DOI:
10.1039/D5RA09088A
(Paper)
RSC Adv., 2026,
16, 5814-5833
Chemical reactivity of 2-methylchromone with some nitrogen-based nucleophiles: reactions, DFT analysis, anticancer activity and molecular docking studies
Received
24th November 2025
, Accepted 22nd January 2026
First published on 28th January 2026
Abstract
The current study investigates the reactivity of 2-methylchromone toward nitrogen-containing heterocyclic amines, with the aim of assessing how substitution pattern and nucleophile type influence chromone ring opening and subsequent heterocycle formation. Reaction of 2-methylchromone (1) with selected heterocyclic amines afforded a series of heterocycle-linked enaminones 5, 8 and 9, which were found to preferentially adopt the Z-configuration based on spectroscopic analysis. In addition, reactions of substrate 1 with 1,3-binucleophilic reagents enabled the construction of novel pyrimidine-fused heterocycles via ring opening followed by intramolecular cyclization. DFT calculations (B3LYP/6-311+G(d,p)) were performed to study the electronic and nonlinear optical properties of the present compounds. Compound 5 was found to be the most stable than compound 3 and compound 4, consistent with experimental results. Theoretical IR and NMR spectra agreed well with experimental data. The high hyperpolarizability values of the prepared compounds suggest potential nonlinear optical applications. The synthesized compounds were evaluated for anticancer activity against HepG-2 cells, revealing compound 9 as the most active derivative, with potency exceeding that of cisplatin. Molecular docking studies against CDK1 supported the observed biological activity, while drug-likeness analysis indicated favorable pharmacokinetic profiles. Overall, this work highlights the utility of 2-methylchromone with diverse nucleophiles in generating structurally varied heterocycles, validated by combined experimental and theoretical studies.
1. Introduction
Chromones constitute an important class of oxygen-containing heterocycles that function not only as bioactive scaffolds but also as highly versatile synthetic intermediates in heterocyclic chemistry.1–3 Beyond their intrinsic biological and photophysical relevance,4–8 chromones are distinguished by their electron-deficient conjugated systems, which impart pronounced electrophilic character and render them particularly susceptible to nucleophilic attack. This reactivity facilitates a wide range of subsequent structural transformations, underscoring the value of chromones as privileged building blocks in organic synthesis.9–12
Among these derivatives, 2-methylchromones represent an important class of oxygen-containing heterocyclic compounds that have attracted considerable interest due to their diverse biological activities and synthetic versatility.13,14 Typically, 2-methylchromones are prepared through the Claisen condensation of 2-hydroxyacetophenones with ethyl acetate, followed by intramolecular acid-catalyzed cyclocondensation, providing efficient access to the chromone framework with structural control.15–17 The reactivity and electronic structure of 2-methylchromones have been systematically examined, especially in the earlier work by Ibrahim et al.,18 who demonstrated that the chromone ring possess two principal electrophilic centers located at C-2 and C-4. These positions play a pivotal role in governing the compound's behavior toward nucleophiles and thus determine the synthetic pathways available for functionalizing the core.18 Owing to the electron-deficient character of the chromone scaffold and the strong conjugation with carbonyl group, the C-2 position represents the most reactive electrophilic center toward nucleophiles. As a result, nucleophilic attack predominantly occurs at this site, often generating subsequent opening of the pyran ring. This ring-opening pathway is a well-established mechanistic feature in chromone chemistry and has been extensively documented in the literature.19
Over the past decade, theoretical studies have gained increasing importance, with computational and experimental chemistry being recognized as complementary approaches in modern research. Among computational methods, density functional theory (DFT) has emerged as a powerful tool, offering profound insights into both the kinetic stability and chemical reactivity of molecular systems.20,21 Herein, DFT calculations were employed not to predict unknown outcomes, but to quantitatively rationalize experimental observations. These include charge distribution, electrophilicity at the C-2 position, preferences for ring opening, and the relative stability of the resulting enaminone and heterocyclic structures. Such calculations provide a molecular-level insight that complements the experimental data and enables meaningful comparison with previously reported chromone systems.8–10
The aim and novelty of the present work differ fundamentally from our earlier studies. Whereas previous investigations focused on 3-substituted chromones bearing electron-withdrawing groups that markedly enhance the electrophilicity at C-2,8–12 the current study concise on 2-methylchromone, in which the methyl substituent introduces both electron-donating and steric effects. This distinct substitution pattern creates a substantially different electronic environment, enabling a systematic evaluation of how reduced electrophilicity and altered charge distribution affect reaction pathways. Moreover, while the previous work on 2-methylchromones primarily involved simple nitrogen nucleophiles, the use of amino-functionalized nitrogen heterocycles as nucleophiles is explored here for the first time. This choice was motivated by the objective of generating enaminone systems incorporating heterocyclic fragments, which are synthetically valuable motifs with recognized relevance in medicinal chemistry and molecular recognition.
In this study, we report the synthesis of a series of novel heterocyclic derivatives via the reaction of 2-methylchromone (1) with selected heterocyclic amines and 1,3-binucleophilic reagents, followed by evaluation of their anticancer activity against HepG-2 cells. To gain deeper insight into their structural and electronic properties, DFT calculations were carried out at the B3LYP/6-311++G(d,p) level, where global reactivity descriptors were derived from HOMO–LUMO energies, and experimental NMR and FT-IR data were validated through GIAO-based theoretical simulations. Additionally, nonlinear optical (NLO) properties and molecular electrostatic potential (MEP) surfaces were examined. Molecular docking studies were performed against the CDK1 protein (PDB ID: 4Y72) to explore possible binding interactions, and drug-likeness assessment was conducted according to Lipinski, Egan, Veber, and Ghose rules.
2. Experimental
Melting points were determined on a digital Stuart SMP3 apparatus. Elemental microanalyses were performed on a PerkinElmer CHN-2400 analyzer. Infrared spectra were measured on FTIR Nicolet IS10 spectrophotometer (cm−1), using KBr disks. 1H NMR spectra were measured on mercury-400BB (400 MHz), using DMSO-d6 as a solvent and TMS (δ) as the internal standard. 13C NMR spectra were measured on mercury-400BB (100 MHz), using DMSO-d6 as a solvent and TMS (δ) as the internal standard. Mass spectra were obtained using GC-2010 Shimadzu Gas chromatography instrument mass spectrometer (70 eV). The purity of the synthesized compounds was tested using TLC. 1-Amino-4,6-dimethyl-2-oxo-1,2-dihydropyridine-3-carbonitrile (2),22 4-amino-5-methyl-2,4-dihydro-3H-1,2,4-triazole-3-thione (6)23 and 4-amino-6-methyl-3-thioxo-3,4-dihydro-1,2,4-triazin-5(2H)-one (7)24 were synthesized according to literature.
2.1. Synthesis and characterization of compounds
2.1.1. 1-{[(2Z)-4-(2-Hydroxyphenyl)-4-oxobut-2-en-2-yl]amino}-4,6-dimethyl-2-oxo-1,2-dihydropyridine-3-carbonitrile (5). A mixture of 2-methylchromone (1) (0.32 g, 2 mmol) and 1-amino-4,6-dimethyl-2-oxo-1,2-dihydropyridine-3-carbonitrile (2) (0.32 g, 2 mmol) in absolute ethanol (10 mL) containing DBU (0.2 mL) was heated under reflux for 2 h. The pale yellow crystals obtained during heating were filtered and crystallized from EtOH, mp 205–206 °C, yield (0.47 g, 73%). IR (KBr, cm−1): 3406 (OH), 3279 (NH), 2217 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1676 (C
N), 1676 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Opyridone), 1642 (COenaminone), 1588 (CC). 1H NMR (DMSO-d6, δ): 2.10 (s, 3H, CH3), 2.31 (s, 3H, CH3), 2.39 (s, 3H, CH3), 6.20 (s, 1H, H-αolefinic), 6.34 (s, 1H, H-5pyridine), 7.38 (t, 1H, J = 7.2 Hz, Ar–H), 7.61 (d, 1H, J = 7.2 Hz, Ar–H), 7.81 (t, 1H, J = 7.5 Hz, Ar–H), 8.12 (d, 1H, J = 7.5 Hz, Ar–H), 9.80 (bs, 1H, NH exchangeable with D2O), 12.22 (bs, 1H, OH exchangeable with D2O). 13C NMR (DMSO-d6, δ): 15.9 (CH3), 17.5 (CH3), 19.4 (CH3), 94.2 (C-αolefinic), 106.2 (C-3pyridone), 108.3 (C-5pyridone), 116.4 (CN), 122.2, 124.9, 126.8, 128.4, 129.5 (5Ar–C), 132.8 (C-6pyridone), 142.3 (C-4pyridone), 148.2 (C-βolefinic), 152.1, (C–OH), 163.4 (COpyridone), 182.6 (COketone). Mass spectrum, m/z (Ir%): 323 (M+, 11), 308 (17), 215 (16), 176 (100), 161 (34), 146 (16), 121 (22), 93 (62), 77 (42), 65 (13). Anal. calcd for C18H17N3O3 (323.34); C, 66.86; H, 5.30; N, 13.00%. Found: C, 66.58; H, 5.12; N, 12.84%.
Opyridone), 1642 (COenaminone), 1588 (CC). 1H NMR (DMSO-d6, δ): 2.10 (s, 3H, CH3), 2.31 (s, 3H, CH3), 2.39 (s, 3H, CH3), 6.20 (s, 1H, H-αolefinic), 6.34 (s, 1H, H-5pyridine), 7.38 (t, 1H, J = 7.2 Hz, Ar–H), 7.61 (d, 1H, J = 7.2 Hz, Ar–H), 7.81 (t, 1H, J = 7.5 Hz, Ar–H), 8.12 (d, 1H, J = 7.5 Hz, Ar–H), 9.80 (bs, 1H, NH exchangeable with D2O), 12.22 (bs, 1H, OH exchangeable with D2O). 13C NMR (DMSO-d6, δ): 15.9 (CH3), 17.5 (CH3), 19.4 (CH3), 94.2 (C-αolefinic), 106.2 (C-3pyridone), 108.3 (C-5pyridone), 116.4 (CN), 122.2, 124.9, 126.8, 128.4, 129.5 (5Ar–C), 132.8 (C-6pyridone), 142.3 (C-4pyridone), 148.2 (C-βolefinic), 152.1, (C–OH), 163.4 (COpyridone), 182.6 (COketone). Mass spectrum, m/z (Ir%): 323 (M+, 11), 308 (17), 215 (16), 176 (100), 161 (34), 146 (16), 121 (22), 93 (62), 77 (42), 65 (13). Anal. calcd for C18H17N3O3 (323.34); C, 66.86; H, 5.30; N, 13.00%. Found: C, 66.58; H, 5.12; N, 12.84%.
2.1.2. (2Z)-1-(2-Hydroxyphenyl)-3-[(3-methyl-5-thioxo-1,5-dihydro-4H-1,2,4-triazol-4-yl)amino]but-2-en-1-one (8). A mixture of 2-methylchromone (1) (0.32 g, 2 mmol) and 4-amino-5-methyl-2,4-dihydro-3H-1,2,4-triazole-3-thione (6) (0.26 g, 2 mmol) in absolute ethanol (10 mL) containing DBU (0.2 mL) was heated under reflux for 2 h. The yellow crystals obtained during heating were filtered and crystallized from isopropanol, mp 236–237 °C, yield (0.46 g, 77%). IR (KBr, cm−1): 3409 (OH), 3248, 3211 (2NH), 1644 (COenaminone), 1613 (CN), 1587 (CC), 1223 (CS). 1H NMR (DMSO-d6, δ): 2.27 (s, 3H, CH3), 2.37 (s, 3H, CH3), 6.32 (s, 1H, H-αolefinic), 7.54 (t, 1H, J = 7.5 Hz, Ar–H), 7.72 (d, 1H, J = 7.8 Hz, Ar–H), 7.82 (t, 1H, J = 7.5 Hz, Ar–H), 8.14 (d, 1H, J = 7.5 Hz, Ar–H), 9.94 (bs, 1H, NH exchangeable with D2O), 10.74 (bs, 1H, NH exchangeable with D2O), 11.81 (bs, 1H, OH exchangeable with D2O). 13C NMR (DMSO-d6, δ): 15.6 (CH3), 16.4 (CH3), 96.1 (C-αolefinic), 122.6, 123.3, 126.4, 125.1, 128.6 (5Ar–C), 144.3 (C-3triazole), 148.1 (C-βolefinic), 150.8, (C–OH), 182.3 (COketone), 194.5 (CS). Mass spectrum, m/z (Ir%): 290 (M+, 21), 245 (13), 219 (18), 176 (100), 163 (31), 148 (19), 122 (27), 105 (54), 94 (76), 77 (49), 65 (22). Anal. calcd for C13H14N4O2S (290.34); C, 53.78; H, 4.86; N, 19.30, S, 11.04%. Found: C, 53.61; H, 4.79; N, 19.13, S, 10.86%.
2.1.3. 4-{[(2Z)-4-(2-Hydroxyphenyl)-4-oxobut-2-en-2-yl]amino}-6-methyl-3-thioxo-3,4-dihydro-1,2,4-triazin-5(2H)-one (9). A mixture of 2-methylchromone (1) (0.32 g, 2 mmol) and 4-amino-6-methyl-3-thioxo-3,4-dihydro-1,2,4-triazin-5(2H)-one (7) (0.48 g, 2 mmol) in absolute ethanol (10 mL) containing DBU (0.2 mL) was heated under reflux for 2 h. The pale yellow crystals obtained during heating were filtered and crystallized from EtOH, mp 251–252 °C, yield (0.48 g, 75%). IR (KBr, cm−1): 3402 (OH), 3237, 3198 (2NH), 1692 (COtriazine), 1646 (COenaminone), 1570 (CC), 1226 (CS). 1H NMR (DMSO-d6, δ): 2.18 (s, 3H, CH3), 2.40 (s, 3H, CH3), 6.21 (s, 1H, H-αolefinic), 7.40 (t, 1H, J = 7.2 Hz, Ar–H), 7.55 (d, 1H, J = 7.2 Hz, Ar–H), 7.84 (t, 1H, J = 7.5 Hz, Ar–H), 8.06 (d, 1H, J = 7.5 Hz, Ar–H), 9.81 (bs, 1H, NH exchangeable with D2O), 11.65 (bs, 1H, NH exchangeable with D2O), 12.36 (bs, 1H, OH exchangeable with D2O). 13C NMR (DMSO-d6, δ): 15.7 (CH3), 17.5 (CH3), 95.9 (C-αolefinic), 122.5, 124.7, 126.3, 128.6, 130.5 (5Ar–C), 144.8 (C-6triazine), 148.6 (C-βolefinic), 151.7 (C–OH), 166.5 (COtriazine), 182.7 (COketone), 195.8 (CS). Mass spectrum, m/z (Ir%): 318 (M+, 15), 303 (27), 275 (23), 231 (24), 176 (100), 142 (39), 120 (36), 93 (45), 77 (34), 65 (23). Anal. calcd for C14H14N4O3S (318.35); C, 52.82; H, 4.43; N, 17.60, S, 10.07%. Found: C, 52.67; H, 4.35; N, 17.41, S, 9.83%.
2.1.4. 6-(2-Hydroxyphenyl)-4-methyl-1,2-dihydro-3H-pyrazolo[3,4-b]pyridin-3-one (10). A mixture of 2-methylchromone (1) (0.32 g, 2 mmol) and 5-amino-2,4-dihydro-3H-pyrazol-3-one (0.25 g, 2 mmol), in sodium ethoxide (prepared by adding 0.2 g sodium in 20 mL absolute ethanol) was heated under reflux for 2 h. After cooling, the reaction mixture was neutralized with conc. HCl. The solid obtained were filtered and crystallized from DMF/H2O as yellow crystals, mp 296–297 °C, yield (0.37 g, 77%). IR (KBr, cm−1): 3405 (OH), 3291 (2NH), 1659 (COpyrrazole), 1609 (CN), 1577 (CC). 1H NMR (DMSO-d6, δ): 2.09 (s, 3H, CH3), 7.27 (t, 1H, J = 7.2 Hz, Ar–H), 7.42 (d, 1H, J = 7.5 Hz, Ar–H), 7.76 (t, 1H, J = 7.5 Hz, Ar–H), 8.00 (d, 1H, J = 7.2 Hz, Ar–H), 8.37 (s, 1H, H-3pyridine), 9.84 (bs, 1H, NH exchangeable with D2O), 10.72 (bs, 1H, NH exchangeable with D2O), 11.90 (bs, 1H, OH exchangeable with D2O). 13C NMR (DMSO-d6, δ): 15.6 (CH3), 112.2 (C-3a), 122.3 (C-3), 123.7, 125.4, 126.4, 128.7, 129.9 (5Ar–C), 142.2 (C-4), 144.5 (C-6), 146.3 (C-7a), 151.6 (C–OH), 165.4 (C-3 as CO). Mass spectrum, m/z (Ir%): 241 (M+, 66), 226 (22), 214 (15), 198 (35), 184 (27), 148 (36), 120 (100), 93 (63), 77 (55), 64 (38). Anal. calcd for C13H11N3O2 (241.24); C, 64.72; H, 4.60; N, 17.42%. Found: C, 64.68; H, 4.45; N, 17.38%.
2.1.5. 6-(2-Hydroxyphenyl)-3,8-dimethyl-4H-pyrimido[2,1-c][1,2,4]triazin-4-one (12). A mixture of 2-methylchromone (1) (0.32 g, 2 mmol) and 3-amino-6-methyl-1,2,4-triazin-5(4H)-one (11) (0.25 g, 2 mmol), in sodium ethoxide (prepared by adding 0.2 g sodium in 20 mL absolute ethanol) was heated under reflux for 2 h. After cooling, the reaction mixture was neutralized with conc. HCl. The solid obtained were filtered and crystallized from AcOH as pale-yellow crystals, mp > 300 °C, yield (0.35 g, 65%). IR (KBr, cm−1): 3405 (OH), 1655 (COtriazine), 1608 (CN), 1566 (CC). 1H NMR (DMSO-d6, δ): 2.15 (s, 3H, CH3), 2.29 (s, 3H, CH3), 7.38 (t, 1H, J = 7.5 Hz, Ar–H), 7.58 (d, 1H, J = 7.5 Hz, Ar–H), 7.82 (t, 1H, J = 7.2 Hz, Ar–H), 8.01 (d, 1H, J = 7.5 Hz, Ar–H), 8.24 (s, 1H, H-5pyrimidine), 11.04 (bs, 1H, OH exchangeable with D2O). 13C NMR (DMSO-d6, δ): 16.7 (CH3), 18.5 (CH3), 108.7 (C-7), 122.7, 124.3, 126.4, 127.2, 128.6 (5Ar–C), 142.3 (C-6), 144.3 (C-3), 145.8 (C-8), 147.4 (C-9a), 151.6 (C–OH), 170.5 (C-3 as CO). Mass spectrum, m/z (Ir%): 268 (100), 240 (62), 225 (20), 199 (29), 175 (43), 120 (56), 91 (38), 77 (26), 65 (13). Anal. calcd for C14H12N4O2 (268.27): C, 62.68; H, 4.51; N, 20.88%. Found: C, 62.57; H, 4.50; N, 20.73%.
2.1.6. 5-(2-Hydroxyphenyl)-7-methyl[1,2,4]triazolo[4,3-a]pyrimidine (13). A mixture of 2-methylchromone (1) (0.32 g, 2 mmol) and 3-amino-1,2,4-triazole (0.17 g, 2 mmol) in sodium ethoxide (prepared by adding 0.2 g sodium in 15 mL absolute ethanol) was heated under reflux for 2 h. After cooling, the reaction mixture was poured onto crushed ice (50 g) and neutralized with conc. HCl. The solid obtained filtered and crystallized from DMF/H2O, mp 268–269 °C, yield (0.32 g, 71%). IR (KBr, cm−1): 3416 (OH), 1608 (CN), 1587 (CC). 1H NMR (DMSO-d6, δ): 2.25 (s, 3H, CH3), 7.35 (t, 1H, J = 7.2 Hz, Ar–H), 7.53 (d, 1H, J = 7.2 Hz, Ar–H), 7.69 (t, 1H, J = 7.2 Hz, Ar–H), 7.99 (d, 1H, J = 7.2 Hz, Ar–H), 8.38 (s, 1H, H-5pyrimidine), 9.12 (s, 1H, H-3triazole), 11.75 (bs, 1H, OH exchangeable with D2O). 13C NMR (DMSO-d6, δ): 18.4 (CH3), 121.7, 123.5, 125.9, 127.3, 128.7 (5Ar–C), 132.3 (C-5pyrimidine), 140.4 (C-4pyrimidine), 140.9 (C-6pyrimidine), 141.2 (C-3), 142.5 (C-8a), 150.3, (C–OH). Mass spectrum, m/z (Ir%): 226 (M+, 44), 214 (11), 195 (27), 148 (16), 133 (40), 107 (21), 94 (100), 77 (56), 64 (39). Anal. calcd for C12H10N4O (226.23); C, 63.71; H, 4.46; N, 24.76%. Found: C, 63.58; H, 4.28; N, 24.54%.
2.1.7. 4-(2-Hydroxyphenyl)-2-methylpyrimido[1,2-a]benzimidazole (14). A mixture of 2-methylchromone (1) (0.32 g, 2 mmol) and 2-aminobenzimidazole (0.27 g, 2 mmol) in sodium ethoxide (prepared by adding 0.2 g sodium in 15 mL absolute ethanol) was heated under reflux for 2 h. After cooling, the reaction mixture was poured onto crushed ice (50 g) and neutralized with conc. HCl. The solid obtained filtered and crystallized from AcOH, mp > 300 °C, yield (0.40 g, 74%). 13C NMR (DMSO-d6, δ): 17.5 (CH3), 122.7, 123.2, 124.1, 125.3, 126.2, 126.7, 127.2, 128.5, 129.1, 129.7, 130.4, (Ar–C), 131.5 (C-5pyrimidine), 140.1 (C-6pyrimidine), 140.9 (C-4pyrimidine), 142.2 (C-2pyrimidine), 150.6 (C–OH). IR (KBr, cm−1): 3410 (OH), 1615 (CN), 1583 (CC). 1H NMR (DMSO-d6, δ): 2.26 (s, 3H, CH3), 7.29–7.40 (m, 4H, Ar–H), 7.54 (t, 1H, J = 7.8 Hz, Ar–H), 7.74 (d, 1H, J = 7.8 Hz, Ar–H), 7.83 (t, 1H, J = 7.5 Hz, Ar–H), 8.08 (d, 1H, J = 7.8 Hz, Ar–H), 8.37 (s, 1H, H-5pyrimidine), 11.39 (bs, 1H, OH exchangeable with D2O). Mass spectrum, m/z (Ir%): 275 (M+, 100), 260 (25), 244 (16), 183 (46), 169 (32), 117 (26), 93 (70), 77 (45), 65 (23). Anal. calcd for C17H13N3O (275.30); C, 74.17; H, 4.76; N, 15.26%. Found: C, 74.02; H, 4.73; N, 15.15%.
2.2. Computational methods
Computational chemistry analyses for the synthesized compounds were carried out using DFT at the B3LYP level with the standard 6-311++G(d,p) basis set, employing the GAUSSIAN 09W software. This method is among the most reliable for assessing compound stability and reactivity.25,26 Molecular visualizations were generated using GaussView 5.0.27–29 No symmetry constraints were applied during geometry optimization. Quantum chemical calculations were performed to evaluate the molecular electrostatic potential (MEP), optimized geometries, and molecular orbital (MO) energy levels. Additionally, chemical shift predictions for 1H and 13C NMR were performed using the GIAO method using the same basis set at the B3LYP level,30 and the results were compared with the experimental data. Further, vibrational frequencies were computed for all new compounds. Polarizability, first hyperpolarizability and electronic dipole moment were also determined at the same DFT level of theory.31
2.3. Biological evaluation
2.3.1. Antitumor activity. The antitumor activity of the synthesized compounds was tested against HepG2 cells using the published procedure of evaluating the effect of the test samples on cell morphology and viability.32–34 Mammalian cell lines: HepG-2 cells (human Hepatocellular carcinoma) were obtained from the American Type Culture Collection (ATCC, Rockville, MD).
2.3.2. ADME analysis. A computational ADME analysis was conducted using SwissADME to evaluate the physicochemical and drug-likeness properties of the studied compounds.35,36
2.3.3. Molecular docking study. Molecular docking studies were performed using AutoDock Vina37 to explore the interaction profiles of the synthesized compounds with cyclin-dependent kinase 1 (CDK1; PDB ID: 4Y72).38 The crystal structure of the target protein was obtained from the Protein Data Bank. Prior to docking, the receptor was prepared by removing all crystallographic water molecules, co-crystallized ligands, and heteroatoms, followed by the addition of polar hydrogens and Kollman charges to ensure proper protonation and electrostatic configuration.39,40Ligand structures were sketched in ChemDraw, subjected to energy minimization, and subsequently converted into PDBQT format. Docking was carried out using a grid box centered at coordinates (X = 29.509, Y = −70.550, Z = 185.362) with dimensions (X = 13.738 Å, Y = 12.295 Å, Z = 11.474 Å), ensuring coverage of the active binding region. Binding energies (kcal mol−1) were used as the primary criterion for evaluating ligand–protein interactions and predicting binding affinity.41,42
3. Results and discussion
3.1. Characterization of the synthesized compounds
The present work focuses on exploring the reactivity of 2-methylchromone (1) toward a variety of heterocyclic amines. Initially, its behavior was examined with 1-amino-4,6-dimethyl-2-oxo-1,2-dihydropyridine-3-carbonitrile (2)22 in absolute ethanol using DBU as a base (Scheme 1). The structure of the obtained product was established through spectroscopic analyses combined with theoretical calculations. Formation of the condensation product 3 was excluded, since the carbonyl group is generally unreactive toward condensation reactions.18 Instead, the most plausible pathway involves nucleophilic attack at the C-2 position, which is activated by both the mesomeric electron-withdrawing effect of the carbonyl group and the inductive effect of the adjacent oxygen atom.19 Following the ring opening of substrate 1, two possible conformers could be generated: 4 (E-isomer) and 5 (Z-isomer). Among these, the Z-conformer 5 is favored due to intramolecular hydrogen bonding between the carbonyl oxygen and the NH proton. The predominance of structure 5 was further validated through spectral evidence and theoretical calculations, both of which play a crucial role in confirming its identity as the Z-conformer. In this context, DFT calculations were employed as a complementary tool to support the experimental assignment and to rationalize the relative stability of the Z and E isomers, rather than to replace NMR-based structural elucidation (vide infra).
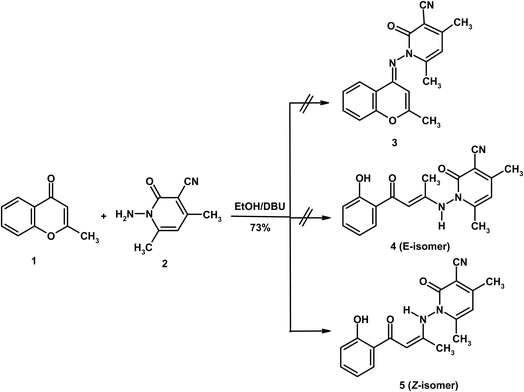 |
| | Scheme 1 Reaction of 2-methylchromone (1) and aminopyridine derivative 2. | |
The Z-configuration of compound 5 was confirmed by spectroscopic evidence. In the IR spectrum, the CO stretching vibration was observed at a relatively low frequency (1642 cm−1), indicative of intramolecular hydrogen bonding with the NH proton. This hydrogen-bonding interaction is further supported by the pronounced downfield shift of the NH signal in the 1H NMR spectrum, appearing at δ 9.80 and reflecting deshielding associated with hydrogen-bond-induced bond elongation. Such stabilization effectively clarifies for the preferential formation of the Z-isomer over the E-isomer. Further, in the 1H NMR spectrum, three singlet signals were recorded in the upfield region at δ 2.10, 2.31, and 2.39 correspond to the three methyl groups. Two additional singlets at δ 6.20 and 6.34 were assigned to H-α (olefinic) and H-5 (pyridine), respectively, while the aromatic protons resonated as expected at δ 7.38–8.12. The 13C NMR spectrum exhibited three methyl carbons at δ 15.9, 17.5, and 19.4. The olefinic α-carbon appeared at δ 94.2, shifted upfield by shielding by the electron donating mesomeric effect of the NH group, while the β-carbon resonated at δ 148.2 due to deshielding by electron withdrawing mesomeric effect of the carbonyl group and the inductive effect adjacent nitrogen atoms. The mass spectrum confirmed the proposed structure, showing a molecular ion peak at m/z 323 (11%) and a base peak at m/z 176 (100%), the latter resulting from facile cleavage of the N–N bond.
Similarly, treatment of 2-methylchromone (1) with 4-amino-5-methyl-2,4-dihydro-3H-1,2,4-triazole-3-thione (6)23 and 4-amino-6-methyl-3-thioxo-3,4-dihydro-1,2,4-triazin-5(2H)-one (7)24 in absolute ethanol containing DBU afforded the enaminone derivatives 8 and 9, respectively (Scheme 2). Both compounds gave a red coloration with FeCl3 solution, indicating the presence of a free phenolic OH group and supporting the pathway of ring-opening. Their mass spectra exhibited molecular ion peaks at m/z 290 (21%) and 318 (15%) supporting the molecular formula of C13H14N4O2S (290.34) and C14H14N4O3S (318.35), that consistent with the proposed structures. The IR spectra of 8 and 9 displayed characteristic absorptions for COenaminone at 1646 and 1644 cm−1, respectively, while compound 9 also showed a distinct absorption band due to COtriazine at 1692 cm−1. The 1H NMR spectra of compounds 8 and 9 revealed singlets for the olefinic proton at δ 6.32 and 6.21, respectively. Signals for NH (δ 9.94/9.81) and OH (δ 11.81/12.36) were also observed, both shifted downfield due to hydrogen bonding with carbonyl groups. In the 13C NMR spectrum of compound 9, the olefinic α-carbon resonated at δ 95.9, whereas the β-carbon resonated at δ 148.6.
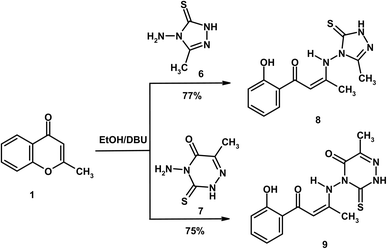 |
| | Scheme 2 Formation of enaminone derivatives 8 and 9. | |
On the other hand, reaction of 2-methylchromone (1) with 5-amino-2,4-dihydro-3H-pyrazol-3-one in sodium ethoxide solution afforded pyrazolo[3,4-b]pyridine 10. Herein, sodium ethoxide was employed to generate enolate species from active methylene compounds and to ensure effective ring opening of the γ-pyrone system. The mechanism proceeds via γ-pyrone ring opening by the deprotonated CH2 group (intermediate A), followed by intramolecular heterocyclization (Scheme 3). The mass spectrum of compound 10 exhibited a molecular ion peak at m/z 241, that consistent with the molecular formula C13H11N3O2. The IR spectrum displayed absorption bands at 3405 (OH), 3291 (2NH), 1659 (COpyrazole), and 1609 cm−1 (CN). The 1H NMR spectrum revealed singlets at δ 2.09 and 8.37, assigned to CH3 and H-3pyridine, respectively. D2O-exchangeable signals due to 2NH and OH were recorded at δ 9.84, 10.72 and 11.90, respectively.
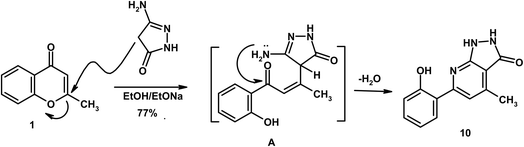 |
| | Scheme 3 Formation of pyrazolo[3,4-b]pyridine derivative 10. | |
Similarly, reaction of 2-methylchromone (1) with 3-amino-6-methyl-1,2,4-triazin-5(4H)-one (11),24 in sodium ethoxide solution under reflux, afforded pyrimido[2,1-c][1,2,4]triazine 12; via γ-pyrone ring opening (intermediate B) followed by heterocyclization (Scheme 4). The mass spectrum displayed a molecular ion peak, as the base peak, at m/z 268 (C14H12N4O2). The IR spectrum showed characteristic absorption bands at 3405 (OH), 1655 (COtriazine) and 1608 (CN) cm−1. The 1H NMR spectrum exhibited two singlets at δ 2.15 and 2.29, attributed to the methyl groups, along with a distinctive singlet at δ 8.24 corresponding to H-5 of the pyrimidine ring.
 |
| | Scheme 4 Formation of pyrimido[2,1-c][1,2,4] triazine 12. | |
Finally, reaction of substrate 1 with 3-amino-1,2,4-triazole and 2-aminobenzimidazole in ethanolic sodium ethoxide solution afforded the novel heterocycles triazolo[4,3-a]pyrimidine 13 and pyrimido[1,2-a]benzimidazole 14, respectively (Scheme 5). Both compounds produced a red coloration with FeCl3 solution, indicating the presence of a free OH group arising from γ-pyrone ring opening. The IR spectra of products 13 and 14 confirmed the absence of the carbonyl absorption band (COγ-pyrone) and exhibited new absorption bands due to CN at 1608 and 1615 cm−1, respectively. Their mass spectra supported the proposed structures, displaying molecular ion peaks at m/z 226 (C12H10N4O, 226.23) for compound 13 and m/z 275 (C17H13N3O, 275.30) for compound 14. In the 1H NMR spectra, both compounds showed characteristic singlets for H-5 of the pyrimidine ring at δ 8.38 (compound 13) and 8.37 (compound 14). Additionally, compound 13 displayed a further singlet at δ 9.12, assigned to H-3 of the triazole moiety.
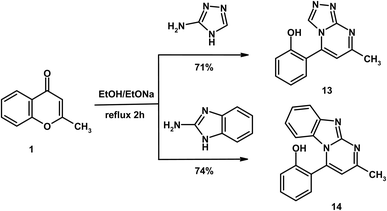 |
| | Scheme 5 Formation of triazolo[4,3-a]pyrimidine 13 and pyrimido[1,2-a]benzimidazole 14. | |
3.2. Theoretical studies
The primary objective of the theoretical investigation was to complement experimental studies. Accordingly, DFT calculations were carried out to rationalize the experimentally obtained products, providing molecular-level insight into charge distribution, preferred reaction pathways, and the relative stability of the resulting enaminone and heterocyclic systems, rather than to predict new reactions.
3.2.1. Frontier molecular orbital energies and chemical reactivity. The molecular structures of the synthesized compounds were optimized using DFT at the B3LYP/6-311+G(d,p) level. The frontier molecular orbital (FMO) analysis (Fig. 1, S1 and S2) revealed extensive π-electron delocalization across the molecular frameworks, consistent with the conjugated enaminone and fused heterocyclic systems.43 The HOMO–LUMO gap provides a useful indicator of electronic mobility and chemical reactivity: smaller gaps correspond to higher reactivity and lower kinetic stability, while larger gaps reflect greater hardness and reduced reactivity.44 In this series, compound 1 exhibits the widest gap (5.067 eV), indicating lower reactivity, whereas compound 9 has the narrowest gap (3.666 eV), consistent with enhanced electron-transfer potential and observed experimental reactivity trends (Table 1).45
 |
| | Fig. 1 Molecular modeling and the electron density of HOMO and LUMO of compounds 1, 5 and 8. | |
Table 1 Calculated chemical reactivity descriptors of the studied compounds
| Compound no. |
ET (au) |
HOMO (au) |
LUMO (au) |
E (HOMO) |
E (LUMO) |
I (eV) |
A (eV) |
ΔE (eV) |
χ (eV) |
Pi (eV) |
η (eV) |
S (eV−1) |
ω (eV) |
ε (eV−1) |
ΔN |
| 1 |
−536.5 |
−0.247 |
−0.060 |
−6.709 |
−1.643 |
6.709 |
1.643 |
5.067 |
4.176 |
−4.176 |
2.533 |
0.395 |
3.442 |
0.291 |
1.648 |
| 5 |
−1086.3 |
−0.228 |
−0.078 |
−6.207 |
−2.117 |
6.207 |
2.117 |
4.090 |
4.162 |
−4.162 |
2.045 |
0.489 |
4.235 |
0.236 |
2.035 |
| 8 |
−1271.7 |
−0.217 |
−0.060 |
−5.907 |
−1.628 |
5.907 |
1.628 |
4.279 |
3.767 |
−3.767 |
2.140 |
0.467 |
3.317 |
0.302 |
1.761 |
| 9 |
−1385.0 |
−0.223 |
−0.088 |
−6.072 |
−2.407 |
6.072 |
2.407 |
3.666 |
4.240 |
−4.240 |
1.833 |
0.546 |
4.903 |
0.204 |
2.313 |
| 10 |
−816.9 |
−0.225 |
−0.066 |
−6.125 |
−1.808 |
6.125 |
1.808 |
4.316 |
3.966 |
−3.966 |
2.158 |
0.463 |
3.645 |
0.274 |
1.838 |
| 12 |
−910.4 |
−0.229 |
−0.084 |
−6.236 |
−2.295 |
6.236 |
2.295 |
3.941 |
4.266 |
−4.266 |
1.971 |
0.508 |
4.617 |
0.217 |
2.165 |
| 13 |
−757.7 |
−0.233 |
−0.075 |
−6.350 |
−2.046 |
6.350 |
2.046 |
4.304 |
4.198 |
−4.198 |
2.152 |
0.465 |
4.095 |
0.244 |
1.951 |
| 14 |
−895.4 |
−0.219 |
−0.076 |
−5.965 |
−2.074 |
5.965 |
2.074 |
3.891 |
4.020 |
−4.020 |
1.945 |
0.514 |
4.153 |
0.241 |
2.066 |
Selected global descriptors [eqn (1)–(9)] derived from the FMO energies further clarify these trends. Compound 1 shows the highest hardness (η), while compound 9 is the softest (S) and most reactive. Electrophilicity (ω) and maximum charge-transfer capacity (ΔNmax) highlight the electron-accepting ability of the molecules, with compound 9 exhibiting the largest values (ω = 4.903 eV, ΔNmax = 2.313), consistent with its pronounced electrophilic character. Compound 12 displays the highest electronegativity (χ), indicating strong electron affinity. Fig. 2 summarizes the calculated global reactivity parameters for compounds 1, 5, 8–10, and 12–14.46–48
| |
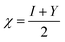 | (3) |
| |
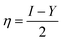 | (4) |
| |
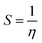 | (5) |
| |
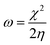 | (6) |
| |
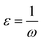 | (7) |
| |
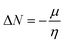 | (9) |
 |
| | Fig. 2 A graphical representation of the global reactivity indices. | |
Finally, in quantum chemical investigations, the total energy of a molecule represents the sum of all electronic and nuclear interactions within the system and serves as a fundamental criterion for evaluating molecular stability.49 Generally, lower total energy values indicate a more stable configuration, as the system requires less energy to maintain its optimized geometry. In the present theoretical study, the calculated total energies of compounds 3–5 were found to be −1101.23, −1164.44 and −1873.59 au, respectively.50 Comparison of these values shows that compound 5 is significantly more stable than compound 3, with an energy difference of 772.36 au, and more stable than compound 4, with a difference of 709.15 au. These theoretical results are consistent with the experimental findings, further confirming the higher stability of compound 5.51–53
3.2.2. Molecular electrostatic potential (MEP). Molecular electrostatic potential (MEP) analysis was employed to visualize charge distribution relevant to chemical reactivity and experimental observations, enabling identification of regions susceptible to electrophilic or nucleophilic attack and providing insight into potential intermolecular interactions.54 The electrostatic potential is represented using a color scale, where positive regions (blue) indicate electron-deficient sites and negative regions (red) correspond to electron-rich areas.55Herein, the MEP maps were used to rationalize the preferred site of nucleophilic attack in the starting chromone derivative and to interpret the electronic features of the resulting enaminone products. For the parent compound 1, the MEP surface (Fig. 3) shows a pronounced positive potential at the C-2 position, indicating its electrophilic character and enhanced susceptibility toward nucleophilic attack. In contrast, the carbonyl carbon at C-4 exhibits a negative potential, consistent with higher electron density at this site. This clear differentiation supports the experimentally observed preference for nucleophilic attack at C-2 followed by ring opening, rather than condensation at the carbonyl group; supporting the experimental results.
 |
| | Fig. 3 Molecular electrostatic potential of compounds 1, 5 and 8. | |
In the MEP maps of the representative products 5, 8, and 9 (Fig. 3, S3 and S4), the charge distribution is dominated by the enaminone fragment. Electron-rich regions are localized at the C-α positions, while the corresponding C-β carbons exhibit electron-deficient character. This polarization of the enaminone system is consistent with conjugation and intramolecular charge delocalization. Importantly, these features correlate well with the experimental NMR data, where C-α carbons resonate upfield relative to the more deshielded C-β carbons. As a result, the MEP analysis directly supports the proposed reaction mechanism and provides a coherent electronic explanation for the observed regioselectivity and NMR characteristics.
3.2.3. FT- IR vibrational analysis. Both experimental and theoretical approaches were employed to compare the vibrational properties of the synthesized compounds. Theoretical IR spectra were obtained using DFT calculations at the B3LYP/6-311++G(d,p) level of theory.56 A comparison of experimental and computed FT-IR spectra is shown in Fig. S14, S18, S22, S26, S30, S34 and S38. Because the theoretical calculations were performed in the gas phase, while the experimental measurements were carried out in the solid state, a scaling factor of 0.99 was applied to the calculated frequency values. The experimental and theoretical wavenumbers and the corresponding functional groups assignments are summarized in Tables 2 and 3.
Table 2 Experimental and theoretical frequencies and corresponding vibrational assignments of compounds 5, 8 and 9 at the B3LYP/6-311++G (d,p)
| Compound 5 |
Compound 8 |
Compound 9 |
| υexp. (cm−1) |
υthe. (cm−1) |
Assignment |
υexp. (cm−1) |
υthe. (cm−1) |
Assignment |
υexp. (cm−1) |
υthe. (cm−1) |
Assignment |
| 3406 |
3415 |
OH |
3409 |
3424 |
OH |
3402 |
3410 |
OH |
| 3279 |
3283 |
NH |
3248, 3211 |
3289, 3220 |
2NH |
3237, 3198 |
3280, 3217 |
2NH |
| 2217 |
2233 |
CN |
1644 |
1651 |
COenaminone |
1692 |
1705 |
COtriazine |
| 1676 |
1680 |
COpyridone |
1613 |
1620 |
CN |
1646 |
1660 |
COenaminone |
| 1642 |
1650 |
COenaminone |
1587 |
1595 |
CC |
1570 |
1600 |
CC |
| 1588 |
1607 |
CC |
1223 |
1244 |
CS |
1226 |
1244 |
CS |
Table 3 Experimental and theoretical frequencies and corresponding vibrational assignments of compounds 10–13 at the B3LYP/6-311++G (d,p)
| Compound 10 |
Compound 12 |
Compound 13 |
Compound 14 |
| υexp. (cm−1) |
υthe. (cm−1) |
Assignment |
υexp. (cm−1) |
υthe. (cm−1) |
Assignment |
υexp. (cm−1) |
υthe. (cm−1) |
Assignment |
υexp. (cm−1) |
υthe. (cm−1) |
Assignment |
| 3405 |
3416 |
OH |
3405 |
3420 |
OH |
3416 |
3433 |
OH |
3410 |
3424 |
OH |
| 3291 |
3320, 3304 |
2NH |
1655 |
1664 |
COtriazine |
1608 |
1620 |
CN |
1615 |
1624 |
CN |
| 1659 |
1672 |
COpyrrazole |
1608 |
1620 |
CN |
1587 |
1592 |
CC |
1583 |
1600 |
CC |
| 1609 |
1620 |
CN |
1566 |
1570 |
CC |
|
|
|
|
|
|
| 1577 |
1608 |
CC |
|
|
|
|
|
|
|
|
|
The IR spectra of the synthesized compounds confirmed the presence of characteristic absorption bands consistent with theoretical predictions. For the free OH group, the calculated values appeared at 3410–3433 cm−1, while the experimental bands were observed at 3402–3416 cm−1. Similarly, the NH groups showed theoretical absorptions within 3217–3320 cm−1, compared with experimental values of 3198–3291 cm−1. The CO stretching of the enaminone moiety was detected experimentally at 1642–1646 cm−1, in close agreement with the calculated range of 1650–1660 cm−1. For the heterocyclic carbonyl groups in compounds 5 and 9–11, the experimental absorption bands were observed at 1655–1692 cm−1, whereas the corresponding theoretical values ranged from 1664 to 1705 cm−1. The CN group exhibited theoretical stretching frequencies of 1620–1624 cm−1, slightly higher than the experimental bands at 1608–1615 cm−1. In compound 8, the stretching vibration cyano function (CN) appeared experimentally at 2217 cm−1, compared to a computed value of 2233 cm−1. Additionally, the CC stretching bands were observed in the experimental range of 1566–1587 cm−1, close to the theoretical values of 1570–1608 cm−1. The correlation between the experimental and theoretical wavenumbers for these functional groups, illustrated in Fig. 4 and S5–S7, was excellent with a correlation coefficient (R2) of 0.99.
 |
| | Fig. 4 The correlation relationships of the experimental versus calculated IR wavenumbers of compounds 5 and 8. | |
3.2.4. 1H NMR and 13C NMR spectroscopy. In recent years, theoretical calculations of 1H and 13C NMR chemical shifts have received considerable attention as a means of verifying the structural and functional features of newly synthesized compounds.57 Among the available methods, the gauge-including atomic orbital (GIAO) approach is the most widely employed DFT-based technique for NMR shift predictions.58 In this study, the chemical shift calculations were performed using Gaussian 09 software at the B3LYP/6-311++G(d,p) level of theory. The computed values were then compared with the experimental NMR data, which were obtained in deuterated DMSO using TMS as the internal standard.The observed and calculated 1H NMR chemical shifts for compounds 5, 8–10, and 12–14 are summarized in Tables 4 and 5, while the spectra (experimental and theoretical) are shown in Fig. S15, S19, S23, S27, S31, S35 and S39. In the upfield region, the CH3 protons were experimentally observed as singlets at δ 2.09–2.40 ppm, whereas the corresponding calculated signals appeared at δ 1.60–2.87 ppm. Theoretically, three distinct signals were predicted for each methyl group, which may be attributed to differences in the spatial orientation of the protons, leading to slightly varied electronic environments and thus different chemical shifts.
Table 4 Calculated and experimental 1H NMR chemical shifts of compounds 5, 8 and 9 on B3LYP/6-311++G(d,p) basis set
| Compound 5 |
Compound 8 |
Compound 9 |
| Atoms |
Calculated |
Experimental |
Atoms |
Calculated |
Experimental |
Atoms |
Calculated |
Experimental |
| 20-H |
1.652009 |
2.10 |
20-H |
1.641953 |
2.27 |
20-H |
1.601549 |
2.18 |
| 19-H |
1.746833 |
2.10 |
19-H |
1.757529 |
2.27 |
21-H |
1.822439 |
2.18 |
| 32-H |
2.054319 |
2.31 |
21-H |
2.144781 |
2.27 |
19-H |
1.828355 |
2.18 |
| 21-H |
2.096794 |
2.10 |
29-H |
2.180696 |
2.37 |
31-H |
1.959994 |
2.40 |
| 35-H |
2.309049 |
2.39 |
30-H |
2.241001 |
2.37 |
33-H |
2.272638 |
2.40 |
| 34-H |
2.318255 |
2.39 |
28-H |
2.264705 |
2.37 |
32-H |
2.329728 |
2.40 |
| 36-H |
2.332206 |
2.39 |
17-H |
6.778829 |
6.32 |
17-H |
6.67227 |
6.21 |
| 31-H |
2.353999 |
2.31 |
13-H |
6.835984 |
7.54 |
13-H |
6.823773 |
7.4 |
| 30-H |
2.521359 |
2.31 |
14-H |
7.040542 |
7.72 |
14-H |
7.031603 |
7.55 |
| 17-H |
6.347872 |
6.20 |
12-H |
7.470823 |
7.82 |
12-H |
7.452422 |
7.84 |
| 28-H |
6.515025 |
6.34 |
11-H |
7.551808 |
8.14 |
11-H |
7.550952 |
8.06 |
| 13-H |
6.82574 |
7.38 |
26-H |
9.40444 |
9.94 |
23-H |
9.287014 |
9.81 |
| 14-H |
7.02641 |
7.61 |
23-H |
11.06389 |
10.74 |
26-H |
12.34601 |
11.65 |
| 12-H |
7.443389 |
7.81 |
10-H |
12.5549 |
11.81 |
10-H |
12.48793 |
12.36 |
| 11-H |
7.550339 |
8.12 |
|
|
|
|
|
|
| 23-H |
9.218664 |
9.80 |
|
|
|
|
|
|
| 10-H |
12.52881 |
12.22 |
|
|
|
|
|
|
Table 5 Calculated and experimental 1H NMR chemical shifts of compounds 10–13 on B3LYP/6-311++G(d,p) basis set
| Compound 10 |
Compound 12 |
Compound 13 |
Compound 14 |
| Atoms |
Calculated |
Experimental |
Atoms |
Calculated |
Experimental |
Atoms |
Calculated |
Experimental |
Atoms |
Calculated |
Experimental |
| 26-H |
2.209143 |
2.09 |
26-H |
2.330597 |
2.29 |
27-H |
2.303778 |
2.25 |
31-H |
2.344838 |
2.26 |
| 24-H |
2.774632 |
2.09 |
20-H |
2.409817 |
2.15 |
25-H |
2.835709 |
2.25 |
33-H |
2.855805 |
2.26 |
| 25-H |
2.847352 |
2.09 |
18-H |
2.447843 |
2.15 |
26-H |
2.851774 |
2.25 |
32-H |
2.869418 |
2.26 |
| 21-H |
7.249949 |
7.27 |
19-H |
2.501868 |
2.15 |
22-H |
7.139134 |
7.53 |
19-H |
7.133265 |
7.29 |
| 28-H |
7.470519 |
7.42 |
24-H |
2.782585 |
2.29 |
20-H |
7.267872 |
7.35 |
23-H |
7.269587 |
7.33 |
| 20-H |
7.680868 |
7.76 |
25-H |
2.820074 |
2.29 |
21-H |
7.666869 |
7.69 |
25-H |
7.302267 |
7.38 |
| 19-H |
8.088426 |
8.00 |
30-H |
6.819517 |
7.38 |
19-H |
7.711719 |
7.99 |
27-H |
7.411334 |
7.40 |
| 22-H |
8.167953 |
8.37 |
28-H |
7.198545 |
7.58 |
23-H |
8.179033 |
8.38 |
24-H |
7.47455 |
7.54 |
| 14-H |
9.845704 |
9.84 |
27-H |
7.502773 |
7.82 |
17-H |
8.698514 |
9.12 |
22-H |
7.671507 |
7.74 |
| 27-H |
10.33798 |
10.72 |
29-H |
7.579888 |
8.01 |
18-H |
11.75176 |
11.75 |
26-H |
7.825796 |
7.83 |
| 18-H |
12.17287 |
11.90 |
32-H |
8.7873 |
8.24 |
|
|
|
18-H |
8.026008 |
8.08 |
| |
|
|
31-H |
11.83166 |
11.04 |
|
|
|
29-H |
8.833845 |
8.37 |
| |
|
|
|
|
|
|
|
|
28-H |
11.23864 |
11.39 |
For enaminone derivatives (compounds 5, 8, and 9), the experimental H-α (olefinic) appeared at δ 6.20–6.32 ppm, closely matching the computed range of δ 6.35–6.78 ppm. The aromatic protons of the synthesized compounds were recorded at δ 7.27–8.14 ppm, in good agreement with the calculated range of δ 6.82–8.09 ppm. In compounds 12–14, the H-5 of the pyrimidine ring was calculated at δ 8.18–8.83 ppm and experimentally detected at δ 8.24–8.38 ppm. Similarly, the H-3 of pyridine and triazole moieties in compounds 10 and 13 showed calculated signals at δ 8.17 and δ 9.12 ppm, compared with experimental values of δ 8.37 and δ 8.18 ppm, respectively. For heteroatom-bound protons, the NH and OH signals were theoretically predicted at δ 9.21–12.35 and δ 11.24–12.55 ppm, respectively, while the experimental values were observed at δ 9.80–11.64 and δ 11.04–12.36 ppm. The relatively downfield chemical shift of the triazole proton can be attributed to the strong electron-withdrawing inductive and mesomeric effects of the adjacent nitrogen atoms, which increase deshielding.
The 13C NMR spectra of compounds 5, 8–10, and 12–14 are presented in Fig. S16, S20, S24, S28, S32, S36 and S40, while the theoretical and experimental chemical shift values are summarized in Tables 6 and 7.
Table 6 Calculated and experimental 13C NMR chemical shifts of compounds 5, 8 and 9 on B3LYP/6-311++G(d,p) basis set
| Compound 5 |
Compound 8 |
Compound 9 |
| Atoms |
Calculated |
Experimental |
Atoms |
Calculated |
Experimental |
Atoms |
Calculated |
Experimental |
| 18-C |
18.31434 |
15.9 |
27-C |
11.65242 |
15.6 |
18-C |
17.6735 |
15.7 |
| 33-C |
21.43412 |
17.5 |
18-C |
18.83939 |
16.4 |
30-C |
18.96897 |
17.5 |
| 29-C |
21.57802 |
19.4 |
9-C |
106.7458 |
96.1 |
9-C |
103.5993 |
95.9 |
| 9-C |
101.2112 |
94.2 |
5-C |
118.593 |
122.6 |
5-C |
118.4184 |
122.5 |
| 25-C |
101.9339 |
106.2 |
1-C |
122.5349 |
123.3 |
1-C |
122.5228 |
124.7 |
| 26-C |
111.3303 |
108.3 |
2-C |
125.011 |
125.1 |
3-C |
128.5142 |
126.3 |
| 37-C |
119.7519 |
116.4 |
3-C |
128.0264 |
126.4 |
2-C |
130.9209 |
128.6 |
| 5-C |
120.4737 |
122.2 |
6-C |
130.3065 |
128.6 |
6-C |
135.9791 |
130.5 |
| 1-C |
122.4745 |
124.9 |
25-C |
147.2659 |
144.3 |
25-C |
148.5132 |
144.8 |
| 3-C |
130.063 |
126.8 |
16-C |
149.7669 |
148.1 |
16-C |
150.2136 |
148.6 |
| 2-C |
131.8666 |
128.4 |
4-C |
151.2982 |
150.8 |
4-C |
155.9602 |
151.7 |
| 6-C |
132.1563 |
129.5 |
7-C |
191.1075 |
182.3 |
27-C |
167.8004 |
166.5 |
| 24-C |
133.3026 |
132.8 |
33-C |
200.7159 |
194.5 |
7-C |
186.2596 |
182.7 |
| 27-C |
144.9511 |
142.3 |
|
|
|
24-C |
198.6326 |
195.8 |
| 16-C |
150.7324 |
148.2 |
|
|
|
|
|
|
| 4-C |
156.1305 |
152.1 |
|
|
|
|
|
|
| 39-C |
163.9764 |
163.4 |
|
|
|
|
|
|
| 7-C |
196.1162 |
182.6 |
|
|
|
|
|
|
Table 7 Calculated and experimental 13C NMR chemical shifts of compounds 10 and 12–14 on B3LYP/6-311++G(d,p) basis set
| Compound 10 |
Compound 12 |
Compound 13 |
Compound 14 |
| Atoms |
Calculated |
Experimental |
Atoms |
Calculated |
Experimental |
Atoms |
Calculated |
Experimental |
Atoms |
Calculated |
Experimental |
| 23-C |
19.75128 |
15.6 |
17-C |
20.36131 |
16.7 |
24-C |
26.68692 |
18.4 |
30-C |
20.87679 |
17.5 |
| 13-C |
110.2318 |
112.2 |
23-C |
21.06106 |
18.5 |
5-C |
119.3801 |
121.7 |
17-C |
118.3667 |
122.7 |
| 8-C |
123.9155 |
122.3 |
8-C |
109.0970 |
108.7 |
3-C |
121.6778 |
123.5 |
5-C |
120.1977 |
123.2 |
| 5-C |
124.8377 |
123.7 |
5-C |
120.3025 |
122.7 |
1-C |
124.1116 |
125.9 |
3-C |
122.6261 |
124.1 |
| 1-C |
125.7894 |
125.4 |
1-C |
123.2929 |
124.3 |
2-C |
128.0574 |
127.3 |
16-C |
123.383 |
125.3 |
| 3-C |
128.5005 |
126.4 |
3-C |
127.5388 |
126.4 |
6-C |
129.6411 |
128.7 |
1-C |
123.992 |
126.2 |
| 6-C |
130.9126 |
128.7 |
2-C |
130.6697 |
127.2 |
8-C |
133.1513 |
132.3 |
20-C |
124.5535 |
126.7 |
| 2-C |
132.4519 |
129.9 |
6-C |
132.0601 |
128.6 |
10-C |
137.9544 |
140.4 |
21-C |
129.4937 |
127.2 |
| 10-C |
145.6349 |
142.2 |
7-C |
143.1455 |
142.3 |
7-C |
138.143 |
140.9 |
13-C |
130.0049 |
128.5 |
| 7-C |
146.0137 |
144.5 |
14-C |
146.4146 |
144.3 |
14-C |
142.4465 |
141.2 |
2-C |
131.3841 |
129.1 |
| 11-C |
148.2719 |
146.3 |
10-C |
147.9013 |
145.8 |
11-C |
144.9029 |
142.5 |
6-C |
132.465 |
129.7 |
| 4-C |
155.9500 |
151.6 |
11-C |
153.2130 |
147.4 |
4-C |
155.7184 |
150.3 |
15-C |
132.6225 |
130.4 |
| 17-C |
171.5148 |
165.4 |
4-C |
154.8726 |
151.6 |
|
|
|
8-C |
133.2795 |
131.5 |
| |
|
|
21-C |
172.2610 |
170.5 |
|
|
|
10-C |
141.4853 |
140.1 |
| |
|
|
|
|
|
|
|
|
7-C |
142.8493 |
140.9 |
| |
|
|
|
|
|
|
|
|
11-C |
143.8656 |
142.2 |
| |
|
|
|
|
|
|
|
|
4-C |
150.0403 |
150.6 |
As expected, the methyl carbons appeared in the upfield region, with theoretical signals in the range δ 11.6–26.7 ppm and experimental values between δ 15.6–19.4 ppm. The aromatic carbons of the benzene rings were observed computationally at δ 118.4–133.9 ppm, closely matching the experimental range of δ 121.7–131.5 ppm. While, the C–OH carbons were calculated at δ 150.0–156.1 ppm, in good agreement with the experimental values at δ 150.3–152.1 ppm. For compounds 8 and 9, the CS signals were predicted at δ 198.6–200.7 ppm, consistent with the experimental resonances at δ 194.5–195.8 ppm. C-α C-β.
In the case of enaminone derivatives (compounds 5, 8, and 9), the C-α resonate theoretically at δ 101.2–106.7 ppm, compared to experimental values at δ 94.2–96.1 ppm, while the C-β appeared computationally at δ 150.2–151.3 ppm, in agreement with experimental signals at δ 148.1–148.6 ppm. The carbonyl carbons of the enaminone moieties were calculated at δ 186.3–196.1 ppm, consistent with the experimental values at δ 182.3–182.7 ppm. The relatively high chemical shift of C-β may be attributed to the combined electron-withdrawing mesomeric effect of the carbonyl group and the inductive effect of the adjacent nitrogen atom. Conversely, the lower chemical shift of C-α may result from the electron-donating influence of the NH group, which increases the electron density at these carbons, leading to shielding and reduced δ values.
To evaluate the reliability of the theoretical calculations, correlation plots of experimental versus calculated chemical shifts for both 1H and 13C NMR are provided in Fig. 5, 6 and S8–S13. The excellent correlation coefficients (R2 = 0.98–0.99 for both 1H and 13C NMR) confirm the strong agreement between the experimental and theoretical chemical shift values.
 |
| | Fig. 5 The correlation relationships of the calculated versus experimental 1H NMR chemical shifts of compounds 5 and 8. | |
 |
| | Fig. 6 The correlation relationships of the calculated versus experimental 13C NMR chemical shifts of compounds 5 and 8. | |
3.2.5. Nonlinear optical (NLO) properties. As a complementary outcome of the electronic structure calculations, the nonlinear optical (NLO) properties of the synthesized compounds were evaluated. Organic π-conjugated systems with donor–acceptor characteristics are known to facilitate intramolecular charge transfer, which can give rise to NLO responses.59,60The key NLO parameters, including dipole moment (µ), polarizability (α), polarizability anisotropy (Δα), and total hyperpolarizability (βtot), were calculated in the gas phase at the B3LYP/6-311+G(d,p) level and are summarized in Table 8.61,62 The dipole moments range from 2.884 to 9.792 D, while the polarizabilities fall between 0.996 × 10−23 and 1.969 × 10−23 esu. The calculated βtot values (0.398 × 10−30–2.358 × 10−30 esu) exceed that of urea, a commonly used reference compound, by approximately 1.1–6.3 times. These findings suggest that the synthesized compounds possess promising potential as candidates for NLO material development.63,64 The principal NLO parameters are evaluated using the following equations:
| | |
µ = (µx2 + µy2 + µz2)1/2
| (10) |
| | |
α = (αxx + αyy + αzz)/3
| (11) |
| | |
Δα = (2)−0.5[(αxx − αyy)2 + (αyy − αzz)2 + (αzz − αxx)2 + 6(αyz)2 + 6(αxy)2 + 6(αxz)2]0.5
| (12) |
| | |
βtot = [(βxxx + βxyy + βxzz)2 +(βyyy + βyzz + βyxx)2 + (βzzz + βzxx +βzyy)2]0.5
| (13) |
Table 8 The dipole moment (µ), mean polarizability (α), anisotropy of the polarizability (Δα) and first-order hyperpolarizability (β) for the studied compounds
| Compound no. |
µx |
µy |
µz |
µtotal |
〈α〉 (au) |
〈α〉 (esu) × 10−23 |
Δα (au) |
Δα (esu) × 10−24 |
βtotal (au) |
βtotal (esu) × 10−30 |
| 1 |
0.510 |
−3.885 |
0.0002 |
3.919 |
67.22 |
0.996 |
20.77 |
3.078 |
46.12 |
0.398 |
| 5 |
1.304 |
−6.600 |
−0.632 |
6.757 |
132.86 |
1.969 |
66.12 |
9.799 |
272.91 |
2.358 |
| 8 |
−1.164 |
−3.577 |
−0.429 |
3.786 |
115.61 |
1.713 |
32.69 |
4.844 |
122.19 |
1.056 |
| 9 |
−1.349 |
1.456 |
2.092 |
2.884 |
122.88 |
1.821 |
36.01 |
5.337 |
133.62 |
1.154 |
| 10 |
−3.983 |
−0.047 |
−0.594 |
4.027 |
98.730 |
1.463 |
30.58 |
4.532 |
131.43 |
1.136 |
| 12 |
−6.134 |
2.395 |
0.982 |
6.658 |
112.92 |
1.674 |
25.69 |
3.808 |
149.79 |
1.294 |
| 13 |
8.257 |
5.230 |
0.591 |
9.792 |
98.90 |
1.466 |
24.03 |
3.561 |
229.73 |
1.985 |
| 14 |
4.450 |
−0.796 |
0.743 |
4.581 |
119.79 |
1.775 |
20.41 |
3.025 |
86.15 |
0.744 |
3.3. Biological evaluation
3.3.1. Vitro anticancer assay. The newly synthesized compounds were evaluated for their anticancer using the MTT colorimetric assay against hepatocellular carcinoma (HepG-2) cell lines after 24 hours of incubation.32–34 The results demonstrated that all of the tested compounds exhibited growth inhibitory effects on HepG-2 cells (Fig. 7). The cytotoxic activity was quantified in terms of IC50 values, defined as the concentration (µM) required to inhibit 50% of cell growth (Fig. 8, and Table 9). Cisplatin, a clinically established anticancer drug, was used as the reference standard. Overall, the synthesized derivatives displayed a broad spectrum of cytotoxic activity, with IC50 values ranging from 7.88 to 32.43 µM, indicating activities that varied from strong to moderate potency.
 |
| | Fig. 7 Relation between cell viability and concentration of all synthesized compounds on the proliferation of HepG2 cell line (cis-platin is the standard drug). | |
 |
| | Fig. 8 Virtual IC50 values of the target compounds and cis-platin as a standard drug against HepG-2 cell lines. | |
Table 9 IC50 values of the prepared compounds against hepatocellular carcinoma cells lines (HepG-2) for 24 hours
| Compounds no. |
(HepG-2 cells) IC50 (µM L−1) |
| 1 |
32.43 ± 1.44 |
| 5 |
14.65 ± 0.88 |
| 8 |
17.17 ± 0.99 |
| 9 |
7.88 ± 0.52 |
| 10 |
26.60 ± 1.17 |
| 12 |
10.66 ± 0.79 |
| 13 |
20.94 ± 1.04 |
| 14 |
8.76 ± 0.67 |
| Cisplatin |
17.86 ± 0.93 |
Among the synthesized product, compound 9 displayed the strongest activity (IC50 = 7.88 µM), while compounds 5 and 8 showed comparable or slightly better activity than cisplatin (Table 9). Compounds 12 and 14 also demonstrated notable activity (IC50 = 10.66 and 8.76 µM, respectively). Moderate cytotoxicity was observed for compounds 1, 10, and 13. The observed activity may be influenced by structural features such as conjugated enaminone groups, thioxo substituents, and fused heterocycles.
These results provide initial insight into the cytotoxic effects of the synthesized compounds; however, they are limited to a single cancer cell line (HepG2), and no selectivity data against normal cells were assessed. Therefore, while the compounds show promising growth–inhibitory activity, further studies across multiple cancer and non-cancer cell lines are required to fully evaluate their anticancer potential and therapeutic relevance.
A preliminary linear regression analysis was conducted using the experimental IC50 values as the dependent variable and selected DFT-derived global reactivity descriptors in the gas phase as independent variables (Table 1). The analysis suggested the following trends:
(1) A reduction in the energy gap (Egap eV−1) of the studied compounds enhanced their anticancer activity, as reflected by the positive slope of IC50 versus Egap, expressed as IC50 = −64.55 + 19.53 Egap eV−1, r = 0.94, n = 8. This positive slope suggests that decreasing the energy gap reduces IC50 values, thereby improving anticancer potential (Fig. 9).
 |
| | Fig. 9 Linear correlation between IC50 versus energy gap, hardness and softness. | |
(2) The stability of the compounds, represented by hardness, showed the relation IC50 = −64.55 + 39.07 η eV−1, r = 0.94, n = 8. The positive slope indicates that lowering hardness decreases IC50, which enhances inhibitory activity (Fig. 9). Conversely, softness displayed an inverse relationship with IC50. This was confirmed by the negative slope of softness against IC50, expressed as IC50 = 105.73 − 183.75 S eV−1, r = 0.94, n = 8 (Fig. 9).
These observations suggest that smaller energy gaps and lower hardness may correlate with higher cytotoxic activity. However, the dataset is limited (n = 8), and therefore, the statistical reliability of these correlations is low. Consequently, these trends should be considered exploratory and interpreted cautiously rather than as definitive predictors of biological activity.
3.3.2. Drug-likeness and in silico ADME anticipation. SwissADME is a widely used computational tool for predicting absorption, distribution, metabolism, and excretion (ADME) parameters, as well as evaluating drug-likeness based on physicochemical properties.65 It provides insight into whether a compound is likely to exhibit favorable oral bioavailability. Compounds that violate more than one of the commonly applied rules—Lipinski's,66 Veber's,67 Egan's,68 or Ghose's69—are generally considered less suitable for oral administration.The results indicated that all studied compounds complied with Lipinski's rule of five, suggesting favorable oral bioavailability. Specifically, molecular weights ranged from 160 to 323 g mol−1, M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logP values from 0.51 to 2.85, hydrogen bond donors between 0 and 3, and hydrogen bond acceptors within the acceptable range (Table 10).
logP values from 0.51 to 2.85, hydrogen bond donors between 0 and 3, and hydrogen bond acceptors within the acceptable range (Table 10).
Table 10 Estimations of ADME and physicochemical properties of the studied compounds
| Compound no. |
MW |
MLOGP |
HBA |
HBD |
TPSA |
nRotB |
WLOGP |
XLOGP3 |
Log S |
Fraction Csp3 |
MR |
Lipinski violations |
Veber violations |
Egan violations |
Ghose violations |
| 1 |
160.17 |
1.14 |
2 |
0 |
30.21 |
0 |
2.10 |
2.27 |
−2.54 |
0.10 |
47.45 |
0 |
0 |
0 |
0 |
| 5 |
323.35 |
1.49 |
4 |
2 |
95.12 |
4 |
2.18 |
2.98 |
−4.64 |
0.17 |
91.27 |
0 |
0 |
0 |
0 |
| 8 |
290.34 |
0.75 |
3 |
3 |
115.03 |
4 |
2.09 |
2.35 |
−4.41 |
0.15 |
78.30 |
0 |
0 |
0 |
0 |
| 9 |
318.35 |
0.51 |
4 |
3 |
132.10 |
4 |
1.45 |
2.23 |
−4.64 |
0.14 |
84.57 |
0 |
0 |
1 |
0 |
| 10 |
241.25 |
1.80 |
3 |
3 |
81.77 |
1 |
1.93 |
2.09 |
−3.44 |
0.08 |
69.14 |
0 |
0 |
0 |
0 |
| 12 |
268.27 |
1.62 |
5 |
1 |
80.38 |
1 |
1.47 |
0.85 |
−2.12 |
0.14 |
74.44 |
0 |
0 |
0 |
0 |
| 13 |
226.23 |
1.60 |
4 |
1 |
63.31 |
1 |
1.81 |
2.23 |
−3.19 |
0.08 |
63.21 |
0 |
0 |
0 |
0 |
| 14 |
275.30 |
2.85 |
3 |
1 |
50.42 |
1 |
3.56 |
3.43 |
−4.17 |
0.06 |
82.92 |
0 |
0 |
0 |
0 |
Veber's rule, which emphasizes total polar surface area (TPSA ≤ 140 Å2) and rotatable bonds (<10), was satisfied, with TPSA values of 30–132 Å2 and an acceptable number of rotatable bonds. Egan's rule (TPSA ≤ 131.6 Å2 and WLOGP ≤ 5.88) and the Ghose filter (molecular weight 160–323 g mol−1, molar refractivity 47–91, WLOGP 1.45–3.56, and total atom count 20–70) were also fully satisfied (Table 10). Collectively, compliance with these rules indicates favorable physicochemical properties and drug-likeness, supporting the potential for oral activity.
The bioavailability radar provides a visual assessment of six key physicochemical parameters: lipophilicity (XLOGP3 −0.7 to +5.0), size (MW 150–500 g mol−1), polarity (TPSA 20–130 Å2, except compound 9), solubility (logS ≤ 6), saturation (fraction Csp3 0.25–1), and flexibility (≤9 rotatable bonds). As shown in Fig. 10 and S42, all compounds met these criteria, with minor deviations observed for saturation (INSATU). Overall, the in silico ADME and drug-likeness results support the potential oral bioavailability of the studied compounds at a screening level. However, these predictions are preliminary and should be interpreted with caution, as they do not replace experimental pharmacokinetic or metabolism studies.
 |
| | Fig. 10 The bioavailability radars of compounds 1, 5 and 8. | |
3.3.3. Molecular docking. Docking simulations are a widely used tool in drug discovery to predict interactions between small molecules and target proteins, providing insight into potential mechanisms of action.70–73 In this study, cyclin-dependent kinase 1 (CDK1) was selected as a relevant anticancer target because it plays a central role in regulating the G2/M phase of the cell cycle and is often overexpressed in hepatocellular carcinoma and other cancers.38,39The docking protocol was validated by re-docking the co-crystallized ligand, [((2,6-difluorophenyl)carbonyl]amino)-N-(4-fluorophenyl)-1H-pyrazole-3-carboxamide, into the CDK1 active site (PDB ID: 4Y72). The re-docked pose closely reproduced the crystallographic binding mode (Fig. S43), confirming the reliability of the docking methodology.74
The synthesized compounds (1, 5, 8–10 and 12–14) were docked into the CDK1 active site, and their interactions were analyzed (Fig. 11, S44, Tables 11, and S1). Across the series, recurring interaction motifs were observed, including hydrogen bonding with GLU12, ASP86, or TYR15, and Pi–anion or Pi–alkyl interactions with residues such as LYS88 and LYS89. Compounds showing the most favorable binding energies (e.g., 9, 14, and 5) generally engaged in multiple hydrogen bonds and Pi–anion interactions, suggesting these contacts are key contributors to binding affinity.
 |
| | Fig. 11 3D representation of the hydrogen bonding between the studied compounds (5, 8, 9 and 14) with the amino acids residues of the target protein (PDB ID: 4Y72). | |
Table 11 Various interactions between the studied compounds 5, 8, 9, and 14 with the amino acids residues of the target protein (PDB ID: 4Y72)
| Receptor |
Distance |
Interaction type |
Receptor |
Distance |
Interaction type |
| Compound 5 (binding energy−8.3 kcal mol−1) |
Compound 8 (binding energy−8.1 kcal mol−1) |
| TYR15 |
1.94 |
H-bond |
VAL18 |
3.82 |
Alkyl |
| LYS88 |
4.36 |
Alkyl |
LYS88 |
3.38 |
Alkyl |
|
| Compound 9 (binding energy−8.6 kcal mol−1) |
Compound 14 (binding energy−8.5 kcal mol−1) |
| ASP86 |
2.18 |
H-bond |
GLU12 |
2.33 |
H-bond |
| LYS89 |
5.31 |
Pi–alkyl |
ASP86 |
3.85 |
Pi–anion |
| |
|
|
GLY11 |
3.93 |
Pi-Sigma |
| |
|
|
VAL18 |
5.05 |
Pi–alkyl |
A comparison of docking results with in vitro cytotoxicity (HepG2 IC50 values) suggests a general trend in which stronger binding energies correspond to lower IC50 values. For example, compounds 9 and 14, which exhibited the most favorable binding energies (−8.6 and −8.5 kcal mol−1, respectively), also displayed the highest cytotoxic activity (IC50 = 7.88 and 8.76 µM, respectively). Conversely, compounds with weaker binding energies, such as 1 (−7.6 kcal mol−1), showed higher IC50 values (32.43 µM). Although this trend is consistent with the proposed mode of action, it should be interpreted cautiously, as in vitro activity may also be influenced by other pharmacokinetic or cellular factors not captured in docking simulations.
Overall, these results indicate that hydrogen bonding and Pi–anion interactions are recurring motifs likely contributing to the observed cytotoxicity, supporting CDK1 as a plausible target. The docking data complement the experimental findings but are presented as a preliminary theoretical evaluation rather than definitive proof of mechanism.
4. Conclusions
In conclusion, the chemical reactivity of 2-methylchromone (1) with different nitrogen-based nucleophiles enabled the synthesis of Z-configured enaminones and a series of novel pyrimidine fused heterocycles, highlighting its utility as a versatile precursor in heterocyclic construction. In addition, computational studies at the B3LYP/6-311++G(d,p) level revealed strong agreement between calculated and experimental IR vibrational frequencies and NMR (1H and 13C) chemical shifts, with correlation coefficients (R2) = 0.99. FMO analysis and global reactivity descriptors indicated that compound 9 possesses the highest softness (S = 0.546 eV), compound 12 showed the greatest electronegativity (χ = 4.266 eV), and compound 9 exhibited the strongest electrophilicity index (ω = 4.903 eV). The MEP mapping identified negative potential regions around sulphur, oxygen and nitrogen atoms, favoring electrophilic attack, and positive regions around hydrogen and carbon atoms, favoring nucleophilic attack. The first hyperpolarizability values (0.398 × 10−30–2.358 × 10−30 esu) suggest promising non-linear optical (NLO) activity. Biologically, compounds 9 and 14 displayed notable anticancer activity compared with cisplatin. Moreover, the binding energy computations have confirmed the experimental data in anticancer evaluation. Further, drug-likeness evaluation showed that all synthesized compounds comply with Lipinski's, Ghose's, Egan's, and Veber's rules, supporting their potential for oral administration.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
Data are available upon request from the authors.
Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5ra09088a.
Acknowledgements
This work was supported and funded by the Deanship of Scientific Research at Imam Mohammad Ibn Saud Islamic University (IMSIU) (grant number IMSIU-DDRSP2601).
References
- N. A. Alshaye, M. A. Ibrahim and A. Badran, Nucleophilic transformation of 3-substituted-6,8-dimethylchromones with phenylhydrazine under various reaction conditions: Theoretical, spectroscopic characterization and in silico ADME studies, J. Mol. Struct., 2024, 1297, 137006, DOI:10.1016/j.molstruc.2023.137006.
- M. A. Ibrahim, M. Abdel-Megid, O. Farouk, N. M. El-Gohary, A. I. Nabeel, M. M. A. Attai and A. Badran, Nucleophilic Reactions with 3-formylchromones: A decade update, Synth. Commun., 2024, 54(18), 1495–1522, DOI:10.1080/00397911.2024.2387134.
- A. Badran, Z. Hussain, N. S. Abdelshafi, S. S. Ibrahim, A. Ahmed, M. Abdel-Megid and M. A. Ibrahim, Ring opening and recyclization reactions with 3-functionalized chromones: Recent synthetic approaches for five, six and seven membered heterocycles, Synth. Commun., 2025, 55(10), 693–716, DOI:10.1080/00397911.2025.2463603.
- M. Sobiesiak, N. Fatyga, A. Brzozowski, A. Sikora, B. Kupcewicz and M. Maj, Chroman-one derivatives: Evaluating selective anticancer activity across human cell lines, Eur. J. Med. Chem., 2025, 295, 117771, DOI:10.1016/j.ejmech.2025.117771.
- F. Bonvicini, L. Menegaldo, R. Orioli, F. Belluti, G. A. Gentilomi, S. Gobbi and A. Bisi, Extended Antimicrobial Profile of Chromone–Butenafine Hybrids, Molecules, 2025, 30, 2973, DOI:10.3390/molecules30142973.
- J. H. Lu, C. H. Zhao, Y. Q. Qiu, L. Q. Shen, M. Zhang and H. Zhu, Design, synthesis, and multi-target evaluation of Chromone-based derivatives as promising anti-Alzheimer's disease agents, Bioorg. Chem., 2026, 168, 109350, DOI:10.1016/j.bioorg.2025.109350.
- X.-H. Li, H. Wang, W.-L. Mei, W.-H. Dong, J. Zeng, S.-Z. Huang, H.-Q. Chen, Y.-M. Wei, F. Wu, Z.-Y. Guo and H.-F. Dai, Two unprecedented 2-(2-phenethyl)chromone derivatives with anti-Helicobacter pylori and anti-inflammatory activities from agarwood of Aquilaria crassna, Phytochem. Lett., 2025, 69, 103039, DOI:10.1016/j.phytol.2025.103039.
- A. A. M. Farag, N. Roushdy, S. Abdel Halim, N. M. El-Gohary, M. A. Ibrahim and S. Said, Synthesis, molecular, electronic structure, linear and non-linear optical and phototransient properties of 8-methyl-1,2-dihydro-4H-chromeno[2,3-b]quinoline-4,6(3H)-dione (MDCQD): Experimental and DFT investigations, Spectrochim. Acta, Part A, 2018, 191, 478–490, DOI:10.1016/j.saa.2017.10.014.
- M. A. Mostafa, M. A. Ibrahim and A. Badran, Spectroscopic elucidation, quantum chemical computations (FMO, HOMO–LUMO, MEP, NLO), and biological activity on some novel heterocyclic compounds using 3-substituted-6,8-dimethylchromones, Synth. Commun., 2024, 54(18), 1523–1550, DOI:10.1080/00397911.2024.2394833.
- A. Badran and M. A. Ibrahim, Recyclization of 3-substituted-6,8-dimethylchromones with some heterocyclic enamines: Spectroscopic, Quantum calculations (HOMO–LUMO, MEP, NLO), Biological evaluation and ADME investigation, J. Heterocycl. Chem., 2024, 61, 1224–1247, DOI:10.1002/jhet.4827.
- N. A. Alshaye and M. A. Ibrahim, Synthetic strategies and anticancer evaluation for the novel 3-(6-heteroaryltriazolo[3,4-b][1,3,4]thiadiazol-3-yl)chromeno[2,3-b]pyridines, Synth. Commun., 2024, 54(9), 735–745, DOI:10.1080/00397911.2024.2330617.
- F. M. Alshareef, Z. M. Alamshany, S. A. Al-Harbi, E. S. Allehyani and M. A. Ibrahim, Novel annulated chromeno[3',2':5,6]pyrido[2,3-d][1,3,4]thiadiazolo[3,2-a]pyrimidines and chromeno[3'',2'':5',6']pyrido[2',3':4,5]pyrimido[2,1-b][1,3,4]thiadiazines: Synthetic approaches and antimicrobial efficiency, Synth. Commun., 2024, 54(10), 843–852, DOI:10.1080/00397911.2024.2346565.
- N. A. Alshaye and M. A. Ibrahim, Recyclization Reactions of 2-Methylchromone-3-Carbonitrile with Active Methylene Nucleophiles: Synthesis and Reactions of 4-Methylchromeno[2,3-b] Pyridines, Polycycl. Aromat. Compd., 2023, 43(10), 9105–9117, DOI:10.1080/10406638.2022.2158885.
- B. V. Safitri, A. N. Kristanti, H. Suwito and K. U. Haq, D. Hendriyanto Synthesis of 2-styrylchromones: In vitro and in silico
anticancer activity evaluation, J. Appl. Pharm. Sci., 2021, 11(01), 121–128, DOI:10.7324/JAPS.2021.110114.
- C. D. A. León, L. A. R. Guerrero, P. M. B. Valladares, G. A. Echeverría, O. E. Piro, S. E. Ulic, J. L. Jios and P. Langer, Synthesis and structural study of 2-(haloalkyl)-3-methylchromones, Monatsh. Chem., 2019, 150, 1929–1940, DOI:10.1007/s00706-019-02512-5.
- K. Rajasekhar, C. J. Achar and T. Govindaraju, A red-NIR emissive probe for the selective detection of albumin in urine samples and live cells, Org. Biomol. Chem., 2017, 15, 1584–1588, 10.1039/C6OB02760A.
- J. Azimvand, Synthesis of some new derivatives of 2-methyl-4H-4-chromenone, J. Chem. Pharm. Res., 2012, 4, 3929–3933 CAS.
- M. A. Ibrahim, T. E. Ali, Y. A. Gabr and Y. A. Alnamer, Synthesis and chemical reactivity of 2-methylchromones, ARKIVOC, 2010, i, 98–135, DOI:10.3998/ark.5550190.0011.103.
- S. S. Ibrahim, H. M. El-Shaaer and A. Hassan, Phosphorus Sulfur Silicon Relat. Elem., 2002, 177, 151–172, DOI:10.1080/10426500210228.
- Z. Nawaz, S. Aslam, S. A. Al-Hussain, M. T. Muhammed, S. Rania, B. B. K. Abbasi, A. Irfan, N. Latif, S. Perveen and M. E. A. Zaki, Synthesis, spectral characterization, antioxidant and antidiabetic activities of transition metal complexes of 4-aminoantipyrine: experimental and theoretical studies, J. Mol. Struct., 2025, 1339, 142224, DOI:10.1016/j.molstruc.2025.142224.
- L. Adjissi, N. Chafai, K. Benbouguerra, I. Kirouani, A. Hellal, H. Layaida, M. Elkolli, C. Bensouici and S. Chafaa, Synthesis, characterization, DFT, antioxidant, antibacterial, pharmacokinetics and inhibition of SARS-CoV-2 main protease of some heterocyclic hydrazones, J. Mol. Struct., 2022, 1270, 134005, DOI:10.1016/j.molstruc.2022.134005.
- W. Ried and A. Meyer, Chem. Ber., 1957, 90, 2841–2848 CrossRef CAS.
- A. Dornow and M. P. Menzel, Chem. Ber., 1964, 97, 2173–7278 CrossRef CAS.
- H. A. Oskooie, M. M. Heravi, N. Nami and A. Nazari, Heterocycl. Commun., 2005, 11, 101 CrossRef CAS.
- Y. El Bakri, D. A. Safin, S. K. Mohamed, I. S. Marae, E. A. Bakhite, E. Khamies, A. A. E. Soliman, S. Abuelhassan, H. A. Abuelizzg, R. Al-Salahi, J. T. Mague and C. H. Lai, A highly substituted isoquinolinethione: Synthesis, crystal structure, DFT analysis and molecular docking studies against a series of the SARS-CoV-2 proteins, J. Mol. Struct., 2025, 1331, 141527, DOI:10.1016/j.molstruc.2025.141527.
- V. Shalini, D. C. V. Kumar, D. Gowda, B. S. Chethan, K. B. Harsha, S. M. Rajesh and K. S. Rangappa, Unveiling the structural and theoretical properties of 6-(2-fluoro-3-methylpyridin-4-yl)-2-(4-methoxyphenyl)-N-phenylquinoline-4-carboxamide compound as Sonic Hedgehog protein inhibitor: Synthesis, SCXRD, HSA, DFT, Docking and ADMET studies, J. Mol. Struct., 2025, 1330, 141495, DOI:10.1016/j.molstruc.2025.141495.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, B. E, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. J. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, D. 0109, Revision D. 01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- R. D. Dennington, T. A. Keith, and J. M. Millam, Gauss View 5.0.8, Gaussian Inc, 2008 Search PubMed.
- V. Shalini, D. C. V. Kumar, D. Gowda, B. S. Chethan, K. B. Harsha, S. M. Rajesh and K. S. Rangappa, Unveiling the structural and theoretical properties of 6-(2-fluoro-3-methylpyridin-4-yl)-2-(4-methoxyphenyl)-N-phenylquinoline-4-carboxamide compound as Sonic Hedgehog protein inhibitor: Synthesis, SCXRD, HSA, DFT, Docking and ADMET studies, J. Mol. Struct., 2025, 1330, 141495, DOI:10.1016/j.molstruc.2025.141495.
- K. Wolinski, J. F. Hinton and P. Pulay, Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations, J. Am. Chem. Soc., 1990, 112(23), 8251–8260, DOI:10.1021/ja00179a005.
- F. Hussain, R. Hussain, M. Adnan, S. Muhammad, Z. Irshad, M. U. Khan, J. Yaqoob and K. Ayub, Insights into the nonlinear optical (NLO) response of pure Aum (2 ≥ m ≤ 7) and copper-doped Aum–xCux clusters, RSC Adv., 2022, 12, 25143–25153, 10.1039/D2RA03664A.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65, 55–63, DOI:10.1016/0022-1759(83)90303-4.
- A. Sabt, W. M. Eldehna, T. Al-Warhi, O. J. Alotaibi, M. M. Elaasser, H. Suliman and H. A. Abdel-Aziz, Discovery of 3,6-disubstituted pyridazines as a novel class of anticancer agents targeting cyclin-dependent kinase 2: Synthesis, biological evaluation and in silico insights, J. Enzyme Inhib, Med. Chem., 2020, 35, 1616–1630, DOI:10.1080/14756366.2020.1806259.
- S. M. Gomha, S. M. Riyadh, E. A. Mahmmoud and M. M. Elaasser, Synthesis and Anticancer Activities of Thiazoles, 1,3-Thiazines, and Thiazolidine Using Chitosan-Grafted-Poly(vinylpyridine) as Basic Catalyst, Heterocycles, 2015, 91, 1227–1243, DOI:10.1002/chin.201538150.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 1–13, DOI:10.1038/srep42717.
- V. Pogaku, K. Gangarapu, S. Basavoju, K. K. Tatapudi and S. B. Katragadda, Design, synthesis, molecular modelling, ADME prediction and anti-hyperglycemic evaluation of new pyrazole-triazolopyrimidine hybrids as potent α-glucosidase inhibitors, Bioorg. Chem., 2019, 93, 103307, DOI:10.1016/j.bioorg.2019.103307.
- O. Trott and A. J. Olson, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31(2), 455–461, DOI:10.1002/jcc.21334.
- S. Narra and N. Munirathinam, Synthesis, structure analysis, Hirshfeld surface studies, molecular docking studies against cyclin-dependent kinases (CDKs) of sulfonamide decorated N6-benzyl aminopurines for cancer treatment, Results Chem., 2024, 8, 101592, DOI:10.1016/j.rechem.2024.101592.
- B. Parveen, S. Shahzadi, S. Ali, M. Feizi-Dehnayebi, K. S. Munawar, M. Ashfaq and M. N. Tahir, Synthesis, spectral characterizations, computational studies and biological investigation of 4-(4-(2-hydroxyethyl)phenylamino)-4-oxobutanoic acid and its trimethyltin(IV) complex, J. Mol. Struct., 2024, 1315, 138851, DOI:10.1016/j.molstruc.2024.138851.
- D. Sahin, R. A. Kepekci, M. Feizi-Dehnayebi, S. Akkoc, J. V. Cuevas-Vicario and N. Micale, Biological Activities, DFT Calculations, and Molecular Docking Simulation of Thymol-Based Compounds, ChemistrySelect, 2024, 9(23), e202304572, DOI:10.1002/slct.202304572.
- R. K. Mohapatra, A. Mahal, A. Ansari, M. Kumar, J. P. Guru, A. K. Sarangi, A. Abdou, S. Mishra, M. Aljeldah and B. M. AlShehail, Comparison of the binding energies of approved mpox drugs and phytochemicals through molecular docking, molecular dynamics simulation, and ADMET studies: An in silico approach, J. Biosaf. Biosecur., 2023, 5(3), 118–132, DOI:10.1016/j.jobb.2023.09.001.
- A. M. El-Saghier, A. Abdou, M. A. Mohamed, H. M. Abd El-Lateef and A. M. Kadry, Novel 2-acetamido-2-ylidene-4-imidazole derivatives (El-Saghier reaction): Green synthesis, biological assessment, and molecular docking, ACS Omega, 2023, 8(33), 30519–30531, DOI:10.1021/acsomega.3c03767.
- U. O. Ozdemir, B. Tuzun, E. B. Ayan and B. S. Cevrimli, Eco-friendly and potential colin esterase enzyme inhibitor agent sulfonylhydrazone series: synthesis, bioactivity screening, DFT, ADME properties, and molecular docking study, J. Mol. Struct., 2023, 1286, 135514, DOI:10.1016/j.molstruc.2023.135514.
- L. Wang, J. Ding, L. Pan, D. Cao, H. Jiang and X. Ding, Quantum chemical descriptors in quantitative structure activity relationship models and their applications, Chemom. Intell. Lab. Syst., 2021, 217, 104384, DOI:10.1016/j.chemolab.2021.104384.
- E. Bilen, U. O. Ozmen, S. Cete, S. Alyar and A. Yasar, Bioactive sulfonyl hydrazones with alkyl derivative: characterization, ADME properties, molecular docking studies and investigation of inhibition on choline esterase enzymes for the diagnosis of Alzheimer's disease, Chem.-Biol. Interact., 2022, 360, 109956, DOI:10.1016/j.cbi.2022.109956.
- C. Kucuk, S. Celik, S. Yurdakul and E. Coteli, A new Ag(I)-complex of 5-chloroquinolin-8-ol ligand: Synthesis, spectroscopic characterization, and DFT investigations, in vitro antioxidant (DPPH and ABTS), α-glucosidase, α-amylase inhibitory activities with protein-binding analysis, J. Mol. Struct., 2025, 1325, 141285, DOI:10.1016/j.molstruc.2024.141285.
- L. Feng, H. Yang, X. Cui, D. Chen and G. Li, Experimental and theoretical investigation on corrosion inhibitive properties of steel rebar by a newly designed environmentally friendly inhibitor formula, RSC Adv., 2018, 8, 6507–6518, 10.1039/c7ra13045g.
- M. A. Mostafa, M. A. Ibrahim, S. S. Ibrahim, N. Mohamed and A. Badran, Synthetic approaches for novel 3-heteroaryl-4-hydroxy-1-methylquinoline-2(1H)one: spectroscopic characterization, molecular docking and DFT investigations, RSC Adv., 2025, 15, 6718–6736, 10.1039/d5ra00325c.
- Z. Zhou and R. G. Parr, Activation hardness: new index for describing the orientation of electrophilic aromatic substitution, J. Am. Chem. Soc., 1990, 112, 5720–5724, DOI:10.1021/ja00171a007.
- L. Pauling, The nature of the chemical bond and the structure of molecules and crystals: an introduction to modern structural chemistry, Nature, 1961, 189, 3, DOI:10.1038/189003a0.
- L. Pauling, The nature of the chemical bond. IV. The energy of single bonds and the relative electronegativity of atoms, J. Am. Chem. Soc., 1932, 54, 3570–3582, DOI:10.1021/ja01348a011.
- H. M. Abd El-Lateef, M. M. Khalaf, A. A. Amer, A. A. Abdelhamid and A. Abdou, Antibacterial, antifungal, anti-inflammatory evaluation, molecular docking, and density functional theory exploration of 2-(1H-benzimidazol-2-yl)guanidine mixed-ligand complexes: Synthesis and characterization, Appl. Organomet. Chem., 2024, 38, e7299, DOI:10.1002/aoc.7299.
- R. G. Parr, L. V. Szentpaly and S. Liu, Electrophilicity Index, J. Am. Chem. Soc., 1999, 121(9), 1922–1924, DOI:10.1021/ja983494x.
- M. Alieva Qudrat, M. N. Tahir, K. S. Munawar, M. Feizi-Dehnayebi, M. Ashfaq, S. H. Saadat, M. H. Ulviyya, Q. K. Tahira, A. I. Simuzer, C. A. Shebnem and M. E. Muhammed, One-dimensional polymer of copper with salicylic acid and pyridine linkers: synthesis, characterizations, solid state assembly investigation by hirshfeld surface analysis, and computational studies, J. Mol. Struct., 2024, 1279, 136956, DOI:10.1016/j.molstruc.2023.136956.
- M. S. Elkotamy, M. K. Elgohary, S. T. Al-Rashood, H. Almahli, W. M. Eldehna and H. A. Abdel-Aziz, Novel imidazo[2,1-b]thiazoles and imidazo[1,2-a]pyridines tethered with indolinone motif as VEGFR-2 inhibitors and apoptotic inducers: Design, synthesis and biological evaluations, Bioorg. Chem., 2024, 151, 107644, DOI:10.1016/j.bioorg.2024.107644.
- R. A. Omer, K. M. Ahmed, K. A. Omar, W. M. Hamad and D. M. Mamad, N,N-Bis (2,4-dihydroxybenzaldehyde)benzidine: Synthesis, characterization, DFT, and theoretical corrosion study, J. Mol. Struct., 2024, 1300, 137279, DOI:10.1016/j.molstruc.2023.137279.
- Ö. Tamer, M. Şimşek, D. Avcı and Y. Atalay, Static/dynamic first and second order hyperpolarizabilities, optimized structures, IR, UV–Vis, 1H and 13C NMR spectra for effective charge transfer compounds: a DFT study, Spectrochim. Acta, Part A, 2023, 286, 122005, DOI:10.1016/j.saa.2022.122005.
- A. Zochedh, K. Reni, A. Shunmuganarayanan, S. Ansar, Y. A. Kumar and A. B. Sultan, Synthesis, characterization, in silico and in vitro assessment of 2-aminopyridine nicotinamide cocrystal as a new agent for breast cancer therapy, J. Mol. Struct., 2025, 1322, 140315, DOI:10.1016/j.molstruc.2024.140315.
- M. Evecen, H. Tanak, A. A. Agar, S. Meral and N. Ozdemir, Comparative structural, spectroscopic and nonlinear optical analysis of a Schiff base compound with experimental and theoretical methods (HF, B3LYP and WB97X-D), Optik, 2021, 228, 166133, DOI:10.1016/j.ijleo.2020.166133.
- A. M. Abu-Dief, R. M. El-Khatib, T. El-Dabea, S. A. Abdel-Latif, I. O. Barnawi, F. S. Aljohani, K. Al-Ghamdi and M. A. A. A. El-Remaily, Design, synthesize, physicochemical characterization, nonlinear optical properties structural elucidation, biomedical studies, and DNA interaction of some new mixed ligand complexes incorporating 4,6-dimethylpyrimidine derivative and imidazole ligand, Appl. Organomet. Chem., 2024, 38, e7463, DOI:10.1002/aoc.7463.
- J. Kulhanek, F. Bures, O. Pytela, T. Mikysek, J. Ludvík and A. Ruzicka, Push-pull molecules with a systematically extended π-conjugated system featuring 4,5-dicyanoimidazole, Dyes Pigm., 2010, 85, 57–65, DOI:10.1016/j.dyepig.2009.10.004.
- M. A. Ibrahim, A. Badran, M. M. A. Attai, N. M. El-Gohary, Z. Hussain and O. Farouk, Synthesis, structural characterization, fukui functions, DFT calculations, molecular docking and biological efficiency of some novel heterocyclic systems, J. Mol. Struct., 2024, 1314, 138815, DOI:10.1016/j.molstruc.2024.138815.
- L. K. Zaitri and S. M. Mekelleche, Computational study of linear and nonlinear optical properties of substituted thiophene imino dyes using long-range corrected hybrid DFT methods, Mol. Phys., 2020, 118, 1618508, DOI:10.1080/00268976.2019.1618508.
- A. Zochedh, K. Reni, A. Shunmuganarayanan, S. Ansar, Y. A. Kumar and A. B. Sultan, Synthesis, characterization, in silico and in vitro assessment of 2-aminopyr idine nicotinamide cocrystal as a new agent for breast cancer therapy, J. Mol. Struct., 2025, 1322, 140315, DOI:10.1016/j.molstruc.2024.140315.
- V. Pogaku, K. Gangarapu, S. Basavoju, K. K. Tatapudi and S. B. Katragadda, Design, synthesis, molecular modelling, ADME prediction and anti-hyperglycemic evaluation of new pyrazole-triazolopyrimidine hybrids as potent α-glucosidase inhibitors, Bioorg. Chem., 2019, 93, 103307, DOI:10.1016/j.bioorg.2019.103307.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 2012, 64, 4–17, DOI:10.1016/j.addr.2012.09.019.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, Molecular properties that influence the oral bioavailability of drug candidates, J. Med. Chem., 2002, 45, 2615–2623, DOI:10.1021/jm020017n.
- W. J. Egan, K. M. Merz and J. J. Baldwin, Prediction of drug absorption using multivariate statistics, J. Med. Chem., 2000, 43, 3867–3877, DOI:10.1021/jm000292e.
- A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases, J. Comb. Chem., 1999, 1, 55–68, DOI:10.1021/cc9800071.
- T. Lanez, M. Feizi-Dehnayebi and E. Lanez, Assessment of the electrostatic binding of ferrocenylmethyl-nitroaniline derivatives to DNA: A combined experimental and theoretical study, J. Mol. Struct., 2024, 1308, 138386, DOI:10.1016/j.molstruc.2024.138386.
- A. M. Abu-Dief, O. A. Omran, M. Feizi-Dehnayebi, A. Alqurashi, I. Omar, D. Alhashmialameer and A. D. M. Mohamad, Fabrication, structural elucidation, and DFT calculation of some new hydrophilic metal chelates based on N N′-(1-methyl-2-oxoindolin-3-ylidene)benzohydrazide ligand: Pharmaceutical studies and molecular docking approach, Appl. Organomet. Chem., 2024, 38(9), e7593, DOI:10.1002/aoc.7593.
- H. M. Abd El-Lateef, A. M. Ali, M. M. Khalaf and A. Abdou, New iron (III), cobalt (II), nickel (II), copper (II), zinc (II) mixed-ligand complexes: Synthesis, structural, DFT, molecular docking and antimicrobial analysis, Bull. Chem. Soc. Ethiop., 2024, 38(1), 147–166, DOI:10.4314/bcse.v38i1.12.
- A. M. El-Saghier, S. S. Enaili, A. M. Kadry, A. Abdou and M. A. Gad, Green synthesis, biological and molecular docking of some novel sulfonamide thiadiazole derivatives as potential insecticidal against Spodoptera littoralis, Sci. Rep., 2023, 13(1), 19142, DOI:10.1038/s41598-023-46602-1.
- M. Malumbres and M. Barbacid, Cell cycle, CDKs and cancer: a changing paradigm, Nat. Rev. Cancer, 2009, 9(3), 153–166 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *c and
Magdy A. Ibrahimc
*c and
Magdy A. Ibrahimc





















