(
Q,
r) is the form factor of spherical particles and can be written as below
| |
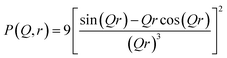 | (6) |
D(r) is the lognormal distribution of the pores.
| |
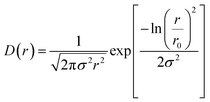 | (7) |
r0 is the median pore radius and
σ is the polydispersity index which vary between 0 and 1. The average pore radius
Rav =
r0exp(
σ2/2).
The estimated pore radius distribution is shown in Fig. 3A, while the fitting of eqn (4) is given in the inset. The average radius and polydispersity index of the intragranular pore is 4.6 nm and 0.54 and average radius and polydispersity of the intergranular pore is 65.5 nm and 0.52, respectively. The pore size distribution demonstrated that the material is nanoporous in nature. The small pores would provide significant interfacial area and pore volume, resulting in a high adsorption capacity. In contrast, the larger pores would improve accessibility and permeability for the adsorbate ([169Er]Er3+).
 |
| | Fig. 3 (A) Pore size distribution obtained from the analysis of the MSANS–SAXS data. The inset of image (A) shows combined MSANS and SAXS profile of nanoporous HA MPs plotted on a double logarithmic scale and the solid line represents model fitting to the data (B) emission spectra of Er-HA and Er2O3 on excitation with 980 nm. | |
In order to establish the strong interaction between Er3+ and nanoporous HA MPs, PL measurements were carried out. The emission spectra of Er2O3 on excitation with 980 nm are depicted in Fig. 3B. The emission spectrum showed an intense broad spectral band around 1550 nm in NIR-B region (∼1100–1700 nm) could be clearly seen which is split into several peaks (Fig. 3B). Such NIR-B emission peak is attributed to 4I13/2 → 4I15/2 transition of Er3+. Moreover, some additional bands detected on the shoulder of these broad bands were assigned to the 4S3/2 → 4I9/2 transitions. Interestingly it could be seen that Er-HA samples show entirely distinct NIR-B peaks in terms of spectral profile, stark splitting, peak width and intensity. Emission spectrum of Er-HA showed reduced intensity, lower number of Stark components and enhanced line width suggesting efficient sorption of Er3+ in HA and strong interaction between them.
3.2 Production of 169Er
The thermal neutron irradiation of enriched (98.2% in 168Er) Er2O3 produced 169Er with >99.99% radionuclidic purity and >99.9% radiochemical purity. The production yield of 169Er in five different batches are summarized in Table S2. The average specific activity of 169Er was found to be 10.2 ± 1.1 mCi mg−1 (370.4 ± 40.7 MBq mg−1) after 24 h from the end of bombardment (EOB).
3.3 Optimization of conditions for loading of 169Er in nanoporous HA MPs
In order to achieve maximum loading of 169Er in nanoporous HA MPs, the involved parameters were systematically optimized. It was observed that 169Er loading yield increased with the increase of concentration of nanoporous HA MPs and reached a state of saturation (Fig. 4A). The loading yield also increased with the pH of the solution and beyond pH ∼ 5, there was no significant improvement in the yield (Fig. 4B). The optimization studies were performed within the pH range of 3 ≤ pH ≥ 7 because below this range dissolution of HA would occur while above this pH range Er3+ would precipitate as Er(OH)3.
 |
| | Fig. 4 Determination of 169Er loading yield at (A) different amounts of nanoporous HA MPs and (B) various pH (error bars indicate standard deviations). | |
3.4 Investigation of sorption mechanism of Er3+ on the surface of nanoporous HA MPs using DFT study
Periodic DFT studies were carried out to explore the adsorption mechanism of Er over the HA surface. To study the surface adsorption of Er, (0001) surface of HA has been considered similar to our previous study and the surface structure was optimized.13
By considering a 2 × 2 × 1 supercell of (0001) surface, Er ion adsorption in two different ways, viz. hydrated Er3+ ion adsorption over the HA surface and the Er3+ ion exchanged with the surface Ca2+ ion is considered and the corresponding optimized geometries are shown in Fig. 5A and B respectively. In the hydrated Er-complex adsorbed system, the interaction of the complex is found to be through strong hydrogen bonding interactions between the hydrogen atoms of water molecules bound to the Er3+ cation and the HA surface oxygen atoms. Adsorption energies for both the cases were measured as:
| | |
HA + Er3+(H2O)9 → [Er(H2O)9@HA]3+ ΔE = −10.07 eV
| (8) |
| | |
HA + Er3+(H2O)9 → [Er_Ca@HA]1+ + Ca2+(H2O)9 ΔE = −11.09 eV
| (9) |
where, [Er(H
2O)
9@HA]
3+ and [Er_Ca@HA]
1+ refer to hydrated Er
3+ complex adsorbed over HA surface and Er
3+ ion exchanged with the surface Ca
2+ ion of the HA surface respectively. The calculated energetics indicate that both the reactions considered are energetically favourable with Er
3+ ion exchange with the surface Ca
2+ ion as more preferred over the hydrated Er
3+ ion adsorption. The DFT results mainly indicate that between the cation exchange and simple hydrated cation adsorption, the cation exchange is energetically preferred. However, the simulation studies did not consider any specific pH.
 |
| | Fig. 5 Optimized structure of (A) Er3+ ion exchanged with HA surface Ca2+ ion and (B) hydrated Er3+ ion adsorbed over the HA surface. | |
3.5 Experimental determination of sorption isotherms and kinetics for loading of 169Er onto nanoporous HA MPs
The sorption capacity of nanoporous HA for 169Er was determined by batch equilibrium method under different concentration of Er3+ solution (feed solution). It was observed that sorption capacity increased with the increase in concentration of feed solution and then reached a level of saturation (Fig. S3). The maximum sorption capacity of nanoporous HA MPs for 169Er was found to be 285 ± 8 mg g−1.
The superiority of the synthesized nanoporous HA MPs over commercially available bulk HA MPs of similar size range (Fig. S4) was established by comparing the sorption capacity of these matrices for 169Er in four batches and the results are summarized in Table S3. It is evident from the results that our synthesized nanoporous HA MPs demonstrated ∼3 times higher sorption capacity for [169Er]Er3+ as compared to commercially available bulk HA. This could be attributed to the high surface area and porous structure of the synthesized nanoporous HA MPs. This is relevant especially for the preparation of [169Er]Er-HA MPs using low specific activity 169Er produced by (n, γ) route in medium flux nuclear research reactors. It is known that amount of 169Er radioactivity required in a clinical dose of 169Er based RSV agent is ∼37 MBq (∼1 mCi).41 The amount bulk HA MPs and synthesized nanoporous HA MPs required for preparation of clinically relevant dose of [169Er]Er-HA MPs using various specific activities of 169Er are shown in Table S4. It is evident that for higher specific activity of 169Er, there is no significant difference in amount of bulk HA and synthesized nanoporous HA required for preparation of clinically relevant dose of the radioactive formulation. However, for 169Er produced at relatively lower flux positions and irradiated for shorter time periods, amount of bulk HA required is significantly higher than synthesized nanoporous HA for the preparation of equivalent dose of [169Er]Er-HA MPs. As mentioned earlier that for our study, 98.2% enriched [168Er]Er2O3 was used to produce 169Er and the practical yield of the 169Er radioactivity was ∼10 mCi mg−1 at 24 h of EOB. Using this activity, clinically relevant dose of [169Er]Er-HA were produced in different batches using commercially available bulk HA and synthesized nanoporous HA. The results were summarized in the Table S5. It was demonstrated that average weight of bulk HA required to prepare clinically relevant dose of [169Er]Er-HA was ∼1.5 mg while the same dose of [169Er]Er-HA MPs could be prepared using ∼400 µg of nanoporous HA MPs. It is worth mentioning that because of the tiny volume of the digital joints, there is an obvious limitation on the amount of radioactive microparticles that can be administered for the RSV procedure. Owing to lower loading capacity, when commercially available bulk HA MPs are used for formulation of clinically relevant doses of [169Er]Er-HA, more amount of the material (HA MPs) would be required. This might pose as an impediment towards clinical use in several patients, especially when low specific activity 169Er is used for preparation of the radioactive formulation using bulk HA MPs. This issue can be addressed by utilizing nanoporous HA MPs, as it necessitates a much-reduced quantity for preparation of a clinically relevant dose of [169Er]Er-HA MPs due to much higher loading capacity of nanoporous HA MPs.
The sorption capacity was determined as a function of time and indicated that an incubation period of 40 min was required to attain the sorption equilibrium (Fig. S5).
From the experimental data, the variation in sorption capacity (qe) under equilibrium condition was plotted as a function of equilibrium concentration of adsorbate (Ce) and then fitted with following sorption isotherms after converting to the suitable algebraic form.
3.5.1 Langmuir adsorption isotherm. The Langmuir adsorption isotherm is written as| |
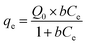 | (10) |
where Q0 is the monolayer sorption capacity (mg g−1) and b (L mg−1) is the reciprocal of the concentration at which half saturation of the adsorbent is reached. The linear form of the Langmuir isotherm could be expressed as:| |
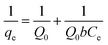 | (11) |
The graph was obtained by plotting 1/qe against 1/Ce (Fig. S6A) and the correlation coefficient (R2) value obtained from the plot was 0.78 which discarded the monolayer adsorption of Er3+ on the surface of nanoporous HA.
3.5.2 Freundlich adsorption isotherm. According to Freundlich model, the variation of sorption capacity with the equilibrium concentration of adsorbate is given by:where KF (L g−1) and n (dimensionless) are Freundlich constant. The linear form of Freundlich isotherm could be expressed as below:| |
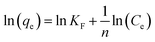 | (13) |
The ln(qe) values were plotted against ln(Ce) values and R2 value was determined (Fig. S6B). The poor R2 value (0.92) indicated that sorption of Er3+ on the surface of nanoporous HA did not follow Freundlich isotherm model.
3.5.4 Pseudo-first-order reaction. The pseudo-first-order reaction also known as Lagergren equation is given below:where, qe and qt are the adsorption capacity at equilibrium and at any given time t and k1 is pseudo-first order rate constant. The linear form of the equation could be expressed as:| |
 | (17) |
From the linear plot of log(qe − qt) vs. log qe, the co-relation coefficient (R2) value of the pseudo-first-order reaction model was determined to be 0.83 [Fig. S7]. Hence, pseudo-first-order reaction model for the adsorption of Er3+ ions on nanoporous HA was discarded.
3.5.5 Pseudo-second-order reaction. The equation of pseudo-second-order reaction is given as:| |
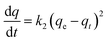 | (18) |
where k2 (g mg−1 min−1) is the rate constant for the pseudo-second-order reaction. The linear form of this equation could be obtained by using proper boundary condition i.e., at t = 0; qt = 0 and integrating within the same limit. Then the linear form would be| |
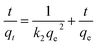 | (19) |
The linear graph could be obtained by plotting t/qt vs. t and the correlation coefficient (R2) value was determined to be 0.99 which ascertained that the sorption followed pseudo-second-order kinetics. Subsequently, the value of adsorption capacity (qe) of nanoporous HA MPs at the equilibrium was evaluated from the slope of the plot (Fig. 6B) and found to be 156 ± 8 mg g−1 which is very close to the value obtained from the experiment (Fig. S3). The adherence of L–F isotherm and pseudo-second-order kinetics essentially indicated that 169Er loading on nanoporous HA MPs was by chemisorption and in the rate determining step there was an involvement of sharing or exchange of electrons between nanoporous HA MPs and Er3+ ions.53 This observation corroborated with the findings obtained from DFT studies as discussed above.
3.6 In vitro stability [169Er]Er-HA MPs under physiological conditions
Under the optimal conditions, the 169Er loaded nanoporous HA MPs ([169Er]Er-HA MPs) was prepared and its stability was investigated in PBS and human serum media at 37 °C. It was observed that [169Er]Er-HA MPs retained their radiochemical integrity >98% in both the media for a period of 14 days (Fig. 7).
 |
| | Fig. 7 Determination of radiochemical stability of [169Er]Er-HA MPs in PBS and human serum over period of 14 days (error bars indicate standard deviations). | |
3.7 SPECT/CT imaging and biodistribution studies after administration of [169Er]Er-HA MPs in the ankle joints of healthy Wistar rats
The SPECT/CT images recorded at different time points (2 and 24 h p.i.) in healthy Wistar rats after intra-articular administration of [169+171Er]Er-HA MPs in one of the ankle joints (Fig. 8A and B) demonstrated homogeneous distribution of the radioactive MPs formulation inside the joint cavity. Further, [169+171Er]Er-HA MPs retained their integrity and did not release free [169+171Er]Er3+ in vivo. Leaching of free [169+171Er]Er3+ from the nanoporous HA MPs matrix would lead to uptake in the liver and bones.54 Also, there was no significant uptake of this formulation in any other organ/tissue. The same conclusion was further established by ex vivo biodistribution studies performed over a prolonged period of time (Fig. 8C).
 |
| | Fig. 8 (A and B) Demonstrate whole body SPECT images of normal Wistar rat at different time points post-injection of [169+171Er]Er-HA MPs. (C) Result of ex vivo biodistribution of normal Wistar rats at different points after intra-articular injection of [169+171Er]Er-HA MPs in one of the ankle joints (error bars indicate standard deviations). | |
4. Conclusions
In summary, we have developed a facile solid-state mechanochemical strategy for the synthesis of biocompatible, nanoporous HA MPs suitable for incorporation of therapeutic radionuclides such as 169Er, with potential application in the management of inflammatory disorders of small digital joints. The nanoporous HA MPs demonstrated significantly enhanced sorption capacity compared to conventional bulk HA, enabling the preparation of therapeutically relevant doses of [169Er]Er-HA even using low-specific-activity 169Er produced in medium-flux nuclear research reactors. The radiolabeled MPs exhibited high radiochemical purity and excellent in vitro stability over 14 days. Furthermore, preliminary in vivo evaluation following intra-articular administration in the ankle joint of healthy Wistar rats indicated favorable retention within the synovial cavity. These findings suggest that [169Er]Er-HA MPs are promising candidates for radiosynovectomy of small joints. However, it is important to recognize certain limitations of the present study. While 169Er is well-suited for treatment of small digital joints, extension of this approach to medium and large joints would necessitate the use of alternative radiolanthanides with higher beta energies. Although similar radiolabeling chemistry is anticipated, systematic investigations into the incorporation of other clinically relevant radionuclides are required. In addition, comprehensive preclinical evaluation in appropriate animal models of arthritis is essential to establish therapeutic efficacy and safety under disease conditions. For eventual clinical translation, it is imperative that the synthesis of HA MPs and subsequent radiolabeling processes are carried out under fully compliant Good Manufacturing Practice (GMP) conditions in accordance with regulatory guidelines. Overall, the mechanochemical approach described herein offers considerable potential not only for HA-based systems but also for the scalable development of a wider class of porous biomaterials for targeted delivery of therapeutic radionuclides and drugs.
Conflicts of interest
The authors have no competing interests to declare that are relevant to the content of this article.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5ra09444e.
Acknowledgements
The authors gratefully acknowledge that the research work carried out at Bhabha Atomic Research Centre (BARC) is part of the ongoing research programs of the Department of Atomic Energy (DAE), Government of India. This work is fully supported through internal funding provided from BARC. The authors are thankful to Dr Y. K. Bhardwaj, Associate Director, Radiochemistry and Isotope Group, BARC, Dr T. Das, Head, Radiopharmaceuticals Division (RPhD), BARC, Dr S. Basu, Head, Radiation Medicine Centre (Medical), BARC, and Dr N. S. Baghel, Radiation Medicine Centre (General), BARC for their support to this work. Mr Jaideep Mor, Radiochemistry Division, BARC is acknowledged for providing XRD data and Mr P. Banerjee, Post Irradiation Examination Division, BARC for providing the TGA data. The help rendered by the staff members of RPhD during production and radiochemical processing of 169Er is also gratefully acknowledged.
References
- M. E. Davis, Nature, 2002, 417, 813–821 CrossRef CAS PubMed.
- T. Kumeria, ACS Biomater. Sci. Eng., 2022, 8, 4025–4027 CrossRef CAS PubMed.
- X. Liu, N. Song, D. Qian, S. Gu, J. Pu, L. Huang, J. Liu and K. Qian, ACS Biomater. Sci. Eng., 2021, 8, 4092–4109 CrossRef PubMed.
- H. Thakar, S. M. Sebastian, S. Mandal, A. Pople, G. Agarwal and A. Srivastava, ACS Biomater. Sci. Eng., 2019, 5, 6320–6341 CrossRef CAS PubMed.
- A. Huang, H. Dai, X. Wu, Z. Zhao and Y. Wu, J. Mater. Res. Technol., 2019, 8, 3158–3166 CrossRef CAS.
- S. Li and K. Du, Chem. Eng. J., 2024, 492, 152404 CrossRef CAS.
- K. V. Vimalnath, A. Rajeswari, S. Patra, K. K. Kamaleshwaran and S. Chakraborty, Appl. Radiat. Isot., 2025, 217, 111644 CrossRef CAS PubMed.
- S. Patra, R. Chakravarty, J. Bahadur, A. Guleria, A. Rajeswari, K. Vimalnath, K. Kamaleshwaran, D. Sen and S. Chakraborty, Mater. Chem. Phys., 2023, 295, 127115 CrossRef CAS.
- C. Zhang, S. Shan, T. Hu, G. Wang, Y. Zhi, H. Su, L. Jiang and Y. Ni, Ceram. Int., 2021, 47, 13552–13571 CrossRef CAS.
- R. Kumar and S. Mohanty, J. Inorg. Organomet. Polym. Mater., 2022, 32, 4461–4477 CrossRef CAS.
- B. Sandhöfer, M. Meckel, J. M. Delgado-López, T. Patrício, A. Tampieri, F. Rösch and M. Iafisco, ACS Appl. Mater. Interfaces, 2015, 7, 10623–10633 CrossRef PubMed.
- G. Verma, K. Barick, N. G. Shetake, B. Pandey and P. Hassan, RSC Adv., 2016, 6, 77968–77976 RSC.
- S. Patra, S. Kancharlapalli, A. Chakraborty, K. Singh, C. Kumar, A. Guleria, S. Rakshit, A. Damle, R. Chakravarty and S. Chakraborty, Ind. Eng. Chem. Res., 2023, 62, 3194–3205 CrossRef CAS.
- Y. Wei, M. Chen, M. Li, D. Wang, K. Cai, Z. Luo and Y. Hu, ACS Appl. Mater. Interfaces, 2022, 14, 42915–42930 CrossRef CAS PubMed.
- M. Roy, G. A. Fielding, H. Beyenal, A. Bandyopadhyay and S. Bose, ACS Appl. Mater. Interfaces, 2012, 4, 1341–1349 CrossRef CAS PubMed.
- A. A. Vu and S. Bose, ACS Appl. Mater. Interfaces, 2020, 12, 52383–52392 CrossRef CAS PubMed.
- R. Akbarzadeh and A. M. Yousefi, J. Biomed. Mater. Res., Part B, 2014, 102, 1304–1315 CrossRef PubMed.
- A. Wang, Y. Ma and D. Zhao, ACS Nano, 2024, 18, 22829–22854 CrossRef CAS PubMed.
- Y.-C. Chiu, J. C. Larson, A. Isom Jr and E. M. Brey, Tissue Eng., Part C, 2010, 16, 905–912 CrossRef CAS PubMed.
- X. Jing, H.-Y. Mi, T. Cordie, M. Salick, X.-F. Peng and L.-S. Turng, Ind. Eng. Chem. Res., 2014, 53, 17909–17918 CrossRef CAS.
- C. Zhang, J. W. Palko, G. Rong, K. S. Pringle, M. T. Barako, T. J. Dusseault, M. Asheghi, J. G. Santiago and K. E. Goodson, ACS Appl. Mater. Interfaces, 2018, 10, 30487–30494 CrossRef CAS PubMed.
- F. Miculescu, A. Maidaniuc, S. I. Voicu, V. K. Thakur, G. Stan and L. Ciocan, ACS Sustain. Chem. Eng., 2017, 5, 8491–8512 CrossRef CAS.
- E. Doustkhah, R. N. Zare, Y. Yamauchi, A. Taheri-Kafrani, H. Mohtasham, M. Esmat, Y. Ide, N. Fukata, S. Rostamnia and M. H. Sadeghi, J. Mater. Chem. B, 2019, 7, 7228–7234 RSC.
- E. Babaie and S. B. Bhaduri, ACS Biomater. Sci. Eng., 2018, 4, 1–39 CrossRef CAS PubMed.
- R. Chakravarty, J. Bahadur, S. Lohar, H. D. Sarma, D. Sen, R. Mishra, S. Chakraborty and A. Dash, Microporous Mesoporous Mater., 2019, 287, 271–279 CrossRef CAS.
- B. Szczęśniak, S. Borysiuk, J. Choma and M. Jaroniec, Mater. Horiz., 2020, 7, 1457–1473 RSC.
- T. F. Grigorieva, A. P. Barinova and N. Z. Lyakhov, Russ. Chem. Rev., 2001, 70, 45–63 CrossRef CAS.
- A. P. Amrute, J. De Bellis, M. Felderhoff and F. Schüth, Chem.–Eur. J., 2021, 27, 6819–6847 CrossRef CAS PubMed.
- J. F. Reynes, F. Leon and F. García, ACS Org. Inorg. Au, 2024, 4, 432–470 CrossRef CAS PubMed.
- Q. Ain, M. Zeeshan, S. Khan and H. Ali, J. Biomed. Mater. Res., Part A, 2019, 107, 2595–2600 CrossRef CAS PubMed.
- S. Chakraborty, T. Das, S. Banerjee, H. D. Sarma and M. Venkatesh, Nucl. Med. Commun., 2006, 27, 661–668 CrossRef PubMed.
- A. Rizaludin, I. Mahendra, M. B. Febrian, A. Kurniawan, B. Abbas, Y. Warastuti, A. A. Sutanningsih, W. Y. Rahman, F. Lukitowati and A. Hardian, J. Radioanal. Nucl. Chem., 2024, 1–10 Search PubMed.
- K. Vimalnath, S. Chakraborty, A. Rajeswari, H. Sarma, J. Nuwad, U. Pandey, K. Kamaleshwaran, A. Shinto and A. Dash, Nucl. Med. Biol., 2015, 42, 455–464 CrossRef CAS PubMed.
- D. Hnatowich, R. Kramer, C. Sledge, J. Noble and S. Shortkroff, J. Nucl. Med., 1978, 19, 303–308 CAS.
- M. Argüelles, I. Luppi Berlanga, E. Torres and V. Pereyra Molina, World J. Nucl. Med., 2002, 1, 300 Search PubMed.
- E. Deutsch, J. Brodack and K. Deutsch, Eur. J. Nucl. Med., 1993, 20, 1113–1127 CrossRef CAS PubMed.
- A. F. Magalhães, L. C. O. de Oliveira, F. A. Pitella, L. Wichert-Ana, E. E. Engel and C. H. Barbieri, Hematol. Transfus. Cell Ther., 2021, 43, 15–20 CrossRef PubMed.
- J. Zhou, M. Kirk, P. Baldo and F. Lu, J. Nucl. Mater., 2021, 557, 153271 CrossRef CAS.
- F. Van der Zant, R. Boer, J. Moolenburgh, Z. Jahangier, J. Bijlsma and J. Jacobs, Clin. Exp. Rheumatol., 2009, 27, 130–139 CAS.
- F. Knapp, A. Dash, F. Knapp and A. Dash, Radiopharmaceuticals for Therapy, 2016, pp. 265–278 Search PubMed.
- J. Farahati, S. Kazek, I. Maric, T. Soestwoehner, P. Kalle, P. F. Costa, W. Jentzen, L. Stein, A. Jalilian and D. Kumm, Appl. Radiat. Isot., 2019, 154, 108853 CrossRef CAS PubMed.
- R. Chakravarty, S. Chakraborty, V. Chirayil and A. Dash, Nucl. Med. Biol., 2014, 41, 163–170 CrossRef CAS PubMed.
- IAEA Research Reactor Database, Available online at: https://nucleus.iaea.org/rrdb/home, Accessed on: May 6, 2025.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- A. A. Shaltout, M. A. Allam and M. A. Moharram, Spectrochim. Acta, Part A, 2011, 83, 56–60 CrossRef CAS PubMed.
- Y. In, U. Amornkitbamrung, M.-H. Hong and H. Shin, ACS Omega, 2020, 5, 27204–27210 CrossRef CAS PubMed.
- F. Granados-Correa, J. Vilchis-Granados, M. Jiménez-Reyes and L. Quiroz-Granados, J. Chem., 2013, 2013, 751696 CrossRef.
- W. A. Breeman, K. van der Wansem, B. F. Bernard, A. van Gameren, J. L. Erion, T. J. Visser, E. P. Krenning and M. de Jong, Eur. J. Nucl. Med. Mol. Imaging, 2003, 30, 312–315 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence cf,
Santosh Kumar Gupta
cf,
Santosh Kumar Gupta



















