DOI:
10.1039/D5RA09282E
(Paper)
RSC Adv., 2026,
16, 10140-10157
Effects of functional groups on the energetics of CO and CO2 formation during secondary oxidation of coal following pre-treatment
Received
1st December 2025
, Accepted 11th February 2026
First published on 20th February 2026
Abstract
To investigate how functional groups introduced in coal during pre-oxidation influence the release behavior of CO and CO2 during subsequent secondary oxidation, experimental characterization was conducted to identify the types and relative abundances of functional groups formed at varying pre-oxidation temperatures. Concurrently, the apparent activation energy (Eα) for CO and CO2 evolution during secondary oxidation was determined via kinetic analysis. A series of multiple linear regression models were then developed to quantitatively assess the relationships between Eα values for CO/CO2 generation and specific functional group contents – or their combinations-in pre-oxidized coal, across distinct temperature intervals of secondary oxidation. The results indicate that, under all pre-oxidation conditions examined, the Eα for CO evolution during secondary oxidation exceeds that for CO2. However, this gap narrows progressively with increasing pre-oxidation termination temperature. Notably, when pre-oxidation is terminated below 140 °C, the Eα for CO formation during secondary oxidation is lower than that observed during initial (unprioritized) coal oxidation – whereas the Eα for CO2 formation remains consistently higher than in the initial oxidation stage. Further, within the secondary oxidation temperature range of 40–170 °C, elevated concentrations and synergistic interactions between aliphatic –CH2/–CH3 groups and carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) groups significantly reduce the Eα for both CO and CO2 evolution. However, as the secondary oxidation temperature interval is refined into narrower subranges, the dominant functional groups-and their interaction patterns-exhibit marked variation, underscoring the context-dependent nature of functional group reactivity.
O) groups significantly reduce the Eα for both CO and CO2 evolution. However, as the secondary oxidation temperature interval is refined into narrower subranges, the dominant functional groups-and their interaction patterns-exhibit marked variation, underscoring the context-dependent nature of functional group reactivity.
1 Introduction
The generation of COx is continuous throughout the process of coal oxidation. Therefore, the degree of coal oxidation is often characterized by COx-related indicators. Thus, the study of COx plays an important role in ensuring the production safety of coal-producing and coal-consuming units and reducing carbon emissions.1–3 Coal spontaneous combustion (CSC) is a slow oxidation process. Most research has focused on the natural low-temperature and heating stages. CSC typically progresses until the ignition temperature is reached. However, the critical research phase for this process generally focuses on the temperature range up to 170 °C. Once this threshold is attained, in the absence of intervention, the temperature is expected to continuously increase toward the ignition point. During the low-temperature oxidation CSC process, the higher the temperature, the greater the amount of toxic and harmful gases produced. The release of COx is a very critical and significant gas release indicator. However, there is no precise mathematical model to describe the release law of COx during coal oxidation and CSC.4–6
Coal comprises a variety of reactive functional groups, and during the smoldering process, numerous reactions and transformations of these active groups occur. Among these gases, the generation of carbon monoxide and carbon dioxide is strongly associated with oxygen-containing functional groups.7 Zhao8 found that oxygen-containing groups are inherently the most active and reactive among all the functional groups present. Zhang9 proposed that the formation of CO2 and CO is primarily attributed to the decomposition of surface oxides formed during coal oxidation, as well as the oxygen-containing functional groups present within the coal matrix. Under low-temperature conditions, most CO2 emissions result from the pyrolysis of these oxygen-containing functional groups. Moreover, these functional groups significantly influence the relative production rates of CO2 and CO. The primary active groups involved in the coal oxidation process include bridge bonds (such as methylene groups), hydroxyl groups, and aliphatic side chains, with the hydroxyl group content having a particularly pronounced effect on the generation of both carbon dioxide and carbon monoxide.10–12 The activation energy (Eα) associated with COx generation during low-temperature coal oxidation can be divided into three distinct stages based on variations in coal temperature. In general, the Eα increases with rising temperature. This variation in Eα suggests that the mechanism of COx formation gradually evolves as coal temperature increases, a transition closely associated with the presence and behavior of oxygen-containing functional groups.13 Carbonyl and carboxyl groups are key functional groups responsible for the generation of carbon monoxide and carbon dioxide. Other functional groups may also be transformed into carbonyl and carboxyl groups, which subsequently decompose into COx. The variation in the content of these two functional groups across different coal temperature stages directly determines the amount of COx produced.14 Li15 conducted isothermal experiments at various temperatures combined with microscopic characterization to investigate the kinetics of COx generation during low-temperature oxidation. The results showed that the Eα for the formation of CO and CO2 under the oxidation pathway were nearly identical, suggesting that they may originate from the same precursor. It was proposed that the reaction between methyl groups in coal and O2 might govern the generation of carbon oxides, resulting in similar apparent activation energies for COx formation during the stable phase. However, studies by Aizenstat16 and Green17 have demonstrated that CO and CO2 originate from two parallel reaction pathways, with the activation energy for CO consistently higher than that for CO2. Moreover, Wang et al.18,19 conducted isothermal reaction experiments and found that, under constant temperature conditions, the activation energy for CO formation in bituminous coal is approximately 4% greater than that for CO2. Wang noted that during coal oxidation, both CO and CO2 formation require overcoming energy barriers. CO is primarily generated during the low-temperature dynamic stage, while CO2 is predominantly produced during the high-temperature stable stage above 70 °C. These distinct stages can be characterized by parameters such as oxygen uptake, oxygen uptake rate, activation energy, adsorption heat, heat release, mass changes, and gas evolution.20 The concentrations of –CH3/–CH2, –CO, and –COOH in low-rank lignite show a significant correlation with the emissions of CO and CO2. For example, the reaction between CO˙ and ˙OH radicals can occur spontaneously and generate a certain amount of heat, which may promote the CSC.21 According to Xu and Wang,22,23 the variation patterns of functional groups during the oxidation process suggest that the rapid depletion of methyl and methylene groups is primarily attributed to their reaction with O2, resulting in the formation of unstable intermediate products. These intermediates are further converted into thermally stable compounds, such as carbonyl and carboxyl groups, while releasing carbon dioxide gas. Using FTIR spectroscopy, Zhang24 investigated the relative abundance of alkyl chains and oxygen-containing functional groups during low-temperature oxidation, as well as their impact on COx generation. The findings revealed that, apart from water, carboxyl, carbonyl, and side-chain C–O bonds among oxygen-containing functional groups are the main contributors to CO emissions during low-temperature oxidation and serve as key active groups that facilitate CSC. The concentration of CO generated during the low-temperature oxidation of low-rank coal was strongly correlated with the relative content of –CH3/–CH2 groups, whereas CO2 concentration was more closely associated with the relative content of –COOH groups. Overall, the CO and CO2 produced during this process were primarily derived from the oxidation of alkyl chains. Specifically, alkyl chains in coal were first oxidized to carbonyl (–CO) groups, which subsequently led to the formation of CO and carboxyl groups; these carboxyl groups then decomposed to produce CO2. Xin25 investigated the generation patterns of COx and their correlation with functional groups during isothermal autoignition using a combined TG-MS and FT-IR approach. The results indicated that carboxyl and carbonyl groups were the primary functional groups directly responsible for COx generation, while aliphatic hydrocarbons also played a crucial role as key intermediates in the formation of carboxyl and carbonyl groups. During the low-temperature oxidation stage of coal, the decomposition of oxygen-containing functional groups, together with their synergistic interaction with the oxidation of active sites, contributed to the production and release of COx.26 The hydroxyl group content was found to influence the formation and relative distribution of CO2 and CO. However, a unified interpretation of the relationship between active group transformations and COx generation remains lacking.27
Zhang believes that if coal contains more –COOH groups and less –CH2 and –CH3, it may lead to higher thermal stability of coal.28 Chen points out that the decomposition of aldehyde and carboxyl groups generates more CO, and the gas desorption and high-energy oxidation of carboxyl groups cause the generation of CO2 to be delayed compared to CO. Temperature and functional group evolution are the key factors controlling the gas generation pathways during coal oxidation.29 Niu's research found that initial oxidation activates functional groups in coal, such as C–O–C, CO, and –CH3, and during secondary oxidation, these functional groups are further oxidized and decomposed to accelerate the formation of gas products.30 Although the above studies have all clarified the qualitative relationship between functional groups and the generation of CO and CO2, few have studied and clarified the quantitative relationship between functional groups and the generation of CO and CO2, providing theoretical support for predicting and judging the generation of CO and CO2 under different conditions in the future. Therefore, it is necessary to conduct relevant research and detection. Based on the theory and method of multiple linear regression, the types and relative contents of functional groups under different treatments and temperatures are determined through micro-observation, and the generation amounts of CO and CO2 are detected macroscopically. The quantitative model between the generation amounts of CO and CO2 and the relative contents of multiple functional groups is calculated through regression.
Although previous studies have provided valuable insights into the smoldering combustion of coal, the gaseous products of oxidation are predominantly analyzed using gas chromatography (GC), a method associated with relatively low sensitivity and lengthy identification times for reaction products.31 Because of these limitations, GC is often inadequate for the analysis of trace coal samples. As a result, mass spectrometry (MS) or integrated thermogravimetric analysis coupled with mass spectrometry (TG-MS) are frequently utilized to investigate the oxidation behavior of small samples, allowing for a more precise characterization of thermal and mass changes during coal oxidation.32–34 Experiments involving trace samples can promote complete oxidation and enable a detailed analysis of low-temperature oxidation behavior, which holds significant value for elucidating the underlying mechanisms of low-temperature coal oxidation. However, under real-world production conditions, coal typically exhibits a broad particle size distribution. The heating process from low-temperature oxidation to ignition primarily involves the oxidation of fine particles or the surface and near-surface layers of larger particles, which generally do not undergo full oxidation. Therefore, significant discrepancies may exist between the results derived from trace sample testing and those observed in real-world production environments. To address this issue, this study aims to use mixed coal samples with particle sizes of 10 mm or smaller to perform programmed temperature experiments simulating both macroscopic pre-oxidation and secondary oxidation stages. GC will be employed to monitor the increasing concentrations of O2, CO, and CO2 throughout the oxidation process. Additionally, the functional groups of coal samples at different stages of pre-oxidation will be analyzed. The Eα of COx generation during various temperature intervals in secondary oxidation will also be calculated. Furthermore, multiple regression analysis will be conducted to investigate the relationship between the Eα of COx generation and the evolution of functional groups, aiming to clarify the variation patterns of COx Eα during secondary coal oxidation and its correlation with functional group transformation.
2 Experimental materials and methods
The coal sample used in this experiment was obtained from Donggucheng Mine in Shanxi Province and had the following industrial composition: Mad 12.17%, Aad 14.07%, Vad 34.32%, and FCad 39.44%. The samples were crushed to a particle size of no more than 10 mm for the programmed temperature rise experiment and FTIR analysis. In the programmed temperature rise experiment, the coal samples were placed in a pure copper tank with an inner diameter of 39 mm and an outer diameter of 42 mm, with a sample height of approximately 240 mm. Dry air was supplied at a flow rate of 100 ml min−1. The temperature was increased at a rate of 0.5 °C min−1 from ambient temperature to the designated termination temperature. The termination temperatures for pre-oxidation were set at 40, 70, 110, 140, and 170 °C, respectively. Once the coal reached the target pre-oxidation temperature, heating was stopped, and the sample was cooled using pure nitrogen. After the coal temperature decreased too and stabilized at room temperature, the nitrogen flow was terminated. The sample processing procedure and their corresponding numbers are shown in Table 1.
Table 1 Sample processing procedure and numbering
| Whether or not it has undergone initial oxidation |
Pre-oxidation temperature |
Will a secondary oxidation be performed following the pre-oxidation process |
Secondary oxidation termination temperature |
Sample number |
| Yes |
170 |
No |
No |
PO |
| Yes |
40 |
Yes |
170 °C |
POTT-40 |
| Yes |
70 |
Yes |
170 °C |
POTT-70 |
| Yes |
110 |
Yes |
170 °C |
POTT-110 |
| Yes |
140 |
Yes |
170 °C |
POTT-140 |
| Yes |
170 |
Yes |
170 °C |
POTT-170 |
Take the coal sample and mix it thoroughly. Grind approximately 10 g of the coal sample to a particle size below 200 mesh. Dry the ground sample at 120 °C under vacuum for 2 hours and then subject it to FTIR analysis using a Nicolet iS50 Fourier Transform Infrared Spectrometer (Thermo NICOLET Company, USA). Each sample, including the original coal sample below 200 mesh, is measured twice, and the average of the two measurements is taken as the result.
The remaining coal samples were placed back into experimental vessels and subjected to secondary oxidation under a controlled heating rate of 0.5 °C min−1 and a dry air flow rate of 100 mL min−1. The secondary oxidation process was terminated at 170 °C for all samples. Gas samples were collected at 10 °C intervals, starting from 40 °C, throughout the oxidation process. The composition and concentration of the collected gases, including O2, CO, CO2, and CHx, were analyzed using a gas chromatograph (GC9100, Beijing Purui Analytical Instrument Co., Ltd). The gas chromatography column uses an 8-meter-long, 3-millimeter-diameter TDX-01 gas chromatography column. The detector includes a dual FID, TCD, and methane conversion furnace. The temperature control range of the detector is from 5 °C above room temperature to 400 °C. The temperature control accuracy of the column chamber is 0.1 °C, and the temperature control of the conversion furnace is 360 °C. The minimum detection limits for various gas components are as follows: CH4 < 0.5 × 10−6, C2H4 < 0.5 × 10−6, C2H6 < 0.5 × 10−6, C2H2 < 0.5 × 10−6, C3H8 < 1 × 10−6, CO < 1 × 10−6, CO2 < 2 × 10−6, O2 < 0.1%. Before and during the detection of oxidized gas, standard gases produced by Anhui Qiangyuan Gas Co., Ltd are generally used for calibration. The concentrations of the gas components are: CO2, O2, CO, CH4, C2H2, C2H4, C2H6, C3H8, N2, with concentrations of 407 × 10−6, 20.2%, 90.8 × 10−6, 704 × 10−6, 50.8 × 10−6, 211 × 10−6, 52.8 × 10−6, 95.6 × 10−6, and the remaining amounts.
3 Result analysis and discussion
3.1. The variation law of COx concentration
As shown in Fig. 1, during the secondary oxidation (SO) of coal samples pre-oxidized at different temperatures, the consumption of O2 concentration shows minimal variation within the temperature range of 40–100 °C. In the temperature range of 100–130 °C, O2 consumption during SO of coal samples pre-oxidized at different temperatures may increase compared to the pre-oxidation stage. However, differences in O2 concentration during SO still exist depending on the pre-oxidation temperature (POT). Within the experimental temperature range of 130–170 °C, the O2 consumption during secondary oxidation is lower than that observed during the pre-oxidation process. Furthermore, within this temperature range, the O2 concentration during SO decreases as the POT increases.
 |
| | Fig. 1 Variation pattern of O2 concentration with temperature during the oxidation process. | |
As shown in Fig. 2, during the low temperature SO of coal samples that were pre-oxidized at different temperatures, only the sample pre-oxidized at 70 °C exhibits a slight increase in CO concentration within the temperature range of 40–90 °C. No significant changes are observed in samples pre-oxidized at other temperatures. Within the temperature range of 90–170 °C, only the sample pre-oxidized at 110 °C shows a decrease in CO concentration during SO compared to the initial oxidation stage. For all other POTs, the CO concentration during SO is higher than that observed during the initial oxidation process.
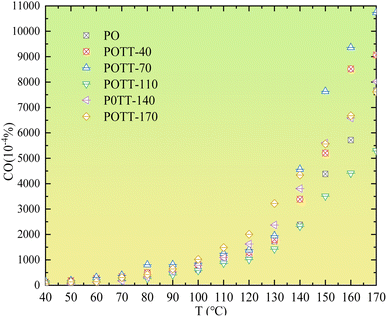 |
| | Fig. 2 Variation pattern of CO concentration with temperature during the oxidation process. | |
As shown in Fig. 3, during the low-temperature SO of coal samples pre-oxidized at different temperatures, the CO2 concentration within the temperature range of 40–110 °C was significantly lower compared to the pre-oxidation stage when the POT is 140 °C or 170 °C. However, for samples pre-oxidized at 40 °C, 70 °C, and 110 °C, no notable differences in CO2 concentration were observed compared to the pre-oxidation process. When the temperature exceeded 110 °C, the variation in CO2 concentration during SO became more pronounced relative to the pre-oxidation stage. Notably, in the case of coal samples pre-oxidized to 170 °C, a significant increase in CO2 concentration was observed during SO once the temperature surpassed 110 °C.
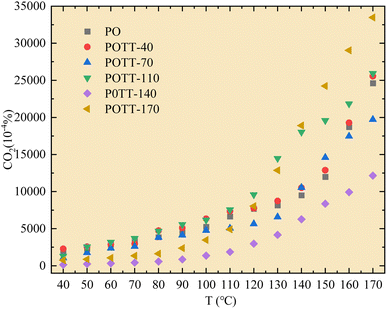 |
| | Fig. 3 Concentration of CO2 during the oxidation process varies with temperature. | |
3.2. The variation law of COx generation rate
Based on the variation trends of O2, CO, and CO2 concentrations with temperature, as illustrated in Fig. 1–3, and combined with eqn (1)–(3), the O2 consumption rate, CO generation rate, and CO2 generation rate of the coal sample can be calculated. By taking the logarithm of both sides of the Arrhenius equation (eqn (4)) and (5) can be derived. According to eqn (5), the Eα for COx generation can be determined through linear regression analysis.| |
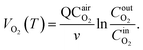 | (1) |
| |
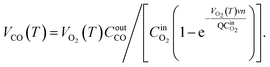 | (2) |
| |
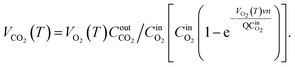 | (3) |
| |
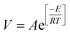 | (4) |
| |
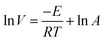 | (5) |
Note: in eqn (1)–(11), VO2(T)is the O2 consumption rate of the coal in the air environment or for an O2 concentration of 21% (mol mL−1 s−1); and CO2airis the O2 concentration in the fresh air environment or for an O2 concentration of 21%. The CO2out and CO2in are the O2 concentrations at the outflow and inflow, respectively (%), the O2 concentration of the intake air is 20.96%. v is the volume of the coal sample (cm3).Q is the rate inflow of the airflow (mL min−1).
The CO generation rate is shown in Fig. 4. As can be observed, when the coal temperature is below 110 °C, the CO generation rates of different coal samples remain relatively consistent, irrespective of whether they undergo primary or SO. However, once the coal temperature surpasses 110 °C, the differences in CO generation rates among the samples become progressively more significant. Overall, the sample that underwent SO after being pre-oxidized to 70 °C exhibits the highest CO generation rate. The rates subsequently decreased in the following order, samples pre-oxidized to 40 °C, 140 °C, and 170 °C. These values are consistently higher than those observed during primary oxidation. Notably, only the sample subjected to SO after pre-oxidation to 110 °C displays the lowest CO generation rate, which is even lower than that of the primary oxidation process.
 |
| | Fig. 4 Variation of CO generation rate with temperature. | |
The variation in CO2 concentration with temperature differs slightly from that of CO. While a detectable amount of CO appears at the onset of oxidation, the abrupt increase in CO2 generation rate becomes evident only after the coal temperature exceeds 110 °C, as shown in Fig. 5. During the oxidation stage characterized by a relatively high CO2 generation rate, the coal samples subjected to SO following initial oxidation to termination temperatures of 170 °C and 110 °C exhibit the highest CO2 generation rates. The next highest rates are observed in samples undergoing primary oxidation only. Subsequently, lower CO2 generation rates are found in samples that undergo SO after initial oxidation (IO) to 70 °C and 40 °C. The lowest CO2 generation rate is observed in the sample subjected to SO after initial oxidation to 140 °C.
 |
| | Fig. 5 Variation of CO2 generation rate with temperature. | |
3.3. Variation law of activation energy for COx generation
3.3.1. Variation law of activation energy for CO generation. The Eα fitting process for CO generation throughout the entire experimental stage, conducted at coal temperatures ranging from 40 to 170 °C, is presented in Fig. 6, with the corresponding fitting results summarized in Table 2. The Eα for CO generation varies across coal samples subjected to different termination temperatures of the IO. Compared to the IO phase, the Eα of CO generation during SO decreases by 9–10% in coal samples with IO termination temperatures of 40, 70, and 110 °C. In contrast, for samples terminated at 140 and 170 °C, the Eα increases by 16% and 3%, respectively. Specifically, when the IO temperature is below 110 °C, the Eα for CO generation during SO decreases significantly. However, when the IO temperature exceeds 140 °C, the Eα increases during SO.
 |
| | Fig. 6 Linear fitting of the activation energy for CO generation in the coal temperature range of 40–170 °C. | |
Table 2 Activation energy of CO generation in different coal samples at 40–170 °C
| Temperature |
Coal sample |
Intercept |
Slope |
R2 |
E |
Rate of change to PO |
| 40–170 °C |
PO |
19.59 |
−5820.37 |
0.9703 |
48.39 |
0.0% |
| POTT-40 |
18.37 |
−5243.60 |
0.9421 |
43.60 |
−10% |
| POTT-70 |
18.61 |
−5269.71 |
0.9243 |
43.81 |
−9% |
| POTT-110 |
18.08 |
−5313.13 |
0.9610 |
44.17 |
−9% |
| P0TT-140 |
22.04 |
−6744.70 |
0.9940 |
56.08 |
16% |
| POTT-170 |
20.27 |
−5980.04 |
0.9938 |
49.72 |
3% |
As outlined above, the entire experimental testing phase encompassing secondary coal oxidation over a temperature range of 40–170 °C was partitioned into four characteristic temperature intervals, as summarized in Table 3, to support a more rigorous and systematic comparative analysis.
Table 3 Division and numbering of different coal temperature stages of COx
| The stage of secondary oxidation of coal temperature (°C) |
40–170 |
40–70 |
70–110 |
110–140 |
140–170 |
| Numbers of activation energies for CO activation in different temperature ranges |
SOP-CO |
S1-CO |
S2-CO |
S3-CO |
S4-CO |
| Numbers of activation energies for CO2 activation in different temperature ranges |
SOP-CO2 |
S1-CO2 |
S2-CO2 |
S3-CO2 |
S4-CO2 |
Fig. 7 illustrates the fitting process of Eα for CO generation at various temperature stages during the SO of coal. Based on these fitting results, the Eα for CO generation during the SO of different coal samples within the temperature ranges of 40–70 °C, 70–110 °C, 110–140 °C, and 140–170 °C were calculated and are summarized in Tables 2 and 4–6, respectively.
 |
| | Fig. 7 The fitting process of CO generation activation energy at different temperature stages (a) coal temperature stage of 40–70 °C, (b) coal temperature stage of 70–110 °C, (c) coal temperature stage of 110–140 °C, (d) coal temperature stage of 140–170 °C. | |
The Eα for the generation of CO during the IO at 40–70 °C is 52.54 kJ mol−1, as shown in Table 4. When the coal samples that have undergone IO at 40, 70, and 110 °C are subjected to SO, the Eα for CO generation in the 40–70 °C stage decreases by 42%, 45%, and 58% respectively indicates that when the IO is terminated at a temperature below 110 °C, the coal samples undergo SO, and the Eα for CO generation in the 40–70 °C stage is significantly lower than that of the IO under the same conditions. Moreover, as the termination temperature of the IO increases, the reduction in the Eα also increases. This suggests that the energy required to produce CO is lower after the initial oxidation. However, when the IO is terminated at 140 °C and SO occurs, the Eα for CO generation in the 40–70 °C stage is only slightly higher than that of the IO by 2%. When the IO is terminated at 170 °C and SO occurs, the Eα for CO generation in the 40–70 °C stage is 14% lower than that of the IO.
Table 4 Activation energy of CO generation in the 40–70 °C stage of secondary oxidation
| Temperature |
Coal sample |
Intercept |
Slope |
R2 |
E |
Rate of change to PO |
| 40–70 °C |
PO |
21.20 |
−6318.92 |
0.8786 |
52.54 |
0% |
| POTT-40 |
13.76 |
−3669.20 |
0.9898 |
30.51 |
−42% |
| POTT-70 |
13.26 |
−3461.25 |
0.9230 |
28.78 |
−45% |
| POTT-110 |
10.17 |
−2671.10 |
0.8846 |
22.21 |
−58% |
| P0TT-140 |
21.20 |
−6452.07 |
0.9720 |
53.64 |
2% |
| POTT-170 |
18.68 |
−5438.84 |
0.8728 |
45.22 |
−14% |
As shown in Table 5, during the coal temperature range of 70–110 °C, except for the coal sample with the IO termination temperature of 70 °C, which has an 8% reduction in the Eα for the generation of CO during the SO, the Eα for the formation of CO during the SO of the other coal samples with different IO termination temperatures all significantly increase.
Table 5 Activation energy of CO generation in the 70–110 °C stage of secondary oxidation
| Temperature |
Coal sample |
Intercept |
Slope |
R2 |
E |
Rate of change to PO |
| 70–110 °C |
PO |
13.69 |
−3686.75 |
0.9294 |
30.65 |
0% |
| POTT-40 |
14.79 |
−4031.55 |
0.9892 |
33.52 |
9% |
| POTT-70 |
13.21 |
−3382.25 |
0.8004 |
28.12 |
−8% |
| POTT-110 |
17.52 |
−5186.89 |
0.9938 |
43.12 |
41% |
| P0TT-140 |
20.98 |
−6383.33 |
0.9871 |
53.07 |
73% |
| POTT-170 |
19.16 |
−5600.23 |
0.9916 |
46.56 |
52% |
As shown in Table 6, during the coal temperature range of 110–140 °C, the Eα for the generation of CO through SO of all coal samples significantly increases. This indicates that the energy required for the SO of coal to produce CO at this temperature stage is significantly higher than that at the same temperature stage during the primary oxidation.
Table 6 Activation energy of CO generation in the 110–140 °C stage of secondary oxidation
| Temperature |
Coal sample |
Intercept |
Slope |
R2 |
E |
Rate of change to PO |
| 110–140 °C |
PO |
19.51 |
−5916.07 |
0.9865 |
49.19 |
0% |
| POTT-40 |
22.01 |
−6832.59 |
0.8913 |
56.81 |
15% |
| POTT-70 |
24.03 |
−7597.78 |
0.7814 |
63.17 |
28% |
| POTT-110 |
20.02 |
−6167.68 |
0.9149 |
51.28 |
4% |
| P0TT-140 |
23.37 |
−7339.66 |
0.9887 |
61.02 |
24% |
| POTT-170 |
22.03 |
−6703.56 |
0.9873 |
55.73 |
13% |
As shown in Table 7, during the coal temperature stage of 140–170 °C, the Eα for the generation of CO during the SO of all coal samples decreases significantly except for the coal sample that undergo SO after pre-oxidation to 40 °C. Moreover, the proportion of reduction increases significantly as the termination POT increases.
Table 7 Activation energy of CO generation in the 140–170 °C stage of secondary oxidation of coal samples
| Temperature |
Coal sample |
Intercept |
Slope |
R2 |
E |
Rate of change to PO |
| 140–170 °C |
PO |
31.94 |
−11030.47 |
0.9905 |
91.71 |
0% |
| POTT-40 |
32.84 |
−11250.57 |
0.9992 |
93.54 |
2% |
| POTT-70 |
29.67 |
−9813.45 |
0.9710 |
81.59 |
−11% |
| POTT-110 |
25.99 |
−8584.38 |
0.9940 |
71.37 |
−22% |
| P0TT-140 |
25.73 |
−8281.20 |
0.9762 |
68.85 |
−25% |
| POTT-170 |
22.04 |
−6708.03 |
0.9957 |
55.77 |
−39% |
3.3.2. Variation law of activation energy for CO2 generation. The fitting process and results of the Eα of CO2 production in different coal samples during the 40–170 °C stage are shown in Fig. 8 and Table 8. The results indicate that, compared with the IO, except for the coal sample that underwent SO at 40 °C after the IO, where the Eα of CO2 production decreased by 2%, the Eα of CO2 production during the SO after the IO at 70 °C, 110 °C, 140 °C, and 170 °C increased by 11%, 14%, 75%, and 61% respectively. This suggests that the energy consumed for CO2 production during the SO of coal samples after different degrees of IO will significantly increase.
 |
| | Fig. 8 The fitting process of activation energy for CO2 generation in the temperature range of 40–170 °C. | |
Table 8 The activation energy for CO2 generation in the coal temperature range of 40–170 °C stage
| Temperature |
Coal sample |
Intercept |
Slope |
R2 |
E |
Rate of change to PO |
| 0–170 °C |
PO |
14.69002 |
−3223.3 |
0.90553 |
26.80 |
0% |
| POTT-40 |
14.61403 |
−3159.19 |
0.91142 |
26.27 |
−2% |
| POTT-70 |
15.44661 |
−3562.62 |
0.92006 |
29.62 |
11% |
| POTT-110 |
16.06605 |
−3663.17 |
0.96159 |
30.46 |
14% |
| P0TT-140 |
19.79523 |
−5638.56 |
0.97476 |
46.88 |
75% |
| POTT-170 |
19.6792 |
−5179.74 |
0.94659 |
43.06 |
61% |
The fitting process of the Eα for the generation of CO2 in coal samples during the IO and SO at different degrees of IO at the temperature stages of 40–70, 70–110, 110–140, and 140–170 °C is shown in Fig. 9(a–d), and the results of the Eα solution are presented in Tables 9–12.
 |
| | Fig. 9 The fitting process of activation energy for CO2 generation at different temperature stages (a) coal temperature stage of 40–70 °C, (b) coal temperature stage of 70–110 °C, (c) coal temperature stage of 110–140 °C, (d) coal temperature stage of 140–170 °C. | |
As shown in Table 9, the Eα of CO2 generated during the 40–70 °C stage of SO after the IO to 40, 70, 110, 140, and 170 °C is compared with the temperature stage of the IO. The Eα of CO2 generated during this temperature stage of the coal sample after the IO to 40 and 170 °C is reduced by 62% and 18% respectively compared to the IO stage. The Eα of CO2 generated during the IO to 70, 110, and 140 °C is increased by 28%, 41%, and 55% respectively compared to the IO stage. As shown in Table 10, the Eα of CO2 generated by coal samples undergoing SO after IO to different temperatures during the 70–110 °C stage decreased by 5% compared to the IO stage at 70 °C. However, other coal samples with different IO termination temperatures showed varying degrees of decrease compared to the IO stage. As shown in Table 11, the Eα of CO2 generated by coal samples undergoing SO during the coal temperature range of 110–140 °C is significantly increased compared to the IO stage. As shown in Table 12, during the coal temperature stage of 140–170 °C, the Eα of CO2 generated by SO decreased by 13%, 23%, 41%, 15%, and 28% respectively compared to the same temperature stage of primary oxidation. This indicates that in the process of coal undergoing different degrees of primary oxidation and then undergoing SO, when the coal temperature rises to a higher temperature stage of 140–170 °C, the Eα of CO2 generated will be significantly lower than that of IO, and the proportion of Eα reduction in this temperature stage where SO occurs after IO to 110 °C is the largest.
Table 9 The activation energy for CO2 generation in the coal temperature of 40–70 °C stage
| Temperature |
Coal sample |
Intercept |
Slope |
R2 |
E |
Rate of change to PO |
| 40–70 °C |
PO |
12.88487 |
−2582.24 |
0.99348 |
21.47 |
0% |
| POTT-40 |
8.08965 |
−989.713 |
0.95854 |
8.23 |
−62% |
| POTT-70 |
14.79108 |
−3305.89 |
0.89879 |
27.49 |
28% |
| POTT-110 |
16.1617 |
−3653.71 |
0.84951 |
30.38 |
41% |
| P0TT-140 |
14.99959 |
−4013.89 |
0.96478 |
33.37 |
55% |
| POTT-170 |
10.56224 |
−2125.8 |
0.97456 |
17.67 |
−18% |
Table 10 The activation energy for CO2 generation in the coal temperature of 70–110 °C stage
| Temperature |
Coal sample |
Intercept |
Slope |
R2 |
E |
Rate of change to PO |
| 70–110 °C |
PO |
12.19121 |
−2368.14 |
0.95618 |
19.69 |
0% |
| POTT-40 |
14.13502 |
−3040.68 |
0.93685 |
25.28 |
28% |
| POTT-70 |
11.74744 |
−2261.51 |
0.89532 |
18.80 |
−5% |
| POTT-110 |
12.63168 |
−2472.87 |
0.99112 |
20.56 |
4% |
| P0TT-140 |
18.24551 |
−5167.16 |
0.98749 |
42.96 |
118% |
| POTT-170 |
17.83019 |
−4642.29 |
0.97498 |
38.60 |
96% |
Table 11 The activation energy for CO2 generation during the coal temperature of 110–140 °C stage
| Temperature |
Coal sample |
Intercept |
Slope |
R2 |
E |
Rate of change to PO |
| 110–140 °C |
PO |
13.11572 |
−2698.24 |
0.97298 |
22.43 |
0% |
| POTT-40 |
14.20797 |
−3096.31 |
0.94946 |
25.74 |
15% |
| POTT-70 |
17.96904 |
−4705.84 |
0.81814 |
39.12 |
74% |
| POTT-110 |
21.15388 |
−5746.96 |
0.98178 |
47.78 |
113% |
| P0TT-140 |
23.40879 |
−7137.73 |
0.99532 |
59.34 |
165% |
| POTT-170 |
26.6729 |
−8009.98 |
0.99992 |
66.60 |
197% |
Table 12 The activation energy for CO2 generation in the coal temperature range of 140–170 °C stage
| Temperature |
Coal sample |
Intercept |
Slope |
R2 |
E |
Rate of change to PO |
| 140–170 °C |
PO |
29.85032 |
−9637.54 |
0.96891 |
80.13 |
0% |
| POTT-40 |
26.93024 |
−8350.48 |
0.99268 |
69.43 |
−13% |
| POTT-70 |
24.68235 |
−7410.11 |
0.99379 |
61.61 |
−23% |
| POTT-110 |
20.91729 |
−5673.86 |
0.95258 |
47.17 |
−41% |
| P0TT-140 |
26.05457 |
−8229.45 |
0.98956 |
68.42 |
−15% |
| POTT-170 |
23.9705 |
−6900.32 |
0.99785 |
57.37 |
−28% |
In the complete experimental stage of 40–170 °C, except for a slight decrease of 2% in the overall Eα of CO2 generated during the SO process of coal samples with an IO termination temperature of 40 °C, the overall Eα of CO2 generated by coal samples with different oxidation termination temperatures significantly increased. After dividing the complete experimental stage of 40–170 °C into four different reaction stages, only when the coal temperature increased to 140–170 °C, the Eα of CO2 generated by the SO temperature significantly decreased to varying degrees (decreased by 13–41%), and other temperature stages significantly increased.
Fig. 10 shows the Eα for the generation of CO and CO2 from different coal samples at different temperature stages. CO requires higher energy to break the C–O bond or reorganize oxygen-containing functional groups, while CO2 may come from more easily dissociated carboxyl or carbonyl groups, resulting in the corresponding Eα for CO generation always being higher than that for CO2 generation under the same coal sample conditions (except for a few cases). During the experimental process in this paper, the Eα for CO generation at the coal temperature stage of 40–170 °C was significantly higher than that for CO2 generation. The Eα for CO2 generation during the pre-oxidation stage was 55% of the Eα for CO generation. After SO of the coal samples with POT of 40, 70, 110, 140, and 170 °C, the Eα for CO2 generation was 60%, 68%, 69%, 84%, and 87% of the Eα for CO generation, respectively. That is, as the termination temperature of the IO increases, the difference in Eα for the generation of CO and CO2 during SO gradually decreases.
 |
| | Fig. 10 The activation energy of CO and CO2 produced by coal at different temperature stages. Note: SOP-CO, S1-CO, S2-CO, S3-CO, S4-CO respectively represents the average activation energy for the generation of CO during the complete stage of secondary oxidation at coal temperatures 40–170, 40–70 °C, 70–110, 110–140, 140–170 °C. SOP-CO2, S1-CO2, S2-CO2, S3-CO2, S4-CO2 these numbers represent the activation energy of CO2, and the temperature stages correspond to those of the previous CO. | |
In conclusion, when coal samples pre-oxidized to the same termination temperature undergo SO under identical temperature conditions, the Eα for CO generation is consistently higher than that for CO2. However, this difference decreases as the POT increases. In the coal temperature range of 40–170 °C, 140 °C serves as a critical threshold. If the POT is below 140 °C, the Eα for CO during SO is about 10% lower than during IO. In contrast, for samples pre-oxidized above 140 °C, the Eα for CO2 during SO is generally 11–75% higher than during IO. Among all samples, the one pre-oxidized at 140 °C shows the highest Eα for CO2 during SO. During the SO of pre-oxidized coal samples, Eα for CO and CO2 generation is generally higher than during pre-oxidation, except in specific temperature ranges. Specifically, when pre-oxidation ends at 40 °C and 170 °C, the Eα for CO2 generation decreases by 62% and 18%, respectively, compared to the corresponding pre-oxidation stages. In the 40–70 °C, 70–110 °C, and 110–140 °C ranges, Eα remains higher during SO. However, in the 140–170 °C range, Eα for both CO and CO2 is lower during SO than during pre-oxidation.
3.3.3. Correlation between the activation energy of COx generation and functional groups. The FTIR scanning curves of the functional groups in raw coal and coal samples oxidized to different temperatures are shown in Fig. 11(a). The infrared spectra of the coal samples were analyzed by PeakFit V4.2 software to obtain the different classifications of functional groups (the classification basis of functional groups is shown in Table 13) and their area data in the coal samples.35–37 Considering that the CC structure in coal basically does not change during the low-temperature oxidation process, the relative contents of different functional groups in the coal samples were obtained by dividing the area of other functional groups detected in each sample by the area of CC, as shown in Fig. 11(b–d).
 |
| | Fig. 11 Infrared spectra of coal samples and relative contents of functional groups (a) FTIR scanning curve, (b) relative content of oxygen-containing functional groups, (c) relative content of aliphatic functional groups, (d) relative content of hydroxyl functional groups. | |
Table 13 Functional group classification in FTIR detection data
| Classification |
Wave number |
Functional group |
Serial number |
| Oxygen-containing functional groups |
1030–1330 |
Ar–C–O– |
B1 |
| 1365–1465 |
–CH2, –CH3 |
B2 |
| 1465–1590 |
CC |
B3 |
| 1595–1635 |
CC |
B4 |
| 1640–1690 |
quin. CO |
B5 |
| 1690–1700 |
car. CO |
B6 |
| 1705–1800 |
R2 CO, ν C = O, RCOOH, ArCOOH, RCOO Ar, etc. CO |
B7 |
| Aliphatic functional groups |
2350–2780 |
–COOH |
C1 |
| 2830–2855 |
sym. –CH2 |
C2 |
| 2862–2882 |
sym. –CH3 |
C3 |
| 2900 |
–CH |
C4 |
| 2918–2935 |
asym. –CH2 |
C5 |
| 2950–2975 |
asym. –CH3 |
C6 |
| Hydroxyl functional groups |
3050–3150 |
aro. C–H |
D1 |
| 3200 |
rh. C–H |
D2 |
| 3300–3315 |
OH⋯OR |
D3 |
| 3400–3440 |
OH⋯OH |
D4 |
| 3530–3545 |
OH⋯π |
D5 |
| 3610–3800 |
fr. –OH |
D6 |
Prior to constructing the multi-dimensional numerical model, the relevant data were normalized to the interval [−1, 1] using eqn (6). This normalization serves three principal purposes: (i) eliminating scale disparities arising from heterogeneous units across indicators-such as relative abundances of functional groups and activation energies-thereby enabling equitable comparison and integration; (ii) mitigating order-of-magnitude differences that could otherwise bias model outcomes toward numerically dominant variables; and (iii) enhancing both the numerical stability and convergence rate of the subsequent computational procedure. The results are presented in Table 14. (Notably, B3 and B4 correspond to CC bonds and were therefore excluded from the relative content analysis. Similarly, D2 and D3, which represent aro. C–H and rh. C–H bonds respectively, were only detected in the POTT-110 sample and not in others. Therefore, they should not be included in the normalization process and stepwise regression either).
| |
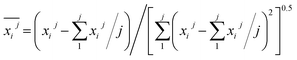 | (6) |
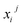
denotes the
j-th observed value of the
i-th indicator.
| |
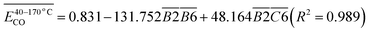 | (7) |
Table 14 The normalized processing results of the relative content of functional groups and activation energy
| Planned variables |
POTT-40 |
POTT-70 |
POTT-110 |
P0TT-140 |
POTT-170 |
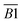 |
0.31 |
0.25 |
0.32 |
0.14 |
0.45 |
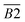 |
−0.07 |
−0.08 |
−0.09 |
−0.05 |
−0.03 |
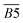 |
−0.14 |
−0.04 |
−0.04 |
−0.07 |
0.11 |
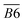 |
−0.16 |
−0.15 |
−0.16 |
−0.06 |
−0.23 |
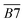 |
0.13 |
0.09 |
0.08 |
0.03 |
0.51 |
 |
−0.11 |
0 |
−0.14 |
−0.06 |
0.25 |
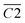 |
−0.11 |
−0.13 |
−0.17 |
−0.11 |
0.12 |
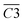 |
−0.11 |
−0.13 |
−0.17 |
−0.1 |
−0.17 |
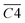 |
−0.16 |
−0.18 |
−0.17 |
−0.16 |
−0.32 |
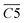 |
−0.16 |
−0.13 |
−0.17 |
−0.1 |
−0.17 |
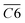 |
−0.06 |
−0.13 |
−0.17 |
−0.16 |
−0.17 |
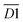 |
−0.11 |
−0.07 |
0.4 |
−0.04 |
−0.32 |
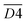 |
−0.1 |
−0.12 |
−0.22 |
−0.09 |
−0.14 |
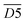 |
0.01 |
−0.06 |
0 |
−0.09 |
0.23 |
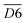 |
0.85 |
0.88 |
0.69 |
0.93 |
−0.13 |
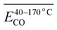 |
−0.37 |
−0.35 |
−0.32 |
0.77 |
0.19 |
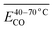 |
−0.28 |
−0.34 |
−0.56 |
0.5 |
0.21 |
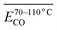 |
−0.26 |
−0.5 |
0.18 |
0.63 |
0.33 |
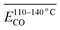 |
0.05 |
0.58 |
−0.41 |
0.4 |
−0.04 |
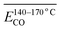 |
0.5 |
0.14 |
−0.18 |
−0.25 |
−0.66 |
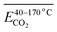 |
−0.38 |
−0.21 |
−0.17 |
0.66 |
0.47 |
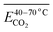 |
−0.72 |
0.21 |
0.35 |
0.49 |
−0.26 |
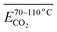 |
−0.1 |
−0.38 |
−0.3 |
0.65 |
0.47 |
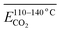 |
−0.45 |
−0.11 |
0.11 |
0.4 |
0.58 |
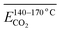 |
0.21 |
−0.1 |
−0.67 |
0.17 |
−0.26 |
During the complete low-temperature SO stage of coal at 40–170 °C in the coal SO process, the average Eα of CO generation 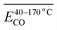 has a relationship with
has a relationship with 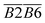 (–CH2/–CH3 & car. CO) and
(–CH2/–CH3 & car. CO) and 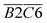 (–CH2/–CH3 & asym. –CH3) as shown in eqn (7). The product of the relative contents of –CH2/–CH3 and car. CO before SO is negatively correlated with the Eα and has the effect of reducing the Eα. The main reason is that the –CH2 adjacent to the carbonyl group is prone to oxidation, and the energy barrier is reduced by forming a six-membered ring transition state. The product of the relative contents of –CH2/–CH3 and asym. –CH3 is positively correlated with the Eα for CO formation at this temperature stage, which has the effect of increasing the Eα. The main reason is that the steric hindrance effect makes the oxidation of the asymmetric –CH3 require higher energy.38–41 The main process of eqn (7) is shown in Fig. 12.
(–CH2/–CH3 & asym. –CH3) as shown in eqn (7). The product of the relative contents of –CH2/–CH3 and car. CO before SO is negatively correlated with the Eα and has the effect of reducing the Eα. The main reason is that the –CH2 adjacent to the carbonyl group is prone to oxidation, and the energy barrier is reduced by forming a six-membered ring transition state. The product of the relative contents of –CH2/–CH3 and asym. –CH3 is positively correlated with the Eα for CO formation at this temperature stage, which has the effect of increasing the Eα. The main reason is that the steric hindrance effect makes the oxidation of the asymmetric –CH3 require higher energy.38–41 The main process of eqn (7) is shown in Fig. 12.
| |
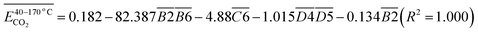 | (8) |
 |
| | Fig. 12 The main reaction pathway of eqn (7). | |
During the complete low-temperature SO stage of coal at 40–170 °C in the coal SO process, the average Eα of CO2 generation 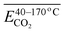 is related to
is related to  (–CH2/–CH3 & car. CO),
(–CH2/–CH3 & car. CO), 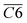 (asym. –CH3)
(asym. –CH3) 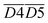 (OH⋯OH & OH⋯π) and
(OH⋯OH & OH⋯π) and 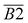 (–CH2/–CH3) as shown in eqn (8). The relative content of the functional groups exhibits a negative correlation with the Eα for CO2 generation. An increase in their relative content contributes to a reduction in the Eα required for CO2 formation during the SO process. The main reaction pathways are illustrated in Fig. 13.
(–CH2/–CH3) as shown in eqn (8). The relative content of the functional groups exhibits a negative correlation with the Eα for CO2 generation. An increase in their relative content contributes to a reduction in the Eα required for CO2 formation during the SO process. The main reaction pathways are illustrated in Fig. 13.
| |
 | (9) |
 |
| | Fig. 13 The main reaction pathway of eqn (8). | |
During the 40–70 °C stage of the SO process of coal, the average Eα for CO generation 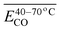 is predominantly influenced by the functional group combinations
is predominantly influenced by the functional group combinations 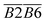 (–CH2/–CH3 & car. CO),
(–CH2/–CH3 & car. CO), 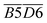 (quin. CO & fr. –OH),
(quin. CO & fr. –OH), 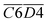 (asym. –CH3 & OH⋯OH), and
(asym. –CH3 & OH⋯OH), and 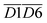 (aro. C–H & fr. –OH). Among these, the relative content of (–CH2/–CH3 & car. CO) exerts the most significant effect on the Eα for CO generation at this low-temperature stage, exhibiting a negative correlation. A higher product of the two functional groups prior to SO results in lower Eα. This phenomenon primarily arises from the homolytic cleavage of C–H bonds in –CH2/–CH3, followed by hydrogen transfer, which leads to the formation of unstable free radical intermediates. These intermediates facilitate the cleavage of CO bonds and the generation of new free radicals. After the break, the C–R′ free radicals recombine or combine with H , eventually forming CO. This process can significantly reduce the Eα for CO generation and promote its formation. Similarly, asym. –CH3 & OH⋯OH also have a negative correlation, but their influence is relatively minor. The primary reason is that asym. –CH3 (asymmetric methyl) groups are susceptible to free radical formation, whereas OH⋯OH (hydrogen bond networks) may stabilize the molecular structure, thereby increasing the strength of C–H bonds, impeding free radical diffusion, and raising the energy barrier for CO generation. Additionally, quin. CO & fr. –OH as well as aro. C–H & fr. –OH exhibit a weak positive correlation with the Eα for CO formation at this temperature stage. An increase in their relative content exerts a minor elevating effect on the Eα required for CO generation. This phenomenon can be attributed to the fact that quin. CO (quinone carbonyl) exhibits a strong electron-withdrawing capability, whereas fr. –OH (free hydroxyl) functions as an electron-donating group. These two groups may engage in hydrogen bonding or undergo electron transfer, leading to the detachment of the ˙CO+ fragment and the subsequent formation of CO. The presence of hydrogen bonds increases the energy barrier for CO release, thereby retarding the CO generation process.
(aro. C–H & fr. –OH). Among these, the relative content of (–CH2/–CH3 & car. CO) exerts the most significant effect on the Eα for CO generation at this low-temperature stage, exhibiting a negative correlation. A higher product of the two functional groups prior to SO results in lower Eα. This phenomenon primarily arises from the homolytic cleavage of C–H bonds in –CH2/–CH3, followed by hydrogen transfer, which leads to the formation of unstable free radical intermediates. These intermediates facilitate the cleavage of CO bonds and the generation of new free radicals. After the break, the C–R′ free radicals recombine or combine with H , eventually forming CO. This process can significantly reduce the Eα for CO generation and promote its formation. Similarly, asym. –CH3 & OH⋯OH also have a negative correlation, but their influence is relatively minor. The primary reason is that asym. –CH3 (asymmetric methyl) groups are susceptible to free radical formation, whereas OH⋯OH (hydrogen bond networks) may stabilize the molecular structure, thereby increasing the strength of C–H bonds, impeding free radical diffusion, and raising the energy barrier for CO generation. Additionally, quin. CO & fr. –OH as well as aro. C–H & fr. –OH exhibit a weak positive correlation with the Eα for CO formation at this temperature stage. An increase in their relative content exerts a minor elevating effect on the Eα required for CO generation. This phenomenon can be attributed to the fact that quin. CO (quinone carbonyl) exhibits a strong electron-withdrawing capability, whereas fr. –OH (free hydroxyl) functions as an electron-donating group. These two groups may engage in hydrogen bonding or undergo electron transfer, leading to the detachment of the ˙CO+ fragment and the subsequent formation of CO. The presence of hydrogen bonds increases the energy barrier for CO release, thereby retarding the CO generation process.
| |
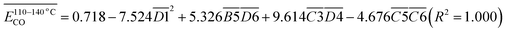 | (10) |
The average Eα for CO formation in the 110–140 °C stage 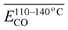 has a certain correlation with
has a certain correlation with 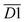 (the relative content of aro. C–H),
(the relative content of aro. C–H), 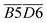 (the product of the relative contents of quin. CO and fr. –OH),
(the product of the relative contents of quin. CO and fr. –OH), 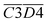 (the product of the relative contents of sym. –CH3 and OH⋯OH), and
(the product of the relative contents of sym. –CH3 and OH⋯OH), and 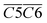 (the product of the relative contents of asym. –CH2 and asym. –CH3). Among them, aro. C–H, asym. –CH2 and asym. –CH3 have an enhancing effect on the Eα for CO formation at this temperature stage. The aromatic C–H bond tends to undergo homolytic cleavage upon heating, resulting in the formation of aromatic radicals (Ar˙), which subsequently react with O2 to generate peroxide intermediates that ultimately decompose into CO. However, the fact that this term in eqn (10) is
(the product of the relative contents of asym. –CH2 and asym. –CH3). Among them, aro. C–H, asym. –CH2 and asym. –CH3 have an enhancing effect on the Eα for CO formation at this temperature stage. The aromatic C–H bond tends to undergo homolytic cleavage upon heating, resulting in the formation of aromatic radicals (Ar˙), which subsequently react with O2 to generate peroxide intermediates that ultimately decompose into CO. However, the fact that this term in eqn (10) is 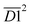 indicates that when the content of aro. C–H is low, its influence on the Eα for the formation of CO is relatively weak. With increasing aro. C–H content, the rate of free radical chain reactions enhances, thereby significantly lowering the Eα. Additionally, asymmetric aliphatic chains (asym. –CH2/CH3) are susceptible to β-cleavage, leading to the formation of aldehyde intermediates that further oxidize to CO. This pathway facilitates low-temperature oxidation and contributes to a reduction in Eα. The quinone carbonyl (quin. CO) is a strong electron-withdrawing group. It forms hydrogen bonds or charge transfer complexes with free hydroxyl groups (fr. –OH), temporarily stabilizing the structure and delaying oxidation. Oxidation requires higher energy to break the hydrogen bonds, thus slightly increasing the Eα. Symmetrical methyl groups (sym. –CH3) are difficult to form free radicals in the hydrogen bond network (OH⋯OH), and the hydrogen bonds enhance the intramolecular cohesion, significantly increasing the oxidation Eα and inhibiting the generation of CO.
indicates that when the content of aro. C–H is low, its influence on the Eα for the formation of CO is relatively weak. With increasing aro. C–H content, the rate of free radical chain reactions enhances, thereby significantly lowering the Eα. Additionally, asymmetric aliphatic chains (asym. –CH2/CH3) are susceptible to β-cleavage, leading to the formation of aldehyde intermediates that further oxidize to CO. This pathway facilitates low-temperature oxidation and contributes to a reduction in Eα. The quinone carbonyl (quin. CO) is a strong electron-withdrawing group. It forms hydrogen bonds or charge transfer complexes with free hydroxyl groups (fr. –OH), temporarily stabilizing the structure and delaying oxidation. Oxidation requires higher energy to break the hydrogen bonds, thus slightly increasing the Eα. Symmetrical methyl groups (sym. –CH3) are difficult to form free radicals in the hydrogen bond network (OH⋯OH), and the hydrogen bonds enhance the intramolecular cohesion, significantly increasing the oxidation Eα and inhibiting the generation of CO.
| |
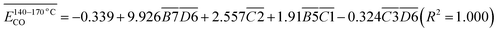 | (11) |
The average Eα for CO formation during the 140–170 °C stage is 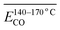 correlates with the relative contributions of
correlates with the relative contributions of 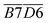 (CO & fr. –OH),
(CO & fr. –OH), 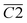 (sym. –CH2),
(sym. –CH2), 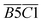 (quin. CO & –COOH), and
(quin. CO & –COOH), and 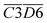 (sym. –CH3 & fr. –OH), as described by eqn (11). Among these functional group combinations, CO & fr. –OH, sym. –CH2, and quin. CO & –COOH tend to moderately increase the Eα required for CO generation at this temperature range. In contrast, only the sym. –CH3 & fr. –OH interaction demonstrates a slight reducing effect on the Eα for CO formation. The carbonyl group (CO) and the free hydroxyl group (fr. –OH) can form intramolecular hydrogen bonds, leading to the formation of a stable six-membered ring transition state. The enhanced stability of this structure necessitates a higher energy input for its cleavage, thereby significantly increasing the Eα. The symmetrical methylene group (sym. –CH2), characterized by its structural regularity and high C–H bond dissociation energy, exhibits resistance to homolytic cleavage. Consequently, an increase in its content results in a corresponding elevation of the overall Eα. The quinone carbonyl (quin. CO) and the carboxyl group (–COOH) can establish a conjugated system, forming a stabilized structure represented as O = quin. CO⋯HOOC–, which requires additional energy to disrupt. In contrast, the interaction between the methyl group (sym. –CH3) and the free hydroxyl group (fr. –OH) introduces steric hindrance (CH3⋯HO–), which weakens the hydrogen bond network (OH⋯OH interactions) and slightly facilitates the generation of free radicals, thereby reducing the Eα.
(sym. –CH3 & fr. –OH), as described by eqn (11). Among these functional group combinations, CO & fr. –OH, sym. –CH2, and quin. CO & –COOH tend to moderately increase the Eα required for CO generation at this temperature range. In contrast, only the sym. –CH3 & fr. –OH interaction demonstrates a slight reducing effect on the Eα for CO formation. The carbonyl group (CO) and the free hydroxyl group (fr. –OH) can form intramolecular hydrogen bonds, leading to the formation of a stable six-membered ring transition state. The enhanced stability of this structure necessitates a higher energy input for its cleavage, thereby significantly increasing the Eα. The symmetrical methylene group (sym. –CH2), characterized by its structural regularity and high C–H bond dissociation energy, exhibits resistance to homolytic cleavage. Consequently, an increase in its content results in a corresponding elevation of the overall Eα. The quinone carbonyl (quin. CO) and the carboxyl group (–COOH) can establish a conjugated system, forming a stabilized structure represented as O = quin. CO⋯HOOC–, which requires additional energy to disrupt. In contrast, the interaction between the methyl group (sym. –CH3) and the free hydroxyl group (fr. –OH) introduces steric hindrance (CH3⋯HO–), which weakens the hydrogen bond network (OH⋯OH interactions) and slightly facilitates the generation of free radicals, thereby reducing the Eα.
| |
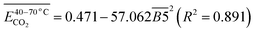 | (12) |
The average Eα for CO2 generation during the 40–70 °C stage is 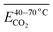 exhibits a negative correlation with the square of the relative content of quin. CO (
exhibits a negative correlation with the square of the relative content of quin. CO (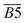 ). As the relative content of quin. CO increases, the Eα decreases. This phenomenon can be primarily attributed to the fact that the quinone carbonyl group functions as a strong electron-withdrawing moiety. Through conjugation effects, it reduces the electron cloud density along adjacent C–C bonds, thereby weakening their bond strength. This structural modification facilitates the formation of carboxylic acid groups (–COOH), which serve as precursors for CO2 generation. Furthermore, the quinone carbonyl group participates in synergistic oxidation reactions with neighboring aliphatic chains, ultimately leading to the production of CO2. Throughout this progressive oxidation process, the reaction energy barriers are systematically lowered, resulting in a continuous decrease in the Eα required for CO2 formation within this temperature range. However, in eqn (12), the quin. CO term is represented by the square of its relative content
). As the relative content of quin. CO increases, the Eα decreases. This phenomenon can be primarily attributed to the fact that the quinone carbonyl group functions as a strong electron-withdrawing moiety. Through conjugation effects, it reduces the electron cloud density along adjacent C–C bonds, thereby weakening their bond strength. This structural modification facilitates the formation of carboxylic acid groups (–COOH), which serve as precursors for CO2 generation. Furthermore, the quinone carbonyl group participates in synergistic oxidation reactions with neighboring aliphatic chains, ultimately leading to the production of CO2. Throughout this progressive oxidation process, the reaction energy barriers are systematically lowered, resulting in a continuous decrease in the Eα required for CO2 formation within this temperature range. However, in eqn (12), the quin. CO term is represented by the square of its relative content 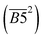 , indicating that at low relative contents of quin. CO, its impact on the Eα for CO2 generation during this temperature stage is negligible. A substantial reduction in the Eα for CO2 formation is only observed when the relative content of quin. CO reaches a relatively high level.
, indicating that at low relative contents of quin. CO, its impact on the Eα for CO2 generation during this temperature stage is negligible. A substantial reduction in the Eα for CO2 formation is only observed when the relative content of quin. CO reaches a relatively high level.
| |
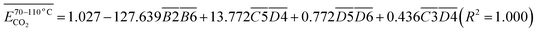 | (13) |
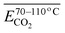
is the average
Eα of CO
2 generation in the 70–110 °C temperature range,
eqn (13) reveals that only the functional group combination
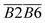
(–CH
2–CH
3 & car. C
O) exhibits a significant negative correlation, with a relatively high regression coefficient of 127.639. This suggests that even a minor increase in the relative content of (–CH
2–CH
3 & car. C
O) can lead to a notable reduction in the
Eα required for CO
2 formation. The underlying mechanism is likely attributed to the synergistic oxidation process between the aliphatic chain and the conjugated carbonyl group. Specifically, the aliphatic chain serves as a hydrogen donor, while the carbonyl group activates the adjacent C–H bond, facilitating the formation of peroxides and subsequent generation of R–COOH species. As a result, the energy barrier associated with this reaction pathway is relatively low, indicating that this process constitutes a primary route for CO
2 production. (R–CH
2–CH
3 + car. C
O + O
2 → R–CH
2–C(OOH)–CH
3 → R–COOH + CH
3COOH → CO
2)Other indicators, such as
(
asym. –CH
2 & OH⋯OH),
(OH⋯π & fr. –OH), and
(
sym. –CH
3 & OH⋯OH), exhibit a positive correlation with
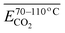 Eα
Eα. That is, an increase in their relative contents may lead to a corresponding increase in
Eα. For instance, the
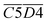
interaction involving
asym. –CH
2 may elevate
Eα primarily due to the inhibitory effect of the hydrogen bond network on oxidation. The intermolecular hydrogen bonding between hydroxyl groups (OH⋯OH) contributes to molecular structural stability. The
asym. –CH
2 bonds themselves require higher energy for cleavage, and any interaction involving these bonds necessitates prior disruption of hydrogen bonds. Therefore, their coexistence and simultaneous increase in relative content result in a higher
Eα required for CO
2 generation through oxidation. Additionally, OH⋯π interactions compete with fr. –OH groups, where the OH⋯π hydrogen bonds disperse radical attack sites, requiring the prior dissociation of π bonds. Similarly, the oxidation of
sym. –CH
3 groups must overcome steric hindrance. Collectively, these processes demand relatively high
Eα.
| |
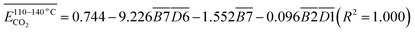 | (14) |
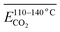
denotes the average activation energy for CO
2 generation during the temperature stage of 110–140 °C (
eqn (14)). In this process, the product of the relative contents of
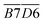
(–C
O & fr. –OH) serves to supply protons from free hydroxyl groups (fr. –OH), while the carbonyl group (–C
O) activates the β position C–C bond, synergistically promoting the decarboxylation reaction through the following pathway: R–CO–CH
2– + fr. –OH → R–CO–CH– + H
2O → R–COOH → CO
2;
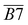
represents the relative content of isolated carbonyl groups (C
O), which undergo direct cleavage, as well as the low-temperature decarboxylation of quinone-like structures, resulting in a reaction rate enhancement of up to 10
3 fold.
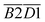
represents the product of the relative contents of –CH
2\–CH
3 and aro. C–H, indicating the cooperative effect between aliphatic chains and aromatic rings in facilitating oxidation reactions. Ar–H + –CH
2–CH
3 → Ar– + –CH
2–CH
2– → Peroxide → Aldehyde → CO
2; The relative content of the relevant functional groups involved in the above process has a negative correlation with the
Eα. As the relative content increases, the
Eα decreases.
| |
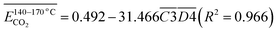 | (15) |
The average Eα of CO2 generation in the 140–170 °C stage is denoted as 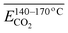 . Eqn (15) shows that
. Eqn (15) shows that 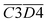 (sym. –CH3 & OH⋯OH) has a negative correlation with
(sym. –CH3 & OH⋯OH) has a negative correlation with 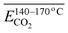 indicating that an increase in the relative content of sym. –CH3 & OH⋯OH can reduce the Eα of CO2 generation at this temperature stage. The primary mechanism underlying this phenomenon involves the activation of methyl groups via proton transfer within the hydrogen bond network, leading to the formation of the free radical CH2–R. The OH⋯OH hydrogen bonding structure contributes to the stabilization of the transition state and enhances the efficiency of proton transfer, thereby significantly reducing the Eα required for methyl dehydrogenation (C–H bond cleavage). The spatial symmetry of the sym. –CH3 group facilitates its interaction with the hydrogen bond network, further accelerating the dehydrogenation process and promoting free radical generation. Subsequently, peroxide chain reactions and deep oxidation pathways proceed through low-energy-barrier mechanisms, enabling the efficient conversion of carbon chains into CO2. These sequential and synergistic processes collectively contribute to a substantial reduction in the Eα for CO2 generation.
indicating that an increase in the relative content of sym. –CH3 & OH⋯OH can reduce the Eα of CO2 generation at this temperature stage. The primary mechanism underlying this phenomenon involves the activation of methyl groups via proton transfer within the hydrogen bond network, leading to the formation of the free radical CH2–R. The OH⋯OH hydrogen bonding structure contributes to the stabilization of the transition state and enhances the efficiency of proton transfer, thereby significantly reducing the Eα required for methyl dehydrogenation (C–H bond cleavage). The spatial symmetry of the sym. –CH3 group facilitates its interaction with the hydrogen bond network, further accelerating the dehydrogenation process and promoting free radical generation. Subsequently, peroxide chain reactions and deep oxidation pathways proceed through low-energy-barrier mechanisms, enabling the efficient conversion of carbon chains into CO2. These sequential and synergistic processes collectively contribute to a substantial reduction in the Eα for CO2 generation.
4 Conclusions
(1) During secondary oxidation (SO) over 40–170 °C, the apparent activation energy (Eα) for CO evolution is predominantly governed by two opposing functional group interactions: (i) the –CH2/–CH3 & carbonyl (CO) pair, which significantly lowers Ea via formation of a six-membered cyclic transition state-facilitated by facile oxidation of the α-carbon adjacent to the carbonyl group; and (ii) the –CH2/–CH3 & asymmetric –CH3 pair, which elevates Eα due to steric congestion that impedes H-abstraction from the hindered methyl site. In contrast, Eα for CO2 evolution across the same temperature interval is jointly modulated by –CH2/–CH3 & CO, asymmetric –CH3, and intermolecular hydrogen-bonded motifs (OH⋯OH and OH⋯π). All exhibit strong negative correlations with CO2 Eα, with the –CH2/–CH3 & CO interaction exerting the largest suppressive effect-consistent with its dual role in promoting both decarbonylation and carboxylate decomposition pathways.
(2) Temperature-resolved analysis reveals distinct structure-reactivity regimes: (i) in the low-temperature SO stage (40–70 °C), the average Eα for CO generation is most sensitively suppressed by elevated –CH2/–CH3 & CO content, while quinonoid CO & free-OH, asymmetric –CH3 & OH⋯OH, and aromatic C–H & free –OH combinations also contribute significantly. (ii) In the mid-temperature range (110–140 °C), Eα exhibits strong negative correlations with aromatic C–H, asymmetric –CH2, and asymmetric –CH3-indicating enhanced radical initiation from these sites-yet shows positive correlations with quinonoid CO & free –OH and symmetric –CH3 & OH⋯OH, suggesting competitive stabilization or kinetic inhibition under these conditions. (iii) In the high-temperature window (140–170 °C), Eα is strongly and positively correlated with CO & free –OH, symmetric –CH2, and quinonoid CO & –COOH, but weakly and negatively correlated with symmetric –CH3 & free–OH–highlighting a shift toward acid-catalyzed and symmetry-facilitated dehydrogenation mechanisms.
(3) For CO2 evolution, functional group effects are markedly temperature-dependent: (i) at 40–70 °C, Eα scales inversely with the square of quinonoid CO concentration-indicating second-order kinetic dependence and pointing to quinone-mediated electron-transfer catalysis in early-stage carboxylation. (ii) Between 70–110 °C, Eα is strongly suppressed by –CH2/–CH3 and carbonyl (CO) groups, while exhibiting weak positive correlations with asymmetric –CH2, OH⋯OH, OH⋯π & free –OH, and symmetric –CH3 & OH⋯OH–suggesting competing roles of H-bonding networks in either stabilizing intermediates or retarding proton-coupled electron transfer. (iii) In the 110–140 °C regime, the dominant factor is the CO & free –OH interaction: free-OH acts as a Brønsted proton donor, while adjacent CO polarizes and activates the β–C–C bond-synergistically lowering the decarboxylation barrier through concerted proton transfer and bond cleavage. (iv) At 140–170 °C, Eα is primarily dictated by symmetric –CH3 & OH⋯OH interactions. The extended hydrogen-bond network enables efficient proton relay, stabilizing dehydrogenation transition states and enhancing methyl radical formation. Critically, the spatial symmetry of –CH3 improves geometric compatibility with the H-bond lattice, accelerating initial C–H homolysis and enabling rapid propagation of peroxide chain reactions-thereby facilitating deep oxidative cleavage of aliphatic chains into CO2 at substantially reduced energetic cost.
Conflicts of interest
The authors declare no competing financial interest.
Data availability
All relevant data utilized in this article are encompassed within the figures and data tables presented in the manuscript.
Acknowledgements
This research was funded by the National Natural Science Foundation of China (grant number 51804107), Specialty Found of Zhejiang Institute of Tianjin University (Grant No. ZITJU2024-ZYHT005).
References
- A. K. Singh, R. V. K. Singh, M. P. Singh, H. Chandra and N. K. Shukla, Mine fire gas indices and their application to Indian underground coal mine fires, Int. J. Coal Geol., 2007, 69, 192–204 CrossRef CAS.
- J. N. Carras, S. J. Day, A. Saghaff and D. J. Williams, Greenhouse gas emissions from low-temperature oxidation and spontaneous combustion at open-cut coal mines in Australia, Int. J. Coal Geol., 2009, 78, 161–168 CrossRef CAS.
- C. P. Wang, X. Y. Zhao, Z. J. Bai, J. Deng, C. M. Shu and M. Zhang, Comprehensive index evaluation of the spontaneous combustion capability of different ranks of coal, Fuel, 2021, 291, 120087 CrossRef CAS.
- X. Huang and G. Rein, Smouldering combustion of peat in wildfires: inverse modelling of the drying and the thermal and oxidative decomposition kinetics, Combust. Flame, 2014, 161, 1633–1644 CrossRef CAS.
- G. Rein, Smoldering combustion phenomena and coal fires, in Coal and Peat Fires: A Global Perspective, Elsevier, 2011, pp. 307–315 Search PubMed.
- H. Yuan, F. Richter and G. Rein, A multi-step reaction scheme to simulate self-heating ignition of coal: effects of oxygen adsorption and smouldering combustion, Proc. Combust. Inst., 2021, 38, 4717–4725 CrossRef CAS.
- D. M. Wang, H. H. Xin, X. Y. Qi, G. I. Dou, G. S. Qi and L. Y. Ma, Reaction pathway of coal oxidation at low temperatures: a model of cyclic chain reactions and kinetic characteristics, Combust. Flame, 2016, 163, 447–460 CrossRef CAS.
- J. Zhao, J. Deng, L. Chen, T. Wang, J. Song and Y. Zhang, Correlation analysis of the functional groups and exothermic characteristics of bituminous coal molecules during high-temperature oxidation, Energy, 2019, 181, 136–147 CrossRef CAS.
- Y. Zhang, J. Wu, L. Chang, J. Wang, S. Xue and Z. Li, Kinetic and thermodynamic studies on the mechanism of low-temperature oxidation of coal: a case study of Shendong coal (China), Int. J. Coal Geol., 2013, 120, 41–49 CrossRef CAS.
- J. L. Li, L. Wei, Y. T. Liang, G. S. Qi, B. Kong and X. M. Hu, Variation of CO2/CO ratio during pure-oxidation of feed coal, Fuel, 2010, 262, 116588 CrossRef.
- L. Yuan and A. C. Smith, CO and CO2 emissions from spontaneous heating of coal under different ventilation rates, Int. J. Coal Geol., 2011, 88, 24–30 CrossRef CAS.
- L. Yuan and A. C. Smith, Experimental study on CO and CO2 emissions from spontaneous heating of coals at varying temperatures and O2 concentrations, J. Loss Prev. Process Ind., 2013, 26, 1321–1327 CrossRef CAS PubMed.
- Y. L. Zhang, J. F. Wang, J. M. Wu, S. Xue, Z. F. Li and L. P. Chang, Modes and kinetics of CO2 and CO production from low-temperature oxidation of coal, Int. J. Coal Geol., 2015, 140, 1–8 CrossRef CAS.
- H. H. Xin, Z. F. Qi, W. J. Tian, L. Lu, P. C. Zhang, B. H. Zhou, J. F. Wu, Z. Y. Xu, H. Z. Wang and D. M. Wang, The disaster-causing characteristics of CO and CO2 rapid generation in coal's high-temperature oxidation: the effect law of oxygen concentration, J. Therm. Anal. Calorim., 2024, 149, 2825–2842 CrossRef CAS.
- J. L. Li, H. Lu, W. Lu, J. H. Li, Q. S. Zhang and H. Zhuo, Study on the kinetic characteristics and control steps of gas production in coal spontaneous combustion under the oxidation path, Energy, 2024, 295, 130991 CrossRef CAS.
- Z. Aizenstat, U. Green and S. Stark, Modes of formation of carbon oxides (COx, x = 1, 2) from coals during atmospheric storage: Part I effect of coal rank, Energy Fuels, 2010, 24, 6374 CrossRef.
- Z. Aizenstat, U. Green and J. C. Hower, Modes of formation of carbon oxides [COx (x = 1 or 2)] from coals during atmospheric storage. Part 2: effect of coal rank on the kinetics, Energy Fuels, 2011, 25, 5626–5631 CrossRef.
- H. Wang, B. Z. Dlugogorski and E. M. Kennedy, Examination of CO2, CO and H2O formation during low-temperature oxidation of a bituminous coal, Energy Fuels, 2002, 16, 586–592 CrossRef CAS.
- H. Wang, B. Z. Dlugogorski and E. M. Kennedy, Pathways for production of CO2 and CO in low-temperature oxidation of coal, Energy Fuels, 2003, 17, 150–158 CrossRef CAS.
- Y. Wang, Y. Sun, L. Dai, K. Wang and G. Cheng, Mechanisms of CO and CO2 production during the low-temperature oxidation of coal: molecular simulations and experimental research, Fire, 2023, 6, 475 CrossRef.
- D. X. Xu, L. J. Zhang, W. J. He, Y. L. Xu, Y. B. Zhao, J. Zhu and Q. K. Qi, The generation mechanism of CO and CO2 in coal spontaneous combustion by mathematical statistical and other methods, Fuel, 2023, 350, 128747 CrossRef CAS.
- T. Xu, D. M. Wang, H. H. Xin, G. L. Dou, X. X. Zhong and X. Y. Qi, In-situ series diffuse reflection FTIR used in studying the oxidation process of coal, Energy Sources, Part A, 2016, 38, 1756–1763 CrossRef.
- D. Wang, X. Zhong, J. Gu and X. Qi, Changes in active functional groups during low-temperature oxidation of coal, Int. J. Min. Sci. Technol., 2010, 20, 35–40 CAS.
- D. Zhang, X. Cen, W. Wang, J. Deng, H. Wen and Y. Xiao, Gray correlation analysis of CO generation pattern during process of low-temperature spontaneous combustion of Jurassic coal, Combust. Sci. Technol., 2023, 195, 2133–2149 CrossRef CAS.
- H. H. Xin, W. J. Tian, B. H. Zhou, J. F. Wu, Z. Y. Xu, L. Lu and D. M. Wang, Characteristics of CO, CO2 generation and reactive group conversion in isothermal smoldering combustion of coal, Fuel, 2023, 332, 126175 CrossRef CAS.
- J. Li, Z. Li, Y. Yang, Y. Duan, J. Xu and R. Gao, Examination of CO, CO2 and active sites formation during isothermal pyrolysis of coal at low temperatures, Energy, 2019, 185, 28–38 CrossRef CAS.
- K. Jayaraman, M. V. Kok and I. Gokalp, Thermogravimetric and mass spectrometric (TG-MS) analysis and kinetics of coal-biomass blends, Renewable Energy, 2017, 101, 293–300 CrossRef CAS.
- X. Zhang, R. Sun, B. Lu, G. Huang, L. Qiao and H. Liang, et al., Study on the characteristics of secondary oxidation and the key reactive groups of fire area coal by different cooling rates, Case Stud. Therm. Eng., 2025, 69, 106014 CrossRef.
- L. Chen, J. Zhao, W. Lu, Z. Liang, J. Li and X. Qi, et al., Reaction pathways and functional group transformation in CO/CO2 generation from low-rank coal at low temperature oxidation, Case Stud. Therm. Eng., 2025, 74, 106773 CrossRef.
- K. Niu, B. Shi, Y. Song and H. Wang, et al., Regulatory mechanism of oxidation temperature on secondary spontaneous combustion activity of coal: correlation between functional group evolution and indicator gas generation, ACS Omega, 2025, 10, 35217–35229 CrossRef CAS PubMed.
- Q. Liu, S. Yang, Z. Liu, Q. Liu, L. Shi and W. Han, Comparison of TG-MS and GC-simulated distillation for determination of the boiling point distribution of various oils, Fuel, 2021, 301, 121088 CrossRef CAS.
- H. Zhang, S. Xian, Z. Zhu and X. Guo, Release behaviors of sulfur-containing pollutants during combustion and gasification of coals by TG-MS, J. Therm. Anal. Calorim., 2020, 143, 377–386 CrossRef.
- T. Wang, H. Rong, S. Chen, Y. Zhou and J. Li, TG-MS study on in-situ sulfur retention during the co-combustion of reclaimed asphalt binder and wood sawdust, J. Hazard. Mater., 2021, 403, 123911 CrossRef CAS PubMed.
- X. Song, J. Wei, Y. Bai, J. Wang, W. Su and M. Yao, Investigation on gas release characteristics of catalytic coal pyrolysis using thermogravimetric analyzer-mass spectrometry, Energy Sources, Part A, 2021, 43, 1805–1817 CrossRef.
- Y. Zhang, C. Wang and B. Zhao, In situ FTIR study on the evolution of functional groups during coal oxidation at low-temperature gradients, Fuel Process. Technol., 2023, 241, 107603 CrossRef.
- H. Li, X. Liu and J. Deng, Functional group responses in pre-oxidized coal under programmed temperature oxidation: a FTIR peak deconvolution approach, Energy, 2024, 288, 129712 CrossRef.
- R. Chen, Y. Tang and S. Xue, Quantitative characterization of oxygen-containing functional groups in coal by FTIR coupled with spectral subtraction and deconvolution, Spectrochim. Acta, Part A, 2022, 271, 120893 CrossRef PubMed.
- B. Zhao, Y. Zhang and H. Wang, Synergistic effects between carbonyl and alkyl groups in low-temperature coal oxidation: evidence from DFT calculations and in situ FTIR, Fuel, 2023, 343, 128039 CrossRef.
- X. Liu, J. Deng and Q. Li, Steric hindrance of methyl groups in coal oxidation: kinetic implications for CO production during secondary oxidation, Energy Fuels, 2024, 38, 5567–5578 Search PubMed.
- R. Chen, Y. Tang and S. Xue, Predicting activation energy of CO emission based on functional group interaction networks during coal re-oxidation, Proc. Combust. Inst., 2023, 39, 4183–4192 Search PubMed.
- G. Huang, Y. Z. Zhang, F. W. Dai and X. Zhang, Research on the intrinsic correlation mechanism between stage oxidation characteristics and spontaneous combustion tendency of coals with different metamorphic degrees, Chem. Eng. Sci., 2025, 314, 121800 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Ziwen Dong*ab and
Keyu Wanga
a,
Ziwen Dong*ab and
Keyu Wanga

















 denotes the j-th observed value of the i-th indicator.
denotes the j-th observed value of the i-th indicator.

























 has a relationship with
has a relationship with  (–CH2/–CH3 & car. C
(–CH2/–CH3 & car. C (–CH2/–CH3 & asym. –CH3) as shown in eqn (7). The product of the relative contents of –CH2/–CH3 and car. C
(–CH2/–CH3 & asym. –CH3) as shown in eqn (7). The product of the relative contents of –CH2/–CH3 and car. C

 is related to
is related to  (–CH2/–CH3 & car. C
(–CH2/–CH3 & car. C (asym. –CH3)
(asym. –CH3)  (OH⋯OH & OH⋯π) and
(OH⋯OH & OH⋯π) and  (–CH2/–CH3) as shown in eqn (8). The relative content of the functional groups exhibits a negative correlation with the Eα for CO2 generation. An increase in their relative content contributes to a reduction in the Eα required for CO2 formation during the SO process. The main reaction pathways are illustrated in Fig. 13.
(–CH2/–CH3) as shown in eqn (8). The relative content of the functional groups exhibits a negative correlation with the Eα for CO2 generation. An increase in their relative content contributes to a reduction in the Eα required for CO2 formation during the SO process. The main reaction pathways are illustrated in Fig. 13.

 is predominantly influenced by the functional group combinations
is predominantly influenced by the functional group combinations  (–CH2/–CH3 & car. C
(–CH2/–CH3 & car. C (quin. C
(quin. C (asym. –CH3 & OH⋯OH), and
(asym. –CH3 & OH⋯OH), and  (aro. C–H & fr. –OH). Among these, the relative content of (–CH2/–CH3 & car. C
(aro. C–H & fr. –OH). Among these, the relative content of (–CH2/–CH3 & car. C
 has a certain correlation with
has a certain correlation with  (the relative content of aro. C–H),
(the relative content of aro. C–H),  (the product of the relative contents of quin. C
(the product of the relative contents of quin. C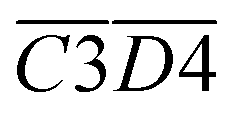 (the product of the relative contents of sym. –CH3 and OH⋯OH), and
(the product of the relative contents of sym. –CH3 and OH⋯OH), and  (the product of the relative contents of asym. –CH2 and asym. –CH3). Among them, aro. C–H, asym. –CH2 and asym. –CH3 have an enhancing effect on the Eα for CO formation at this temperature stage. The aromatic C–H bond tends to undergo homolytic cleavage upon heating, resulting in the formation of aromatic radicals (Ar˙), which subsequently react with O2 to generate peroxide intermediates that ultimately decompose into CO. However, the fact that this term in eqn (10) is
(the product of the relative contents of asym. –CH2 and asym. –CH3). Among them, aro. C–H, asym. –CH2 and asym. –CH3 have an enhancing effect on the Eα for CO formation at this temperature stage. The aromatic C–H bond tends to undergo homolytic cleavage upon heating, resulting in the formation of aromatic radicals (Ar˙), which subsequently react with O2 to generate peroxide intermediates that ultimately decompose into CO. However, the fact that this term in eqn (10) is  indicates that when the content of aro. C–H is low, its influence on the Eα for the formation of CO is relatively weak. With increasing aro. C–H content, the rate of free radical chain reactions enhances, thereby significantly lowering the Eα. Additionally, asymmetric aliphatic chains (asym. –CH2/CH3) are susceptible to β-cleavage, leading to the formation of aldehyde intermediates that further oxidize to CO. This pathway facilitates low-temperature oxidation and contributes to a reduction in Eα. The quinone carbonyl (quin. C
indicates that when the content of aro. C–H is low, its influence on the Eα for the formation of CO is relatively weak. With increasing aro. C–H content, the rate of free radical chain reactions enhances, thereby significantly lowering the Eα. Additionally, asymmetric aliphatic chains (asym. –CH2/CH3) are susceptible to β-cleavage, leading to the formation of aldehyde intermediates that further oxidize to CO. This pathway facilitates low-temperature oxidation and contributes to a reduction in Eα. The quinone carbonyl (quin. C
 correlates with the relative contributions of
correlates with the relative contributions of  (C
(C (sym. –CH2),
(sym. –CH2),  (quin. C
(quin. C (sym. –CH3 & fr. –OH), as described by eqn (11). Among these functional group combinations, C
(sym. –CH3 & fr. –OH), as described by eqn (11). Among these functional group combinations, C
 exhibits a negative correlation with the square of the relative content of quin. C
exhibits a negative correlation with the square of the relative content of quin. C ). As the relative content of quin. C
). As the relative content of quin. C , indicating that at low relative contents of quin. C
, indicating that at low relative contents of quin. C
 is the average Eα of CO2 generation in the 70–110 °C temperature range, eqn (13) reveals that only the functional group combination
is the average Eα of CO2 generation in the 70–110 °C temperature range, eqn (13) reveals that only the functional group combination  (–CH2–CH3 & car. C
(–CH2–CH3 & car. C (asym. –CH2 & OH⋯OH),
(asym. –CH2 & OH⋯OH),  (OH⋯π & fr. –OH), and
(OH⋯π & fr. –OH), and  (sym. –CH3 & OH⋯OH), exhibit a positive correlation with
(sym. –CH3 & OH⋯OH), exhibit a positive correlation with  Eα. That is, an increase in their relative contents may lead to a corresponding increase in Eα. For instance, the
Eα. That is, an increase in their relative contents may lead to a corresponding increase in Eα. For instance, the  interaction involving asym. –CH2 may elevate Eα primarily due to the inhibitory effect of the hydrogen bond network on oxidation. The intermolecular hydrogen bonding between hydroxyl groups (OH⋯OH) contributes to molecular structural stability. The asym. –CH2 bonds themselves require higher energy for cleavage, and any interaction involving these bonds necessitates prior disruption of hydrogen bonds. Therefore, their coexistence and simultaneous increase in relative content result in a higher Eα required for CO2 generation through oxidation. Additionally, OH⋯π interactions compete with fr. –OH groups, where the OH⋯π hydrogen bonds disperse radical attack sites, requiring the prior dissociation of π bonds. Similarly, the oxidation of sym. –CH3 groups must overcome steric hindrance. Collectively, these processes demand relatively high Eα.
interaction involving asym. –CH2 may elevate Eα primarily due to the inhibitory effect of the hydrogen bond network on oxidation. The intermolecular hydrogen bonding between hydroxyl groups (OH⋯OH) contributes to molecular structural stability. The asym. –CH2 bonds themselves require higher energy for cleavage, and any interaction involving these bonds necessitates prior disruption of hydrogen bonds. Therefore, their coexistence and simultaneous increase in relative content result in a higher Eα required for CO2 generation through oxidation. Additionally, OH⋯π interactions compete with fr. –OH groups, where the OH⋯π hydrogen bonds disperse radical attack sites, requiring the prior dissociation of π bonds. Similarly, the oxidation of sym. –CH3 groups must overcome steric hindrance. Collectively, these processes demand relatively high Eα.
 denotes the average activation energy for CO2 generation during the temperature stage of 110–140 °C (eqn (14)). In this process, the product of the relative contents of
denotes the average activation energy for CO2 generation during the temperature stage of 110–140 °C (eqn (14)). In this process, the product of the relative contents of  (–C
(–C represents the relative content of isolated carbonyl groups (C
represents the relative content of isolated carbonyl groups (C represents the product of the relative contents of –CH2\–CH3 and aro. C–H, indicating the cooperative effect between aliphatic chains and aromatic rings in facilitating oxidation reactions. Ar–H + –CH2–CH3 → Ar– + –CH2–CH2– → Peroxide → Aldehyde → CO2; The relative content of the relevant functional groups involved in the above process has a negative correlation with the Eα. As the relative content increases, the Eα decreases.
represents the product of the relative contents of –CH2\–CH3 and aro. C–H, indicating the cooperative effect between aliphatic chains and aromatic rings in facilitating oxidation reactions. Ar–H + –CH2–CH3 → Ar– + –CH2–CH2– → Peroxide → Aldehyde → CO2; The relative content of the relevant functional groups involved in the above process has a negative correlation with the Eα. As the relative content increases, the Eα decreases.
 . Eqn (15) shows that
. Eqn (15) shows that  (sym. –CH3 & OH⋯OH) has a negative correlation with
(sym. –CH3 & OH⋯OH) has a negative correlation with  indicating that an increase in the relative content of sym. –CH3 & OH⋯OH can reduce the Eα of CO2 generation at this temperature stage. The primary mechanism underlying this phenomenon involves the activation of methyl groups via proton transfer within the hydrogen bond network, leading to the formation of the free radical CH2–R. The OH⋯OH hydrogen bonding structure contributes to the stabilization of the transition state and enhances the efficiency of proton transfer, thereby significantly reducing the Eα required for methyl dehydrogenation (C–H bond cleavage). The spatial symmetry of the sym. –CH3 group facilitates its interaction with the hydrogen bond network, further accelerating the dehydrogenation process and promoting free radical generation. Subsequently, peroxide chain reactions and deep oxidation pathways proceed through low-energy-barrier mechanisms, enabling the efficient conversion of carbon chains into CO2. These sequential and synergistic processes collectively contribute to a substantial reduction in the Eα for CO2 generation.
indicating that an increase in the relative content of sym. –CH3 & OH⋯OH can reduce the Eα of CO2 generation at this temperature stage. The primary mechanism underlying this phenomenon involves the activation of methyl groups via proton transfer within the hydrogen bond network, leading to the formation of the free radical CH2–R. The OH⋯OH hydrogen bonding structure contributes to the stabilization of the transition state and enhances the efficiency of proton transfer, thereby significantly reducing the Eα required for methyl dehydrogenation (C–H bond cleavage). The spatial symmetry of the sym. –CH3 group facilitates its interaction with the hydrogen bond network, further accelerating the dehydrogenation process and promoting free radical generation. Subsequently, peroxide chain reactions and deep oxidation pathways proceed through low-energy-barrier mechanisms, enabling the efficient conversion of carbon chains into CO2. These sequential and synergistic processes collectively contribute to a substantial reduction in the Eα for CO2 generation.