DOI:
10.1039/D5RA08786D
(Review Article)
RSC Adv., 2026,
16, 27937-27959
Advanced oxidation processes based on non-radical pathways in persulfate systems: a comprehensive review of their characteristics, detection methods, and catalyst design
Received
14th November 2025
, Accepted 21st April 2026
First published on 22nd May 2026
Abstract
Peroxysulfate-based advanced oxidation processes (AOPs) are increasingly employed for the degradation of organic contaminants in wastewater. However, conventional AOPs primarily rely on highly oxidative free radicals, which are often scavenged by inorganic anions and dissolved organic matter, leading to low selectivity. Recently, non-radical oxidation technologies, particularly those utilizing persulfate, have emerged as promising alternatives due to their enhanced selectivity and stability in complex wastewater environments. Despite their potential, the mechanisms underlying non-radical oxidation are not yet fully understood, and challenges related to catalytic processes and catalyst regulation hinder their broader application. This review provides a comprehensive analysis of recent advancements in persulfate-based non-radical oxidation technologies, focusing on the generation mechanisms, characteristics, and identification of non-radical species, including singlet oxygen and high-valent metal oxides, as well as electron transfer processes. Additionally, the review examines the role of catalysts in regulating non-radical oxidation processes. Finally, it outlines future research directions aimed at advancing non-radical oxidation technologies, with a particular emphasis on oxidation mechanisms, active species identification, and catalyst design.
1. Introduction
Rapid urbanization and industrialization have introduced a wide range of synthetic chemicals into aquatic environments, many of which persist as recalcitrant organic pollutants. In recent years, advanced oxidation processes (AOPs) have shown significant potential in decontaminating refractory organic pollutants, attracting considerable interest from researchers.1 The activation of peroxymonosulfate (PMS) or peroxydisulfate (PDS) through catalytic decomposition or external energy input can break the O–O bond, generating highly reactive species with high redox potentials, such as hydroxyl radicals (˙OH, E0 = 1.9–2.7 V vs. NHE) and sulfate radicals (SO4˙−, E0 = 2.6–3.1 V vs. NHE).2,3 However, due to the strong oxidative capacity of these free radicals, background natural organic matter (NOM), halides, and carbonates react with them, reducing their effectiveness in targeting pollutants.4,5 These side reactions significantly diminish persulfate utilization, often necessitating chemical doses far exceeding theoretical requirements. Furthermore, free radicals can react with halides, producing toxic by-products such as trichloromethane (TCM). Thus, there is an urgent requirement for the development of selective catalytic oxidation technologies based on persulfate to minimize oxidant consumption and the formation of toxic by-products.
In the 1990s, a non-radical catalytic pathway was proposed, with Fe(IV) identified as the dominant oxidative species in the Fe(II)/H2O2 system.6 However, due to its lower redox potential (E0 (Fe(IV)/Fe(III)) = 1.8–2.0 V vs. NHE) compared to that of ˙OH, this pathway garnered limited attention. A breakthrough occurred in 2014, when CuO was shown to selectively degrade 2,4-dichlorophenol (2,4-DCP) by activating PDS through non-radical mechanisms in AOPs.7 Since then, research into non-radical oxidation processes, including those involving 1O2 and high-valence metal species and direct electron transfer, has gained momentum. Non-radical oxidation is increasingly recognized for its higher selectivity, greater anti-interference capability, and enhanced efficacy toward electron-rich compounds, sparking significant interest in non-radical AOPs.8–10
Although non-radical persulfate activation has emerged as a promising alternative to traditional radical-based AOPs, significant challenges remain before it can be widely implemented in real wastewater treatment.11 In particular, the selective nature of non-radical pathways makes their performance highly dependent on the precise identity of active species, which are often short-lived, difficult to detect, and susceptible to misinterpretation due to the limitations of current diagnostic techniques.12–14 Furthermore, the mechanistic complexity—stemming from the concurrent generation of multiple oxidative species, catalyst–pollutant interfacial interactions, and the influence of water matrix constituents—remains insufficiently understood, hindering process optimization.15,16 Catalyst design for non-radical pathways also lacks a unified framework linking structural features (e.g., defect sites, coordination environments, and electronic structure) with specific activation modes and oxidative selectivity.
While some articles have summarized the application of non-radical oxidation technology in wastewater treatment, a comprehensive discussion that systematically integrates its classification, characteristics, active species characterization techniques, catalyst design strategies, and potential mechanisms remains lacking.4–6,11 Furthermore, the design of catalysts targeting ideal non-radical pathways requires further investigation. Most importantly, the mechanisms underlying non-radical catalytic oxidation are not yet fully understood and warrant more in-depth and detailed research for clarification.
Although several recent reviews have examined non-radical oxidation in advanced oxidation systems, their scopes and oxidant frameworks differ.17,18 Wu et al. focused on mechanistic uncertainty and practical implementation in heterogeneous persulfate activation;19 Yao et al. reviewed radical and non-radical processes in peracetic acid-based systems;20 and Liu et al. emphasized electron-transfer regulation at carbon–iron–oxidant interfaces and its role in radical/non-radical switching in iron-mediated systems.21 In contrast, the present review specifically addresses persulfate-based non-radical oxidation systems for wastewater treatment. Within a unified framework, it integrates the classification of major pathways, oxidation characteristics, methods for active-species identification, catalyst design principles, and engineering implications. It further combines quantitative bibliometric analysis, systematic pathway classification, catalyst design principles linked to mechanistic outcomes, a hierarchical multi-method framework for rigorous pathway identification, and quantitative comparisons between radical and non-radical systems. Collectively, these elements provide a more systematic and integrated perspective that complements and extends the existing literature.
This article systematically reviews research published over the past decade on persulfate-based non-radical oxidation systems for wastewater treatment. First, this review employs bibliometric methods to identify the research hotspots and gaps within the field. Subsequently, it examines the functional characteristics and identification methods of three typical non-radical reaction pathways: electron transfer pathways (ETP) and those involving singlet oxygen (1O2), and high-valent metal species. The focus is on exploring the three strategies for achieving non-radical pathway oxidation of pollutants through catalyst design: modifying surface characteristics, adjusting active site dimensions, and incorporating non-metallic atoms. Finally, the article addresses the challenges and future development prospects of this field. This work aims to provide guidance for advancing non-radical oxidation technology from laboratory applications to practical engineering in wastewater treatment.
2. Literature review
This paper conducts a bibliometric analysis of research on non-radical oxidation processes within persulfate systems, focusing on publications from the Web of Science (WOS) and CNKI Core Collection from January 1, 2014, to December 31, 2025. The analysis utilized keyword searches across titles, abstracts, and keywords, employing terms such as “non-radical”, “non-radical AND persulfate”, “non-radical AND catalyst”, and “non-radical AND catalyst AND persulfate”. The literature was filtered to include only articles and reviews, and the retrieved data underwent statistical analysis. The bibliometric approach provided insights into the trends and development of non-radical oxidation pathways, both globally and domestically, with a particular emphasis on persulfate-based systems.
The analysis indicates a notable shift in research trends beginning in 2017. Prior to this period, research on non-radical oxidation (topic N) grew slowly, averaging 42 papers annually in WOS and 1.0 in CNKI. After 2017, publication rates surged, with WOS publications averaging 367.1 per year and CNKI averaging 19.5. This increase reflects a growing interest in non-radical oxidation processes, especially those involving persulfate. Publications on non-radical oxidation in persulfate systems (topic NP) gained momentum in WOS from 2019, reaching 328 papers annually by 2025. Catalyst-related non-radical oxidation research (topic NC) began around 2017, yielding approximately 2000 papers across both WOS and CNKI over the past decade. In contrast, research on non-radical catalytic oxidation in persulfate systems (topic NPC) remains limited, with approximately 80.5 papers published in WOS during the last decade (Fig. 1). These findings emphasize the significance of persulfate-based non-radical oxidation and highlight the need for further studies on catalyst mechanisms and optimization.
 |
| | Fig. 1 Annual publication trends in the field of non-radical pathways in persulfate systems: (a) Web of Science database and (b) CNKI database. | |
The statistics on national publications are derived from author data in the Web of Science database, as shown in Table 1. The top five countries by publication volume are China (1372 papers, 80.8%), the United States (79 papers, 4.6%), South Korea (67 papers, 3.9%), India (51 papers, 3.0%), and Australia (39 papers, 2.3%). China's output exceeds 80% of the total, underscoring its dominant role in this field. It is important to note that VOSviewer uses the “full count method” for collaborative papers, meaning that articles with multiple authors are counted separately for each country. As a result, the total publication count for each country exceeds the actual number of distinct articles, reflecting the breadth of international cooperation and highlighting the role of collaboration in advancing the field.
Table 1 Top 10 countries by publication output in research on non-radical pathways in persulfate systems
| Rank |
Country |
Number of publications |
Percentage of total publications |
| 1 |
China |
1372 |
80.8% |
| 2 |
United States |
79 |
4.6% |
| 3 |
South Korea |
67 |
3.9% |
| 4 |
India |
51 |
3.0% |
| 5 |
Australia |
39 |
2.3% |
| 6 |
Iran |
30 |
1.8% |
| 7 |
Pakistan |
29 |
1.7% |
| 8 |
United Kingdom |
22 |
1.3% |
| 9 |
Canada |
21 |
1.2% |
| 10 |
Saudi Arabia |
20 |
1.1% |
Regarding institutional publications, the top five institutions are all from China: Chinese Academy of Sciences (77 papers, 4.5%), Hunan University (70 papers, 4.1%), Harbin Institute of Technology (60 papers, 3.5%), Tongji University (51 papers, 3.0%), and Sichuan University (42 papers, 2.5%), as detailed in Table 2. The Chinese Academy of Sciences stands out with the highest publication count, reinforcing its leadership in this research area.
Table 2 Top 10 institutions by publication output in research on non-radical pathways in persulfate systems
| Rank |
Institution |
Number of publications |
Percentage of total publications |
| 1 |
Chinese Academy of Sciences |
77 |
4.5% |
| 2 |
Hunan University |
70 |
4.1% |
| 3 |
Harbin Institute of technology |
60 |
3.5% |
| 4 |
Tongji University |
51 |
3.0% |
| 5 |
Sichuan University |
42 |
2.5% |
| 6 |
South China University of technology |
38 |
2.2% |
| 7 |
University of Chinese Academy of Sciences |
33 |
1.9% |
| 8 |
Tianjin University |
32 |
1.9% |
| 9 |
Huazhong University of Science and technology |
31 |
1.8% |
| 10 |
Nankai University |
23 |
1.4% |
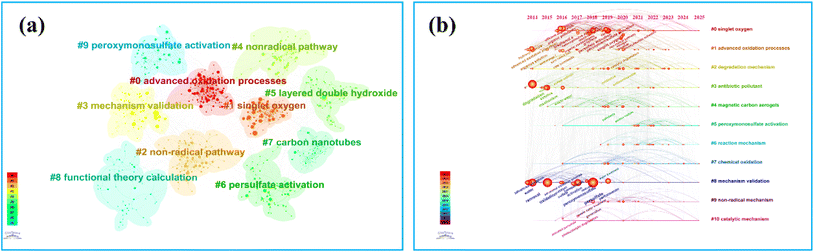
A co-citation clustering analysis of literature related to non-radical oxidation of peroxides was conducted using the CiteSpace software. Based on the co-citation frequency, a network was constructed, and literature correlations were measured through clustering algorithms. Ten clusters were identified, with the knowledge graph highlighting the central role of advanced oxidation processes (cluster #0) in the research network (Fig. 2a). This suggests that non-radical peroxide systems primarily focus on oxidation efficiency and selectivity. Clusters #1 (singlet oxygen) and #2/#4 (non-radical pathway) indicate the growing interest in the generation mechanisms of singlet oxygen and other non-radical species, central to the “non-radical pathway characteristics” of the topic.
 |
| | Fig. 2 (a) Co-citation clustering graph of the literature on non-radical pathways in persulfate systems. (b) Timeline graph of keywords related to non-radical pathways in persulfate systems. | |
The timeline and literature node distribution reveal that the clustering of #3 (mechanism validation) and #8 (functional theory calculation) reflects an increasing trend of integrating experimental validation with theoretical calculations in mechanism research. Quantum chemical calculations and in situ characterization play a crucial role in identifying non-radical reactive oxygen species and enhancing catalyst design precision.
In catalyst research, clusters #5 (layered double hydroxide), #6 (persulfate activation), and #7 (carbon nanotubes) suggest the widespread use of metal oxides and carbon-based materials in non-radical persulfate systems. These materials achieve selective activation of reactive oxygen species by modulating electronic structures and surface functional groups. Cluster #9 (peroxymonosulfate activation) emphasizes the importance of diverse oxidant activation strategies in optimizing non-radical pathways.
This article utilizes the CiteSpace software to analyze the temporal evolution of keywords in the non-radical oxidation of persulfate. Based on keyword collinearity analysis and clustering evaluation indicators, 11 clusters were identified. The horizontal axis position of the keyword nodes indicates their first appearance in the literature, with the node size representing keyword frequency and the thickness of the connecting lines reflecting the strength of relationships between keywords.
Keyword timeline analysis places singlet oxygen (#0) and advanced oxidation processes (#1) at the forefront of the network, highlighting that singlet oxygen, as a non-radical reactive oxygen species, is a primary focus in research on persulfate-based advanced oxidation systems. Cluster analysis of degradation mechanisms (#2) and reaction mechanisms (#6) suggests that researchers are exploring the relationship between non-radical pathways and pollutant degradation efficiency, contributing to a theoretical framework for guiding experimental work.
In catalyst design, clusters #4 (magnetic carbon aerogels), #5 (peroxymonosulfate activation), and #10 (catalytic mechanisms) highlight the importance of structural and functional optimization of carbon-based and metal composite catalysts for achieving non-radical oxidation. Cluster #3 (antioxidant polar) reflects the growing focus on environmental drug pollution control as a key research driver.
Timeline analysis indicates that from 2014 to 2017, research predominantly focused on persulfate activation and traditional oxidants (Fig. 2b). From 2018 to 2022, there was a notable increase in studies on singlet oxygen and non-radical mechanisms. Starting in 2023, the verification of binding mechanisms between carbon-based and metal composite catalysts, combined with computational chemistry, is expected to become a research frontier, creating a logical progression from the discovery of active species to mechanism analysis, catalyst design, and application optimization.
To enhance the quantitative analysis, keyword co-occurrence networks and visual clustering maps were generated using the VOSviewer software to identify thematic clusters and key research areas. Four primary clusters emerged from the keyword co-occurrence network (Fig. 3). The red cluster focused on “peroxymonosulfate”, “oxidation”, and “degradation”, emphasizing oxidants, oxidation mechanisms, and reaction kinetics in aqueous systems. The green cluster concentrated on “persulfate”, “activation”, and “electron transfer”, highlighting persulfate activation and non-radical pathways in wastewater treatment. The blue cluster, featuring “biochar”, “bisphenol A”, and “adsorption”, emphasized catalysts and adsorption in pollutant removal. The yellow cluster dealt with catalyst performance and degradation efficiency.
 |
| | Fig. 3 Co-occurrence network and clustered visualization of high-frequency keywords from the WOS database: (a) topic N; (b) topic NP; (c) topic NC; and (d) topic NPC. | |
Further analysis of persulfate-related non-radical oxidation (NP) revealed three main clusters. The red cluster focused on pollutant removal efficiency and mechanisms, while the green cluster highlighted carbon-based catalysts and activation efficiency. The yellow cluster concentrated on activation mechanisms and degradation processes. Research on catalyst-related processes in non-radical oxidation (NC) was categorized into four clusters: red, emphasizing catalysts in degradation and activation; green and yellow, addressing catalyst-mediated degradation, catalytic efficiency, and reaction mechanisms; and blue, examining catalyst activation, active species transformation, and interfacial adsorption behavior. For the NPC topic, three clusters were identified: red, focusing on pollutant degradation mechanisms; green, on catalyst-driven persulfate systems; and blue, on persulfate activation for bisphenol A degradation.
In summary, non-radical oxidation technology based on persulfate has emerged as a significant research direction in environmental remediation, evidenced by 463 related publications over the past decade. Current studies largely target removal efficiency, activation pathways, degradation mechanisms, and reaction kinetics, whereas the properties of non-radical oxidants, identification of active species, and catalyst design receive comparatively less attention. Comprehensive investigations of non-radical active-species characteristics and reliable identification methods are essential to elucidate reaction mechanisms. In parallel, rational catalyst design is required to realize complete non-radical oxidation pathways suitable for practical wastewater treatment. Future work should prioritize mechanistic clarification, development of high-efficiency catalysts, and advancement of robust diagnostics for active-species identification to close these gaps.
3. Typical non-radical pathways in AOPs
Research on non-radical species in AOPs has intensified in the past decade, highlighting three primary mechanisms: ETP, 1O2 generation, and high-valent metal species formation.
3.1. ETP
In advanced oxidation systems utilizing PDS and PMS, ETP represent the predominant non-radical mechanism. This process comprises three stages: (i) co-adsorption of persulfate and organic pollutants onto the catalyst surface, (ii) activation of persulfate, resulting in the formation of catalyst–persulfate complexes (Cat–PS) that enhance the catalyst's redox potential (Ecat), and (iii) selective oxidation of pollutants through electron transfer from the Cat–PS complex, leading to their reduction to sulfate ions (SO42−) when the catalyst's redox potential surpasses that of the organic compounds (Fig. 4).22
 |
| | Fig. 4 Oxidation mechanism via electron-transfer processes in a persulfate/catalyst system. | |
ETP mechanisms can be classified into inner-sphere and outer-sphere oxidation, depending on the strength of the catalyst–reactant interactions. Inner-sphere reactions are characterized by stronger adsorption and direct chemical bonding of reactants to the catalysts, whereas outer-sphere reactions by weaker interactions with reactants. The persulfate-based ETP process typically occurs in two stages: (i) reactant adsorption and persulfate activation, and (ii) oxidation of the organic compounds accompanied by SO42− release.22,23 Four types of ETP mechanisms are identified based on the interaction strength: Type I involves inner-sphere interactions for both persulfate and organic compounds, while Types II, III, and IV correspond to outer-sphere interactions with the organic compound, persulfate, or both (Fig. 5).
 |
| | Fig. 5 Four non-radical oxidation pathways in the electron transfer mechanism of a persulfate/catalyst system. | |
Metal oxides and transition-metal catalysts significantly influence the ETP by modifying crystal structures, defect sites, and electronic configurations. For instance, the PDS/CuO system activates persulfate through outer-sphere interactions, whereas NiO nanocatalysts facilitate the activation of PMS to form non-radical PMS–NiO complexes, thereby promoting efficient oxidation.7,24 Additionally, carbon-based materials, such as nitrogen-doped carbon nanotubes (NCNTs), demonstrate catalytic efficacy through a synergistic combination of free radical and ETP mechanisms, although the precise activation mechanism remains a subject of ongoing debate.25
3.2. 1O2
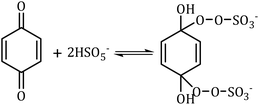
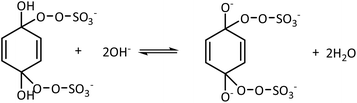
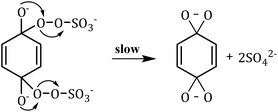
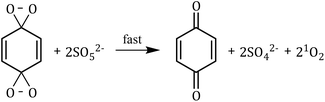
1O2 is a highly reactive molecular oxygen species characterized by a short lifetime (10−3–10−6 s) and a redox potential (E0 = 1.52 V vs. NHE) lower than that of ˙OH and SO4˙−.26–28 It selectively oxidizes organic pollutants with electron-donating groups, primarily generated by (i) self-decomposition of persulfate, (ii) catalytic activation at active sites, or (iii) conversion from intermediates such as superoxide radicals (˙O2−) and hydrogen peroxide (H2O2).29,30 In PS-AOPs, the generation mechanisms differ between PMS and PDS systems. In PMS, 1O2 is produced either through HSO5− decomposition or via catalytic activation at active sites, where HSO5− loses electrons to form ˙SO5−, which subsequently generates 1O2. In the PDS system, 1O2 is produced through the catalytic formation of ˙O2− and ˙HO2 from S2O82− (eqn (1)–(15)).27,31–33 While electron transfer systems predominantly facilitate 1O2 generation in persulfate activation, homogeneous systems can also yield 1O2. Zhou et al. demonstrated this in PMS systems under alkaline conditions (pH 8–10), where p-benzoquinone (p-BQ) reacted with PMS to degrade sulfamethoxazole (SMX), highlighting bis(oxyethane) as a key intermediate (eqn (16)–(20)).29 Overall, 1O2 shows strong promise for degrading persistent organics, though aspects of its formation remain unresolved.| | |
HSO5− + SO52− → HSO4− + SO42− + 1O2
| (1) |
| | |
HSO5− + H2O → HSO4− + H2O2
| (2) |
| | |
H2O2 + OH+ → H2O + HO2−
| (4) |
| | |
HSO5− + HO2− → H2O + SO4˙− + 1O2
| (5) |
| | |
HSO5− → SO5˙− + H+ + e−
| (6) |
| | |
SO5˙− + SO5˙− → 2SO4− + 1O2
| (7) |
| | |
HSO5− → SO5˙− + H+ + 1O2
| (8) |
| |
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O + S2O82− → 4SO42− + 1O2 + 2H2O O + S2O82− → 4SO42− + 1O2 + 2H2O
| (9) |
| |
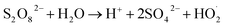 | (10) |
| |
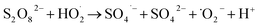 | (11) |
| | |
2˙O2− + 2H+ → 1O2 + H2O2
| (12) |
| | |
˙O2− + 2H2O → 2H+ + 1O2 + H2O2
| (13) |
| |
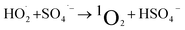 | (14) |
| |
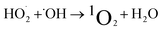 | (15) |
| | |
OH− + HSO5− ⇌ SO52− + H2O
| (16) |
| |
 | (17) |
| |
 | (18) |
| |
 | (19) |
| |
 | (20) |
Carbon-based catalysts, including carbon nanotubes (CNTs) and nitrogen-doped mesoporous carbon, have been demonstrated to activate persulfate for the generation of 1O2. Cheng et al. demonstrated CNT-mediated PDS activation for degrading 2,4-DCP, with surface –CO groups playing a critical role in 1O2 formation.34 Additional studies indicated that surface defects also contribute to 1O2 production, with –CO groups facilitating electron transfer. Long et al. further investigated nitrogen-doped carbon, revealing its capacity to activate PMS for the degradation of rhodamine B, with graphite and pyrrolic nitrogen enhancing the formation of active sites.35 Nevertheless, debates continue regarding the specific active sites and mechanisms within carbon-based systems.
Metal oxides, including manganese oxides (α-, β-, and γ-MnO2), also activate persulfate to generate 1O2, with oxygen vacancies (OVs) playing a crucial role. Zhu et al. demonstrated that β-MnO2 exhibited the highest activity for phenol degradation, generating 1O2 through a semi-stable manganese intermediate (Mn(IV)–O–O–O–SO3).36 This mechanism, involving ˙O2− as 1O2 precursors, has been similarly observed in BiOBr, where surface OVs facilitate 1O2 production. Some studies suggest that OVs directly contribute to 1O2 generation, particularly in PMS/Co/Bi25FeO40 and PMS/CoAl-LDH@CoSx systems, where OVs generate reactive oxygen species (ROS), leading to 1O2 formation (eqn (21)–(26)).37,38 Additionally, zero-valent metals (e.g., Pd0 and Fe0) and single-atom catalysts (SACs) have been explored for persulfate activation and 1O2 generation.27,28,39,40 Although 1O2 is highly resistant to anionic interference in aqueous environments, it remains a selective oxidant, effectively degrading electron-donating pollutants such as phenols and sulfonamides. However, its limited efficacy against inert pollutants like iodinated contrast agents necessitates further investigation into the structure–activity relationships of 1O2 and the characterization of active sites to enhance its applicability in complex water matrices.
| | |
2[≡Mn(IV)–OH]III + HS2O8− → 2[≡Mn(IV)–O–O–SO3]II + 3H+
| (21) |
| | |
2[≡Mn(IV)–O–O–SO3]II + 4H2O + S2O82− → 2[≡Mn(III)–OH]II + 4SO42− + 2˙O2− + 8H+
| (22) |
| | |
[≡Mn(IV)–O–O–SO3]II + ˙O2− + OH+ → [≡Mn(III)–OH]II + SO42− + 1O2
| (23) |
| | |
2˙O2− + 2H2O → 1O2 + H2O2 + 2OH−
| (24) |
| | |
O* + HSO5− → HSO4− + 1O2
| (26) |
3.3. High-valent metal species
High-valent metal species, including Fe(IV)/Fe(V), Co(IV), Mn(V), and Cu(III), have been identified as effective activators of PMS in both homogeneous and heterogeneous systems (Fig. 6).41,42 These species demonstrate selective oxidation capabilities, preferentially reacting with electron-rich pollutants, and exhibit significant resistance to interference from water quality parameters. Their generation generally involves one- or two-step reaction mechanisms, including single- or double-electron transfer processes.
 |
| | Fig. 6 Formation mechanisms of high-valent metals in homogeneous and heterogeneous systems, Reprinted with permission from ref. 41. Copyright 2024 Elsevier. | |
For instance, Fe(IV)O is generated through the heterolysis of O–O bonds in PMS, involving both double-electron transfer and single-oxygen atom transfer. Research conducted by Wang et al. confirmed that Fe(IV) is the predominant active species in the PDS/Fe(II) system, particularly within the pH range of 3–5, thereby challenging the traditional assumption that SO4˙− are the primary oxidants (eqn (27)).43 Furthermore, Bao et al. illustrated the sustained generation of Fe(IV)O in the ZnFe LDH/PMS system, which showed stable degradation performance even under elevated salinity conditions.44
In the cobalt system, Co(II) reacts with PMS to form Co(III) and SO4˙− (eqn (28)).45 Recent studies have indicated that Co(II) can form high-valent cobalt complexes (Co(IV)O2+) through double-electron transfer (eqn (29)).46 However, electron repulsion in Co(IV)O2+ facilitates its reduction to Co(III). Single-atom cobalt catalysts, includingFexCo2−x(OH)2CO3, CoSA@C3N4, Co3O4@CNT, and Fe/Co/N@BC, enhance the generation of Co(IV)O2+ by modulating the electronic distribution of metal d-orbitals via asymmetric O/N coordination.47–49
Manganese and copper are extensively utilized in the activation of PMS. Mn(V) is primarily produced through the oxidation of Mn(II) in the Mn(II)/PMS system, where Mn(II) is first oxidized to Mn(III), which subsequently forms Mn(V) complexes via double-electron transfer (eqn (30) and (31)).50 The generation of Mn(III) is essential for effective degradation. In copper systems, µM-grade Cu(II) reacts with PMS under weakly alkaline conditions to generate Cu(III) (E0 = 1.57–2.3 V vs. NHE), which interacts with water to produce ˙OH, thereby enhancing the degradation of pollutants, exemplified by a 95% removal of 10 µM 2,4-DCP within 15 min.51 Chen et al. found that when Cu(II) complexes with nitrogen-containing organic compounds such as cefoxitin, the ligand's lone pair electrons occupy Cu's d-orbitals, promoting the oxidation of Cu(II) to Cu(III) (eqn (32)–(34)).52 Under conditions of 0.05 mM Cu(II), 1.1 mM PDS, and pH 7, the degradation rate of 0.1 mM cefoxitin achieved 90%.46
Although high-valent metal oxidation systems effectively degrade pollutants under acidic to weakly alkaline conditions (pH 3–8), the stability of active species in complex aqueous matrices remains a significant challenge. Future research should focus on the regulation of electronic structures in catalysts and the synergistic mechanisms of multiple metals, employing in situ characterization techniques such as X-ray absorption near-edge structure (XANES) and Mössbauer spectroscopy to enhance the practical application of this technology.
| | |
Fe(II) + S2O82− + H2O → Fe(IV)O2+ + 2SO42− + 2H+
| (27) |
| | |
Co(II) + HSO5− → Co(III) + SO4˙− + OH−
| (28) |
| | |
Co(II) + HSO5− → Co(IV)O2+ + SO42− + H+
| (29) |
| | |
Mn(II)–L + PMS → Mn(III)–L + caged SO4˙−
| (30) |
| | |
Mn(II)–L + PMS → Mn(V)–L + SO42− + OH−
| (31) |
| | |
Cu(II) + HSO5− → Cu(II)–HSO5− → Cu(I) + SO5˙− + H+
| (32) |
| | |
Cu(I) + HSO5− → Cu(III) + SO42− + OH−
| (33) |
| | |
Cu(II) + ˙OH ⇌ Cu(III) + OH−
| (34) |
3.4. Interplay among non-radical pathways: ETP, 1O2, and high-valent metal species
Non-radical persulfate activation typically involves multiple coexisting pathways. The relative contributions of ETP, 1O2 generation, and high-valent metal-oxo species depend on operating conditions (e.g., oxidant dose, pH, ionic strength, NOM, and halides), catalyst microstructure (e.g., defects, coordination environment, and exposed facets), and substrate electronic structure. This section synthesizes the synergy and competition between these routes and identifies key controls over pathway selectivity.
ETP ↔ 1O2: conditional synergy via peroxy intermediates. On sp2-carbon or oxygen vacancy-rich surfaces, adsorption-coupled ETP reduces persulfate (PMS/PDS) to surface-bound peroxy species (e.g., SO5˙− and 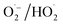 ), which disproportionate or couple to yield 1O2. Carbonyl groups and OVs accelerate this cascade by stabilizing catalyst–persulfate complexes, weakening the O–O bond, and facilitating the one-electron reductions that sustain 1O2 generation. This ETP → 1O2 pathway occurs on CNTs and N-doped carbons (assisted by carbonyl groups) and on MnO2/BiOBr (where OVs route persulfate to 1O2 via superoxide precursors). This synergy is enhanced by increasing the defect density, tuning surface hydrophobicity to strengthen co-adsorption, and using moderate oxidant doses. Competition arises when (i) rapid outer-sphere ETP depletes substrate electrons, limiting the pool of peroxy intermediates, or (ii) 1O2 quenching reveals a dominant ETP contribution. Diagnostic signatures include significant open-circuit potential (OCP) shifts upon persulfate/pollutant addition (suggesting ETP), coupled with accelerated degradation in deuterium oxide (D2O), confirming 1O2 involvement.53
), which disproportionate or couple to yield 1O2. Carbonyl groups and OVs accelerate this cascade by stabilizing catalyst–persulfate complexes, weakening the O–O bond, and facilitating the one-electron reductions that sustain 1O2 generation. This ETP → 1O2 pathway occurs on CNTs and N-doped carbons (assisted by carbonyl groups) and on MnO2/BiOBr (where OVs route persulfate to 1O2 via superoxide precursors). This synergy is enhanced by increasing the defect density, tuning surface hydrophobicity to strengthen co-adsorption, and using moderate oxidant doses. Competition arises when (i) rapid outer-sphere ETP depletes substrate electrons, limiting the pool of peroxy intermediates, or (ii) 1O2 quenching reveals a dominant ETP contribution. Diagnostic signatures include significant open-circuit potential (OCP) shifts upon persulfate/pollutant addition (suggesting ETP), coupled with accelerated degradation in deuterium oxide (D2O), confirming 1O2 involvement.53
ETP ↔ high-valent metal: mechanistic coupling. At Fe, Co, or Mn sites, heterolytic O–O cleavage within an inner-sphere catalyst–persulfate complex generates high-valent metal-oxo species (M(IV/V)O). These species oxidize electron-rich substrates via a two-electron transfer, formally constituting an ETP where the metal-oxo acts as the electron acceptor. Consequently, reactions described as “ETP” are often kinetically mediated by a high-valent metal center. Sustained M(IV)O turnover requires efficient interfacial electron flow to regenerate the lower-valent metal and reactivate persulfate. Synergy occurs when (i) metal centers and adjacent heteroatoms or defects co-stabilize the catalyst–persulfate adduct, lowering the barrier for heterolytic cleavage, and (ii) conductive scaffolds shuttle electrons efficiently, minimizing radical leakage. Competition emerges when strong organic adsorption favors outer-sphere ETP over inner-sphere complex formation, or when high PMS doses induce homolytic cleavage, reducing the high-valent fraction. Contributions can be assigned using electrochemical transients (OCP/chronopotentiometry) in response to oxidant/pollutant pulses and the conversion of sulfoxide to sulfone (PMSO → PMSO2) in the absence of radical signatures.54
High-valent metal ↔ 1O2: convergent oxygen activation. Metal-oxo systems can co-generate 1O2 through several routes: (i) H˙/e− abstraction by M(IV)O forms reactive oxygen atoms that couple to 1O2; (ii)  side-products from high-valent cycles disproportionate to 1O2; and (iii) metal-vacancy ensembles facilitate parallel 1O2 and metal-oxo pathways. Catalysts such as Fe–Mn and Fe–N–C exhibit coexisting 1O2 and Fe(IV) signals and demonstrate strong tolerance to anions and NOM, consistent with complementary selectivity for electron-donating substrates. The pathways compete for oxidant and active sites: stabilizing the metal-oxo intermediate can depress the 1O2 yield, whereas high vacancy density with abundant dissolved O2 favors 1O2 but shortens M(IV) lifetimes. Deconvoluting these channels requires combined diagnostic techniques, including D2O kinetic studies, azide quenching, and time-resolved operando spectroscopy.55
side-products from high-valent cycles disproportionate to 1O2; and (iii) metal-vacancy ensembles facilitate parallel 1O2 and metal-oxo pathways. Catalysts such as Fe–Mn and Fe–N–C exhibit coexisting 1O2 and Fe(IV) signals and demonstrate strong tolerance to anions and NOM, consistent with complementary selectivity for electron-donating substrates. The pathways compete for oxidant and active sites: stabilizing the metal-oxo intermediate can depress the 1O2 yield, whereas high vacancy density with abundant dissolved O2 favors 1O2 but shortens M(IV) lifetimes. Deconvoluting these channels requires combined diagnostic techniques, including D2O kinetic studies, azide quenching, and time-resolved operando spectroscopy.55
4. Characteristics of non-radical pathways oxidation processes
Non-radical oxidation processes exhibit milder oxidative strength and greater controllability compared to free radical systems, facilitating selective pollutant degradation, enhanced persulfate utilization, and reduced formation of toxic by-products.
4.1. Selective oxidation
Free radical oxidation systems exhibit poor selectivity, often leading to competitive reactions between background organic matter and target pollutants. Consequently, oxidant dosages must be 10–100 times higher to maintain treatment efficiency (eqn (35)–(40)).56–58 In contrast, non-radical oxidation processes employ high-valent metals as electropositive oxidants with elevated redox potentials, selectively targeting electron-donating functional groups (e.g., formyl, amino, hydroxyl, and alkoxy) while showing minimal reactivity toward electron-withdrawing groups (e.g., acyl, carboxyl, nitro). Recent quantitative studies demonstrate that non-radical systems can achieve comparable or superior pollutant removal at substantially lower oxidant dosages. For example, in a Fe–N–C/peroxymonosulfate system, the non-radical pathway—dominated by high-valent Fe(IV) and 1O2—achieved 95% tetracycline degradation within 15 min using only 0.2 mM PMS, whereas a radical-dominated system required 1.0 mM PMS under identical conditions. The pseudo-first-order degradation rate constant for the non-radical system was 0.32 min−1, approximately 2.5 times higher than that of the radical system (0.13 min−1), highlighting the enhanced efficiency of non-radical pathways.59 Zhou et al. further demonstrated that an iron-hole-anchored layered double hydroxide (LDH)-activated PMS system transitioned from radical to non-radical pathways in SMX degradation, achieving a 30-fold increase in removal efficiency.60| | |
SO4˙− + HCO3− → CO3˙− + SO42− + H+
| (35) |
| | |
˙OH + HCO3− → CO3˙− + H2O
| (36) |
| | |
SO4˙− + CO32− → CO3˙− + SO42−
| (37) |
| | |
˙OH + CO32− → CO3˙− + OH−
| (38) |
| | |
SO4˙− + NO32− → NO3˙− + SO42−
| (39) |
| | |
˙OH + NO32− → NO3˙− + OH−
| (40) |
The selectivity of ETP is influenced by two factors: (i) the catalyst's adsorption capacity for organic substrates and persulfates, which is determined by molecular size, morphology, and surface hydrophobicity/hydrophilicity; and (ii) the efficiency of electron transfer, which is governed by the redox potential of the persulfate complex, the half-wave oxidation potential of the target molecule, and its electrochemical reversibility.61,62 Yan et al. demonstrated that Ni(II)-doped g-C3N4 (Ni4.60CN) catalysts effectively activate PMS, with Ni–N4–C active sites promoting electron transfer from phenol to PMS, ensuring highly selective degradation.63
1O2, an electrophilic species, exhibits strong reactivity toward electron-rich unsaturated pollutants. Studies have shown that 1O2 selectively degrades compounds such as phenols, sulfonamides, sulfides, thiourea, imidazoles, pyrroles, and indoles via electron transfer and electrophilic addition mechanisms.64 Peng et al. designed a nitrogen-oxygen co-doped, graphene-supported cobalt single-atom catalyst (Co–NO–GC) to activate PMS for 1O2 generation, achieving selective phenol degradation in complex aqueous environments.65
Nevertheless, the selectivity of non-radical oxidation should not be interpreted as an unconditional advantage. In some cases, highly selective pathways may preferentially transform specific electron-rich moieties or induce partial oxygenation/electron-transfer conversion without achieving deep oxidation, aromatic ring cleavage, or mineralization. As a result, partially oxidized intermediates may accumulate, and the apparent disappearance of the parent pollutant may not necessarily correspond to complete detoxification. Therefore, alongside degradation kinetics, future studies should pay greater attention to transformation-product profiling, TOC abatement, carbon mass balance, and toxicity evolution to distinguish selective conversion from truly beneficial contaminant removal.
4.2. Anti-interference oxidation
In free-radical PMS activation, SO4˙− forms concurrently with oxidation of Mn+ to M(n+1)+ (eqn (41)).2 To close the catalytic cycle, M(n+1)+ is reduced back to Mn+ by a second PMS molecule, generating SO5˙− (eqn (42)).14 Because SO5˙− has a low redox potential (E0 = 1.1 V vs. NHE), this branch shows poor oxidative performance and can divert a large fraction (≥50%) of PMS from productive oxidation.5 In contrast, in non-radical pathways a single PMS molecule can, in principle, accept two electrons from the substrate, improving oxidant utilization. Moreover, inorganic anions (e.g., Cl− and CO32−) and NOM strongly scavenge free radicals and consume oxidant, whereas non-radical oxidation is far less susceptible to interference by anions and NOM. Quantitative assessments further substantiate this advantage: in a single-atom Fe-doped carbon nitride/PMS system, the presence of 10 mM Cl− caused only a 5% decline in tetracycline degradation efficiency, whereas a radical-based system experienced a 62% reduction under identical conditions.57 Similarly, in a Co–N–C/PMS system, the degradation rate of bisphenol A remained above 90% even in the presence of 20 mM HCO3−, while the radical-based system showed a 45% decrease.65| | |
HSO5− + Mn+ → M(n+1)+ + SO4˙− + OH−
| (41) |
| | |
HSO5− + M(n+1)+ → Mn+ + SO5˙− + H+
| (42) |
4.3. Inhibition of toxic by-product formation
Halide ions in aqueous solutions react with free radicals to produce halogen radicals (e.g., Cl˙ and Br˙), resulting in the formation of toxic halogenated by-products (eqn (43)–(50)).1,31,66 In free radical-based AOPs, the degradation of NOM frequently generates harmful oxidation by-products, including haloacetic acids, trihalomethanes, haloketones, halogenated acetonitriles, halonitromethanes, and haloacetamides (eqn (51) and (52)).67–69 In contrast, non-radical persulfate oxidation processes suppress the formation of such by-products and significantly diminish the residual toxicity of organic pollutants. Wang et al. developed a single-atom iron-doped carbon nitride catalyst that activates PMS to generate non-radical species such as 1O2 and Fe(IV), facilitating efficient tetracycline degradation.70 In this system, the reaction rate remained largely unaffected by Cl− (0–100 mM), with virtually no toxic by-products detected. In contrast, a free radical-based system exhibited a 62% reduction in degradation rate upon Cl− addition (10 mM), resulting in the formation of chlorinated by-products such as TCM and trichloroacetic acid (TCAA).71 Quantitative analysis revealed that in the non-radical system, the total yield of halogenated by-products was below 5 µg L−1, whereas the radical system produced over 50 µg L−1 of TCM alone under identical conditions, underscoring the superiority of non-radical pathways in minimizing toxic by-product formation.72| | |
SO4˙− + Cl− → Cl˙ + SO42−
| (43) |
| | |
˙OH + Cl− → Cl˙ + OH−
| (44) |
| | |
Cl2˙− + RH → R˙ + 2Cl− + H+
| (46) |
| | |
SO4˙− + Br− → Br˙ + SO42−
| (47) |
| | |
˙OH + Br− → Br˙ + OH−
| (48) |
| | |
Br2˙− + NOM → Bromide products + Br−
| (50) |
| | |
SO4˙− + NOM → products
| (51) |
| | |
˙OH + NOM → products
| (52) |
Despite the advantages of non-radical pathways, achieving a fully non-radical reaction remains a significant challenge. The O–O bond in PMS has a relatively low dissociation energy and requires only a single electron to undergo homolytic cleavage, making free radical formation thermodynamically favorable and kinetically facile. This inherent tendency hinders the complete suppression of radical pathways and limits the transition to purely non-radical mechanisms.
4.4. Advantages and limitations
Non-radical persulfate activation exhibits several advantages: (i) selective oxidation of electron-rich moieties, minimizing over-oxidation of less reactive functional groups; (ii) efficient pollutant removal under relatively mild reaction conditions; (iii) reduced formation of halogenated or radical-derived by-products; and (iv) strong tolerance to coexisting anions, salts, and natural organic matter in water matrices. Nevertheless, several limitations should be noted: (i) some systems require relatively high persulfate dosages to achieve meaningful degradation, increasing chemical cost; (ii) very low concentrations of target pollutants may reduce the reaction efficiency due to limited interaction with catalytic sites; (iii) highly selective oxidation may lead to the accumulation of partially oxidized intermediates; and (iv) pathway selectivity and durability are strongly dependent on catalyst structure, requiring careful design and operando monitoring. Future studies should aim to optimize these trade-offs, balancing treatment efficiency, chemical usage, and environmental safety.
5. Recognition of non-radical pathways
Because multiple non-radical routes often coexist in persulfate systems, the term dominant non-radical pathway in this review is used in an operational rather than absolute sense, referring to the pathway that contributes the largest share of pollutant conversion under a defined set of reaction conditions and whose selective suppression leads to a decrease of more than 50% in the observed degradation rate or conversion. In practice, however, pathway dominance should not be assigned on the basis of a single indicator, because pathway contributions are system- and substrate-dependent and may not be strictly additive; instead, a weight-of-evidence approach should be adopted in which free-radical participation is first assessed by conventional quenching experiments and EPR analysis, followed by the verification of the target non-radical route using route-specific diagnostics, such as electrochemical tests and evidence for catalyst–persulfate complex formation for electron-transfer pathways, TEMP-EPR, D2O substitution and selective probe compounds for 1O2, and oxygen-atom-transfer probes combined with operando spectroscopy or isotope labelling for high-valent metal species. Accordingly, a non-radical route should be considered dominant only when these independent results are mutually consistent and inhibition of the proposed route causes a substantial (>50%) loss of degradation activity under otherwise identical conditions.
5.1. ETP recognition
ETP in heterogeneous catalysis occurs on the catalyst surface, and is primarily studied through electrochemical techniques. Methods such as linear sweep voltammetry (LSV), cyclic voltammetry (CV), chronoamperometry (CP), and OCP are commonly used to capture ETP in non-radical AOPs.17,73,74 Nevertheless, experimental conditions such as substrate concentration and catalyst loading can significantly affect the results.
LSV and CV are widely employed to investigate redox reactions in adsorption-induced interfacial catalytic systems. LSV reveals significant alteration in surface current density upon the introduction of oxidants and pollutants, indicating electron transfer between the catalyst and species. However, it cannot directly measure electron transfer between oxidants and organic compounds, necessitating its combination with CP to clarify the direction of electron transfer. CV assesses electrochemical reversibility and half-wave potential ((φ1/2) (Eps eqn (53) and (54))) of compounds, with oxidation rates determined by the half-wave potential of organic pollutants.17 Compounds with lower oxidation half-wave potentials are more susceptible to oxidation. OCP is used to identify ETP during oxidant addition. For example, Zhang et al. demonstrated a sharp OCP increase upon PMS addition, indicating surface complex formation. Subsequent naproxen addition led to a rapid OCP decline, signifying electron transfer.75,76 Similarly, Peng et al. used CNTs to activate persulfate for phenol degradation, observing a significant OCP change in the CNT-persulfate system.76 It should be noted that OCP can only reflect the trend of potential changes and cannot quantify the number of electron transfers. Therefore, it needs to be combined with other electrochemical methods to obtain more comprehensive mechanism information. In addition, electrochemical response is also influenced by various factors such as catalyst loading mode, background electrolyte composition, and system operation stability, which need to be comprehensively considered in data analysis.77 It is important to note that OCP can only reflect trends in potential changes and cannot quantify the number of electron transfers. Therefore, it should be combined with other electrochemical methods for a more comprehensive understanding of the mechanism. Furthermore, the electrochemical response is influenced by various factors, including catalyst loading mode, background electrolyte composition, and system operational stability, all of which must be considered in data analysis.
where
Epa and
Epc correspond to the anodic and cathodic peak potentials of a reversible system, respectively, while
Ep and
Ep/2 represent the peak and half-peak potentials of an irreversible system.
Density functional theory (DFT) is widely applied to resolve catalyst–pollutant electronic structure and elucidate ETP on a molecular scale. Under the usual sign convention, a more negative persulfate adsorption energy (Eads), an elongated O–O bond length (lO−O), and a moderate charge-transfer propensity (ΔQ) correlate with stronger adsorption and facilitate O–O activation, thereby initiating interfacial electron transfer.17 For example, dimethylformamide-doped g-C3N4 exhibits a more negative Eads value than that of pristine g-C3N4, consistent with its higher electron-transfer efficiency.77 Likewise, PMS binds more strongly at Co–N3 than at Co–N4 coordination sites; adsorption at Co–N3 promotes O–O bond weakening/cleavage and accelerates electron transfer.78 Most current studies screen static, ground-state descriptors using conventional DFT. Future work should integrate constant-potential ab initio molecular dynamics with explicit solvent/electrolyte and nonadiabatic electron-transfer simulations to capture transient interfacial charge transfer and activation barriers. These advances will enable quantitative prediction of ETP kinetics and mechanism-guided catalyst design.
It is important to distinguish the diagnostic roles of electrochemical evidence, isotope labeling, and scavenger experiments when interpreting non-radical pathways. Electrochemical methods (e.g., OCP, CV, LSV, and chronoamperometry) are particularly effective for revealing interfacial charge-transfer behavior, adsorption-induced redox changes, and catalyst–oxidant–pollutant coupling, but they are generally not species-specific and therefore cannot by themselves unambiguously differentiate direct electron transfer from oxidation mediated by 1O2 or high-valent metal species. In contrast, isotope-labeling techniques provide higher mechanistic specificity because they can trace the origin and transfer route of oxygen atoms, thereby offering stronger evidence for oxygen-transfer processes involving 1O2 or metal-oxo intermediates. Scavenger experiments are useful as rapid preliminary screening tools, but they remain indirect and may be affected by non-selective quenching, competitive adsorption, and kinetic perturbation. Therefore, electrochemical evidence combined with scavenger experiments is suitable for preliminary assignment of an interfacial non-radical regime, whereas electrochemical evidence combined with isotope labeling provides a more robust basis for mechanism confirmation. In practice, the most convincing mechanistic identification should rely on a hierarchy of evidence in which electrochemical measurements define interfacial electron flow, scavenger/probe tests screen possible pathways, and isotope labeling or operando spectroscopy provides decisive verification.
A particularly important unresolved issue is how to distinguish outer-sphere ETP from radical-mediated oxidation. Because both pathways may yield pollutant degradation without obvious accumulation of long-lived reactive intermediates, mechanistic assignment should not rely on a single diagnostic result. In principle, outer-sphere ETP is more convincingly supported when the following criteria are simultaneously satisfied: (i) dominant free-radical oxidation is systematically excluded by multiple complementary approaches, including but not limited to selective scavengers, spin-trapping/EPR, and control reactions; (ii) interfacial electron flow is directly evidenced by electrochemical transients (e.g., OCP, chronoamperometry, and CV) or equivalent galvanic-cell-type measurements showing catalyst-mediated charge transfer between the pollutant and persulfate; (iii) degradation kinetics and substrate selectivity correlate with oxidation potential, adsorption behavior, and catalyst–pollutant interfacial coupling rather than with conventional radical reactivity patterns; and (iv) product distribution, isotope-labeling results, operando spectroscopy, and theoretical calculations do not support dominant contributions from free radicals, 1O2, surface-bound radicals, or high-valent metal intermediates. Therefore, outer-sphere ETP should be regarded as a mechanism assigned by converging exclusionary and affirmative evidence, rather than by any single electrochemical, quenching, or probe-based test.
For ETP, dominance is more convincingly established when electrochemical evidence (e.g., OCP shift, chronoamperometric response, or current attenuation after pollutant addition) is accompanied by weak radical signatures, negligible D2O enhancement, and selective oxidation behavior consistent with substrate oxidation potential or electron-donating ability. In practice, OCP or current responses alone indicate interfacial electron flow, but dominance should only be assigned when these signals are consistent with kinetic inhibition results obtained after disrupting the catalyst–persulfate interfacial complex or blocking pollutant adsorption.
5.2. 1O2 recognition
Various methods are available for the detection of 1O2, including fluorescent probes, chemiluminescence probes, electron paramagnetic resonance (EPR), and D2O experiments. Fluorescent probes, such as 9-[2-(3-carboxy-9,10-diphenyl)anthryl]-6-hydroxy-3H-xanthen-3-one (DPAX) and 9-[2-(3-carboxy-9,10-dimethyl)anthryl]-6-hydroxy-3H-xanthen-3-one (DMAX), selectively detect 1O2 through [4 + 2] cycloaddition, resulting in the formation of stable endoperoxides that exhibit strong fluorescence at 530–550 nm when excited at 380–400 nm.79 Probe performance is pH-dependent, with fluorescence intensity decreasing under acidic conditions (pH < 6), necessitating precise pH control within the optimal range of 7–9.80
Chemiluminescent probes detect 1O2 by generating excited-state intermediates that emit photons upon decomposition. This method eliminates interference from excitation light and autofluorescence, thereby enhancing signal-to-noise ratios. Although firefly fluorescein analogs (CLA) are frequently employed, they exhibit limited specificity and react with ˙O2−, necessitating the co-administration of ˙O2− scavengers for selective 1O2 detection. Recent advancements include ethylene oxide-functionalized probes for ultrasensitive 1O2 detection, achieving a limit of 5 × 10−13 mol L−1, which holds promise for real-time environmental monitoring.81,82
EPR spectroscopy is extensively utilized for the indirect detection of 1O2 in AOPs due to its simplicity and sensitivity. 1O2 interacts with spin-trapping agents, such as TEMP, to form a stable TEMP-1O2 adduct, which produces a characteristic three-line EPR signal.83 Liu et al. identified this signal in the Fe–PAC/PMS system, thereby confirming 1O2 as the predominant ROS.84 The intensity of the signal exhibits a linear correlation with 1O2 concentration, facilitating quantitative analysis. However, despite its advantages, EPR-based 1O2 detection is highly susceptible to false positives. For instance, TEMP can be oxidized to TEMPO (yielding the same three-line signal) by direct electron transfer, high-valent metal species, or under alkaline conditions, rather than by 1O2 itself. To rigorously exclude these false positives, researchers must employ a multi-evidence approach. This includes conducting control experiments with specific 1O2 scavengers (e.g., L-histidine, furfuryl alcohol, or sodium azide) to observe signal quenching, utilizing the D2O solvent isotope effect to observe signal enhancement, and cross-validating EPR results with highly specific fluorescent probes or direct near-infrared phosphorescence at 1270 nm.
D2O enhances the half-life of 1O2, thereby improving its oxidative capacity and making it a diagnostic solvent for verifying 1O2-mediated degradation. Luo et al. used D2O to demonstrate dominant 1O2 activity in non-radical Fenton-like systems by achieving a 38% higher bisphenol A degradation rate than that of H2O, confirming 1O2 involvement in pollutant removal.85 Future technological developments should focus on creating novel molecular probes with high specificity, stability, and minimal environmental interference, as well as promoting the integration of multiple detection methods (e.g., EPR and fluorescence probes) to establish reliable 1O2 identification techniques.
It should be emphasized that 1O2 dominance should not be concluded from one probe or one quencher alone. Recent studies have shown that the contribution of 1O2 in PMS activation can be overestimated when based solely on furfuryl alcohol consumption, TEMP-EPR, or isolated scavenging results. Therefore, 1O2 should be considered dominant only when multiple independent observations, such as D2O-enhanced degradation, characteristic TEMP-derived signals, and selective inhibition by 1O2 quenchers, all converge and jointly account for the majority of pollutant removal.
5.3. High-valent metal recognition
Methods for identifying high-valent metals are classified as direct or indirect. Direct detection employs advanced techniques, such as Mössbauer spectroscopy, X-ray absorption fine structure (XAFS), CV, and in situ Raman spectroscopy (Fig. 7).41 Indirect methods use chemical probes that selectively react with high-valent metal-oxo species, generating identifiable products.
 |
| | Fig. 7 Four non-radical oxidation pathways in the electron transfer mechanism of a persulfate/catalyst system. Reprinted with permission from ref. 3, 43, 58 and 86 respectively. Copyright 2021, 2019, 2023 and 2024, Elsevier, respectively. | |
XAFS includes XANES and extended X-ray absorption fine structure (EXAFS).87 XANES reveals oxidation states, bonding environments, and coordination geometry, while EXAFS determines atomic identities, coordination numbers, and bond lengths. In the α-Fe2O3/PMS system, Kang et al. observed energy shifts in XANES spectra, thereby confirming the Fe(IV) formation on the catalyst surface.3 Li et al. identified a Raman-active vibrational mode associated with Fe(IV) centers, correlating its intensity with persulfate concentration.58 Mössbauer spectroscopy complements these findings by quantifying hyperfine parameters, offering insights into valence states and spin configurations. CV detects transient high-valent metal species through redox signatures.86,88
Sulfoxides, such as methyl p-tolyl sulfoxide (TMSO), dimethyl sulfoxide (DMSO), PMSO, and diphenyl sulfoxide (DPSO), are selective probes for high-valent metals, undergoing two-electron oxidation to form sulfones, distinct from single-electron radical oxidation.89 PMSO is particularly useful in non-radical Fenton-like processes due to its ultrasensitive detection via high-performance liquid chromatography (HPLC). Zong et al. confirmed Co(IV) generation in the Co(II)/PMS system by observing quantitative conversion of PMSO to PMSO2.48 However, it is critical to note that sulfoxide probes are not fully selective under all conditions. Recent studies have cautioned that PMSO can be directly oxidized by PMS/PDS at slow rates or by specific reactive intermediates like carbon-centered radicals, potentially leading to the overestimation of high-valent metal contributions.87,88 For instance, in systems with high halide concentrations, the formation of reactive halogen species may also interfere with the PMSO → PMSO2 transformation. The 18O isotope labeling technique facilitates the identification of high-valent metal species by exploiting oxygen transfer mechanisms. Fe(IV)-oxo complexes undergo oxygen exchange with H218O, resulting in the formation of Fe(IV)–18O2+, which subsequently reacts with PMSO to produce 18O-labeled sulfoxide.90 Wang et al. demonstrated this method in the Fe(II)-activated persulfate system, confirming the intermediacy of Fe(IV) through the formation of PMSO18O.41,86 Therefore, the use of sulfoxide probes should ideally be cross-validated with isotopic labeling (18O-exchange) or spectroscopic evidence (e.g., XANES) to ensure the accuracy of high-valent metal identification.43,89
Future research should focus on in situ characterization techniques with higher temporal resolution, such as ultrafast XAFS and transient Raman spectroscopy, to effectively capture the dynamic generation and evolution of these transient high-valent intermediates. Furthermore, the integration of multidimensional techniques—such as chemical probes with isotope labeling and spectroscopic analysis—alongside theoretical calculations will be essential in addressing the limitations of individual methods, facilitating precise quantification of reaction pathway contributions, and ultimately elucidating the true chemical forms and reaction mechanisms.
For high-valent metal species, pathway dominance should be assigned only when chemical-probe results are corroborated by spectroscopic or isotopic evidence. In practice, quantitative or near-quantitative conversion of PMSO to PMSO2 is informative, but it should be interpreted together with operando XANES/EXAFS, Mössbauer or Raman spectroscopy, and, where possible, H218O isotope-labeling experiments, because probe oxidation alone may not fully exclude side reactions. Therefore, a high-valent-metal pathway is regarded as dominant only when these complementary data collectively support that metal-oxo species account for the majority of pollutant oxidation under the given conditions.90–92
Given the well-recognized limitations of individual diagnostic methods, future studies should move toward standardized multi-method validation protocols for confirming non-radical mechanisms. In such a framework, no single technique should be regarded as definitive evidence on its own. Instead, mechanistic assignment should follow a hierarchical workflow: (i) first exclude dominant radical pathways using selective radical quenchers, EPR/spin-trapping, and control experiments; (ii) then identify candidate non-radical routes through pathway-relevant indicators, such as electrochemical transients for interfacial electron transfer, D2O enhancement and selective probes for 1O2, and sulfoxide oxidation signatures for high-valent metal-oxo species; (iii) subsequently apply higher-specificity tools, including isotope labeling and operando/in situ spectroscopy, to verify atom origin, oxygen-transfer pathways, and transient intermediate identity; and (iv) finally cross-check the mechanistic conclusion against substrate selectivity, product distribution, oxidant utilization efficiency, and theoretical calculations.93,94 Such a standardized protocol would improve the robustness, comparability, and reproducibility of non-radical pathway identification across different catalyst systems and laboratories.
Likewise, the dominance of high-valent metal pathways should not be inferred solely from sulfoxide oxidation products. Although PMSO-to-PMSO2 conversion remains a useful indicator of oxygen-atom-transfer reactivity, recent work suggests that PMSO may participate more actively than previously assumed in some PMS-based systems. Therefore, assignment of a dominant high-valent metal pathway should ideally be supported by complementary evidence, such as operando XANES/Raman/Mössbauer spectroscopy or 18O isotope-labeling analysis, together with kinetic suppression experiments.
6. Design and optimization of catalysts
To enhance the application relevance of this section, Table 3 summarizes the major classes of organic contaminants amenable to persulfate-based non-radical oxidation, together with their representative catalytic systems and predominant non-radical pathways. Overall, non-radical oxidation is generally more effective for compounds containing electron-donating substituents, activated aromatic rings, or favorable interfacial adsorption and electron-transfer properties, whereas structurally inert contaminants often require more specifically designed catalysts or synergistic pathways for efficient oxidation.
Table 3 Major classes of organic contaminants amenable to persulfate-based non-radical oxidation and their representative catalytic systems
| Catalysts |
Oxidant |
Non-radical pathway |
Contaminant |
Removal |
References |
| MnOX |
PMS |
1O2 |
SMX |
97% |
91 |
| N-PC |
PMS |
ETP, 1O2 |
Naproxen |
98.8% |
92 |
| NixCu-LDHs |
PMS |
1O2 |
Methyl 4-hydroxybenzoate |
99.07% |
95 |
| NCNTs |
PMS |
1O2 |
Phenol |
100% |
96 |
| Cu-FVvac-LDH |
PMS |
Fe(IV), Fe(V) |
SMX |
98% |
60 |
| δ-MnO2 |
PMS |
ETP |
BPA |
90% |
97 |
| Single cobalt atoms |
PMS |
1O2 |
Sulfisoxazole |
90% |
98 |
| 0.8 N@FeCS |
PDS |
1O2, ETP |
Ciprofloxacin |
100% |
99 |
| Fe-based N-doped carbon catalysts |
PMS |
1O2, Fe(V) |
Tetracycline |
100% |
100 |
| Fe3C/FeN |
PDS |
ETP |
Sulfadiazine |
99% |
101 |
| N/B co-doped biochars |
PDS |
ETP |
Tetracycline |
85.5% |
102 |
| COCN |
PDS |
1O2 |
ACT |
99.9% |
103 |
| CNClS |
PMS |
1O2 |
Tetracycline |
95% |
104 |
| N/P/S co-doping of Fe-SACs |
PMS |
Fe(V) |
Ofloxacin |
100% |
105 |
| N-doped graphene (N-G) |
PMS |
1O2 |
Phenol |
82% |
106 |
6.1. Modification of catalyst surface characteristics
Unlike free-radical oxidation, which occurs largely in the aqueous phase, non-radical oxidation is predominantly surface-confined. Consequently, catalyst physicochemical properties govern pathway selectivity and kinetics. Persulfate can be steered toward non-radical oxidants by engineering defect sites (e.g., oxygen, sulfur, nitrogen, and iodine vacancies), applying chemical etching, creating cation vacancies, and tailoring surface chemistry and exposed facets (Fig. 8).22
 |
| | Fig. 8 Surface physicochemical properties of modified catalysts enabling non-radical oxidation pathways: (a) defect sites, (b) chemical etching, (c) cationic vacancies, and (d) surface properties. Reprinted with permission from ref. 60, 93, 96 and 99 respectively. Copyright 2023 Elsevier, 2022 Elsevier, 2013 Elsevier and 2024 Elsevier, respectively. | |
Defect sites can be introduced through temperature control, atomic substitution, and etching. Xie et al. found that the concentration of OVs increased when the synthesis temperature of amorphous MnO2 increased from 120 °C to 180 °C, and the non-radical activity of catalysts to activate persulfate has correspondingly improved.93 Similarly, Wang et al. observed that the decrease in nitrogen doping in N co-doped porous carbon (N-PC) 1000 due to nitrogen loss during annealing could generate additional vacancies, thereby enhancing persulfate activation.92 Metal doping also facilitates vacancy formation. Zhu et al. showed that copper-doped nickel-based LDHs possessed abundant OVs, leading to more efficient persulfate activation.95
Chemical etching, such as plasma treatment, leaches unstable surface atoms, resulting in the generation of surface defects. Liu et al. synthesized NCNTs with abundant vacancies via plasma etching, and the generation of 1O2 was promoted through oxygen adsorption and OOH*/˙O2− intermediates, and electron trapping facilitated valence state transitions.96 In hydrothermally synthesized amorphous MnOx, the 1O2 yield scales with oxygen-vacancy density, vacancies enable heterolytic persulfate cleavage in which persulfate adsorbed at vacancies reacts with dissolved persulfate to form 1O2.91 Sulfur vacancies in chalcopyrite similarly drive persulfate oxidation without radical intermediates.
Cationic vacancies, such as missing metal ions, also promote non-radical pathways. Zhou et al. reported that iron vacancies in Fe-hole-anchored LDHs served as active sites for persulfate adsorption.60 Adjacent Fe(II) centers form Fe(II)/persulfate complexes that facilitate S–O bond cleavage and generate high-valent iron species (Fe(IV)O or Fe(V)O) of capacity to oxidize organic contaminants like SMX via non-radical mechanisms.
The surface properties of catalysts significantly influence the oxidation pathways. In carbon nanotube materials, persulfate acts as an electron acceptor, binding to sp2-hybridized carbon or ketone (CO) sites and then forming active surface complexes. These complexes help transfer electrons, promoting acetaminophen (ACT) degradation. In metal-based catalysts, exposed crystal planes also influence the oxidation process. Zeng et al. found that exposing the (−111) plane of boromagnesite (δ-MnO2) allowed BPA degradation via electron transfer in the SN-KMO/PMS system at low PMS dosages.97 MnO2 crystal facets also influence non-radical oxidation pathways, with different facets exhibiting distinct behaviors. For example, the 310-M/PMS system facilitates Mn(III) formation, which removes electrons from phenol, while the 110-M/PMS system generates 1O2 via single-electron reduction of O2.66,98
It should be emphasized that defect sites are not necessarily static under persulfate activation conditions. During long-term operation, oxygen/sulfur vacancies, edge defects, and surface oxygen-containing groups may undergo formation, annihilation, healing, re-oxidation, or interconversion because of repeated redox cycling, oxidant attack, adsorbate binding, and matrix-induced surface reconstruction. Such dynamic defect evolution can alter persulfate adsorption strength, interfacial charge distribution, and O–O bond activation mode, thereby shifting the relative contribution of electron-transfer pathways, 1O2 generation, and high-valent metal species, and in some cases even inducing radical leakage. Therefore, defect engineering should be considered not only from a static structure–activity perspective, but also from a time-dependent structure–stability–pathway perspective under realistic operating conditions.
6.2. Adjusting the size of catalyst active sites
In heterogeneous catalysis, the size of the active site dictates activity and selectivity. This size effect is pronounced for supported metals: decreasing particle size increases specific surface area and reshapes the distribution and structure of surface active centers, thereby enhancing performance.
Precise modulation of parameters such as metal precursor concentration, synthesis temperature, and post-synthesis processing controls metal atom aggregation on the support, thus reducing the metal active site size.99 Lower metal precursor concentrations enhance metal dispersion, promoting the formation of SACs, while excessive precursors lead to aggregation into nanoclusters or nanoparticles. Elevated pyrolysis temperatures increase the thermal mobility of metal atoms, facilitating aggregation. Acid etching can remove unstable nanoclusters, which promotes the transformation of nanoparticles into single atoms. Wu et al. demonstrated these strategies by synthesizing cobalt single atoms, clusters, and nanoparticles via varying the Zn/Co ratio (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 4:1, and 2:1, respectively) (Fig. 9a and b).100 Additionally, Li et al. showed that reducing the size of Fe active centers shifts the reaction mechanism from synergistic oxidation involving both free and non-free radicals to complete non-radical oxidation (Fig. 9c).101
:1, 4:1, and 2:1, respectively) (Fig. 9a and b).100 Additionally, Li et al. showed that reducing the size of Fe active centers shifts the reaction mechanism from synergistic oxidation involving both free and non-free radicals to complete non-radical oxidation (Fig. 9c).101
 |
| | Fig. 9 (a) EPR spectra of 1O2 capture by TEMP for the CoSA, CoAC, and CoNP systems. (b) Relative intensity distribution of radical and non-radical mechanisms in the CoSA, CoAC, and CoNP systems. (c) Proportion of 1O2 in different metal–N/C catalyst-activated PMS systems. (d) Relationship between Fe active centers and non-radical pathways. Reprinted with permission from ref. 100 and 101, respectively. Copyright 2023, the American Chemical Society and Copyright 2024, Elsevier, respectively. | |
SACs preferentially activate persulfate for pollutant degradation via non-radical pathways. Atomically dispersed metal sites form covalent bonds with nitrogen or carbon, resulting in metal–N/C coordination structures that act as active sites. Gao et al. synthesized a series of metal–N–C SACs (M = Fe, Co, Mn, Ni, and Cu) to assess catalytic activities and mechanisms. Among them, Fe–SAC exhibited the highest single-site kinetic value, with catalytic activity in the following order: Fe-SAC > Co-SAC > Mn-SAC > Ni-SAC > Cu-SAC (Fig. 9d).100 Quencher experiments and EPR analysis identified 1O2 as the main ROS. DFT revealed that the optimal pathway for 1O2 generation is PMS → OH* → O* → 1O2, with Fe–SAC displaying superior catalytic activity due to moderate Gibbs free energies at each reaction step.102
The metal particle size also influences the adsorption energy of persulfate. Persulfate adsorption on SACs is weaker than that on metal particles, making the O–O bond cleavage less favorable, thus preventing the generation of ˙OH and SO4˙− via O–O bond elongation and scission. Consequently, SACs primarily mediate pollutant oxidation via non-radical pathways.107 Huang et al. calculated persulfate adsorption energies on cobalt single-atom catalysts (Co-SAC, −6.475 eV), cobalt nanocluster catalysts (Co-NCC, −8.170 eV), and cobalt nanoparticle catalysts (Co-NPC, −11.700 eV). The shortest O–O bond was observed on Co-SAC, favoring 1O2 generation.99
Although single-atom catalysts are often described as structurally uniform active centers, their local coordination environments may still evolve during reaction. Under long-term operation, isolated metal atoms can experience valence fluctuation, ligand exchange, coordination-number variation, partial migration, aggregation into subnanoclusters, or metal leaching, especially under strong oxidizing conditions and repeated interfacial electron transfer. These structural changes may modify the adsorption mode of PMS/PDS, the degree of O–O bond polarization, and the electron density of the metal center, thereby changing the dominant non-radical route from direct electron transfer to 1O2 generation or high-valent metal-oxo chemistry, or vice versa. Consequently, the practical design of SACs should emphasize not only initial activity and selectivity, but also anchoring strength, anti-migration/anti-leaching ability, and operando stability of the active coordination structure.
6.3. Non-metallic doping
Non-metallic heteroatoms (N, S, O, P, Cl, and B) can modulate the electronic structure and physicochemical properties of catalysts, then influencing non-radical pathways. Nitrogen-doped biochar materials have been widely used to enhance non-radical generation. Nitrogen doping primarily affects spin density and charge distribution within the carbon lattice and alters morphology and defect density, which can largely increase active sites.108
The impact of heteroatom doping is system-dependent and varies with the type and concentration of the heteroatom. For example, nitrogen doping enhances persulfate adsorption in PMS systems, while its effect is less pronounced in PDS systems. Dou et al. found that nitrogen- and boron-cobalt-doped biochar materials enhanced electron transfer to the highest occupied molecular orbital (HOMO) of metastable biochar/PDS complexes, promoting non-radical degradation.109 Tuan Nguyen et al. showed that PMS adsorption was accelerated on carbon- and oxygen-cobalt-doped graphite carbon nitride (COCN), promoting oxygen generation and electron transfer, leading to superior paracetamol (ACT) degradation compared to COCN/PDS.103 Xu et al. demonstrated that doping with Cl, S, and N atoms sequentially converted dominant reactive species (CN: SO4˙− and ˙OH → CNS: ˙O2− → CNCl: 1O2 → CNClS: 1O2), with nitrogen species and lattice defects playing crucial roles.104
In metal-based catalysts, heteroatom doping exerts similar yet amplified effects owing to unsaturated coordination and isolated single-atom centers. N/P/S co-doping of Fe-SACs enables the formation of high-valent FeO at Fe–N4 sites for efficient ofloxacin oxidation; DFT attributes the activity gains to the dopant-induced tuning of the Fe–N4 electronic structure.105 Both dopant identity and concentration are critical regulators of pathway selectivity. For instance, N-doped graphene (N-G) predominantly activates PMS via non-radical routes, whereas additional S dopants in S, N-G shift activity toward radical pathways. Likewise, excessive S incorporation in Co nanorods suppresses interfacial electron transfer and lowers 1O2 yields.106
7. Conclusions
The design of catalysts for non-radical persulfate activation is crucial for enhancing the efficiency and selectivity of AOPs. However, current methods for identifying and characterizing active species in non-radical AOPs remain immature, leading to ongoing debates regarding their reaction mechanisms and pathways. While non-free radical AOPs are generally perceived to lower oxidative potential compared to conventional radical-based AOPs, this conclusion is based on limited experimental evidence and lacks of systematic quantitative evaluation. Additionally, the selective degradation capability of non-free radical AOPs is contested. Although selectivity is typically attributed to non-radical pathways, the structural diversity of target pollutants in real wastewater raises doubts about the practical advantages of non-free radical AOPs under complex environmental conditions. These controversies highlight the incomplete understanding of non-free radical AOP mechanisms and the potential of non-radical species to drive novel, selective degradation pathways.
To address these challenges, future development of non-free radical AOPs should focus on the following areas:
(I) In-depth study of non-radical oxidation mechanisms of organic pollutants: mechanism-resolved studies are essential for understanding organic-substrate oxidation. Progress requires operando/in situ spectroscopy (e.g., ATR-IR, Raman, XAS, and XPS), isotopic labeling (e.g., 18O and 2H), and multi-scale theory integrating constant-potential ab initio molecular dynamics (MD), explicit solvent/electrolyte models, and nonadiabatic electron-transfer calculations. These tools will help quantify intermediates, map reaction networks, and compute activation barriers under realistic interfacial conditions. In addition, future mechanism-resolved studies should explicitly address the trade-off between selective transformation and deep detoxification. Because non-radical pathways may favor functional-group-selective oxidation, researchers should evaluate whether pollutant removal is accompanied by the accumulation of partially oxidized intermediates. Accordingly, parent-compound conversion should be complemented by LC-MS/MS product analysis, TOC mineralization, carbon balance, and bioassay-based toxicity evolution to assess whether selective oxidation leads to genuine detoxification or merely to intermediate transformation.
(II) High-sensitivity, selective detection and quantification of non-radical oxidants: current techniques such as EPR, XPS, and conventional molecular probes suffer from cross-reactivity and limited sensitivity. The adoption of in situ mass spectrometry, high-resolution NMR, time-resolved optical methods (e.g., direct 1270 nm phosphorescence of 1O2), and validated chemoselective probes—along with kinetic calibration and reporting of detection/quantification limits (LoD/LoQ)—will improve speciation and quantification accuracy. In addition, standardized multi-method validation protocols and reference reactions should be established for consistent cross-laboratory comparisons. A practical protocol should include radical exclusion tests, preliminary pathway screening, isotope-labeling and operando spectroscopic confirmation, and cross-validation using product analysis, selectivity patterns, and theoretical calculations. Such a hierarchy of evidence would reduce false-positive mechanistic assignments and provide a more reproducible basis for distinguishing electron-transfer pathways, 1O2, and high-valent metal routes.
(III) Stable and selective catalysts with minimized radical leakage: in saline or high-chloride environments, radical pathways can generate harmful halogenated by-products. Practical deployment therefore requires catalysts that not only favor non-radical persulfate activation initially but also maintain this pathway during prolonged operation. Future studies should focus on the time-dependent evolution of defect sites and single-atom coordination structures, as vacancy healing/reconstruction, active-site oxidation-state fluctuations, atom migration/aggregation, and metal leaching may induce pathway drift among electron transfer, 1O2, and high-valent metal routes, or even trigger partial radicalization. Long-duration cyclic and continuous-flow experiments, coupled with operando XAS/XANES, EPR, Raman, Mössbauer spectroscopy, and online electrochemical monitoring, are essential to establish direct correlations between active-site evolution and pathway stability. Integrating artificial intelligence/machine learning (AI/ML) with microkinetic modeling will enable mechanism-informed, multi-objective optimization of activity, selectivity, and stability, accelerating the development of deployment-ready non-radical AOP catalysts.
Author contributions
Xin Yang: formal analysis, investigation, methodology, data curation, writing – original draft. Jiping Cao: supervision, methodology, formal analysis. Tao Song: supervision, writing – review & editing. Xingyun Hu: formal analysis; validation. Linghao Kong: formal analysis; validation. Zhilin Xia: project administration, resources, supervision, writing – review & editing, funding acquisition.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability
No new data were generated or analyzed in this study. All information presented in this article is derived from previously published literature, and the sources are cited within the manuscript.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (52500107), Natural Science Basic Research Program of Shaanxi (Program: 2025JC-YBQN-372). We are also grateful to the editors and anonymous reviewers for their valuable comments and suggestions for our paper.
References
- Q. Zou, Q. Shi, M. Cheng, G. Wang, W. Wang, Y. Chen, A. Chen and Y. Ma, Boosting H2O2 activation by Cu and Ce co-doped g-C3N4 for pollutants removal: From the free radical pathway to the non-free radical pathway, J. Environ. Chem. Eng., 2024, 12, 114808 CrossRef CAS.
- X. Bai, C. Li, S. He and J. Zhou, Non-free radicals based advanced oxidation processes: Research progress and future prospects, Chem. Eng. Res. Des., 2024, 211, 21–34 CrossRef CAS.
- H. Kang, D. Lee, K.-M. Lee, H.-H. Kim, H. Lee, M. Sik Kim and C. Lee, Nonradical activation of peroxymonosulfate by hematite for oxidation of organic compounds: A novel mechanism involving high-valent iron species, Chem. Eng. J., 2021, 426, 130743 CrossRef CAS.
- L. Qin, W. Zhu, L. Yang, M. Zheng and G. Liu, Persistent free radicals in the environment, J. Hazard. Mater., 2025, 493, 138332 CrossRef CAS PubMed.
- L. Wu, Y. Guan, C. Li, L. Shi, S. Yang, B. Rajasekhar Reddy, G. Ye, Q. Zhang, R. K. Liew, J. Zhou, R. Vinu and S. S. Lam, Free-radical behaviors of co-pyrolysis of low-rank coal and different solid hydrogen-rich donors: A critical review, Chem. Eng. J., 2023, 474, 145900 CrossRef CAS.
- Z. Wang, L. Huang, Y. Wang, X. Chen and H. Ren, Activation of peroxymonosulfate using metal-free in situ N-doped carbonized polypyrrole: A non-radical process, Environ. Res., 2021, 193, 110537 CrossRef CAS.
- T. Zhang, Y. Chen, Y. Wang, J. Le Roux, Y. Yang and J.-P. Croué, Efficient peroxydisulfate activation process not relying on sulfate radical generation for water pollutant degradation, Environ. Sci. Technol., 2014, 48, 5868–5875 CrossRef CAS.
- L. Zhu, H. Wang, J. Sun, L. Lu and S. Li, Sulfur vacancies in pyrite trigger the path to nonradical singlet oxygen and spontaneous sulfamethoxazole degradation: Unveiling the hidden potential in sediments, Environ. Sci. Technol., 2024, 58, 6753–6762 CrossRef CAS PubMed.
- C. Wang, Y. Zou, X. Wu, X. Zhang, W. Sang, T. Zhou and M. Huang, Citrate-ball milled Cu-Fe oxides activate O2 triggering selective non-radical mechanism: A boosted strategy coupling oxygen vacancies with surface carboxyl, Chem. Eng. J., 2025, 518, 164580 CrossRef CAS.
- X. Di, X. Zeng, T. Tang, D. Liu, Y. Shi, W. Wang, Z. Liu, L. Jin, X. Ji and X. Shao, Non-radical activation of peroxymonosulfate by modified activated carbon for efficient degradation of oxytetracycline: Mechanisms and applications, Sep. Purif. Technol., 2024, 349, 127877 CrossRef CAS.
- F. Dong, C. Fu, M. Feng, D. Wang, S. Song, C. Li, E. Lichtfouse, J. Li, Q. Lin and V. K. Sharma, Simultaneous generation of free radicals, Fe(IV) and Fe(V) by ferrate activation: A review, Chem. Eng. J., 2024, 481, 148669 CrossRef CAS.
- C. Li, L. Kang, S. Wang, B. Gao, X. Gao, W. Shi, S. Ai, J. Wu and Y. Sun, Enhanced PFOA degradation via non-radical PMS pathway through synergistic Fe single-atoms and Fe nanoparticles in Fe–N–C catalysts, Chem. Eng. J., 2025, 512, 162424 CrossRef CAS.
- H. Ming, X. Bian, J. Cheng, C. Yang, Y. Hou, K. Ding, J. Zhang, M. Anpo and X. Wang, Carbon nitride with a tailored electronic structure toward peroxymonosulfate activation: A direct electron transfer mechanism for organic pollutant degradation, Appl. Catal., B, 2024, 341, 123314 CrossRef CAS.
- H. Tian, C. Li, Z. Wang, S. Zhao, Y. Xu and S. Wang, Polycyclic aromatic hydrocarbons degradation mechanisms in methods using activated persulfate: Radical and non-radical pathways, Chem. Eng. J., 2023, 473, 145319 CrossRef CAS.
- H. Wang, H. Xu, W. Cheng, Y. Luo, T. Zhang and J. Ma, Trace Cu(II) as an efficient electron-transfer shuttle for reductive deiodination of refractory iodinated contrast media compounds, Environ. Sci. Technol., 2024, 58, 3495–3505 CAS.
- L. Wang, Q. Li, C. Xu, Y. Fu, Y. Tang, P. Wang, Z. Zhang, Y. Xia, X. Liu, J. Cao, S. Qiu, Y. Xue, J. Chen and Z. Wang, Phosphate-mediated degradation of organic pollutants in water with peroxymonosulfate revisited: Radical or non-radical oxidation?, Water Res., 2024, 255, 121519 CrossRef CAS PubMed.
- Y. Yan, Z. Wei, X. Duan, M. Long, R. Spinney, D. D. Dionysiou, R. Xiao and P. J. J. Alvarez, Merits and limitations of radical vs. nonradical pathways in persulfate-based advanced oxidation processes, Environ. Sci. Technol., 2023, 57, 12153–12179 CrossRef CAS PubMed.
- J. Cai, M. Zhou, Q. Zhang, Y. Tian and G. Song, The radical and non-radical oxidation mechanism of electrochemically activated persulfate process on different cathodes in divided and undivided cell, J. Hazard. Mater., 2021, 416, 125804 CrossRef CAS.
- Y. Wu, D. Huang, D. Li, X. Qian and J. Niu, From ambiguity to application: Deciphering non-radical pathways in heterogeneous persulfate activation, Adv. Mater., 2026, 38, 16166 CrossRef.
- G. Yao, X. Zhou, H. Gao, T. Liu, Y. Zhang and J. Chen, Peracetic acid-driven advanced oxidation processes for wastewater treatment: Demystifying organic radicals and non-radical species, Crit. Rev. Env. Sci. Tech., 2025, 55, 1124–1147 CrossRef CAS.
- J. Liu, J. Huang, M. Lin, X. Zheng, K. Zhao and H. Hu, Electron-transfer management at carbon–iron–oxidant interfaces: carbon architectures bridging radical/non-radical pathways for selective, self-sustained advanced oxidation, Environ. Sci. Nano, 2026, 13, 748–778 RSC.
- W. Ren, C. Cheng, P. Shao, X. Luo, H. Zhang, S. Wang and X. Duan, Origins of electron-transfer regime in persulfate-based nonradical oxidation processes, Environ. Sci. Technol., 2022, 56, 78–97 CrossRef CAS.
- L. Shi, Y. Li, H. Dong, J. Sun, C. Xia, L. Cai and B. Hui, Heterostructural Cu2S@CuO nanoarrays enable peroxymonosulfate activation for sulfamethoxazole degradation through non-free radical pathways, Desalination, 2025, 600, 118527 CrossRef CAS.
- L. Zhang, H. Ma, Y. Li, Z. Pan, Y. Xu, G. Wang, X. Fan, S. Zhao, H. Lu and C. Song, Activating peroxymonosulfate with MOF-derived NiO-NiCo2O4/titanium membrane for water treatment: A non-radical dominated oxidation mechanism, J. Colloid Interface Sci., 2024, 676, 1032–1043 CrossRef CAS.
- M. Mou, Y. Wang, W. Yu, H. Jiang, S. Zhang, Y. Zhao, J. Ma, L. Yan, X. Kong and X. Zhao, General design of self-supported Co-Ni/nitrogen-doped carbon nanotubes array for efficient oxygen evolution reaction, J. Colloid Interface Sci., 2025, 685, 988–997 CrossRef CAS.
- Y. He, H. Qin, Z. Wang, H. Wang, Y. Zhu, C. Zhou, Y. Zeng, Y. Li, P. Xu and G. Zeng, Fe-Mn oxycarbide anchored on N-doped carbon for enhanced Fenton-like catalysis: Importance of high-valent metal-oxo species and singlet oxygen, Appl. Catal., B, 2024, 340, 123204 CrossRef CAS.
- Y. Fang, W. Zhou, Y. Zhang, Y. Cheng, K. Zhang, Z. Xia and S. Song, Activation of peroxymonosulfate for sulfamethoxazole degradation by Fe-Mn bimetallic catalysts prepared based on steel pickling waste liquid: 1O2 and electron transfer dominated non-radical mechanism, J. Environ. Chem. Eng., 2025, 13, 117746 CrossRef CAS.
- Y. Yao, C. Wang, X. Yan, H. Zhang, C. Xiao, J. Qi, Z. Zhu, Y. Zhou, X. Sun, X. Duan and J. Li, Rational regulation of Co–N–C coordination for high-efficiency generation of 1O2 toward nearly 100% selective degradation of organic pollutants, Environ. Sci. Technol., 2022, 56, 8833–8843 CrossRef CAS PubMed.
- Y. Zhou, J. Jiang, Y. Gao, J. Ma, S.-Y. Pang, J. Li, X.-T. Lu and L.-P. Yuan, Activation of peroxymonosulfate by benzoquinone: A novel nonradical oxidation process, Environ. Sci. Technol., 2015, 49, 12941–12950 CrossRef CAS PubMed.
- Y. Wang, J. Zheng, T. Zhou, Q. Zhang, M. Feng and S. Zhang, Confinement-modulated singlet-oxygen nanoreactors for water decontamination, Environ. Sci. Technol., 2025, 59, 6341–6351 CrossRef CAS PubMed.
- X. Zhou, Q. Zhao, J. Wang, Z. Chen and Z. Chen, Nonradical oxidation processes in PMS-based heterogeneous catalytic system: Generation, identification, oxidation characteristics, challenges response and application prospects, Chem. Eng. J., 2021, 410, 128312 CrossRef CAS.
- M. Cheng, Y. Zhang, B. Lai, L. Wang, S. Yang, K. Li, D. Wang, Y. Wu, G.-H. Chen and J. Qian, Nitrogen and phosphorus co-doped porous carbons (NPCs) for peroxydisulfate (PDS) activation towards tetracycline degradation: Defects enhanced adsorption and non-radical mechanism dominated by electron transfer, Chem. Eng. J., 2023, 455, 140615 CrossRef CAS.
- H. Li, X. Zhang, S. Yang, Y. Sun and J. Qian, Discerning the relevance of singlet oxygen in pollutant degradation in peroxymonosulfate activation processes, Environ. Sci. Technol., 2024, 58, 14005–14012 CrossRef CAS PubMed.
- X. Cheng, H. Guo, Y. Zhang, X. Wu and Y. Liu, Non-photochemical production of singlet oxygen via activation of persulfate by carbon nanotubes, Water Res., 2017, 113, 80–88 CrossRef CAS PubMed.
- Y. Long, S. Bu, Y. Huang, Y. Shao, L. Xiao and X. Shi, N-doped hierarchically porous carbon for highly efficient metal-free catalytic activation of peroxymonosulfate in water: A non-radical mechanism, Chemosphere, 2019, 216, 545–555 CrossRef CAS PubMed.
- S. Zhu, X. Li, J. Kang, X. Duan and S. Wang, Persulfate activation on crystallographic manganese oxides: Mechanism of singlet oxygen evolution for nonradical selective degradation of aqueous contaminants, Environ. Sci. Technol., 2019, 53, 307–315 CrossRef CAS PubMed.
- J. Qin, Y. Wei, W. Geng, X. Yu, B. Zhou and M. Long, Simultaneous activation of peroxydisulfate and hydrogen peroxide by sulfidated nanoscale zero-valent iron for efficient MTBE degradation: Significant role of oxygen vacancy, ACS ES&T Water, 2023, 3, 1223–1232 Search PubMed.
- Y. Bu, H. Li, W. Yu, Y. Pan, L. Li, Y. Wang, L. Pu, J. Ding, G. Gao and B. Pan, Peroxydisulfate activation and singlet oxygen generation by oxygen vacancy for degradation of contaminants, Environ. Sci. Technol., 2021, 55, 2110–2120 CrossRef CAS.
- X. Huang, N. Zhu, X. Wei, Y. Ding, Y. Ke, P. Wu and Z. Liu, Mechanism insight into efficient peroxydisulfate activation by novel nano zero-valent iron anchored yCo3O4 (nZVI/yCo3O4) composites, J. Hazard. Mater., 2020, 400, 123157 CrossRef CAS PubMed.
- Y. Gao, Z. Chen, Y. Zhu, T. Li and C. Hu, New insights into the generation of singlet oxygen in the metal-free peroxymonosulfate activation process: Important role of electron-deficient carbon atoms, Environ. Sci. Technol., 2020, 54, 1232–1241 CrossRef CAS PubMed.
- X. Peng, Z. Fan, Q. Wang and B. Yang, Recent advances on the role of high-valent metals formed during persulfate activation, Sep. Purif. Technol., 2024, 345, 127365 CrossRef CAS.
- J. Lin, B. He, X. Li, X. Wang, L. Zhang, P. J. J. Alvarez and M. Long, In-situ formed Zr-phosphonate surface complex promotes peroxymonosulfate activation of single-atom Cu for efficient phosphonate oxidation, Environ. Sci. Technol., 2025, 59, 15547–15557 CrossRef CAS.
- Z. Wang, W. Qiu, S.-Y. Pang, Y. Zhou, Y. Gao, C. Guan and J. Jiang, Further understanding the involvement of Fe(IV) in peroxydisulfate and peroxymonosulfate activation by Fe(II) for oxidative water treatment, Chem. Eng. J., 2019, 371, 842–847 CrossRef CAS.
- Y. Bao, C. Lian, K. Huang, H. Yu, W. Liu, J. Zhang and M. Xing, Generating high-valent iron-oxo≡FeIV=O complexes in neutral microenvironments through peroxymonosulfate activation by Zn–Fe layered double hydroxides, Angew. Chem. Int. Ed., 2022, 61, e202209542 CrossRef CAS.
- Y. Zeng, D. He, J. Sun, A. Zhang, H. Luo and X. Pan, Non-radical oxidation driven by iron-based materials without energy assistance in wastewater treatment, Water Res., 2024, 264, 122255 CrossRef CAS PubMed.
- Y. Huang, S. Zhao, K. Chen, B. Huang and R. Jin, A review of persulfate-based advanced oxidation system for decontaminating organic wastewater via non-radical regime, Front. Environ. Sci. Eng., 2024, 18, 134 CrossRef CAS.
- J. Pei, K. Fu, Y. Fu, X. Liu, S. Luo, K. Yin and J. Luo, Manipulating high-valent cobalt-oxo generation on Co/N codoped carbon beads via PMS activation for micropollutants degradation, ACS ES&T Eng., 2023, 3, 1997–2007 Search PubMed.
- Y. Zong, X. Guan, J. Xu, Y. Feng, Y. Mao, L. Xu, H. Chu and D. Wu, Unraveling the overlooked involvement of high-valent cobalt-oxo species generated from the cobalt(II)-activated peroxymonosulfate process, Environ. Sci. Technol., 2020, 54, 16231–16239 CrossRef CAS PubMed.
- Z. Yang, X. Yang, W. Zhang, J. Ai and D. Wang, Local electric field driving selective formation of high-valent Co(IV)=O species by regulating coordination microenvironment of Co single atom, ACS ES&T Water, 2024, 4, 3297–3308 Search PubMed.
- Y. Gao, Y. Zhou, S.-Y. Pang, J. Jiang, Y.-M. Shen, Y. Song, J.-B. Duan and Q. Guo, Enhanced peroxymonosulfate activation via complexed Mn(II): A novel non-radical oxidation mechanism involving manganese intermediates, Water Res., 2021, 193, 116856 CrossRef CAS PubMed.
- Y. Wei, J. Miao, J. Ge, J. Lang, C. Yu, L. Zhang, P. J. J. Alvarez and M. Long, Ultrahigh peroxymonosulfate utilization efficiency over CuO nanosheets via heterogeneous Cu(III) formation and preferential electron transfer during degradation of phenols, Environ. Sci. Technol., 2022, 56, 8984–8992 CrossRef CAS PubMed.
- J. Chen, X. Zhou, P. Sun, Y. Zhang and C.-H. Huang, Complexation enhances Cu(II)-activated peroxydisulfate: A novel activation mechanism and Cu(III) contribution, Environ. Sci. Technol., 2019, 53, 11774–11782 CrossRef CAS.
- Z. Yu, D. Yu, X. Wang, M. Huang, Y. Hou, W. Lin, M. Anpo, J. C. Yu, J. Zhang and X. Wang, Photoinduced formation of oxygen vacancies on Mo-incorporated WO3 for direct oxidation of benzene to phenol by air, J. Am. Chem. Soc., 2025, 147, 13885–13892 CrossRef CAS.
- X. Wang, S. Xue, M. Huang, W. Lin, Y. Hou, Z. Yu, M. Anpo, J. C. Yu, J. Zhang and X. Wang, Pressure-induced engineering of surface oxygen vacancies on metal oxides for heterogeneous photocatalysis, J. Am. Chem. Soc., 2025, 147, 4945–4951 CrossRef CAS.
- S. Zhong, D. Yu, Y. Ma, Y. Lin, X. Wang, Z. Yu, M. Huang, Y. Hou, M. Anpo, J. C. Yu, J. Zhang and X. Wang, Oxygen vacancy-enhanced selectivity in aerobic oxidation of benzene to phenol over TiO2 photocatalysts, Angew. Chem. Int. Ed., 2025, 64, e202502823 CrossRef CAS PubMed.
- C. Deng, M. Duan, Y. Zhao, Y. Li, J. Yang, S. Yang, H. Ji, H. Sheng, C. Chen and J. Zhao, Non-radical photoelectrochemical production of free chlorine from diluted chloride solutions on BiVO4, Appl. Catal., B, 2025, 361, 124644 CrossRef CAS.
- N. Jiang, H. Xu, L. Wang, J. Jiang and T. Zhang, Nonradical oxidation of pollutants with single-atom-Fe(III)-activated persulfate: Fe(V) being the possible intermediate oxidant, Environ. Sci. Technol., 2020, 54, 14057–14065 CrossRef CAS.
- H. Li, R. Qiu, Y. Tang, X. Duan, Y. Gu, H. Yang, Q. Chen, Z. Shu, L. Xiang, S. Liu and X. Tan, Formation of Fe(IV) over a wide pH range via iron-carbon composite-catalyzed persulfate activation, Chem. Eng. J., 2023, 461, 141951 CrossRef CAS.
- X. Peng, J. Wu, Z. Zhao, X. Wang, H. Dai, Y. Li, Y. Wei, G. Xu and F. Hu, High efficiency degradation of tetracycline by peroxymonosulfate activated with Fe/NC catalysts: Performance, intermediates, stability and mechanism, Environ. Res., 2022, 205, 112538 CrossRef CAS PubMed.
- X. Zhou, R. Yin, J. Kang, Z. Li, Y. Pan, J. Bai, A. J. Li and R. Qiu, Atomic cation-vacancy modulated peroxymonosulfate nonradical oxidation of sulfamethoxazole via high-valent iron-oxo species, Appl. Catal., B, 2023, 330, 122640 CrossRef CAS.
- Y. Tang, L. Wang, Y. Fu and Z. Wang, Temperature-regulated non-radical to radical oxidation of organic pollutants by peroxymonosulfate: A tale of ice and fire, J. Water Process Eng., 2025, 72, 107608 CrossRef.
- H. Zhu, H. Ma, Z. Zhao, L. Xu, M. Li, W. Liu, B. Lai, M. Vithanage and S. Pu, Electron transfer tuning for persulfate activation via the radical and non-radical pathways with biochar mediator, J. Hazard. Mater., 2025, 486, 136825 CrossRef CAS PubMed.
- Y. Yan, Z. Wei, X. Duan, M. Long, R. Spinney, D. D. Dionysiou, R. Xiao and P. J. J. Alvarez, Merits and limitations of radical vs. nonradical pathways in persulfate-based advanced oxidation processes, Environ. Sci. Technol., 2023, 57, 12153–12179 CrossRef CAS PubMed.
- L. Zhang, W. Peng, W. Wang, Y. Cao, G. Fan, Y. Huang and M. Qi, A comprehensive review of the electrochemical advanced oxidation processes: Detection of free radical, electrode materials and application, J. Environ. Chem. Eng., 2024, 12, 113778 CrossRef CAS.
- W. Peng, D. Li, D. Qin, H. Luo, F. Qin, H. Weng, L. Chen and C. Zhang, Maximizing singlet oxygen generation: Cobalt single-atom advanced oxidation system for efficient and selective degradation of phenolic pollutants in actual waters, Chem. Eng. J., 2025, 519, 165195 CrossRef CAS.
- H. Zhao, X. Liu, Y. Liu, D. Wu, W. Hu, X. Shang and M. Lv, Directionally inducing non-radical pathways for peroxymonosulfate activation by regulating the exposed crystal plane of MnO2, Process Saf. Environ., 2023, 177, 947–958 CrossRef CAS.
- H. Zhang, S. Hu, S. Li and L. Zhang, Synergistic mechanism of ultrasonic cavitation and advanced oxidation: Free radical path optimization and advanced treatment of industrial wastewater, J. Environ. Chem. Eng., 2025, 13, 117232 CrossRef CAS.
- K. Yin, R. Wu, Y. Shang, D. Chen, Z. Wu, X. Wang, B. Gao and X. Xu, Microenvironment modulation of cobalt single-atom catalysts for boosting both radical oxidation and electron-transfer process in Fenton-like system, Appl. Catal., B, 2023, 329, 122558 CrossRef CAS.
- S. Yang, S. Sun, Z. Xie, Y. Dong, P. Zhou, J. Zhang, Z. Xiong, C.-S. He, Y. Mu and B. Lai, Comprehensive insight into the common organic radicals in advanced oxidation processes for water decontamination, Environ. Sci. Technol., 2024, 58, 19571–19583 CrossRef PubMed.
- G. Wang, D. Huang, M. Cheng, L. Du, S. Chen, W. Zhou, R. Li, S. Li, H. Huang, W. Xu and L. Tang, The surface confinement of FeO assists in the generation of singlet oxygen and high-valent metal-oxo species for enhanced Fenton-like catalysis, Small, 2024, 20, 2401970 CrossRef CAS PubMed.
- H. Liu, X. Lu, Y. Yue, D. Fang, Z. Zheng, X. Gao, Z. Liu and H. Ma, Peroxymonosulfate activation by Fe N co-doped biochar for enhanced degradation of high concentration tetracycline: Radical and non-radical pathways, J. Water Process Eng., 2024, 67, 106260 CrossRef.
- Y. Yang, X. Huang, M. Dong, Z. Chang, H. Yuan, Y. Huang, X. Liu, J. Zhang and A. Dai, Fe0Ni0 bimetal co-immobilized horseradish peroxidase on ZIF-8@HMON for degradation of trichloroacetic acid, Chem. Eng. J., 2024, 480, 147952 CrossRef CAS.
- C.-W. Luo, L. Cai, C. Xie, G. Li and T.-J. Jiang, Sulfur vacancies on MoS2 enhanced the activation of peroxymonosulfate through the co-existence of radical and non-radical pathways to degrade organic pollutants in wastewater, RSC Adv., 2022, 12, 25364–25376 RSC.
- X. Zhu, X. Chen, J. Wang, C. Ge, M. Guo, Y. Cao and B. Lin, Sulfidation of magnetic CoFeAl-layered double hydroxide material as peroxymonosulfate activator for efficient degradation of norfloxacin, RSC Adv., 2025, 15, 37062–37073 RSC.
- J. Zhang, H. Zeng, L. Bu, S. Zhou, Z. Shi and L. Deng, Cu0 incorporated cobalt/nitrogen doped carbonaceous frameworks derived from ZIF-67 (Cu@Co-N-C) as PMS activator for efficient degradation of naproxen: Direct electron transfer and 1O2 dominated nonradical mechanisms, Chem. Eng. J., 2023, 454, 139989 CrossRef CAS.
- J. Peng, L. Li, S. Deng, H. Zhou, Y. Li, C. Fu, L. Lin, Y. Yuan, W. Wei, G. Lv, G. Yang, X. Lu and B. Lai, Activation of persulfates on carbon nanotubes for water decontamination: Is the non-radical process consistently considered across different pH levels?, J. Hazard. Mater., 2025, 485, 136911 CrossRef CAS PubMed.
- J. Li, Y. Wang, Z. Wang and D. Ding, Triggering nanoconfinement effect in advanced oxidation processes (AOPs) for boosted degradation of organic contaminants: A review, Chem. Eng. J., 2025, 503, 158428 CrossRef CAS.
- J. Dong, W. Xu, S. Liu, L. Du, Q. Chen, T. Yang, Y. Gong, M. Li, X. Tan and Y. Liu, Recent advances in applications of nonradical oxidation in water treatment: Mechanisms, catalysts and environmental effects, J. Clean. Prod., 2021, 321, 128781 CrossRef CAS.
- S. Wu, Y. Zhao, R. Sun, Z. Sun, C. Yang and J. Ma, Chemistry, generation and regulation of reactive species in persulfate-based advanced oxidation processes, Chem. Eng. J., 2025, 515, 163588 CrossRef CAS.
- Y. Jiang, K. Gao, T. Chen, Y. Xiong, Y. Li, A. Addisu, S. C. Pillai, D. D. Dionysiou and D. Wang, Regulating the generation of singlet oxygen (1O2) in Advanced oxidation processes by catalyst design for water treatment, Chem. Eng. J., 2024, 500, 156532 CrossRef CAS.
- H. Wu, Q. Song, G. Ran, X. Lu and B. Xu, Recent developments in the detection of singlet oxygen with molecular spectroscopic methods, TrAC, Trends Anal. Chem., 2011, 30, 133–141 CrossRef CAS.
- H. Zhou, L. Lian, S. Yan and W. Song, Insights into the photo-induced formation of reactive intermediates from effluent organic matter: The role of chemical constituents, Water Res., 2017, 112, 120–128 CrossRef CAS PubMed.
- H. F. V. Victória, D. C. Ferreira, J. B. G. Filho, D. C. S. Martins, M. V. B. Pinheiro, G. D. A. M. Sáfar and K. Krambrock, Detection of singlet oxygen by EPR: The instability of the nitroxyl radicals, Free Radical Biol. Med., 2022, 180, 143–152 CrossRef PubMed.
- Y. Liu, H. Zhou, C. Jin, C. Tang, W. Zhang, G. Liu, L. Zhu, F. Chu and Z. Kong, Bio-porphyrin supported single-atom iron catalyst boosting peroxymonosulfate activation for pollutants degradation: A singlet oxygen-dominated nonradical pathway, Appl. Catal., B, 2023, 338, 123061 CrossRef CAS.
- R. Luo, M. Li, C. Wang, M. Zhang, M. A. Nasir Khan, X. Sun, J. Shen, W. Han, L. Wang and J. Li, Singlet oxygen-dominated non-radical oxidation process for efficient degradation of bisphenol A under high salinity condition, Water Res., 2019, 148, 416–424 CrossRef CAS PubMed.
- W. Li, R. Tang, S. Xiong, L. Li, Z. Zhou, L. Su, D. Gong and Y. Deng, High-valent metal-oxo species in catalytic oxidations for environmental remediation and energy conversion, Coord. Chem. Rev., 2024, 510, 215840 CrossRef CAS.
- J. Yu, L. Tang, Y. Pang, X. Liang, Y. Lu, H. Feng, J. Wang, L. Deng, J. Zou, X. Zhu and J. Tang, Non-radical oxidation in environmental catalysis: Recognition, identification, and perspectives, Chem. Eng. J., 2022, 433, 134385 CrossRef CAS.
- C. Li, H. Wang, X. Xu, M. Liu, Y. Liu, S. He, Y. Qian and Z. Li, Anionic polyelectrolyte modified perovskite composite activated hydrogen peroxide to treat high-salinity organic wastewater: Dual effects of electrostatic interaction, Chem. Eng. J., 2024, 488, 151033 CrossRef CAS.
- Y. Zong, H. Zhang, X. Zhang, W. Liu, L. Xu and D. Wu, High-valent cobalt-oxo species triggers hydroxyl radical for collaborative environmental decontamination, Appl. Catal., B, 2022, 300, 120722 CrossRef CAS.
- B. Huang, Z. Wu, X. Wang, X. Song, H. Zhou, H. Zhang, P. Zhou, W. Liu, Z. Xiong and B. Lai, Coupled surface-confinement effect and pore engineering in a single-Fe-atom catalyst for ultrafast Fenton-like reaction with high-valent iron-oxo complex oxidation, Environ. Sci. Technol., 2023, 57, 15667–15679 CrossRef CAS PubMed.
- Q. Pan, C. Wang, P. Zhan, F. Zhao, H. Dai, Y. Hu, F. Hu and X. Peng, Generation and regulation of high-valent metal species in advanced oxidation processes, Environ. Funct. Mater., 2025, 4, 1–10 Search PubMed.
- J. Yao, N. Wu, X. Tang, Z. Wang, R. Qu and Z. Huo, Methyl phenyl sulfoxide (PMSO) as a quenching agent for high-valent metal-oxo species in peroxymonosulfate based processes should be reconsidered, Chem. Eng. J. Adv., 2022, 12, 100378 CrossRef CAS.
- L. Xie, J. Hao, Y. Wu and S. Xing, Non-radical activation of peroxymonosulfate with oxygen vacancy-rich amorphous MnOX for removing sulfamethoxazole in water, Chem. Eng. J., 2022, 436, 135260 CrossRef CAS.
- J. Wang, X. Duan, J. Gao, Y. Shen, X. Feng, Z. Yu, X. Tan, S. Liu and S. Wang, Roles of structure defect, oxygen groups and heteroatom doping on carbon in nonradical oxidation of water contaminants, Water Res., 2020, 185, 116244 CrossRef CAS PubMed.
- J. Zhu, Y. Zhu and W. Zhou, Cu-doped Ni-LDH with abundant oxygen vacancies for enhanced methyl 4-hydroxybenzoate degradation via peroxymonosulfate activation: key role of superoxide radicals, J. Colloid Interface Sci., 2022, 610, 504–517 CrossRef CAS PubMed.
- S. Liu, S. Yin, Z. Zhang, L. Feng, Y. Liu and L. Zhang, Regulation of defects and nitrogen species on carbon nanotube by plasma-etching for peroxymonosulfate activation: Inducing non-radical/radical oxidation of organic contaminants, J. Hazard. Mater., 2023, 441, 129905 CrossRef CAS PubMed.
- H. Zeng, H. Zhu, J. Deng, B. Liu, S. Zhou, Z. Shi and L. Deng, Tunable peroxymonosulfate activation by (-111) crystal plane exposed δ-MnO2: Oxidant concentration induced intrinsic mechanisms transformation, Chem. Eng. J., 2023, 473, 145222 CrossRef CAS.
- V. L. Pham, D.-G. Kim and S.-O. Ko, Advanced oxidative degradation of acetaminophen by carbon catalysts: Radical vs non-radical pathways, Environ. Res., 2020, 188, 109767 CrossRef CAS PubMed.
- B. Huang, Z. Wu, H. Zhou, X. Wang, Y. Liu, H. Zhang, Z. Xiong and B. Lai, The structure-performance relationships in active center size-dependent Fenton-like catalysis: From nanoparticles to single atoms, Appl. Catal., B, 2024, 355, 124157 CrossRef CAS.
- Z. Wu, Z. Xiong, W. Liu, R. Liu, X. Feng, B. Huang, X. Wang, Y. Gao, H. Chen, G. Yao and B. Lai, Active center size-dependent Fenton-like chemistry for sustainable water decontamination, Environ. Sci. Technol., 2023, 57, 21416–21427 CrossRef CAS PubMed.
- H. Li, N. Wang, H. Li, Z. Ren, W. Ma, J. Li, Y. Du and Q. Xu, Polyvinylpyrrolidone-induced size-dependent catalytic behavior of Fe sites on N-doped carbon substrate and mechanism conversion in Fenton-like oxidation reaction, Appl. Catal., B, 2024, 341, 123323 CrossRef CAS.
- C. Ma, Y. Guo, D. Zhang, Y. Wang, N. Li, D. Ma, Q. Ji and Z. Xu, Metal-nitrogen-carbon catalysts for peroxymonosulfate activation to degrade aquatic organic contaminants: Rational design, size-effect description, applications and mechanisms, Chem. Eng. J., 2023, 454, 140216 CrossRef CAS.
- T. Tuan Nguyen, D. Gun Kim and S. Oh Ko, Catalytic degradation of acetaminophen by C and O co-doped graphitic carbon Nitride: Peroxymonosulfate vs. Peroxydisulfate, Chem. Eng. J., 2024, 480, 148348 CrossRef CAS.
- S. Xu, P. Wang, X. Mi, Y. Bao, H. Zhang, F. Mo, Q. Zhou and S. Zhan, N, S, and Cl tri-doped carbon boost the switching of radical to non-radical pathway in Fenton-like reactions: Synergism of N species and defects, J. Hazard. Mater., 2024, 466, 133321 CrossRef CAS PubMed.
- Y. Li, J. Hu, Y. Zou, L. Lin, H. Liang, H. Lei, B. Li and X.-y. Li, Catalytic activity enhancement by P and S co-doping of a single-atom Fe catalyst for peroxymonosulfate-based oxidation, Chem. Eng. J., 2023, 453, 139890 CrossRef CAS.
- X. Li, J. Wang, X. Duan, Y. Li, X. Fan, G. Zhang, F. Zhang and W. Peng, Fine-tuning Radical/Nonradical pathways on graphene by porous engineering and doping strategies, ACS Catal., 2021, 11, 4848–4861 CrossRef CAS.
- J. Zhao, K. Cao, M. Wang, M. Guo, K. Jiang, C. Chen and D. Wu, Laser induced iron/carbon composites for catalytic fabric towards synchronous PDS activation and antibiotic degradation: mechanism study and fix-bed apparatus, Chem. Eng. J., 2025, 515, 163483 CrossRef CAS.
- X. Fan, Q. Lin, K. Xu, J. Zheng, J. Sun, H. Fu, Y. Liu, Y. Ma and J. He, Activation of peroxydisulfate degradation of ciprofloxacin by nitrogen-doped modified graphitized iron-based carbon materials: The transition from free to nonfree radicals, Sep. Purif. Technol., 2023, 316, 123783 CrossRef CAS.
- J. Dou, J. Cheng, Z. Lu, Z. Tian, J. Xu and Y. He, Biochar co-doped with nitrogen and boron switching the free radical based peroxydisulfate activation into the electron-transfer dominated nonradical process, Appl. Catal., B, 2022, 301, 120832 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b
*b














 ), which disproportionate or couple to yield 1O2. Carbonyl groups and OVs accelerate this cascade by stabilizing catalyst–persulfate complexes, weakening the O–O bond, and facilitating the one-electron reductions that sustain 1O2 generation. This ETP → 1O2 pathway occurs on CNTs and N-doped carbons (assisted by carbonyl groups) and on MnO2/BiOBr (where OVs route persulfate to 1O2 via superoxide precursors). This synergy is enhanced by increasing the defect density, tuning surface hydrophobicity to strengthen co-adsorption, and using moderate oxidant doses. Competition arises when (i) rapid outer-sphere ETP depletes substrate electrons, limiting the pool of peroxy intermediates, or (ii) 1O2 quenching reveals a dominant ETP contribution. Diagnostic signatures include significant open-circuit potential (OCP) shifts upon persulfate/pollutant addition (suggesting ETP), coupled with accelerated degradation in deuterium oxide (D2O), confirming 1O2 involvement.53
), which disproportionate or couple to yield 1O2. Carbonyl groups and OVs accelerate this cascade by stabilizing catalyst–persulfate complexes, weakening the O–O bond, and facilitating the one-electron reductions that sustain 1O2 generation. This ETP → 1O2 pathway occurs on CNTs and N-doped carbons (assisted by carbonyl groups) and on MnO2/BiOBr (where OVs route persulfate to 1O2 via superoxide precursors). This synergy is enhanced by increasing the defect density, tuning surface hydrophobicity to strengthen co-adsorption, and using moderate oxidant doses. Competition arises when (i) rapid outer-sphere ETP depletes substrate electrons, limiting the pool of peroxy intermediates, or (ii) 1O2 quenching reveals a dominant ETP contribution. Diagnostic signatures include significant open-circuit potential (OCP) shifts upon persulfate/pollutant addition (suggesting ETP), coupled with accelerated degradation in deuterium oxide (D2O), confirming 1O2 involvement.53 side-products from high-valent cycles disproportionate to 1O2; and (iii) metal-vacancy ensembles facilitate parallel 1O2 and metal-oxo pathways. Catalysts such as Fe–Mn and Fe–N–C exhibit coexisting 1O2 and Fe(IV) signals and demonstrate strong tolerance to anions and NOM, consistent with complementary selectivity for electron-donating substrates. The pathways compete for oxidant and active sites: stabilizing the metal-oxo intermediate can depress the 1O2 yield, whereas high vacancy density with abundant dissolved O2 favors 1O2 but shortens M(IV) lifetimes. Deconvoluting these channels requires combined diagnostic techniques, including D2O kinetic studies, azide quenching, and time-resolved operando spectroscopy.55
side-products from high-valent cycles disproportionate to 1O2; and (iii) metal-vacancy ensembles facilitate parallel 1O2 and metal-oxo pathways. Catalysts such as Fe–Mn and Fe–N–C exhibit coexisting 1O2 and Fe(IV) signals and demonstrate strong tolerance to anions and NOM, consistent with complementary selectivity for electron-donating substrates. The pathways compete for oxidant and active sites: stabilizing the metal-oxo intermediate can depress the 1O2 yield, whereas high vacancy density with abundant dissolved O2 favors 1O2 but shortens M(IV) lifetimes. Deconvoluting these channels requires combined diagnostic techniques, including D2O kinetic studies, azide quenching, and time-resolved operando spectroscopy.55

