DOI:
10.1039/D5RA08708B
(Paper)
RSC Adv., 2026,
16, 995-1007
Stability and hydrogen storage performance of Na2LiXH6 (X = Zr, V, Cr) double perovskite hydrides via DFT and AIMD
Received
11th November 2025
, Accepted 19th December 2025
First published on 6th January 2026
Abstract
This study aims to provide a comprehensive first-principles investigation, based on density functional theory (DFT) using the GGA-PBE functional, of Na2LiXH6 (X = Zr, V, Cr) double perovskite hydrides that crystallize in the Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (225) space group. The structural, electronic, optical, and thermodynamic properties were systematically explored to evaluate their potential for advanced hydrogen storage and clean energy applications. Phonon dispersion and ab initio molecular dynamics (AIMD) simulations confirm the dynamical and thermal stability of the system at 300 K, without any structural distortion. Among the investigated compounds, Na2LiVH6 shows the highest gravimetric capacity (5.50 wt%) and an optimal desorption temperature (540.23 K), favorable for reversible hydrogen release. Electronic analysis reveals metallic conductivity, while thermodynamic parameters, including heat capacity, enthalpy, entropy, and free energy, exhibit stable temperature-dependent trends. Collectively, these DFT-GGA-PBE results demonstrate that Na2LiVH6 possesses superior mechanical endurance, lattice stability, and multifunctional potential for next-generation hydrogen storage and sustainable energy applications.
m (225) space group. The structural, electronic, optical, and thermodynamic properties were systematically explored to evaluate their potential for advanced hydrogen storage and clean energy applications. Phonon dispersion and ab initio molecular dynamics (AIMD) simulations confirm the dynamical and thermal stability of the system at 300 K, without any structural distortion. Among the investigated compounds, Na2LiVH6 shows the highest gravimetric capacity (5.50 wt%) and an optimal desorption temperature (540.23 K), favorable for reversible hydrogen release. Electronic analysis reveals metallic conductivity, while thermodynamic parameters, including heat capacity, enthalpy, entropy, and free energy, exhibit stable temperature-dependent trends. Collectively, these DFT-GGA-PBE results demonstrate that Na2LiVH6 possesses superior mechanical endurance, lattice stability, and multifunctional potential for next-generation hydrogen storage and sustainable energy applications.
Introduction
The increasing global energy needs and escalating environmental degradation necessitate the rapid advancement of sustainable energy technologies. Among the different alternative fuels, hydrogen has emerged as a highly promising energy carrier due to its exceptional energy density and clean combustion products.1 Nevertheless, its large-scale utilization is still limited by difficulties associated with safe, efficient, and practical storage and transportation.2 Traditional storage techniques, such as high-pressure gas compression, cryogenic liquid storage, and cryo-compressed systems, demand significant energy to maintain extreme pressure or low-temperature conditions and are further challenged by unavoidable boil-off losses.3,4 Alternatively, solid-state hydrogen storage has emerged as a focal area of research owing to its superior volumetric density, enhanced safety, and reversible storage capability.5,6 A wide range of solid materials has been investigated, including intermetallic alloys,7,8 metal hydrides,9–12 graphene-based nanostructures,13 metal–organic frameworks (MOFs),14,15 MXenes,16,17 and liquid organic hydrogen carriers (LOHCs).18 Among these, metal hydrides are particularly attractive because hydrogen is chemically bonded under moderate conditions, offering high volumetric capacities and intrinsic safety. Furthermore, their storage performance can be optimized through compositional engineering and catalytic modification.19,20
Perovskite hydrides have recently emerged as a promising subclass of ternary hydrides with remarkable promise for hydrogen storage applications. These materials possess the ability to absorb and retain hydrogen both on their surfaces and within their crystal frameworks, offering an efficient approach to overcoming the persistent challenges of hydrogen storage and transport.21 Their high gravimetric capacity, excellent thermal and cyclic stability, and favorable reversibility make them highly attractive for next-generation solid-state hydrogen systems. Various perovskite hydrides such as NaMgH3, KAlH3, and NaAlH3 have been synthesized and characterized, exhibiting orthorhombic or cubic crystal symmetries with remarkable thermodynamic stability.22,23 Reported hydrogen storage capacities for AVH3 (A = Na, K, Rb, Cs), XPtH3 (X = Li, Na, K, Rb), and XFeH3 (X = Ca, Sr, Ba) fall within 1–5 wt%, highlighting their competitive storage performance.24–26 Double perovskite systems, including Na2LiAlH6 and K2LiAlH6, show dehydrogenation capacities of approximately 3.09 wt% and 5.2 wt%, respectively.27–29 Li-doped K2NaAlH6 demonstrates an improved storage capacity of 4.91 wt% with increasing Li concentration,30 while substitution of alkaline earth metals in A2KNaH6 (A = Be, Mg, Ca) further enhances capacity to 8.57, 5.19, and 4.1 wt%, respectively.31 Moreover, K2XAlH6 (X = Li, Na) hydrides exhibit wide band gaps and strong mechanical stability under external stress.32 Despite these advances, many double perovskite hydrides remain insufficiently examined, underscoring the need for detailed AIMD investigations to optimize their hydrogen storage characteristics and practical applicability.
In this study, Na2LiXH6 (X = Zr, V, Cr) double perovskite hydrides are investigated using DFT to explore their fundamental structural, electronic, mechanical, optical, and thermodynamic behaviors. The study aims to identify their feasibility and effectiveness as potential prospects for future solid-state H2 storage technologies. Despite the growing interest in complex hydrides, these specific Zr, V, and Cr based Na2LiXH6 (X = Zr, V, Cr) compounds have not yet been systematically explored, either computationally or experimentally. The strategic substitution of X-site cations with varying electronegativities and ionic radii introduces tunable effects on formation energy, stability, electronic structure, and hydrogen release thermodynamics, an aspect that remains largely unreported in existing literature. To bridge this gap, DFT calculations were conducted to elucidate the atomic-level bonding interactions, energetic characteristics, and hydrogen storage potential of the compounds. Complementary AIMD simulations were employed to evaluate their thermal robustness. The results reveal the structural and energetic stability of these previously unexplored perovskite hydrides, providing a theoretical basis for the rational design of advanced materials for H2 storage and energy conversion applications.
Computational methodology
All computational analyses were conducted using DFT within the CASTEP simulation package33 to investigate the ground-state characteristics and optimized structural configurations of Na2LiXH6 (X = Zr, V, Cr) double perovskite hydrides. The Kohn–Sham equations were implemented with a plane-wave basis set, applying a kinetic energy cutoff of 600 eV,34 and ultrasoft pseudopotentials were utilized to model the interactions between electrons and ions accurately. The exchange correlation interactions were described within the framework of the generalized gradient approximation (GGA) using the Perdew–Burke–Ernzerhof (PBE) functional.35 Structural optimization was performed through the Broyden–Fletcher–Goldfarb–Shanno (BFGS) minimization algorithm, applying strict convergence thresholds of 2 × 10−5 eV per atom for total energy, 0.05 eV Å−1 for atomic forces, 0.1 GPa for stress, and 0.002 Å for atomic displacements.36 For Brillouin zone integration, a Monkhorst–Pack k-point mesh of 6 × 6 × 6 was utilized to ensure accurate sampling. After full relaxation, all Na2LiXH6 (X = Zr, V, Cr) double perovskite hydrides were verified to adopt a cubic symmetry within the Fmm (No. 225) space group, characterized by lattice angles α = β = γ = 90°, thereby confirming their crystallographic and structural stability.37 To evaluate the thermal stability, AIMD simulations were carried out using the pw.x module of Quantum ESPRESSO within the canonical (NVT) ensemble. The simulations employed the velocity Verlet integration algorithm coupled with a Berendsen thermostat fixed at 300 K to maintain temperature control. A time step of 0.96756 fs was adopted for 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 iterations, corresponding to a total simulation duration of approximately 9.7 ps. The calculations utilized ultrasoft pseudopotentials in combination with the PBEsol exchange-correlation functional, while the plane-wave and charge density cutoff energies were set to 60 Ry and 400 Ry, respectively. Convergence criteria were defined as 1.0 × 10−8 Ry for self-consistent field (SCF) cycles, 1.0 × 10−5 Ry for total energy, and 1.0 × 10−4 Ry bohr−1 for forces, ensuring high numerical precision throughout the simulations.38 These computational settings ensured robust convergence and high numerical precision across all investigated systems.
000 iterations, corresponding to a total simulation duration of approximately 9.7 ps. The calculations utilized ultrasoft pseudopotentials in combination with the PBEsol exchange-correlation functional, while the plane-wave and charge density cutoff energies were set to 60 Ry and 400 Ry, respectively. Convergence criteria were defined as 1.0 × 10−8 Ry for self-consistent field (SCF) cycles, 1.0 × 10−5 Ry for total energy, and 1.0 × 10−4 Ry bohr−1 for forces, ensuring high numerical precision throughout the simulations.38 These computational settings ensured robust convergence and high numerical precision across all investigated systems.
Results and discussion
Structural stability
The optimized crystal structure of Na2LiXH6 (X = Zr, V, Cr) double perovskite hydrides demonstrates a cubic symmetry classified under the Fmm (225) space group, as illustrated in Fig. 1. The atomic positions within the unit cell are specified as follows: Na atoms reside at the 8c (1/4, 3/4, 3/4) Wyckoff sites, Li atoms are located at 4b (1/2, 0, 0), X = (Zr/V/Cr) atoms reside at 4a (0, 0, 0), and H atoms are positioned at 24e (1/2, 0.718326, 0). The structural motif consists of alternating LiH6 and XH6 (X = Zr, V, Cr) octahedra, forming a three-dimensional framework wherein Na+ ions occupy the cuboctahedral cavities, consistent with the characteristic rock-salt arrangement of perovskite-type hydrides.39
 |
| | Fig. 1 Optimized crystal structure of Na2LiXH6 (X = Zr, V, Cr) perovskite hydrides. | |
Table 1 shows the optimized lattice parameters (a = b = c) and unit cell volumes (V) of Na2LiXH6 (X = Zr, V, Cr) double perovskite hydrides. The results reveal a gradual contraction in lattice dimensions, decreasing from 7.93 Å (Zr) to 7.33 Å (Cr). This gradual reduction in lattice dimension results from the decreasing ionic radii of the X-site cations, which enhances the crystal compactness and strengthens the metal–hydrogen interactions. The bonding environment serves a crucial role in function in controlling hydrogen retention and desorption behavior, where an optimal balance of bond strength facilitates hydrogen release at moderately low temperatures, beneficial to efficient storage-release cycles. In this context, the Goldschmidt tolerance factor (τG), calculated using eqn (1),40 exhibits a slight increase from 0.88 for Na2LiZrH6 to 0.93 for Na2LiCrH6, remaining well within the stability range (0.8–1.0) characteristic of cubic double perovskite frameworks. This gradual rise in τG reflects enhanced structural symmetry and lattice compatibility, which contribute to the observed thermodynamic stability and favorable hydrogen desorption characteristics of the Cr-based compound. All compounds studied have octahedral values (µ) within the optimal range (0.42–0.75). These values confirm that all three compounds possess geometrically stable frameworks without significant distortion.
| |
 | (1) |
| |
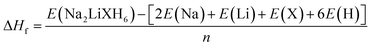 | (2) |
| |
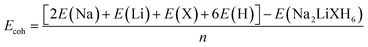 | (3) |
Table 1 Calculated structural parameters of Na2LiXH6 (X = Zr, V, Cr) perovskite hydrides
| Compound |
Lattice constant a = b = c (Å) |
Volume (Å)3 |
τG |
µ |
ΔHf (eV per atom) |
Ecoh (eV per atom) |
| Na2LiZrH6 |
7.93 |
499.35 |
0.88 |
0.53 |
−1.26 |
+1.26 |
| Na2LiVH6 |
7.45 |
414.38 |
0.92 |
0.46 |
−1.47 |
+1.47 |
| Na2LiCrH6 |
7.33 |
393.57 |
0.93 |
0.45 |
−1.61 |
+1.61 |
The enthalpy of formation (ΔHf) further provides insight into the thermodynamic stability of the hydrides, which can be calculated using eqn (2).41 The calculated ΔHf values of −1.26, −1.47, and −1.61 eV per atom for Na2LiZrH6, Na2LiVH6, and Na2LiCrH6, respectively, indicate an increasing exothermic trend, and n represents the total number of atoms. The progressively more negative formation enthalpies imply that substitution of Zr with V and Cr enhances the overall thermodynamic stability of the lattice. Although this trend corresponds with the decrease in the effective ionic size of the X-site cation, it is important to emphasise that the enhanced stability cannot be solely attributed to ionic radii. Instead, the observed exothermic behaviour is primarily governed by changes in electronic structure, bonding characteristics, and metal-hydrogen hybridization induced by the transition-metal substitution. Likewise, the cohesive energy (Ecoh) follows a similar sequence, increasing from +1.26 eV (Zr) to +1.61 eV (Cr), and was calculated using eqn (3),42 confirming stronger interatomic bonding as the X-site cation changes from Zr to Cr. The increase in cohesive strength is indicative of an augmentation in metal-hydrogen bonding interactions and an improvement in charge transfer. Rather than being a simple monotonic dependence on ionic radius alone, this phenomenon leads to enhanced lattice rigidity and a reduction in susceptibility to structural deformation. The combined structural, geometric, and energetic indicators reveal a consistent trend in the stability of the studied hydrides. Among the three, Na2LiCrH6 exhibits the highest structural and thermodynamic stability, followed by Na2LiVH6 and Na2LiZrH6. The enhanced compactness, favorable τG and higher Ecoh collectively confirm the superior integrity of Na2LiCrH6 within the Na2LiXH6 series.
In addition to lattice parameters and formation enthalpies, the calculated M–H bond lengths provide significant insight into the bonding characteristics and hydrogen storage behavior of Na2LiXH6 (X = Zr, V, Cr) perovskite hydrides. The optimized structures exhibit short X–H bond lengths of 1.99 Å for Zr–H, 1.77 Å for V–H, and 1.71 Å for Cr–H, indicating strong M–H interactions within the [XH6] octahedra. The continuing contraction of the X–H bond from Zr to Cr reflects increasing orbital overlap between transition-metal d-states and H-state, consistent with enhanced covalent character and improved framework stability. In contrast, the significantly longer Na–H separations of 2.81 Å in Na2LiZrH6, 2.64 Å in Na2LiVH6, and 2.59 Å in Na2LiCrH6 show weaker ionic interactions between hydrogen and the A-site cations. This pronounced bonding asymmetry suggests that while H atoms coordinated to the X-site ensure structural rigidity, those associated with the A site are more-weakly bound and thus more amenable to thermally activated release. Such coexistence of strong X–H bonding and weaker A–H interactions is a defining structural feature governing hydrogen uptake and release kinetics in perovskite hydrides and has been widely recognized as a key factor in optimizing reversible solid-state H2 storage performance.43,44
Dynamic stability
Dynamic stability is a key factor governing the structural integrity and long-term functionality of materials, particularly in applications such as hydrogen storage, where materials must endure repeated thermal and mechanical cycles without degradation.45 To assess the dynamic stability of Na2LiXH6 (X = Zr, V, Cr) perovskite hydrides, phonon dispersion analysis was performed across the first Brillouin zone, as illustrated in Fig. 2a–c. Phonon dispersion relations serve as a fundamental tool for assessing the vibrational stability of crystalline materials, as they reveal how atomic vibrations propagate through the lattice. The appearance of imaginary (negative) phonon frequencies signifies dynamical instability, suggesting possible lattice distortions or phase transitions, whereas their absence confirms a stable crystal configuration suitable for practical applications. The lack of imaginary modes in the computed phonon band structures indicates that these compounds are dynamically stable under ambient conditions, ensuring their potential for practical applications.46,47
 |
| | Fig. 2 Phonon dispersions describing (a) Na2LiZrH6, (b) Na2LiVH6 and (c) Na2LiCrH6 perovskite hydrides. | |
As depicted in Fig. 2a–c, Na2LiZrH6, Na2LiVH6, and Na2LiCrH6 all exhibit stable phonon dispersions, with no imaginary phonon modes observed across the Brillouin zone. The acoustic branches smoothly converge to zero near the Γ-point, whereas the optical modes remain clearly divided, displaying well-defined oscillatory mode features that are indicative of a structurally stable lattice.48 While the phonon frequencies vary slightly among the compounds, with Na2LiCrH6 showing higher frequencies indicative of a stiffer lattice, all three compounds maintain stable vibration profiles, suggesting that they are not susceptible to lattice distortions. The higher frequencies observed in Na2LiCrH6 indicate a stronger interatomic bond strength and more robust vibrational modes, potentially due to the higher atomic number and stronger bonding of Cr compared to Zr and V. This analysis confirms that the optimized structures of Na2LiZrH6, Na2LiVH6, and Na2LiCrH6 correspond to stable, minimum-energy states rather than saddle points, further supporting their dynamical stability and suitability for applications where long-term material performance is critical. The origin of the differences in phonon frequencies can be attributed to the differing electronic structures and bond strengths between the transition metals Zr, V, and Cr. The stronger metallic bonds in Na2LiCrH6 lead to higher phonon frequencies, reflecting a more rigid and robust lattice structure, while Na2LiZrH6 and Na2LiVH6 exhibit lower frequencies due to the weaker bonds in comparison. This gives rise to the subtle variation in dynamic behavior across these compounds.
The lack of low-frequency vibrational instabilities further verifies the vibrational robustness of these materials, indicating their ability to endure mechanical stresses while maintaining structural integrity. Additionally, the consistent stability observed across all three compounds reinforces their potential as reliable materials for high-temperature applications, such as H2 storage and clean energy technologies.
Ab initio molecular dynamics (AIMD) calculations
The thermal and dynamic stability of Na2LiXH6 (X = Zr, V, Cr) perovskite hydrides were systematically evaluated through AIMD simulations conducted at room temperature, as depicted in Fig. 3a–c. The results reveal strikingly similar behaviors across all three compounds, highlighting their robust thermodynamic stability. Na2LiZrH6, Na2LiVH6, and Na2LiCrH6 exhibit stable total energy profiles, with fluctuations confined within tightly constrained ranges, underscoring a lack of significant lattice degradation or phase transitions throughout the simulation period. Specifically, Na2LiZrH6 fluctuates between −310.86 and −310.71 Ry, Na2LiVH6 between −356.67 and −356.52 Ry, and Na2LiCrH6 between −387.03 and −386.91 Ry. These confined energy oscillations suggest that the systems are thermally stable and do not undergo any energetic instabilities, ensuring that their structural integrity remains intact under thermal perturbations. The slight difference in energy profiles across the compounds can be attributed to the differing electronic states of the transition metals. The higher binding energies in Na2LiCrH6 are likely due to the stronger bonding associated with Cr, contributing to its higher thermodynamic stability. The temperature fluctuations, oscillating around the equilibrium values of 300 K for all compounds, further confirm the sustained thermodynamic equilibrium throughout the simulations, with temperatures fluctuating between 270 and 330 K for Na2LiZrH6, 280 and 340 K for Na2LiVH6, and 270 and 320 K for Na2LiCrH6. These results underscore the durability of Na2LiXH6 compounds under realistic, dynamic conditions, which is crucial for their potential applications in energy storage.
 |
| | Fig. 3 AIMD total-energy traces versus time for (a) Na2LiZrH6, (b) Na2LiVH6, and (c) Na2LiCrH6 perovskite hydrides. | |
In addition to energy and temperature stability, the pressure fluctuations observed across all three compounds remain periodic and within expected bounds, further demonstrating their structural resilience.49 The absence of any abrupt changes in pressure supports the notion that these perovskite hydrides can maintain their crystallographic framework under varying thermal and pressure conditions. Such stability is particularly important for materials intended for high-temperature applications, where they must endure fluctuating conditions without undergoing degradation or structural failure.38,50 These results are of significant importance for the potential application of Na2LiXH6 hydrides in energy storage systems, such as H2 storage, where materials must endure fluctuating temperatures and pressures without compromising their structural integrity. The differences in the pressure fluctuation behavior are minimal across the three compounds, with slight differences indicating the varying degrees of resistance to mechanical stress due to the electronic properties of Zr, V, and Cr. The consistent stability observed across Na2LiZrH6, Na2LiVH6, and Na2LiCrH6 positions these compounds as promising candidates for use in demanding high-temperature applications, reinforcing their potential for advancing clean energy technologies.
Hydrogen storage
Hydrogen is a dynamic, sustainable energy carrier with an unparalleled gravimetric energy density and flawlessly clean combustion, making it an essential part of the shift to carbon-neutral systems as the world looks for the next generation of clean power. However, a significant obstacle to its broad use is still the absence of safe, effective, and compact storage solutions. In this context, complex hydrides, particularly double perovskite-type hydrides, have attracted increasing theoretical interest due to their high hydrogen content and structural tunability. However, Na2LiXH6 (X = Zr, V, Cr) perovskite hydrides remain largely unexplored in the literature, especially with respect to a systematic first-principles assessment of their stability and hydrogen-related properties.
Based on our first-principles calculations, Na2LiXH6 (X = Zr, V, Cr) exhibit favorable structural and dynamical stability, which motivates their consideration as potential candidates for solid-state hydrogen storage rather than established storage materials. As seen in Table 2, the H2 storage capacity of Na2LiXH6 (X = Zr, V, Cr) was evaluated in terms of gravimetric capacity (Cwt%), volumetric capacity (ρvol), and desorption temperature (Tdes). Eqn (4) was used to calculate the gravimetric H2 capacity, which is defined as the ratio of hydrogen mass to the total mass of the hydride.51
| |
 | (4) |
where
mh and
mhost represent the molar masses of hydrogen and the host material, respectively. Na
2LiZrH
6, Na
2LiVH
6, and Na
2LiCrH
6 have determined
Cwt% of 4.03 weight percent, 5.50 weight percent, and 5.45 weight percent, respectively. Among these, Na
2LiVH
6 and Na
2LiCrH
6 show potential as high-capacity H
2 storage materials by getting close to the U.S. DOE 2025 objective of 5.5 weight percent. The decrease in molecular weight and increased hydrogen bonding strength within the lattice, which increases hydrogen uptake efficiency, are the main causes of the rise in
Cwt% from Zr to V and Cr replacement. Another crucial performance parameter for real-world storage applications where compactness is necessary is the volumetric H
2 capacity (
ρvol), which shows the hydrogen content per unit volume. It was evaluated using
eqn (5).
52| |
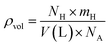 | (5) |
Table 2 The ρvol, Tdes and gravimetric ratios of Na2LiXH6 (X = Zr, V, Cr) perovskite hydrides
| Compound |
Cwt (%) |
ΔHf (kJ per mol per H2) |
ρvol (g per H2 per L) |
Tdes (K) |
References |
| K2NaAlH6 |
4.47 |
— |
74.25 |
484.52 |
56 |
| Cs2NaInH6 |
1.48 |
— |
14.18 |
492.70 |
57 |
| K2LiTiH6 |
4.38 |
−56.96 |
19.12 |
435.80 |
46 |
| Ca2LiCuH6 |
3.86 |
— |
15.68 |
717.20 |
39 |
| Na2LiZrH6 |
4.03 |
−60.54 |
20.11 |
463.16 |
Present study |
| Na2LiVH6 |
5.50 |
−70.61 |
24.24 |
540.23 |
Present study |
| Na2LiCrH6 |
5.45 |
−77.69 |
25.52 |
594.44 |
Present study |
The ρvol were determined to be 20.11 g per H2 per L for Na2LiZrH6, 24.24 g per H2 per L for Na2LiVH6, and 25.52 g per H2 per L for Na2LiCrH6, as indicated in Table 2. Denser atomic packing and stronger lattice cohesion increase hydrogen density per unit volume, as indicated by the increasing trend in ρvol from Zr to Cr replacement. This is beneficial for applications requiring both high energy density and compact storage. When evaluating a storage material's viability, especially for fuel cell integration, hydrogen desorption is represented by the desorption temperature (Tdes) is crucial. Gibbs free energy governs the desorption process thermodynamically and is characterized by (eqn 6) and (7).53,54
| |
 | (7) |
where the enthalpy and entropy changes during desorption are represented by Δ
H and Δ
S. According to the computed results,
Tdes of Na
2LiZrH
6, Na
2LiVH
6, and Na
2LiCrH
6 are 463.16, 540.23, and 594.44 K, respectively. Stronger metal-hydrogen interactions inside the lattice are correlated with a progressive increase in
Tdes, especially for Cr-substituted systems. These temperatures show that these materials are thermodynamically stable and capable of reversible hydrogen release under moderate heating conditions, even if they are somewhat higher than the optimal range for direct PEM fuel cell operation (233–333 K).
55 In brief, both gravimetric and volumetric measurements demonstrate that Na
2LiVH
6 and Na
2LiCrH
6 outperform Na
2LiZrH
6 in terms of H
2 storage capacity and stability. The higher desorption enthalpy and temperature values for Na
2LiCrH
6 confirm stronger M–H bonding and improved thermal resistance, which makes it particularly attractive for high-temperature H
2 storage and transport applications. The present results demonstrate that controlled transition-metal substitution can effectively tune the hydrogen-related properties of Na
2LiXH
6 perovskite hydrides. While the present analyses are based on thermodynamic stability, phonon dispersion, and AIMD simulations, they establish a theoretical foundation that motivates future experimental validation and kinetic studies.
Mechanical properties
The mechanical properties serve as a crucial indicator of the structure-dependent elastic response in perovskite hydrides. Accurate determination of second-order elastic constants (Cij) is fundamental for evaluating mechanical stability and understanding a material's resistance to external deformation and applied stress. In the present study, the elastic constants of Na2LiXH6 (X = Zr, V, Cr) were systematically computed, from which essential mechanical moduli and anisotropic factors were derived to provide a comprehensive assessment of their stability and mechanical resilience. The obtained parameters not only validate the mechanical robustness of these compounds but further elucidate their deformability, rigidity, and bonding nature, which are key determinants of their reliability and efficiency under hydrogen storage conditions. The elastic response of Na2LiXH6 (X = Zr, V, Cr) was characterized by computing the distinct elastic coefficients C11, C12, and C44 corresponding to the cubic symmetry; the full set of derived moduli is reported in Table 3. The calculated C11, C12, and C44 values increase from Zr to Cr, indicating that substitution at the X site produces a progressive stiffening of the lattice. Physically, the rising C11 and C44 reflect enhanced resistance to longitudinal and shear deformations, respectively, as the X-site cation is varied from Zr to Cr.
Table 3 The computed elastic moduli of Na2LiXH6 (X = Zr, V, Cr) perovskite hydrides
| Elastic moduli |
Na2LiZrH6 |
Na2LiVH6 |
Na2LiCrH6 |
| C11 |
47.74 |
71.24 |
93.23 |
| C12 |
15.58 |
22.43 |
28.15 |
| C44 |
18.45 |
26.74 |
30.15 |
| Cp |
−2.87 |
−4.31 |
−2.0 |
| B(GPa) |
26.29 |
38.69 |
49.85 |
| G(GPa) |
17.46 |
25.78 |
31.08 |
| E(GPa) |
42.89 |
63.28 |
77.20 |
| B/G |
1.51 |
1.50 |
1.60 |
| υ |
0.22 |
0.23 |
0.24 |
| C′ |
16.08 |
24.41 |
32.54 |
| AU |
1.15 |
1.10 |
0.93 |
All three compounds satisfy the Born mechanical criteria for cubic symmetry, namely C11 − C12 > 0, C11 + 2C12 > 0, and C44 > 0. In particular, the shear constant (C′ = (C11 − C12)/2) is positive in every case (16.08, 24.41, and 32.54 GPa), confirming mechanical stability against shear modes and indicating that the relaxed structures are mechanically robust (Table 3).
The bulk modulus (B) increases progressively from 26.29 to 38.69 to 49.85 GPa, while the shear modulus (G) rises from 17.46 to 25.78 to 31.08 GPa, and the Young's modulus (E) increases from 42.89 to 63.28 to 77.20 GPa. All elastic moduli were computed using eqn (8)–(12).58–61 This consistent enhancement in elastic moduli demonstrates that Na2LiCrH6 exhibits the greatest stiffness and lowest compressibility within the series. The systematic improvement in elastic stiffness is closely associated with the observed reduction in lattice constants and the increase in cohesive energy, suggesting that the Cr-based compound possesses superior mechanical integrity under both hydrostatic and shear stresses.
Ductility/brittleness was assessed using Pugh's ratio (B/G) and Poisson's ratio (υ) as shown in Fig. 4. The computed B/G values (1.51, 1.50, and 1.60) are all below the empirical ductile threshold of ≈1.75, and υ (0.22, 0.23, and 0.24) remain below 0.26; both criteria therefore classify these materials as predominantly brittle with limited plasticity. Nonetheless, the slight increase of B/G and υ for Na2LiCrH6 indicates a modest shift toward less brittle behavior relative to the Zr- and V-variants. Cauchy pressure, defined as Cp = C12 − C44, is negative for all three compositions (−2.87, −4.31, and −2.00 GPa). A negative Cp generally signals significant directional (covalent-like) bonding contributions; thus, the elastic data imply that the bonding in these hydrides has a notable non-ionic/covalent component, consistent with strong X–H and M–H interactions in the framework. Elastic anisotropy was quantified using the universal anisotropy index AU. The computed AU values (1.15, 1.10, and 0.93) show that Na2LiCrH6 is the most nearly isotropic (AU ≈ 1), whereas Na2LiZrH6 and Na2LiVH6 display modest anisotropy. The presence of finite anisotropy, particularly in the Zr and V compounds, suggests directional dependence of mechanical response that should be considered when designing single crystals, thin films, or polycrystalline components. The elastic data verify that all three Na2LiXH6 perovskites are mechanically stable in the cubic phase. Progressive substitution from Zr to V to Cr strengthens the lattice, as reflected by the higher values of Cij, B, G, and E, and slightly decreases brittleness. Among them, Na2LiCrH6 exhibits the most favorable combination of stiffness and nearly isotropic elastic behavior. These mechanical characteristics, enhanced rigidity and structural stability for the Cr-based compound, are advantageous for practical handling and integration into devices. However, the inherently brittle nature of these materials may necessitate microstructural engineering strategies, such as the development of composites or protective coatings, to enable their use in applications requiring greater mechanical flexibility. Such elastic attributes significantly reinforce the structural stability and adaptability of these compounds, thereby emphasizing their strong potential for efficient hydrogen (H2) storage applications.
| |
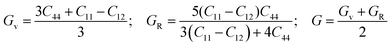 | (8) |
| |
 | (9) |
| |
 | (10) |
| |
 | (11) |
| |
 | (12) |
 |
| | Fig. 4 Graphs of (a) Poisson's ratio (υ), and (b) Pugh's ratio (B/G)of Na2LiXH6 (X = Zr, V, Cr) perovskite hydrides. | |
Electronic properties
Electronic characteristics, especially the energy bandgap (Eg) and density of states (DOS), serve a vital function in controlling the adsorption and desorption behavior, structure stability, as well as catalytic activity of hydrogen storage materials, thereby facilitating efficient hydrogen dissociation and recombination processes. A detailed understanding of the electronic band structure is fundamental to elucidating the electrical conductivity and optical behaviour of crystalline hydrides. It provides insights into energy distribution and bonding characteristics within the material. Fig. 5 presents the calculated band structures of Na2LiXH6 (X = Zr, V, Cr) along the high symmetry path X–R–M–Γ–R, obtained using the GGA-PBE functional. The Fermi level is indicated by a dashed olive line. All three compounds display an overlap at the Fermi level, with the valence band maximum (VBM) and conduction band minimum (CBM) located at distinct symmetry points, confirming the absence of a bandgap and indicating their metallic nature. The valence bands (VB) near the Fermi level appear relatively flat, suggesting localized bonding states, while the conduction bands (CB) show greater dispersion, implying enhanced electron mobility. With substitution from Zr to V to Cr, there is a gradual increase in the dispersion of the conduction bands and a narrowing of the valence bandwidth, reflecting subtle electronic restructuring across the series. These observations are key to understanding the DOS and optical response discussed in the subsequent section.
 |
| | Fig. 5 Computed band structures of describing (a) Na2LiZrH6, (b) Na2LiVH6 and (c) Na2LiCrH6 perovskite hydrides. | |
Fig. 6 presents the total and partial density of states (PDOS) for Na2LiXH6 (X = Zr, V, Cr) hydrides, offering comprehensive insights into their electronic characteristics and bonding nature. In all three systems, the Fermi level (EF) intersects prominent peaks, indicating metallic characteristics and significant electronic delocalization. For Na2LiZrH6, the VB region from about −5 eV up to the EF is mainly formed by the hybridized Zr-d and H-s orbitals, with minor contributions from Na-s and Li-s states. The CB is primarily governed by Zr-d states, indicating pronounced metal-hydrogen interactions near the EF. In the case of Na2LiVH6, the top of the VB primarily originates from V-d and H-s orbitals, while the CB reveals further hybridization between V-d and H-s states, pointing to strong covalent interactions within the VH6 octahedra. Likewise, Na2LiCrH6 exhibits intense mixing between Cr-d and H-s orbitals near the EF, where the partially filled Cr-d states play a significant role in verifying the electronic character. This pronounced d-s hybridization suggests a comparatively more covalent Cr–H bonding character than in the Zr- and V-based counterparts.
 |
| | Fig. 6 TDOS and PDOS for Na2LiXH6 (X = Zr, V, Cr) perovskite hydrides. | |
As the transition metal changes from Zr to V and then to Cr, the increasing covalent contribution reduces the purely ionic stabilization of H−, which may facilitate hydrogen mobility and lower the dehydrogenation barrier. Consequently, while Na2LiCrH6 exhibits strong metal–hydrogen interactions and high thermal stability, its hydrogen storage behavior should be viewed as a balance between enhanced bonding strength and potentially improved hydrogen release kinetics rather than exclusively superior high-temperature storage performance. Overall, the PDOS analysis confirms that Na2LiXH6 (X = Zr, V, Cr) exhibits metallic behavior at the EF, enabling rapid charge transport and promoting efficient hydrogen storage-release dynamics.
Optical properties
Optical properties offer valuable information on the interaction between materials and incident electromagnetic waves, revealing the bandgap nature, electronic transition mechanisms, and hydrogen adsorption and desorption behavior. These attributes are fundamental for assessing the potential of hydride-based compounds in advanced functional and energy-related applications. In the context of hydrogen storage, the metallic or insulating nature of a material critically influences thermal and electronic transport during hydrogen uptake and release.62–65 Metallic behavior, associated with enhanced electronic and thermal conductivity, can promote efficient heat management and improved cycling kinetics, whereas insulating or wide-bandgap characteristics may reduce electronic losses and contribute to structural stability.66 The optical response discussed herein is fully consistent with this electronic classification, where metallic systems exhibit pronounced low-energy absorption without a distinct optical band edge, while insulating or semiconducting compounds show a clear absorption onset arising from interband transitions. This correlation establishes a coherent link between optical behavior, electronic structure, and hydrogen storage performance. Fig. 7 shows the computed optical properties of Na2LiXH6 (X = Zr, V, Cr) in the photon energy range 0–10 eV, including the real [ε1(ω)] and imaginary [ε2(ω)] parts of the dielectric function, absorption coefficient [α(ω)], and reflectivity R(ω). The trends in these spectra are consistent with the previously discussed electronic structures, highlighting the influence of the transition-metal cation on light–matter interaction. Fig. 7a illustrates the real part of the dielectric function, ε1(ω), where Na2LiVH6 displays the highest static dielectric constant, signifying stronger polarization and enhanced electronic screening. Na2LiZrH6 exhibits a comparatively moderate response, whereas Na2LiCrH6 shows the lowest value, indicating reduced polarizability. Hence, the pronounced dielectric response of Na2LiVH6 enhances its suitability for hydrogen storage systems, as efficient polarization promotes effective charge transfer and strengthens the overall adsorption–desorption dynamics. The imaginary part ε2(ω) (Fig. 7b) demonstrates a similar order, where Na2LiVH6 and Na2LiZrH6 possess pronounced low-energy peaks arising from interband transitions near the Fermi level, whereas Na2LiCrH6 displays weaker intensity and a shift of major optical activity toward higher energies. Hence, the distinct interband transitions observed in Na2LiCrH6 enhanced its potential for hydrogen storage applications by promoting light-induced electronic excitation and facilitating efficient hydrogen desorption processes.
 |
| | Fig. 7 Optical response of Na2LiXH6 (X = Zr, V, Cr) perovskite hydrides. | |
The absorption spectra (Fig. 7c) confirm that Na2LiVH6 and Na2LiZrH6 absorb significantly in the visible and near-UV regions, with multiple absorption edges starting near their band gaps, whereas Na2LiCrH6 exhibits a sharp increase only in the ultraviolet domain, consistent with its wider gap. Reflectivity (Fig. 7d) trends complement this observation: Na2LiVH6 and Na2LiZrH6 exhibit high low-energy reflectance (>0.5), indicating reduced transmission, whereas Na2LiCrH6 maintains relatively low reflectivity and a smoother spectral variation. Hence, Na2LiVH6 and Na2LiZrH6 demonstrate strong optical screening and intense interband transitions, making them promising for broadband absorption-based devices, whereas Na2LiCrH6, with its moderate dielectric response and dominant UV absorption, stands out as a potential candidate for ultraviolet optoelectronic or protective coating applications.
Thermal properties
The thermodynamic properties of Na2LiXH6 (X = Zr, V, Cr) perovskite hydrides are crucial in assessing their capabilities for hydrogen storage and release mechanisms. Favorable thermodynamic characteristics in H2 storage materials facilitate effective cycles of hydrogen absorption and desorption, which are essential for preserving reversibility and long-term stability. The structural response of these hydrides to temperature changes and their capacity to efficiently store and release hydrogen can be better understood by examining characteristics like heat capacity, enthalpy, entropy, and free energy. If the hydrides' thermodynamic profile remains constant, they can endure multiple cycles of hydrogenation and dehydrogenation without experiencing noticeable degradation in structure.67 Examining thermodynamic parameters like longitudinal sound velocity (vl), transverse sound velocity (vt), average sound velocity (vm), Debye temperature (θD), and melting temperature (Tm) is essential for understanding the vibrational and lattice dynamics that affect hydrogen diffusion and lattice resilience at high temperatures. Eqn (13)–(15) were used to derive these parameters:68,69| |
 | (13) |
| |
 | (14) |
| | |
Tm = [553 + 5.911(C11)] ± 300
| (15) |
where ρ is the material density, M is the molecular weight, C11 is the elastic constant, and B and G are the bulk and shear moduli. These equations connect the material's thermal and dynamic stability, which is essential for H2 storage devices that operate in temperature-varying environments, to mechanical stiffness and atomic vibrations.
All three compounds (Na2LiZrH6, Na2LiVH6, and Na2LiCrH6) show identical thermodynamic trends with increasing temperature, as shown in Fig. 8a–d and detailed in Table 4. At increasing temperatures, the heat capacity (Fig. 8a) and enthalpy (Fig. 8b) increase monotonically, indicating increased phonon contributions and lattice vibration amplitudes. Notably, Na2LiCrH6 exhibits the highest enthalpy and heat capacity values, indicating improved energy storage and stronger interatomic bonding. The vibrational and configurational entropy contributions grow with temperature, supporting the desorption process by lowering the Gibbs free energy barrier, as the total entropy (Fig. 8c) rises linearly. Fig. 8d shows that all compounds exhibit a consistent decline in free energy as the temperature rises, a typical behavior of thermodynamically stable hydrides that facilitates hydrogen release at higher temperatures.
 |
| | Fig. 8 Thermal parameters of Na2LiXH6 (X = Zr, V, Cr) perovskite hydrides. | |
Table 4 Calculated thermodynamic parameters of Na2LiXH6 (X = Zr, V, Cr) perovskite hydrides
| Parameters |
Na2LiZrH6 |
Na2LiVH6 |
Na2LiCrH6 |
| vl (km s−1) |
3.08 |
3.74 |
4.20 |
| vt (km s−1) |
2.09 |
2.54 |
2.79 |
| vm (km s−1) |
2.28 |
2.77 |
3.05 |
| θD (K) |
369 |
497 |
546 |
| Tm (K) |
835 |
974 |
1104 |
The most reliable thermodynamic performance is shown quantitatively by Na2LiCrH6 with vl = 4.20 km s−1, vt = 2.79 km s−1, vm = 3.05 km s−1, θD = 546 K, and Tm = 1104 K, signifying higher lattice stiffness and strong atomic interactions. Na2LiZrH6 has slightly lower values (θD = 369 K, Tm = 835 K), suggesting a relatively softer lattice, while Na2LiVH6 follows closely (θD = 497 K, Tm = 974 K). By increasing vibrational frequency and phonon transport efficiency, Cr inclusion strengthens lattice rigidity and improves thermal stability, as demonstrated by the observed rise in Debye temperature from Zr to Cr substitution. Na2LiCrH6's mechanical and thermal endurance during hydrogen cycling is improved by its higher Debye temperature, which indicates a higher phonon frequency and a smaller atomic vibration amplitude. These findings demonstrate that Na2LiCrH6 has the strongest lattice among the compounds under investigation, which makes it a viable option for H2 storage applications where effective absorption–desorption performance depends on high thermal and structural stability.
Conclusion
In summary, this in-depth first-principles investigation elucidates the intrinsic correlation between the structural, mechanical, electronic, optical, and thermal characteristics of Na2LiXH6 (X = Zr, V, Cr) double perovskite hydrides. The compounds crystallize to form a cubic Fmm structure, showing negative formation energies and favourable tolerance factors. These factors indicate that these compounds are structurally and thermodynamically stable. All compounds maintain mechanical and thermodynamic stability within the cubic phase, exhibiting progressive improvements in elastic stiffness, cohesive strength, and lattice integrity from Zr to Cr substitution. Among the studied systems, Na2LiVH6 demonstrates the most resilient configuration, supported by its bulk modulus, Debye temperature (597 K), melting point (974 K), and sound velocities, confirming strong atomic bonding and high thermal endurance. Its favorable desorption enthalpy (−70.61 kJ per mol per H2) and volumetric hydrogen density (24.24 g per H2 per L) further verify its superior hydrogen storage capability. In addition, the compound's broad optical absorption and pronounced dielectric response suggest potential for dual hydrogen-optoelectronic functionality. Collectively, the synergistic combination of mechanical rigidity, thermal stability, and optical activity designates Na2LiVH6 as a potential multifunctional material for sustainable hydrogen storage and advanced optoelectronic technologies.
Ethical statement
Approval all authors affirm that this submission is entirely original, has not been published elsewhere, and fully adheres to the established ethical standards for scholarly research and publication.
Author contributions
All authors affirm their substantial contributions to this research and collectively assume full responsibility for its content, encompassing conceptualization, computational analysis, manuscript preparation, and revision processes.
Conflicts of interest
The authors declare that there are no financial or personal relationships that could have influenced the outcomes, interpretation, or conclusions presented in this study.
Data availability
The data will be provided on request by the corresponding author.
Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5ra08708b.
Acknowledgements
The authors sincerely acknowledge the International Islamic University, Islamabad, Pakistan, for extending essential computational facilities and research infrastructure that enabled the successful execution of this study.
References
- O. Faye, J. Szpunar and U. Eduok, A critical review on the current technologies for the generation, storage, and transportation of hydrogen, Int. J. Hydrogen Energy, 2022, 47(29), 13771–13802 CrossRef CAS.
- N. Klopčič, et al., A review on metal hydride materials for hydrogen storage, J. Energy Storage, 2023, 72, 108456 Search PubMed.
- R. Morales-Ospino, A. Celzard and V. Fierro, Strategies to recover and minimize boil-off losses during liquid hydrogen storage, Renewable Sustainable Energy Rev., 2023, 182, 113360 Search PubMed.
- Q. Ain, et al., A precise prediction of structure stability and hydrogen storage capability of KCdH3 perovskite hydride using density functional theory calculations, J. Energy Storage, 2024, 100, 113734 Search PubMed.
- Y. Yang, et al., Recent advances in catalyst-modified Mg-based hydrogen storage materials, J. Mater. Sci. Technol., 2023, 163, 182–211 CrossRef CAS.
- M. Ali, et al., Effective hydrogen storage in Na2 (Be/Mg) H4 hydrides: perspective from density functional theory, Int. J. Hydrogen Energy, 2024, 64, 329–338 CrossRef CAS.
- J. Yan, et al., Intrinsic mechanisms of superior hydrogen storage properties in V–Fe–Ti alloys: a combined experimental and theoretical study, J. Phys. Chem. Solids, 2023, 182, 111582 CrossRef CAS.
- K. Malleswararao, et al., Experimental studies on LaNi4.25Al0.75 alloy for hydrogen and thermal energy storage applications, Int. J. Hydrogen Energy, 2023, 48(69), 26911–26920 CrossRef CAS.
- M. Ali, et al., Enhancement of hydrogen storage characteristics of Na2CaH4 hydrides by introducing the Mg and Be dopant: a first-principles study, Int. J. Hydrogen Energy, 2024, 70, 579–590 CrossRef CAS.
- M. Ali, et al., Atomistic simulations for electronic structure, mechanical stability and optical responses of sodium-based NaAH3 (A = Sc, Ti and V) metal hydride perovskites for hydrogen storage applications, Int. J. Hydrogen Energy, 2025, 128, 749–759 CrossRef CAS.
- M. Ali, et al., A computational investigation of lithium-based metal hydrides for advanced solid-state hydrogen storage, ChemistrySelect, 2024, 9(10), e202304582 CrossRef CAS.
- M. Ali, et al., DFT study of hydrogen-substituted KMgF3 fluoroperovskite: structural, electronic, and storage implications, Inorg. Chem. Commun., 2025, 115186 CrossRef CAS.
- E. Boateng, et al., Functionalization of graphene-based nanomaterials for energy and hydrogen storage, Electrochim. Acta, 2023, 452, 142340 CrossRef CAS.
- Y. Cao, et al., Potential application of metal-organic frameworks (MOFs) for hydrogen storage: simulation by artificial intelligent techniques, Int. J. Hydrogen Energy, 2021, 46(73), 36336–36347 CrossRef CAS.
- C. Y. Wong, et al., Tuning the functionality of metal–organic frameworks (MOFs) for fuel cells and hydrogen storage applications, J. Mater. Sci., 2023, 58(21), 8637–8677 CrossRef CAS.
- Q. Hu, et al., MXene: a new family of promising hydrogen storage medium, J. Phys. Chem. A, 2013, 117(51), 14253–14260 Search PubMed.
- A. Yadav, et al., Study of 2D MXene Cr2C material for hydrogen storage using density functional theory, Appl. Surf. Sci., 2016, 389, 88–95 Search PubMed.
- M. R. Usman, Hydrogen storage methods: review and current status, Renewable Sustainable Energy Rev., 2022, 167, 112743 CrossRef CAS.
- J. B. Von Colbe, et al., Application of hydrides in hydrogen storage and compression: achievements, outlook and perspectives, Int. J. Hydrogen Energy, 2019, 44(15), 7780–7808 CrossRef.
- T. Huang, et al., MOF-derived Ni nanoparticles dispersed on monolayer MXene as catalyst for improved hydrogen storage kinetics of MgH2, Chem. Eng. J., 2021, 421, 127851 CrossRef CAS.
- D. P. Broom, Hydrogen Storage Materials: the Characterisation of Their Storage Properties, Green energy and technology, Springer, London, 2011 Search PubMed.
- A. Bouamrane, et al., Structural characterization of NaMgH2F and NaMgH3, Mater. Res. Bull., 2000, 35(4), 545–549 Search PubMed.
- N. Xu, et al., First-principles study on hydrogen storage properties of the new hydride perovskite XAlH3 (X = Na, K), Int. J. Hydrogen Energy, 2024, 60, 434–440 CrossRef CAS.
- N. Xu, et al., First-principles investigations for the hydrogen storage properties of XVH3 (X = Na, K, Rb, Cs) perovskite type hydrides, J. Mater. Res. Technol., 2023, 26, 4825–4834 CrossRef CAS.
- R. Song, et al., First-principles to explore the hydrogen storage properties of XPtH3 (X = Li, Na, K, Rb) perovskite type hydrides, Int. J. Hydrogen Energy, 2024, 57, 949–957 Search PubMed.
- R. Song, et al., Insight into the mechanical, electronic, kinetic, thermodynamic, and hydrogen storage properties of XFeH3 (X = Ca, Sr, Ba) perovskites for hydrogen storage applications: first-principle calculations, Chin. J. Phys., 2024, 89, 1152–1163 CrossRef CAS.
- X. Fan, et al., Direct synthesis and hydrogen storage behaviors of nanocrystalline Na2LiAlH6, J. Mater. Sci., 2011, 46(10), 3314–3318 CrossRef CAS.
- Y. Liu, et al., Mechanisms for the enhanced hydrogen desorption performance of the TiF4-catalyzed Na2 LiAlH6 used for hydrogen storage, Energy Environ. Sci., 2010, 3(5), 645–653 RSC.
- M. Irfan, et al., Principles-based investigation of lithium-based halide perovskite X2LiAlH6 (X = K, Mn) for hydrogen storage, optoelectronic, and radiation shielding applications, Int. J. Hydrogen Energy, 2024, 91, 775–786 CrossRef CAS.
- T. Tang and Y. Tang, Lithium doping in Na-based double perovskite for hydrogen storage and improving their optoelectronic properties: first-principles investigation, Mater. Chem. Phys., 2024, 316, 129099 CrossRef CAS.
- T. Tang and Y. Tang, First-principles investigations for the structural, optoelectronic and hydrogen storage properties of double perovskite KNaMg2F6-xHx and KNaAe2H6 (Ae = Be, Mg, Ca), Int. J. Hydrogen Energy, 2024, 61, 13–24 CrossRef CAS.
- R.-K. Pan, et al., First principles study on elastic and electronic properties of bialkali alanates M2M′ AlH6, Int. J. Hydrogen Energy, 2018, 43(7), 3862–3870 CrossRef CAS.
- S. J. Clark, et al., First principles methods using CASTEP, Z. Kristallogr. Cryst. Mater., 2005, 220(5–6), 567–570 CrossRef CAS.
- P. Motamarri and V. Gavini, Configurational forces in electronic structure calculations using Kohn-Sham density functional theory, Phys. Rev. B, 2018, 97(16), 165132 CrossRef CAS.
- N. Hernandez-Haro, et al., DFT prediction of band gap in organic-inorganic metal halide perovskites: an exchange-correlation functional benchmark study, Chem. Phys., 2019, 516, 225–231 CrossRef CAS.
- A. Siddique, et al., Structural, electronic, mechanical and dynamical stability properties of LiAH3 (A = Sc, Ti & V) perovskite-type hydrides: a first principle study, Chem. Phys., 2023, 568, 111851 CrossRef CAS.
- W. Khan, Computational screening of BeXH3 (X: Al, Ga, and In) for optoelectronics and hydrogen storage applications, Mater. Sci. Semicond. Process., 2024, 174, 108221 Search PubMed.
- A. N. Khan, et al., Multi-functional DFT and SCAPS-1D analysis of lead-free Z2MgGeI6 (Z = Na, K) double perovskites for optoelectronic, photo-catalytic, and photovoltaic applications, Sol. Energy Mater. Sol. Cells, 2026, 294, 113922 Search PubMed.
- A. Ayyaz, et al., Investigation of hydrogen storage and energy harvesting potential of double perovskite hydrides A2LiCuH6 (A = Be/Mg/Ca/Sr): a DFT approach, Int. J. Hydrogen Energy, 2025, 102, 1329–1339 Search PubMed.
- V. M. Goldschmidt, Die gesetze der krystallochemie, Naturwissenschaften, 1926, 14(21), 477–485 CrossRef CAS.
- S. Fatima, et al., Efficient hydrogen storage in KCaF3 using GGA and HSE approach, Int. J. Hydrogen Energy, 2023, 48(9), 3566–3582 CrossRef CAS.
- S. Al, M. Yortanlı and E. Mete, Lithium metal hydrides (Li2CaH4 and Li2SrH4) for hydrogen storage; mechanical, electronic and optical properties, Int. J. Hydrogen Energy, 2020, 45(38), 18782–18788 CrossRef CAS.
- H. Alkhalidi, Investigation of double perovskite hydrides X2CuGaH6 (X = Li, Na) for hydrogen energy storage, Int. J. Hydrogen Energy, 201, 2025, 152976 Search PubMed.
- P. Schouwink, et al., Structure and properties of complex hydride perovskite materials, Nat. Commun., 2014, 5(1), 5706 CrossRef CAS PubMed.
- Z. Zang, et al., Study of structural, electronic, phonon, thermodynamic, and hydrogen storage properties of hydride Rb2AsSnH6 perovskites: DFT insights, Int. J. Hydrogen Energy, 2025, 133, 225–234 CrossRef CAS.
- M. A. Ullah, et al., An approach towards next-generation hydrogen storage: a DFT study on A 2 LiTiH 6 (A = K, Ca) perovskite hydrides, RSC Adv., 2025, 15(46), 38714–38728 RSC.
- D. Samanta, et al., Phonon anharmonicity and soft-phonon mediated structural phase transition in Cs3Bi2Br9, Phys. Rev. B, 2023, 108(5), 054104 CrossRef CAS.
- A. Hosen, et al., First-principles insights into NaScQH6 (Q = Fe, Ru, Os): promising high-density hydrogen storage materials, Int. J. Hydrogen Energy, 2025, 177, 151392 CrossRef CAS.
- M. Archi, et al., Investigation of structural, phonon, thermodynamic, electronic, and mechanical properties of non-toxic XZnH3 (X = Li, Na, K) perovskites for solid-state hydrogen storage: a DFT and AIMD approach, J. Energy Storage, 2025, 112, 115492 CrossRef.
- A. Hosen, et al., Systematic Computational Screening and Design of Double Perovskites Q2LiMH6 (Q = K, Rb; M = Ga, In, Tl) for Efficient Hydrogen Storage: A DFT and AIMD Approach, Surf. Interfaces, 2025, 106608 CrossRef CAS.
- M. mana Al-Anazy, et al., Study of alkaline metals hydrides RbXH3 (X = Mg/Ca/Sr/Ba) for green energy and hydrogen storage applications, Int. J. Hydrogen Energy, 2024, 78, 927–937 CrossRef.
- L. Charpin and A. Ehrlacher, Estimating the poroelastic properties of cracked materials, Acta Mech., 2014, 225(9), 2501–2519 CrossRef.
- S. Al, Mechanical and electronic properties of perovskite hydrides LiCaH3 and NaCaH3 for hydrogen storage applications, Eur. Phys. J. B, 2021, 94(9), 182 CrossRef CAS.
- A. Züttel, et al., LiBH4 a new hydrogen storage material, J. Power Sources, 2003, 118(1–2), 1–7 CrossRef.
- C. Kürkçü, Computational insights of double perovskite Na2CaCdH6 hydride alloy for hydrogen storage applications: a DFT investigation, Sci. Rep., 2024 DOI:10.1038/s41598-024-76062-0.
- M. Baaddi, et al., The effect of strain on hydrogen storage characteristics in K 2 NaAlH 6 double perovskite hydride through first principle method, Environ. Sci. Pollut. Res., 2024, 31(53), 62056–62064 CrossRef CAS PubMed.
- M. Tarekuzzaman, et al., Cesium-Based Perovskite Hydrides: A Theoretical Insight into Hydrogen Storage and Optoelectronic Characteristics, Solid State Commun., 2025, 116043 CrossRef CAS.
- M. El Akkel and H. Ez-Zahraouy, First-principles study of triaxial strain effect on structural, mechanical, electronic, optical, and photocatalytic properties of K2SeBr6 for solar hydrogen production, Chem. Phys., 2025, 595, 112739 CrossRef CAS.
- Q. Lin and Y. Yan, First-principles study on the physical properties of double perovskite X2NaAlH6 (X= K, Cs) and X2NaInH6 (X= Rb, Cs) for hydrogen storage, Int. J. Hydrogen Energy, 2025, 135, 429–439 CrossRef CAS.
- G. N. Greaves, et al., Poisson's ratio and modern materials, Nat. Mater., 2011, 10(11), 823–837 CrossRef CAS.
- H. Naeem, et al., Exploring novel characteristics of GaBaX3 (X = F, Cl, Br, I, H) for energy harvesting applications: a DFT-based analysis, Int. J. Hydrogen Energy, 2025, 105, 203–213 Search PubMed.
- M. Ali, et al., Structural, optoelectronic and thermodynamical insights into 2H-ZrO2: a DFT investigation, Inorg. Chem. Commun., 2024, 160, 111891 Search PubMed.
- M. Ali, M. Yousaf and J. Munir, Achieving controllable multifunctionality through layer sliding, J. Mol. Graphics Modell., 2024, 126, 108638 CrossRef CAS.
- M. Ali, et al., CO Adsorption on two-dimensional 2H-ZrO2 and its effect on the interfacial electronic properties: implications for sensing, Phys. Scr., 2023, 98(11), 115801 CrossRef CAS.
- M. Ali, et al., First-principles investigation of structural, mechanical, and optoelectronic properties of Hf2AX (A = Al, Si and X = C, N) MAX phases, J. Am. Ceram. Soc., 2024, 107(4), 2679–2692 Search PubMed.
- M. Ali, et al., Exploring the electronic structure, mechanical stability and optoelectronic responses of arsenic-based M2AsX (M = Nb, Mo and X = C, N) MAX phase ceramics, J. Mol. Graphics Modell., 2025, 136, 108965 CrossRef CAS PubMed.
- S. Al-Qaisi, et al., A comprehensive first-principles computational study on the physical properties of lutetium aluminum perovskite LuAlO3, Mater. Chem. Phys., 2020, 250, 123148 CrossRef CAS.
- P. Wachter, M. Filzmoser and J. Rebizant, Electronic and elastic properties of the light actinide tellurides, Phys. B, 2001, 293(3–4), 199–223 CrossRef CAS.
- O. L. Anderson, A simplified method for calculating the Debye temperature from elastic constants, J. Phys. Chem. Solids, 1963, 24(7), 909–917 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Malik Muhammad Asif Iqbal
a,
Malik Muhammad Asif Iqbal



















