DOI:
10.1039/D5RA08970K
(Paper)
RSC Adv., 2026,
16, 8573-8583
Sonochemical synthesis and characterization of Tb2Cu2O5 nanostructures and their application as a nano-photocatalyst for the degradation of water-soluble organic pollutants under visible light
Received
20th November 2025
, Accepted 27th January 2026
First published on 12th February 2026
Abstract
In this research, nanostructured Tb2Cu2O5 was synthesized using ultrasonic-assisted techniques with copper and terbium nitrates (in the presence of tetraethylenepentamine as a chelating and surface-modifying agent). The synthesis conditions were systematically optimized by varying the sonication power and duration. The optimal condition was achieved at a sonication power of 60 W for 15 min, resulting in phase-pure Tb2Cu2O5 nanostructures with a uniform morphology. The FESEM images also indicated the presence of well-dispersed quasi-spherical Tb2Cu2O5 nanostructures with uniform morphology. The photocatalytic performance of the synthesized Tb2Cu2O5 was evaluated using visible-light irradiation in the presence of several organic dyes, including erythrosine (ER), eriochrome black T (ET), methylene blue (MB), methyl orange (MO), rhodamine B (RhB), and methyl violet (MV). The degradation efficiency for ER was the highest (100.0% in 35 min), followed by that for ET (50.0% in 100 min), with lower degradation efficiencies for all other dyes. The photocatalytic mechanism was demonstrated to be influenced by the dye concentration, amount of catalyst, and pH of the solution. Radical scavenger experiments indicated that the radical mechanism was predominantly facilitated by the superoxide (˙O2−) and hydroxyl radicals (˙OH) while photogenerated holes had a lesser contribution. Tb2Cu2O5 exhibited excellent photostability, retaining 95.2% of its degradation efficiency after five reuse cycles. Furthermore, with a narrow bandgap of ∼1.61 eV, high efficiency, and excellent durability, the synthesized Tb2Cu2O5 could be a viable candidate for visible-light-driven photocatalytic water treatment.
1. Introduction
Wastewater discharged from both domestic activities and diverse industrial operations typically contains substantial amounts of hazardous chemicals and toxic substances, such as dyes, which pose serious environmental threats.1,2 The accumulation of toxic and organic pollutants in ecosystems represents a significant risk to both human health and biodiversity.3,4 A variety of methods have been explored to tackle these harmful pollutants.5 Among them, photocatalysis has gained significant attention as a sustainable and environmentally benign strategy that leverages sunlight and photocatalytic materials to break down or oxidize hazardous compounds.6 Its advantages—such as non-toxicity, high efficiency, cost-effectiveness, and capacity to generate harmless by-products—have made it a popular choice for environmental cleanup.7 Numerous photocatalytic materials have already been developed and applied to remove toxic pollutants.8 Nevertheless, designing photocatalysts with optimized bandgaps that are suitable for targeting a wide range of pollutants remains a key challenge. Consequently, there is a growing interest in developing high-performance photocatalytic materials capable of degrading various environmental toxins. Over the past few decades, photocatalysis has seen a rapid advancement due to its outstanding redox stability, excellent recyclability, and green nature, making it widely applicable in processes such as artificial photosynthesis, pollutant degradation, and other energy- and chemistry-related applications.9,10 Lately, the use of solar-driven photocatalysis has expanded into areas like hydrogen generation,11 environmental detoxification,12 hydrogen peroxide production, and carbon dioxide reduction.
In parallel, eco-friendly synthetic techniques based on green chemistry principles have emerged as promising methods for creating nanoscale oxide materials.13 These sustainable techniques rely on safe and non-toxic precursors, reducing the risk of environmental pollution. They are also recognized for being energy-efficient, reproducible, and operationally simple.14
Lanthanide-based double copper oxides (Ln2Cu2O5, where Ln = Lu, Sm, Pr, Tm, Tb, and La) have gained increasing attention due to their unique physicochemical properties, including narrow bandgaps, good redox stability, rich electronic structures arising from f–d orbital interactions, and promising optical and magnetic behaviors.15,16 These compounds typically crystallize in an orthorhombic structure with the space group Pna21.17 However, their synthesis has mostly relied on conventional solid-state or co-precipitation routes,16 which lack control over particle size and morphology. Recently, alternative approaches like sol–gel, hydrothermal, and sonochemical methods have gained interest due to their potential to lower processing temperatures and yield uniform materials with higher surface areas.18 Therefore, there is a need to establish a straightforward, reproducible, energy-efficient, and rapid technique for synthesizing Ln2Cu2O5 with controlled characteristics.
To date, no study has reported a simple and eco-friendly method for producing Tb2Cu2O5 nanostructures. Moreover, achieving high-purity Tb2Cu2O5 remains difficult due to the challenge of removing residual CuO.16 In this study, we report—for the first time—the successful use of a sonochemical approach to synthesize Tb2Cu2O5 nanostructures and evaluate their photocatalytic performance under visible irradiation. Additionally, the effects of sonication power and reaction time on the purity and morphology of the resulting nanostructures were systematically investigated.
2. Materials and methods
2.1. Materials
Copper nitrate trihydrate 99% (Cu(NO3)2·3H2O), terbium(III) nitrate hexahydrate (Tb(NO3)3·6H2O), tetraethylenepentamine (TEPA), benzoic acid (BA), ethylenediaminetetraacetic acid (EDTA), 4-benzoquinone (BQ), rhodamine B (RB), erythrosine (ER), methyl violet (MV), eriochrome black T (ET), methylene blue (MB), and methyl orange (MO) were purchased from Merck Co. and applied for additional processes.
2.2. Preparation of Tb2Cu2O5 nanostructures
Tb2Cu2O5 nanostructures were synthesized through an ultrasonic-assisted reaction involving copper nitrate trihydrate (Cu(NO3)2·3H2O) and terbium(III) nitrate hexahydrate (Tb(NO3)3·6H2O) in a 0.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio. Specifically, 3.8 mmol of Tb(NO3)3 was dissolved in 20 mL of deionized water. This Tb solution was then added to a beaker containing an appropriate amount of copper nitrate (3.0 mmol), and the mixture was stirred for 15 minutes. Subsequently, a diluted solution of TEPA was introduced gradually under continuous sonication until the pH of the mixture reached 10. TEPA, a tetraamine compound, acted as both a precipitation promoter and a surfactant in the reaction medium. The resulting solid was separated by filtration, rinsed thoroughly with water, and then subjected to calcination at 900 °C for 4 hours. Various synthesis conditions for Tb2Cu2O5 are outlined in Table 1.
:1 molar ratio. Specifically, 3.8 mmol of Tb(NO3)3 was dissolved in 20 mL of deionized water. This Tb solution was then added to a beaker containing an appropriate amount of copper nitrate (3.0 mmol), and the mixture was stirred for 15 minutes. Subsequently, a diluted solution of TEPA was introduced gradually under continuous sonication until the pH of the mixture reached 10. TEPA, a tetraamine compound, acted as both a precipitation promoter and a surfactant in the reaction medium. The resulting solid was separated by filtration, rinsed thoroughly with water, and then subjected to calcination at 900 °C for 4 hours. Various synthesis conditions for Tb2Cu2O5 are outlined in Table 1.
Table 1 Preparation conditions of Tb2Cu2O5 nanostructures
| Sample no. |
Power of sonication (W) |
Time of sonication (min) |
Calcination temperature (°C)/time (h) |
Grain size (nm) |
Product |
| TCO-1 |
60 |
15 |
900/4 |
37.7 |
Tb2Cu2O5 |
| TCO-2 |
80 |
27.8 |
Tb2Cu2O5 + Tb2O3 + CuO |
| TCO-3 |
40 |
38.3 |
Tb2Cu2O5 + Tb2O3 + CuO |
| TCO-4 |
60 |
10 |
31.8 |
Tb2Cu2O5 + Tb2O3 + CuO |
| TCO-5 |
20 |
27.3 |
Tb2Cu2O5 + Tb2O3 + CuO |
To evaluate energy transfer efficiency during the process, temperature changes over time were recorded. The rate of temperature increase (dT/dt) was derived from the thermal profile (T vs. t). Based on this, the ultrasonic power input (P) was calculated using the following equation:15
| |
 | (1) |
where
cp is the specific heat capacity of the solvent (J kg
−1 K
−1) and
M is the mass of the solvent (kg). Under the described conditions, with 60 W of input power in distilled water, the effective acoustic power output was calculated to be approximately 16.2 W.
2.3. Photocatalytic test
The photocatalytic performance of Tb2Cu2O5 in breaking down various hazardous organic pollutants was evaluated using visible light (150 W). For each test, 50 mg of the Tb2Cu2O5 catalyst was added to a 10-ppm solution of the targeted dye contaminant. Prior to visible-light irradiation, the catalyst–dye suspension was stirred in the dark for 30 min to establish adsorption–desorption equilibrium. Negligible dye removal during this period confirms that adsorption alone plays a minimal role and that the observed removal is predominantly due to photocatalytic degradation. The suspension was initially kept in the dark for 30 minutes to establish an adsorption–desorption equilibrium between the catalyst surface and the dye molecules. After this period, the respective light source was turned on to initiate the photocatalytic reaction. The pollutant degradation efficiency was calculated using the following relation:19| |
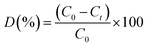 | (2) |
where C0 and Ct represent the pollutant concentrations (based on absorbance) before and after irradiation, respectively.
2.4. Scavenger experiment
To determine the principal reactive species involved in the photocatalytic oxidation of ER dye, separate scavenger tests were conducted. Specific chemical scavengers were used at a concentration of 1 mmol to selectively inhibit individual reactive species: benzoic acid (BA) for hydroxyl radicals (˙OH), ethylenediaminetetraacetic acid (EDTA) for photogenerated holes (h+), and benzoquinone (BQ) for superoxide anion radicals (˙O2−).20
2.5. Physical instruments
To determine the crystalline structure and phase purity of the synthesized nanoparticles, X-ray diffraction (XRD) measurements were carried out using a Philips diffractometer (X'PertPro) equipped with a monochromatic Cu Kα radiation source (λ = 1.54 Å). The morphology, particle dispersion, and elemental composition were analyzed via field-emission scanning electron microscopy (FESEM) using a Mira3 TESCAN instrument coupled with energy-dispersive X-ray spectroscopy (EDS). Transmission electron microscopy (TEM) was conducted using a JEM-2100 microscope to observe the internal structure of the nanoparticles. The UV-visible absorption spectra were recorded using a JASCO V-670 UV-vis spectrophotometer, and the optical bandgap was estimated via diffuse reflectance measurements using a HO-SP-DRS100 spectrometer.
3. Results and discussion
3.1. Crystallographic structure
X-ray diffraction (XRD) analysis was utilized to assess the crystal structure of synthesized Tb2Cu2O5. The XRD patterns of samples produced under various ultrasonic powers and durations are displayed in Fig. 1. The principal aim of this study was to establish the sonication conditions best suited for the pure phase of Tb2Cu2O5. Applying ultrasonic energy at 60 W resulted in a single-phase Tb2Cu2O5 (JCPDS No. 00-034-0385) with an orthorhombic structure (Fig. 1a). The characteristic reflections corresponding to the (202), (203), (210), (211), (031), (401), (412), (413), and (020) planes confirmed the successful formation of phase-pure Tb2Cu2O5 under optimal sonication conditions. However, the samples made at higher (80 W) or lower (40 W) power had impurity phases. The sample that underwent 80-W ultrasonic power (Fig. 1b) showed peaks corresponding to Tb2O3 (hexagonal structure, PDF No. 00-019-1329) and CuO (monoclinic structure, PDF No. 01-080-1917), thereby indicating an incomplete reaction. The sample that underwent 40 W (Fig. 1c) produced peaks indicative of hexagonal Tb2O3 and a small amount of monoclinic CuO and therefore suggested an incomplete transformation of reactants due to insufficient energy. The results presented show that sonication power is critical for phase development. Moderate ultrasonic power was conducive to producing homogeneous crystalline products, whereas excessive or limited energy produced incomplete reactions or defects in the lattice structure. In this case, 60-W power was recognized as the optimal condition for producing Tb2Cu2O5 of high purity. In contrast, samples synthesized at non-optimal ultrasonic power exhibited additional peaks attributed to hexagonal Tb2O3 and monoclinic CuO phases, indicating incomplete reaction or insufficient atomic diffusion. These results clearly demonstrate that ultrasonic power and duration play a decisive role in phase evolution and crystallinity. The influence of synthesis time at a constant ultrasonic power of 60 W was investigated at 10, 15, and 20 minutes. Among these, the sample made at 15 minutes (Fig. 1a) possessed the best-defined and strongest diffraction peaks, which indicate higher crystallinity and higher phase purity than those synthesized at 10 and 20 minutes. An incomplete reaction may be caused by a short time (Fig. 1d), while partial degradation or defect may be generated due to longer exposure (Fig. 1e).
 |
| | Fig. 1 . XRD patterns of Tb2Cu2O5 prepared under different conditions: (a) power = 60 W, time = 15 min, (b) power = 80 W, time = 15 min, (c) power = 40 W, time = 15 min, (d) power = 60 W, time = 10 min, and (e) power = 60 W, time = 20 min. | |
Fourier-transform infrared (FTIR) spectroscopy (Fig. 2) revealed the characteristic absorption bands of metal–oxygen stretching vibrational modes. Absorption bands in the 400–700-cm−1 range were attributed to Tb–O and Cu–O lattice vibrations,21 indicating the formation of the mixed metal oxide.22 The residual organic species bands were trivial, indicating successful decomposition of precursors during synthesis. Two distinct absorption bands appeared at 1621 and 3445 cm−1,23 corresponding to hydroxyl groups from water molecules adsorbed on the surface of Tb2Cu2O5. This indicates that water is present and likely interacts with the material via hydrogen bonding.
 |
| | Fig. 2 FTIR spectrum of Tb2Cu2O5 prepared at power = 60 W and time = 15 min. | |
3.2. Synthesis mechanism
One of the major benefits of ultrasonic devices is that they can effectively control the structure of a wide range of nanomaterials. Through sonication, which replicates the effects of cavitation, it is possible to create nanoscale systems with high uniformity and well-characterized properties.24 According to the hotspot theory, the energy pulses and elevated temperatures formed under bubble collapse can significantly increase the degradation of bulk structures to nanostructures.25 As demonstrated in earlier studies, it is possible to precisely control the morphology and formation of nanostructures by altering the parameters of ultrasonic waves. Ultrasonic energy absorption by water molecules generates hydroxyl radicals, which play a crucial role in the hydrolysis of samarium and copper precursors. Such reactive species are strongly engaged in the ultrasonic-assisted chemical synthesis of the Tb2Cu2O5 nanophotocatalyst.26 The given equations represent an explained mechanism for the sonochemical synthesis of spherical Tb2Cu2O5 nanostructures:| |
 | (3) |
| | |
˙OH + H2N2(CH2CH2NH)4CH2CH2NH2 + 2H2O → NH2(CH2CH2NH)3CH2CH2NH + NH3 + 2OH−
| (4) |
| | |
Tb(NO3)3 + 3OH− → Tb(OH)3 + NO3−
| (5) |
| | |
Cu(NO3)2 + 2OH−→Cu(OH)2
| (6) |
| |
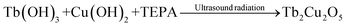 | (7) |
Additionally, the surface hydroxyl groups in Tb2Cu2O5 can establish hydrogen bonds with TEPA that enable adsorption on crystal surfaces. The interaction prevents the development of isolated particles and inhibits the unregulated growth of particles. The core of every particle remains compartmentalized due to the coating agent. Besides, the long carbon chain in TEPA enhances interparticle spacing, which effectively reduces the likelihood of agglomeration. During crystallization of the phase, there is a need to perform a calcination step; however, the sonochemical process contributes greatly to the regulation of the nucleation, homogeneity of precursors, and particle size prior to annealing. Ultrasonic cavitation increases the mass transfer rate and accelerates the kinetics of the reaction, which provides phase purity and morphology enhancements over that of traditional calcination. TEPA functions as both a surface-modifying agent and a chelating agent, thereby regulating the coordination of metal ions to the precursor and preventing uncontrolled particle growth. The long-chain structure of TEPA provides greater distances between the individual particles, thereby preventing agglomeration during the sonochemical process.
3.3. Morphological study
Field-emission scanning electron microscopy (FESEM) micrographs (Fig. 3) showed the surface topography of synthesized Tb2Cu2O5 under different conditions. The sample synthesized at 60-W power and 15 minutes (Fig. 3a) showed well-graded nanoscale particles with fairly good dispersion and moderate agglomeration. By contrast, shorter (10 min, Fig. 3b) or longer (20 min, Fig. 3c) synthesis times yielded less homogeneous topographies. In addition, changing the ultrasonic power to 40 W (Fig. 3d) led to under-refinement of the particles, while 80-W power (Fig. 3e) led to over-fragmentation and spiked edges. These findings indicate that intermediate power and time (60 W, 15 min) offer the best balance between particle size, morphology, and homogeneity. When comparing the appearance of the samples, there were similarities in their morphology, but there were distinct differences in the uniformity of the particles, number of agglomerated particles, and type of surface texture of each. The sample prepared using 60 W of power for 15 minutes had a higher degree of particle homogeneity and a moderate level of agglomeration compared with the previous samples prepared with shorter or longer sonication times. It can be seen that the samples prepared with longer or shorter times contain particles that are not of the same size and therefore have a higher level of aggregation. The presence of agglomerated particles leads to reduced accessibility to available active sites and increased light–matter interactions, leading to poor photocatalytic efficiency. Furthermore, even small changes in morphology can have major effects on photocatalytic efficiency.
 |
| | Fig. 3 FESEM images of Tb2Cu2O4 prepared in different conditions of Tb2Cu2O5 prepared in different conditions (a) power = 60 W, time = 15 min, (b) power = 80 W, time = 15 min, (c) power = 40 W, time = 15 min, (d) power = 60 W, time = 10 min, and power = 60 W, time = 20 min, and (f) EDS spectrum of Tb2Cu2O5 prepared at power = 60 W and time = 15 min. | |
Fig. 3f shows the EDS analysis of Tb2Cu2O5 prepared at optimum sonication time and power. The chemical composition of the prepared sample indicates the existence of the terbium, oxygen, and copper elements in the composition, indicating homogeneous elemental distribution throughout the nanostructures.
The TEM images provided further evidence of the nanostructure and morphology of optimally synthesized Tb2Cu2O5 (TCO–1) samples. The particles had a quasi-spherical morphology and some relatively narrow particle-size distribution, typically in the range of 60–150 nanometers Fig. 4.
 |
| | Fig. 4 (a and b) TEM images of Tb2Cu2O5 prepared at power = 60 W and time = 15 min (TCO–1). | |
3.4. Photocatalytic activity of Tb2Cu2O5
Diffuse reflectance spectroscopy (DRS) and the corresponding Tauc plot (Fig. 5) were used to determine the optical bandgap of Tb2Cu2O5. Indeed, the Tb2Cu2O5 nanostructures were synthesized under different ultrasonic power levels and irradiation times, and these variations slightly influenced the optical bandgap energy. As experimentally determined, the bandgap values varied in a narrow range of 1.56 to 1.63 eV, indicating that the fundamental electronic structure of Tb2Cu2O5 remained preserved while subtle changes occurred due to differences in crystallinity, particle size, and defect density induced by the synthesis conditions. Information regarding the bandgap of Tb2Cu2O5 is lacking in the literature. When Ln = Pr, Nd, Sm, and Gd, the gap energies for Ln2CuO4 were 0.79, 1.06, 1.20, and 1.36 eV, respectively.27 The bandgap discovered in this investigation thus agrees well with that of similar molecules. To further clarify the effect of synthesis conditions on the electronic structure, the conduction band (CB) and valence band (VB) edge potentials were calculated using the Mulliken electronegativity method. Based on the calculated average electronegativity of Tb2Cu2O5 (χ ≈ 4.85 eV) and the experimentally obtained bandgaps, the CB and VB positions were determined. The results are shown in Table 2, which indicate that the CB edge lies in the range of −0.43 to −0.47 eV (vs. NHE) and the VB edge is located between 1.13 and 1.17 eV (vs. NHE). These small shifts in CB and VB positions directly correlate with the observed variations in bandgap values and are attributed to the different synthesis conditions. Importantly, all samples exhibited a sufficiently negative CB potential for effective reduction reactions and a positive VB potential suitable for oxidation processes, confirming their applicability as visible-light-driven photocatalysts. Therefore, although different synthesis conditions slightly affect the bandgap and band-edge positions, the overall band alignment remains favorable for photocatalytic applications, and the variations can contribute to differences in photocatalytic performance through the modulation of charge separation efficiency and redox ability. It is noteworthy that the determined bandgap value indicates favorable optical properties for the visible-spectrum photocatalytic process. When utilized for photocatalysis, Tb2Cu2O5 can effectively absorb visible light. Because visible light accounts for a large portion of solar radiation, its utilization is highly beneficial, making it a powerful option for sunlight conversion applications. Furthermore, a smaller bandgap suggests that Tb2Cu2O5 can absorb lower energy photon levels, increasing the material's productivity in generating electron–hole pairs during photocatalysis. The breakdown of dye must be accelerated by these electron–hole pairs in order to initiate chemical reactions. The bandgap energy of 1.61 eV is suitable for visible light-driven photocatalytic activity, further increasing the material's utility in environmental remediation.
 |
| | Fig. 5 DRS spectrum and Tauc plot of Tb2Cu2O5 prepared under different conditions: (a and b) power = 60 W and time = 15 min (TCO–1), (c and d) power = 80 W and time = 15 min (TCO–2), (e and f) power = 40 W and time = 15 min (TCO–3), (g and h) power = 60 W and time = 10 min (TCO–4), and (i and j) power = 60 W and time = 20 min (TCO–5). | |
Table 2 Optical bandgap and the calculated band-edge positions of Tb2Cu2O5 nanostructures synthesized under different ultrasonication conditions
| Samples |
Bandgap, Eg (eV) |
CB edge, ECB (eV vs. NHE) |
VB edge, EVB (eV vs. NHE) |
| TCO-1 |
1.61 |
−0.455 |
1.155 |
| TCO-2 |
1.60 |
−0.450 |
1.150 |
| TCO-3 |
1.56 |
−0.430 |
1.130 |
| TCO-4 |
1.63 |
−0.465 |
1.165 |
| TCO-5 |
1.61 |
−0.455 |
1.155 |
The photocatalytic performance of Tb2Cu2O5 nanoparticles was systematically researched under visible-light irradiation for the degradation of various organic dyes (Fig. 6). The results showed that the synthesized material exhibited excellent photocatalytic performance, with an exceptional performance for the degradation of erythrosine (ER), which reached 95.3% at 30 min. The rest of the dyes came in the order of eriochrome black T (ET) at 50.0%, methylene blue (MB) at 20.1%, rhodamine B (RhB) at 6.7%, methyl orange (MO) at 20.2%, and methyl violet (MV) at 16.0% after 100 min (Fig. 6a). ER's stronger degradation activity would be attributed to its higher interaction with the surface of the catalyst and better orientation of their absorption spectrum towards the visible-light activity of Tb2Cu2O5. Differences in efficiency for the photocatalytic degradation of various dyes can be attributed to their differing molecular structures, charge distributions, and affinities for attaching to the catalyst surface. Erythrosine is an example of a dye that has a stronger ability to interact with Tb2Cu2O5 because of the electrostatic attraction and improved spectral overlap between the visible light being absorbed by Tb2Cu2O5 and the absorption spectrum of this dye. On the other hand, dyes that have either lower affinity for the catalyst or higher steric hindrance (due to bulky substituents) will exhibit reduced rates of degradation. Therefore, the structure dependence of the photocatalytic selectivity for Tb2Cu2O5 is clearly evident.
 |
| | Fig. 6 The photocatalytic degradation diagrams of Tb2Cu2O5 under different conditions (a) different organic contaminants, (b) different concentration of ER, and (c) different Tb2Cu2O5 dosages, (d) different pHs, (e) different scavengers, and (f) reusability test after 35 min under visible light. | |
To gain better insight into the degradation behavior, the effect of the initial dye concentration was explored using ER as a model contaminant (Fig. 6b). The degradation efficiency showed a slight decrease with increasing dye concentration: 100.0% for 10 ppm, 100.0% for 15 ppm, and 95.5% for 20 ppm. The loss can be explained in terms of reduced light penetration and saturation of the catalyst surface at high dye concentrations, which hinders the creation of reactive oxygen species.
The impact of catalyst dosage was also evaluated (Fig. 6c). The optimum dosage was 50 mg in 100-mL solution, which resulted in 100.0% degradation of ER. Decreasing the dosage to 30 mg decreased efficiency to 83.4%, which could be due to the insufficient active sites. Doubling the dose to 70 mg did not change the degradation percentage (100.0%). The influence of pH on the photocatalytic process was also investigated. As shown in Fig. 6d, the catalyst operated well in weakly acidic to neutral pH. The degradation was 100.0% at pH = 4, and at neutral pH = 7, it was 85.6%. It reduced to 62.1% at basic pH = 10. All these observations suggest that the surface charge of the catalyst and the extent of dye ionization influence the adsorption and degradation processes. The strong dependence of photocatalytic activity on solution pH suggests that surface charge characteristics play a crucial role in dye adsorption and degradation. Although the point of zero charge (PZC) was not experimentally determined, the observed behavior indicates favorable electrostatic interactions under acidic and neutral conditions.
Radical scavenger experiments were performed to identify the active species involved in the photocatalytic reaction. The degradation efficiency without any scavenger was 100.0%. When a hydroxyl radical scavenger, benzoic acid, was added, the degradation fell to 79.3%. For benzoquinone (a superoxide radical scavenger), the efficiency dropped to 47.3%. On the other hand, when EDTA (a hole scavenger) was added, a lower reduction level was achieved with 92.4% degradation (Fig. 6e). These findings indicate that superoxide radical (˙O2−) and hydroxyl radicals (˙OH) are the predominant active species for dye degradation, while photogenerated holes (h+) play a secondary role.
Finally, the reusability of the Tb2Cu2O5 photocatalyst was also examined over five consecutive degradation cycles. The material exhibited high stability, with degradation efficiency decreasing only marginally from 100.0% in cycle one to 95.2% in cycle five (Fig. 6f). Such a limited degradation confirms that the structural and photocatalytic integrity of Tb2Cu2O5 is still largely intact under repeated cycles of application, which is appropriate for real environmental remediation applications.
3.2.1. Proposed mechanism of Tb2Cu2O5. Both benzoquinone (BQ) and benzoic acid (BA), known scavengers for superoxide radical (˙O2−) and hydroxyl radical (˙OH), respectively, significantly suppressed Tb2Cu2O5 photocatalytic activity, based on the radical scavenging experiments. This suggests that both reactive oxygen species play a vital role in the degradation process. Upon visible-light irradiation, Tb2Cu2O5 absorbs photons due to its narrow bandgap (∼1.61 eV), promoting the electrons from the valence band (VB) to the conduction band (CB), thereby forming photogenerated electrons (e−) and holes (h+):| | |
Tb2Cu2O5 + hν → e− (CB) + h+ (VB)
| (8) |
The electrons generated in the conduction band can reduce the surface-adsorbed oxygen molecules (O2) to form superoxide radicals:
Concurrently, valence-band photogenerated holes may oxidize water molecules or surface hydroxyl groups to form highly reactive hydroxyl radicals:
These reactive oxygen species, i.e., ˙OH and ˙O2−, destroy and decompose the organic dye molecules to innocuous products such as CO2 and H2O. The very strong inhibitory effect of BA confirms that ˙OH is one of the most dominating oxidative species, and partial inhibition by BQ indicates that ˙O2− plays a significant role but less dominantly. Thus, the photocatalytic degradation mechanism of Tb2Cu2O5 involves both oxidative and reductive pathways, where ˙OH radicals generated as a result of hole-mediated water oxidation are the predominant oxidizers, while ˙O2− radicals generated as a result of electron–oxygen interaction provide additional degradation support.28
The photocatalytic efficiency of Tb2Cu2O5 was determined using pseudo-first-order kinetic modeling, which is illustrated in Fig. 7. The degradation kinetics of the organic dye (Fig. 7a) indicated differences in reaction rates for each type of dye based on geometric structure. Erythrosine (ER) has a rate constant of k = 0.08958 min−1, providing the best reaction rate among the other dyes, which had much slower reaction rates, implying little to no adsorption and limited interaction with the surface of the catalyst, indicating the selectivity of Tb2Cu2O5 toward specific dye structures next to geometric adsorption and degradation characteristics. The effect of initial dye concentration (10, 15, and 20 ppm) on degradation rate is illustrated in Fig. 7b. When the concentration increased, the rate constant decreased slightly (k = 0.14246 min−1 at 10 ppm, k = 0.13286 min−1 at 20 ppm). This was attributed to the saturation of the active sites and the limited light penetration through the catalyst surface in concentrated dye solutions.
 |
| | Fig. 7 Kinetics plots of Tb2Cu2O5 under different conditions (a) different organic contaminants, (b) different concentration of ER, (c) different and (d) different pHs after 35 min under visible light. | |
The kinetics of the photocatalytic degradation were investigated using the Langmuir–Hinshelwood pseudo-first-order model. In general, ln(C0/Ct) versus irradiation time will provide a straight line; however, a small deviation from linearity was observed at the end of the irradiation period. This small deviation is most likely due to the extremely low absorbance (i.e., 0.001), which is very close to the detection limit of the UV-vis spectrophotometer, such that even minor instrument noise or fluctuations in the UV-vis spectrophotometer can produce a substantial alteration in the ln(C0/Ct), thereby lowering the R2 value. Similar kinetic deviations at near-complete dye removal have been widely reported in photocatalytic studies and are considered an experimental limitation rather than a change in the reaction mechanism or kinetic order.29–31 The effect of catalyst dosage supplemented by other test results from above (Fig. 7c) indicates a clear increase in photocatalytic efficiency with an increasing amount of Tb2Cu2O5. The rate constant increased from k = 0.05129 min−1 at 30 mg to k = 0.21417 min−1 at 70 mg due to the numerous active sites available and the preferred formation of reactive oxygen species. As demonstrated by the test, increasing the amount of catalyst used to conduct the experiment resulted in deviations from pure linearity with respect to the relationship between reactant concentration and kinetic rate. Specifically, if a large amount of catalyst is added to a sample, the increase will cause several problems, including light scattering, shielding effects, particle agglomeration, and mass transfer limitations, which will negatively impact the observed kinetic fit. Therefore, the result showed that the deviation observed at the higher level of the catalyst is not indicative of a change in the order of reaction but instead reveals what limitations exist for the photocatalytic system that are inherent in having too high a concentration of solids present.32–34 The solution pH significantly influenced photocatalytic activity, as shown in Fig. 7d. The catalyst demonstrated the greatest activity in an acidic medium (pH = 3) with a rate constant k = 0.14246 min−1; neutral and basic mediums demonstrated significantly lower performance (k = 0.05697 min−1 at pH = 7 and k = 0.02783 min−1 at pH = 10). The increase in activity in acidic medium could be attributed to favorable surface charge interactions and stability of photogenerated radicals. The photocatalytic degradation kinetics generally follow a pseudo-first-order model. Deviations from linearity observed at higher catalyst loadings are attributed to light scattering, shielding effects, and surface saturation rather than a change in reaction order. These phenomena are commonly reported in heterogeneous photocatalytic systems at elevated catalyst concentrations.
3.2.3. Comparison investigation. The photocatalytic activity of the prepared Tb2Cu2O5 was fully evaluated and compared with the reported materials for water pollutant degradation, as presented in Table 3. It can be seen that Tb2Cu2O5 exhibits excellent photocatalytic activity for water treatment, with performance similar to those of other structurally analogous photocatalysts.
Table 3 Comparison of the photocatalytic performances of different compounds
| Nanocatalyst |
Preparation method |
Dye |
Efficiency (%) |
Duration (min) |
Ref. |
| Tb2Cu2O5 |
Sonochemical |
Erythrosine |
100.0 |
35 |
Current work |
| La2CuO4–ZnO |
Green synthesis |
Malachite green |
91.0 |
120 |
35 |
| Sm2CuO4 |
Sonochemical |
Methyl orange |
91.4 |
100 |
36 |
| La2Cu2O5 |
Sonochemical |
Acid black |
80.1 |
120 |
37 |
| Co2CuO4 |
Co-precipitation |
Acid brown 14 |
79.0 |
90 |
38 |
| Lu2Cu2O5/Lu2O3 |
Auto combustion |
Thymol blue |
98.5 |
120 |
39 |
| ZnO/La2O3/NiO |
Co-precipitation |
Methylene blue |
98.0 |
150 |
40 |
| La2Ti2O7 |
Sol–gel |
Reactive red 22 |
99.0 |
180 |
41 |
| CuO |
Sol–gel |
Methylene blue |
85.0 |
50 |
42 |
| Ce2Sn2O7 |
Co-precipitation |
Methylene blue |
80.0 |
60 |
43 |
| La2Sn2O7 |
Hydrothermal |
Methyl orange |
98.0 |
50 |
44 |
| Cu3B2O6 |
Co-precipitation |
Acid violet 7 |
86.0 |
90 |
45 |
| Sm2Ti2O7 |
Co-precipitation |
Rhodamine B |
94.0 |
80 |
46 |
| Dy2Cu2O5 |
Sonochemical |
Phenol red |
96.4 |
120 |
47 |
| CoFe2O4 |
Co-precipitation |
Congo red |
91.0 |
90 |
48 |
4. Conclusions
The preparation of Tb2Cu2O5 nanostructures was executed successfully using an ultrasonic-assisted method that provided good control of the composition, morphology, and crystallinity of the final product. The synthesis condition determined to achieve the best results was at a 60-W power for 15 min, which provided highly crystalline, well-distributed nanoparticles with minimal agglomeration. After surface and structural characterization, the development of pure Tb2Cu2O5 phase and the desired nanoscale morphology were confirmed separately. In terms of photocatalytic evaluation, Tb2Cu2O5 had a high activity under visible light for the degradation of a range of organic dyes, where ER degradation exceeded 95% within 30 min. Operations such as initial dye concentration, catalyst dosage, and pH affected the photocatalytic efficiency (the pH between neutral and slightly acidic levels was most applicable). The scavenger studies confirmed that hydroxyl and superoxide radicals were primarily responsible for pollutant degradation. The synthesized Tb2Cu2O5 photocatalyst offers great promise towards environmental remediation, particularly visible-light-driven wastewater purification. With competitive activity against the other reported photocatalysts, its inclusion in real-world applications is highly plausible.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article. Additional data are available from the corresponding author upon reasonable request.
Acknowledgements
The authors appreciate the council of the Iran National Science Foundation, INSF (4046368), and the University of Kashan for supporting this work through Grant No. 159271/FU4. During the development of this manuscript, the authors utilized ChatGPT to assist with grammar refinement and enhancing the clarity of the text. Following the use of this tool, the authors thoroughly reviewed, revised, and edited the manuscript to ensure accuracy and appropriateness. The authors assume full responsibility for the content and integrity of the final work.
References
- N. H. Mohamad Idris, J. Rajakumar, K. Y. Cheong, B. J. Kennedy, T. Ohno, A. Yamakata and H. L. Lee, ACS Omega, 2021, 6, 14493–14503 Search PubMed.
- Y. Zhu, M. T. Chen, Y. Feng, Q. Ai, Y. Liu, Y. Yan, Q. Li and J. Lou, Small Struct., 2025, 6, 2400389 CrossRef CAS.
- M. Avais and S. Chattopadhyay, ACS Appl. Polym. Mater., 2021, 3, 789–800 Search PubMed.
- M. Jothibas, C. Manoharan, S. Johnson Jeyakumar, P. Praveen, I. Kartharinal Punithavathy and J. Prince Richard, Sol. Energy, 2018, 159, 434–443 Search PubMed.
- R. Salazar, E. Brillas and I. Sirés, Appl. Catal., B, 2012, 115–116, 107–116 CrossRef CAS.
- M. Pirhashemi and A. Habibi-Yangjeh, J. Colloid Interface Sci., 2017, 491, 216–229 CrossRef CAS PubMed.
- Y. Gao and T. Wang, J. Mol. Struct., 2021, 1224, 129049 CrossRef CAS.
- J. Guo, C.-H. Shen, J. Sun, X.-J. Xu, X.-Y. Li, Z.-H. Fei, Z.-T. Liu and X.-J. Wen, Sep. Purif. Technol., 2021, 259, 118109 CrossRef CAS.
- L. Liang, L. Shi, F. Wang, H. Wang, P. Yan, Y. Cong, L. Yao, Z. Yang and W. Qi, Appl. Catal., A, 2020, 599, 117618 CrossRef CAS.
- K. Fang, L. Shi, L. Cui, C. Shi and W. Si, CrystEngComm, 2021, 23, 4475–4485 RSC.
- L. Liang, L. Shi, F. Wang, L. Yao, Y. Zhang and W. Qi, Int. J. Hydrogen Energy, 2019, 44, 16315–16326 CrossRef CAS.
- K. Fang, L. Shi, L. Yao and L. Cui, Mater. Res. Bull., 2020, 129, 110888 Search PubMed.
- P. Naresh Kumar Reddy, D. P. M. D. Shaik, V. Ganesh, D. Nagamalleswari, K. Thyagarajan and P. Vishnu Prasanth, Appl. Surf. Sci., 2021, 561, 150092 CrossRef CAS.
- A. Zaleska-Medynska, M. Marchelek, M. Diak and E. Grabowska, Adv. Colloid Interface Sci., 2016, 229, 80–107 Search PubMed.
- S. Milad Tabatabaeinejad, M. Ghanbari, Z. Mohsen Najm, M. N. Abdul-Fattah, N. M. Hameed and M. Salavati-Niasari, J. Mol. Liq., 2022, 362, 119718 Search PubMed.
- R. Guo, J. You, F. Han, C. Li, G. Zheng, W. Xiao and X. Liu, Appl. Surf. Sci., 2017, 396, 1076–1084 Search PubMed.
- L. Li, J. Wang, K. Su, D. Huo and Y. Qi, J. Alloys Compd., 2016, 658, 500–504 CrossRef CAS.
- S. M. Tabatabaeinejad, O. Amiri, M. Ghanbari and M. Salavati-Niasari, J. Mol. Liq., 2021, 117883 Search PubMed.
- M. R. Saadati-Gullojeh, M. Ghanbari and M. Salavati-Niasari, Sol. Energy, 2024, 268, 112316 Search PubMed.
- M. Hosseini, M. Ghanbari, E. A. Dawi, A. M. Aljeboree, M. H. S. Alubiady, A. M. Al-Ani and M. Salavati-Niasari, Surf. Interfaces, 2024, 46, 104162 Search PubMed.
- S. Sagadevan, K. Pal and Z. Z. Chowdhury, J. Mater. Sci.: Mater. Electron., 2017, 28, 12591–12597 Search PubMed.
- M. Majumder, R. B. Choudhary, A. K. Thakur and U. Kumar, J. Electroanal. Chem., 2017, 804, 42–52 CrossRef CAS.
- M. Ghanbari, M. Salavati-Niasari and F. Mohandes, Arabian J. Chem., 2022, 15, 103730 Search PubMed.
- K. Prasad, D. V. Pinjari, A. B. Pandit and S. T. Mhaske, Ultrason. Sonochem., 2011, 18, 1128–1137 Search PubMed.
- L. Yin, Y. Wang, G. Pang, Y. Koltypin and A. Gedanken, J. Colloid Interface Sci., 2002, 246, 78–84 Search PubMed.
- D. P. Dutta, V. Sudarsan, P. Srinivasu, A. Vinu and A. K. Tyagi, J. Phys. Chem. C, 2008, 112, 6781–6785 Search PubMed.
- B. Marzougui, A. Marzouki, Y. B. Smida and R. Marzouki, 2023.
- F. Yousefzadeh, Q. A. Yousif, M. Ghanbari and M. Salavati-Niasari, J. Mol. Liq., 2022, 118443 Search PubMed.
- J.-M. Herrmann, Catal. Today, 1999, 53, 115–129 Search PubMed.
- M. N. Chong, B. Jin, C. W. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 Search PubMed.
- P. V. Kamat, Journal, 2002, 106, 7729–7744 CAS.
- A. Mills and S. Le Hunte, J. Photochem. Photobiol., A, 1997, 108, 1–35 Search PubMed.
- A. Houas, H. Lachheb, M. Ksibi, E. Elaloui, C. Guillard and J.-M. Herrmann, Appl. Catal., B, 2001, 31, 145–157 CrossRef CAS.
- N. Daneshvar, D. Salari and A. Khataee, J. Photochem. Photobiol., A, 2003, 157, 111–116 Search PubMed.
- Y. Yulizar, D. O. B. Apriandanu and R. I. Ashna, Chem. Phys. Lett., 2020, 755, 137749 Search PubMed.
- F. Yousefzadeh, M. Ghanbari and M. Salavati-Niasari, Chemosphere, 2023, 338, 139564 CrossRef CAS PubMed.
- S. M. Tabatabaeinejad, M. Ghanbari, Z. M. Najm, M. N. Abdul-Fattah, N. M. Hameed and M. Salavati-Niasari, J. Mol. Liq., 2022, 362, 119718 CrossRef.
- R. Rahmatolahzadeh, M. Mousavi-Kamazani and S. A. Shobeiri, J. Inorg. Organomet. Polym. Mater., 2017, 27, 313–322 CrossRef CAS.
- S. M. Tabatabaeinejad, S. Zinatloo-Ajabshir, O. Amiri and M. Salavati-Niasari, RSC Adv., 2021, 11, 40100–40111 Search PubMed.
- J. Shubha, N. Sushma, S. Adil, M. Khan, M. E. Assal, M. R. Hatshan and B. Shaik, J. King Saud Univ. Sci., 2022, 34, 101738 CrossRef.
- W.-M. Hou and Y. Ku, J. Alloys Compd., 2011, 509, 5913–5918 Search PubMed.
- A. Muthuvel, M. Jothibas and C. Manoharan, Nanotechnol. Environ. Eng., 2020, 5, 1–19 Search PubMed.
- V. Jayaraman, B. Palanivel, C. Ayappan, M. Chellamuthu and A. Mani, Sep. Purif. Technol., 2019, 224, 405–420 Search PubMed.
- J. Zeng, H. Wang, Y. Zhang, M. K. Zhu and H. Yan, J. Phys. Chem. C, 2007, 111, 11879–11887 Search PubMed.
- R. Rahmatolahzadeh, S. A. Shobeiri and K. Motevalli, J. Mater. Sci.: Mater. Electron., 2018, 29, 4327–4333 CrossRef CAS.
- K. Kaviyarasu, C. M. Magdalane, D. Jayakumar, Y. Samson, A. Bashir, M. Maaza, D. Letsholathebe, A. H. Mahmoud and J. Kennedy, J. King Saud Univ. Sci., 2020, 32, 1516–1522 CrossRef.
- S. M. Tabatabaeinejad, O. Amiri, M. Ghanbari and M. Salavati-Niasari, J. Mol. Liq., 2021, 344, 117883 CrossRef.
- N. Ali, A. Said, F. Ali, F. Raziq, Z. Ali, M. Bilal, L. Reinert, T. Begum and H. M. Iqbal, Water, Air, Soil Pollut., 2020, 231, 1–16 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Asaad H. Alzaidyb,
Layth S. Jasim
a,
Asaad H. Alzaidyb,
Layth S. Jasim









