DOI:
10.1039/D5RA08679E
(Paper)
RSC Adv., 2026,
16, 13369-13380
Synthesis and biological evaluation of N/O-propargylated diarylpyrimidines as dual inhibitors of acetylcholinesterase and monoamine oxidase
Received
10th November 2025
, Accepted 16th February 2026
First published on 10th March 2026
Abstract
Alzheimer's disease is a complex neurological disorder and is becoming a global health concern as the population ages. Considering the complex aetiology of the disease and ineptness of single-targeted drugs, the development of multi-targeted drugs emerges as the most effective strategy for the treatment of the disease. Cholinesterases and monoamine oxidases are amongst the most widely explored targets in Alzheimer’s disease, and their dual inhibition offers a promising approach for achieving multipotent therapeutic effects. Herein, we designed and synthesized a series of N/O-propargylated diaryl pyrimidines and evaluated their inhibitory activity against acetylcholinesterase (AChE) and monoamine oxidase (MAO) enzymes. Most of the compounds were found to be active against AChE, MAO-A and MAO-B. Amongst the synthesised derivatives of the series, compounds, compounds NV-1 and NV-9 exhibited a balanced multipotent activity profile against both the targets i.e. acetylcholinesterase and monoamine oxidase. Compounds NV-1 and NV-9 displayed IC50 values of 1.30 µM and 0.88 µM against AChE, 0.232 µM and 9.31 µM against MAO-A and 0.949 µM and 9.23 µM against MAO-B, respectively. In the reversibility inhibition studies, both the compounds were found to be reversible in nature. In kinetic inhibition studies, both NV-1 and NV-9 showed non-competitive inhibition for AChE. Additionally, NV-1 and NV-9 were found to be moderately neuroprotective in nature and exhibit no cytotoxicity at lower compound concentrations. In the partition coefficient studies (octanol/water), the compound NV-9 was found to be lipophilic in nature. Molecular docking studies illustrate their stability within the active cavity of both enzymes. Simulation studies confirmed the thermodynamic stability of these compounds within the cavity for up to 100 ns. Thus, the N/O-propargylated diarylpyrimidines have the potential to be developed as multipotent drugs for the treatment of Alzheimer's disease.
Introduction
Alzheimer's disease (AD) is a severe neurological condition,1 and more than 50 million people are affected worldwide. It is the most common cause of dementia that causes memory loss and cognitive impairment2 by the degeneration of the neuronal cells, causes continuous declining of thinking, communication, and social skills, and affects the normal functioning of daily life.3 The available treatments give only symptomatic relief, and there is no drug available for the complete eradication of the disease.4 The aetiology of Alzheimer's disease is complex in nature,5 and many pathological targets are involved in the initiation and progression of the disease, including increased acetylcholinesterase (AChE) inhibitor concentration, overactivation of monoamine oxidase (MAO) enzyme, excess formation of amyloid plaques and tangled clumps of tau protein, disruption of glutaminergic and adrenergic systems, metal ion dyshomeostasis and increased reactive oxygen species (ROS) levels.6,7 Because of the complex aetiology of Alzheimer's disease and the involvement of many pathological targets, the development of multi-target-directed ligands (MTDLs) for the simultaneous inhibition of two or more targets is proposed as an effective treatment strategy.8
Pyrimidine scaffold is an important part of DNA and RNA involved in almost all the biological systems.9 Many pyrimidine-based derivatives have been designed, synthesized, and evaluated as anti-Alzheimer agents.10 Aryl substitution at the different positions of the pyrimidine scaffold have shown different biological activities.11,12 S. Ahmad et al. designed and synthesized dihydropyrimidine derivatives as dual binding (AChE and BuChE) inhibitors with good binding affinity to the key amino acid residues of the catalytic active site (CAS) and peripheral anionic site (PAS) of AChE.13 Manzoor and group reported phenyl-sulfonyl substituted pyrimidine carboxylate derivatives as potential multi-target leads for the inhibition of AChE and AChE-induced amyloid-β aggregation. Almost all the compounds of the series showed selective AChE inhibition over BuChE in higher nanomolar range.14 Pyrimidine-triazolopyrimidine and pyrimidine-pyridine-based hybrid scaffolds were synthesized and screened against cholinesterase enzymes, showing IC50 values in the nanomolar range and interactions with both the CAS and PAS of AChE.15 Blood–brain barrier (BBB) permeability is one of the major challenges in AD drug discovery. A series of novel propargylamine-modified aminoalkyl imidazole-substituted pyrimidinylthiourea derivatives were screened as multifunctional agents for potential drug-likeness, low cytotoxicity, and enhanced BBB permeability. The lead compound from this series was reported to ameliorate scopolamine-induced cognitive impairment in a mouse model of Alzheimer's disease.16 Nadeem et al. reported variation in several functionalities and linker lengths in indole-containing 2-arylidine derivatives of thiazolopyrimidine that showed enhanced cholinesterase and monoamine oxidase inhibition activities along with improved BBB permeability and no cytotoxicity to the neuronal cells.17 Javed et al. synthesised and biologically evaluated a series of pyrimidine/pyrrolidine-sertraline hybrids and improved short and long-term memory, and enhanced learning behavior in AD mouse model was observed.18 Recently, substituted pyrimidine scaffolds were synthesized and studied as potent anti-AD agents using in vivo mouse model by Swati and group. The lead molecule showed promising activities comparable to that of the standard.19
For the past few years, our laboratory has been working on the design, synthesis, and screening of small organic molecules as multi-target-directed ligands for the treatment of AD. A number of pyrimidine derivatives were synthesized, and a structure–activity relationship profile was developed for the identification of active leads with a balanced multipotent profile against different targets of AD. The propargyl group has been identified as an important pharmacophore20,21,22 for its interaction with monoamine oxidases, and we are further exploring this moiety in the pyrimidine scaffold. We synthesized diphenyl-substituted pyrimidine derivatives as dual inhibitors of AChE and MAO enzymes. With variation in the substitution pattern, these compounds showed varying activity profiles and potent leads were identified, showing a balanced activity profile against both the enzymes. The identified leads exhibited good neuroprotective potential against SH-SY5Y cells in the 6-OHDA and H2O2-induced neurotoxicity.22,23 Additionally, the lead molecule were evaluated in vivo using scopolamine-induced neurotoxicity in mice model of Alzheimer's disease.24 Recently, a series of phenylstyrylpyrimidine derivatives was synthesized and evaluated as anti-AD agents, exhibiting multipotent profile against AChE, MAO, and self-induced amyloid-β aggregation.20 Computational studies revealed that the propargyl group strongly interacts with the MAO enzyme, probably through the covalent interaction between its terminal alkyne and the FAD co-factor.25
Taking leads from the previous research work done in our laboratory, the computational studies revealed that the pyrimidine moiety usually orients towards the FAD co-factor of the MAO enzyme. In our previous studies,22,23,24 the propargyl moiety was substituted at the diaryl rings attached to the pyrimidine ring, and many times it orients away from the FAD pocket. The propargyl group is a classified MAO pharmacophore, which is well known for developing mechanism-based inhibitors. Propargylation at the pyrimidine core was designed to generate an MAO-reactive warhead with a rigid heteroaromatic scaffold, exhibiting improved active-site positioning, enabling mechanism-based FAD interaction, and enhanced overall binding and activity toward the MAO enzyme. Thus, in the present study, we incorporated the propargyl moiety on the pyrimidine ring and investigated its impact on the inhibition activity of the target enzymes. The attachment of two phenyl rings to the pyrimidine nucleus provided an elongated structure, which can effectively bind to the PAS and CAS of the AChE enzyme. Most of the synthesized compounds showed potent inhibitory activities against AChE and MAO enzymes at lower to sub-micromolar concentrations. The neuroprotection potential and cytotoxicity of the potent compounds were evaluated against SH-SY5Y neuronal cells. In addition, molecular docking and dynamics simulation studies were performed to assess the binding interactions and stability of the ligands and enzyme–ligand complexes.
Results and discussion
Chemistry
All the compounds were synthesized based on the reaction procedures described in Scheme 1. Briefly, substituted acetophenone and benzaldehydes were reacted in the presence of urea and iodine to produce pyrimidinone, which was chlorinated using POCl3 in toluene. Finally, the chlorinated derivative was substituted with propargyl amine/alcohol in DMF to get the final product. The formation of the product was confirmed by 1H, 13C, and HMRS data.
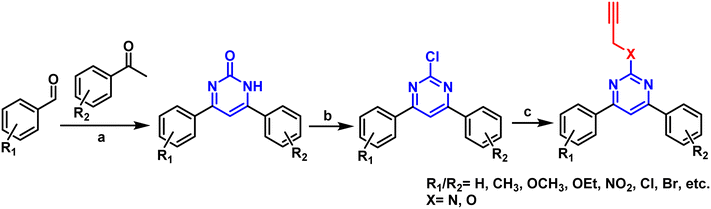 |
| | Scheme 1 Synthetic route for diaryl-substituted propargylated pyrimidine derivatives. | |
Biological results
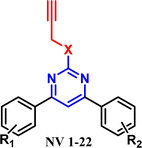
hMAO and AChE inhibition studies. The evaluation of MAO and AChE inhibition studies of the synthesized compounds is performed through fluorometric absorption methods using Amplex Red26 and Ellman's assay27 kits from Molecular Probes (Invitrogen), Life Technologies. The results of these inhibition assays are reported in the form of IC50 (µM) values summarised in Table 1. It has been observed that some of the compounds were found to inhibit both MAO and AChE enzymes in the low micromolar range concentration. The aryl rings attached to the pyrimidine scaffold were alternatively substituted (R1 and R2) with different groups, including electron-withdrawing and electron-donating substituents, to develop a structure–activity relationship (SAR) profile. As evident from Table 1, compound NV-1 was found to be the most potent inhibitor of MAO-A and MAO-B isoforms with IC50 values of 0.232 µM and 0.949 µM, respectively. It displayed IC50 value of 1.30 µM against the AChE enzyme. Similarly, compound NV-9 was found to be the most potent inhibitor of the AChE enzyme with IC50 value of 0.88 µM, and it showed IC50 values of 9.31 µM and 9.23 µM against MAO-A and MAO-B isoforms, respectively.
Table 1 Results of the biological inhibition studies of MAO-A, MAO-B and AChEa
|
| Entry name |
X |
R1 |
R2 |
IC50 values (mean ± S.E., µM) |
| hMAO-A |
hMAO-B |
AChE |
| nd = not determined. |
| NV-1 |
N |
4-CH3 |
H |
0.232 ± 0.04 |
0.949 ± 0.001 |
1.30 ± 0.01 |
| NV-2 |
N |
H |
3-Br |
1.667 ± 0.05 |
4.429 ± 0.06 |
8.70 ± 0.02 |
| NV-3 |
N |
4-Cl |
4-Br |
0.996 ± 0.03 |
9.728 ± 0.02 |
3.48 ± 0.012 |
| NV-4 |
N |
4-Cl |
H |
>10 |
2.528 ± 0.04 |
7.00 ± 0.005 |
| NV-5 |
N |
4-Br |
4-OMe |
>10 |
12.371 ± 0.1 |
3.28 ± 0.006 |
| NV-6 |
N |
4-OMe |
H |
>10 |
10.459 ± 0.02 |
4.23 ± 0.006 |
| NV-7 |
N |
4-CH3 |
4-OMe |
0.805 ± 0.02 |
13.191 ± 0.1 |
9.16 ± 0.009 |
| NV-8 |
N |
4-Cl |
4-OMe |
>10 |
7.547 ± 0.02 |
6.30 ± 0.01 |
| NV-9 |
N |
3,4,5-OMe |
3-NO2 |
9.31 ± 0.1 |
9.231 ± 0.02 |
0.88 ± 0.013 |
| NV-10 |
N |
4-CH3 |
3-Br |
8.80 ± 0.02 |
13.115 ± 0.17 |
23.41 ± 0.004 |
| NV-11 |
O |
H |
3-Br |
9.815 ± 0.02 |
14.275 ± 0.03 |
6.56 ± 0.015 |
| NV-12 |
O |
4-OMe |
4-OMe |
4.90 ± 0.07 |
>25 |
26.24 ± 0.015 |
| NV-13 |
O |
4-OMe |
4-Cl |
>10 |
5.947 ± 0.02 |
1.38 ± 0.003 |
| NV-14 |
O |
4-OMe |
H |
0.808 ± 0.009 |
>25 |
6.99 ± 0.007 |
| NV-15 |
O |
H |
2,4-Cl |
>10 |
>25 |
3.26 ± 0.006 |
| NV-16 |
O |
4-OMe |
4-Br |
>10 |
8.602 ± 0.048 |
6.96 ± 0.014 |
| NV-17 |
O |
4-CH3 |
3-Br |
6.68 ± 0.02 |
>25 |
7.75 ± 0.042 |
| NV-18 |
O |
4-CH3 |
4-OMe |
>10 |
>25 |
5.46 ± 0.023 |
| NV-19 |
O |
4-Cl |
H |
4.38 ± 0.024 |
>25 |
8.73 ± 0.038 |
| NV-20 |
O |
3-NO2 |
H |
7.69 ± 0.08 |
>25 |
3.55 ± 0.045 |
| NV-21 |
O |
3-Br |
3,4-OMe |
15.26 ± 0.31 |
>25 |
19.46 ± 0.025 |
| Clorgyline |
0.049 ± 0.002 |
nd |
nd |
| Pargyline |
nd |
0.072 ± 0.001 |
nd |
| Donepezil |
nd |
nd |
0.026 ± 0.003 |
Reversibility inhibition studies. Reversible inhibition studies were conducted to assess whether the compounds were reversible or irreversible inhibitors28 of AChE and MAO-B enzymes. At IC50 concentration, NV-1 showed 57.43% and 78.92% activity, while NV-9 showed 65.30% and 55.27% inhibitory activity against AChE and MAO-B, respectively. Inhibition was increased by increasing the concentrations to 10 × IC50 and 100 × IC50. The maximum inhibition was achieved at 100 × IC50, where only 7.45% and 7.49% activities were observed for AChE, while 43.92% and 37.39% activities were observed for MAO-B by NV-1 and NV-9, respectively. The activity was recovered by diluting the compounds to a concentration of 0.1 × IC50. A total of 96.56% and 97.45% activity was recovered for AChE and 85.38% and 75.44% activity for the MAO-B enzyme by NV-1 and NV-9, respectively (Fig. 1). This study demonstrates the reversible nature of both the inhibitors against AChE and MAO-B enzymes.
 |
| | Fig. 1 Reversibility inhibition study of NV-1 and NV-9 for AChE and MAO-B enzymes. | |
Kinetic studies of VAChE inhibition. Kinetic studies were performed with the lead compounds NV-1 and NV-9 against AChE to determine the mechanism of inhibition.28 From the reciprocal Lineweaver–Burk plots, it was observed that with an increase in the concentration of the compound, reduced Vmax and unaffected Km values are obtained, as shown in Fig. 2. The intercept of the double reciprocal Lineweaver–Burk plot is on the x-axis, showing that the reaction follows non-competitive inhibition. Therefore, the lead compounds NV-1 and NV-9 have an affinity to bind to free enzymes on a site other than the active site (allosteric site) and thus reduce the enzyme's ability to catalyse the reaction regardless of the substrate concentration.
 |
| | Fig. 2 Kinetic studies on the AChE inhibition mechanism by NV-1 and NV-9. Lineweaver–Burk plots of AChE initial velocities at increasing substrate concentrations (0.25–6 mM) in the presence and absence of the inhibitors. | |
Neuroprotective effects. The most potent compounds (NV-1 and NV-9) were evaluated for their neuroprotection potential using neurotoxin 6-hydroxydopamine (6-OHDA) on human neuroblastoma SH-SY5Y cells. Compound NV-9 did not show any neuroprotective effects, while NV-1 displayed moderate neuroprotective potential against the 6-OHDA neurotoxin. As the concentration of the inhibitor increases from 0.1 µM to 10 µM, the percentage cell viability also increases. As depicted in Fig. 3, compound NV-1 showed a maximum percentage cell viability of 63.97%, while NV-9 showed a maximum cell viability of 51.57%. Therefore, the compounds exhibited only moderate neuroprotective potential.
 |
| | Fig. 3 Neuroprotective studies of NV-1 and NV-9 against SH-SY5Y cells. | |
Cytotoxicity studies. To assess the cell viability, the lead compounds (NV-1 and NV-9) were evaluated for their cytotoxicity against neuronal cells (SH-SY5Y). The test compounds at 0.1 µM, 1 µM and 10 µM concentrations were incubated for 24 h with SH-SY5Y cells, and the viability of the cells was determined using the MTT assay, as shown in Fig. 4. With the increase in the concentration of the test compounds, the percentage cell viability decreases. The compounds were found to be non-toxic at lower concentrations. Compound NV-1 showed a minimum cell viability of 51.38% at 10 µM, whereas it showed a maximum cell viability of 61.36% at the lowest (0.1 µM) compound concentration. As more than 50% cell viability was observed even with the highest compound concentration (10 µM), so, the lower micromolar IC50 values of the synthesized compounds suggest minimal cytotoxic effects on the neuronal cell lines.
 |
| | Fig. 4 Cytotoxicity studies of NV-1 and NV-9 against SH-SY5Y cells after 24 h of treatment. | |
Molecular docking. Molecular docking is an important tool for understanding the interactions between target proteins and test molecules. It provides information about the orientation of the test molecules and their interactions with amino acids in the active cavity of the enzyme.29 All the synthesized compounds of the series were docked against AChE (PDB ID-1EVE co-crystallised with donepezil) to know the binding interactions with the active sites of AChE. The docking studies were performed using Maestro 12.8 (Schrodinger LLC) software. The interactions and alignments of the most active compounds, NV-1 and NV-9, from in vitro studies are depicted in Fig. 5. The active site of AChE is divided into two compartments, namely, the catalytic active site (CAS) and peripheral anionic site (PAS). The CAS is mainly composed of Trp84, Tyr130, Phe330, Phe331, and the PAS consists of Tyr70, Asp72, Tyr121, Trp279, and Tyr334 amino acid residues at its active sites.30 The pyrimidine ring of both NV-1 and NV-9, with an N-propargyl moiety, is accommodated in the PAS, where one of the phenyl rings containing the methyl group of compound NV-1 shows π–π stacking interactions with Tyr70. Another phenyl ring of the compound is accommodated in the CAS, where it displays strong π–π stacking interactions with the benzene ring of Phe330 and the 5-membered indole ring of Trp84. NO2-containing phenyl ring of compound NV-9 showed π–π and cationic–π interactions with Phe330 and Trp84, respectively, in the CAS region. It confirmed that the test compounds interact with both the CAS and PAS of AChE, thus making them more potent against enzymatic inhibition.
 |
| | Fig. 5 (A) 2D interactions of NV-1 with amino acid residues of AChE, (B) 3D binding pattern of NV-1 with amino acid residues at the active site of AChE, (C) 2D interactions of NV-9 with amino acid residues of AChE, and (D) 3D binding pattern of NV-9 with amino acid residues at the active site of AChE. | |
Similarly, both the compounds were docked at hMAO-B (PDB ID-2BYB co-crystallised with pargyline) to determine the binding interactions at the active site of the enzyme. The active site of MAO-B is mainly divided into two cavities, namely, the entrance cavity and the substrate cavity. The side chain of Ile199 separates both the cavities and acts as a gate between them.31 The pyrimidine ring with the N-propargyl moiety of NV-1 was accommodated in the substrate cavity and was surrounded by Phe99, Phe103, Thr202, and Ser200, while the pyrimidine ring of NV-9 was situated in the entrance cavity, which was surrounded by Ile198, Ile199, and Tyr326 (Fig. 6). The methyl-containing phenyl ring of the compound NV-1 was at the entrance cavity, while the other phenyl ring was situated deep within the substrate cavity. Similarly, the trimethoxy phenyl ring of the compound NV-9 was lying deep within the active substrate cavity, while the nitrophenyl ring showed a cationic–π interaction between the benzene ring of Tyr435 and the nitrogen atom of NO2 group. Thus, it is evident that the compounds are well accommodated within the cavities and show similar interactions to the standard inhibitor pargyline.
 |
| | Fig. 6 (A) 2D interactions of NV-1 with amino acid residues of MAO-B, (B) 3D binding pattern of NV-1 with amino acid residues at the active site of MAO-B, (C) 2D interactions of NV-9 with amino acid residues of MAO-B, and (D) 3D binding pattern of NV-9 with amino acid residues at the active site of MAO-B. | |
Molecular dynamics simulation studies. Understanding the protein-ligand binding interaction is crucial in the drug design and development process, and molecular dynamics simulation can be used as an effective tool for the identification of potent leads.32 It is an important tool for understanding the stability and conformational changes of ligands within the active cavity of an enzyme. Therefore, the most potent compounds, NV-1 and NV-9, were subjected to MD simulation for 100 ns using GROMACS 2021.3. Fig. 7 depicts the results of the analysis from the MD trajectory of the protein–ligand complex of NV-9. Initially, there was a fluctuation in the RMSD plot (Fig. 7A) of the protein between 0.075 and 0.225 nm. Protein RMSD stabilizes between 0.08 and 0.15 nm, and the ligand's RMSD value was found to be extremely stable throughout the simulation ranging from 0.15 to 0.20 nm. The protein and ligand RMSD values were found to be in the acceptable range for both the test compounds, suggesting that the protein and ligand complex was stable throughout the time-period of MD simulation. Additionally, the radius of gyration (Fig. 7B) was calculated to determine the degree of compactness in the structure and to gain insights into the overall dimensions of the protein. The rGy value remained constant throughout the entire trajectory time at 2.32 nm, indicating the high stability of the complex. The stability and flexibility in the protein structure were recorded using RMSF throughout the simulation study to predict the dynamic behavior of the amino acid residues. The analysis of the root mean square fluctuation (RMSF) plot showed that there were no internal fluctuations throughout the simulations, and the interaction RMSF values stayed well within the permissible range of 0.05 to 0.4 nm. The accommodation and stability of the ligand within the catalytic pocket are further supported by the lower RMSF values of the ligand in the range of 0.02–0.18 nm with minimal fluctuation during the simulation (Fig. 7C and D).
 |
| | Fig. 7 Molecular dynamics trajectory analysis of the NV9-protein complex. (A) Protein–ligand RMSD; (B) radius of gyration for the complex, (C) RMSF for the protein and (D) RMSF for the ligand. | |
Fig. 8 shows the results of the analysis from the MD trajectory of the protein–ligand complex of NV-1. Initially, there was a fluctuation in the RMSD plot (Fig. 8A) of the protein between 0.1 and 0.4 nm for 18 ns. After that, it stabilized between 0.4 and 0.42 nm, and the ligand's RMSD value was found to be extremely stable throughout the simulation from 0.075 to 0.12 nm. The protein and ligand RMSD values were within the acceptable range for both the ligands, suggesting that the protein and the ligand were stable throughout the time-period of MD simulation. Additionally, the radius of gyration (Fig. 8B) was calculated to determine the degree of compactness in the structure and to gain insights into the overall dimensions of the protein. This revealed that the complex is highly stable, as the rGy value remains constant throughout the entire trajectory time at 2.33–2.36 nm. The stability and flexibility in the protein structure were recorded using RMSF throughout the simulation study to predict the dynamic behavior of the amino acid residues. The analysis of the root mean square fluctuation (RMSF) plot showed that there were no internal fluctuations throughout the simulations, and their RMSF values fell well within the permissible range of 0.07 to 0.6 nm. The accommodation and stability of the ligand within the catalytic pocket are further supported by the lower RMSF values of the ligand in the range of 0.02–0.22 nm without much fluctuation (Fig. 8C and D).
 |
| | Fig. 8 Molecular dynamics trajectory analysis of the NV1-protein complex. (A) Protein–ligand RMSD; (B) radius of gyration of the complex, (C) RMSF for the protein and (D) RMSF for the ligand. | |
Physicochemical properties. For the evaluation of the drug-like characteristics of NV-1 and NV-9, various physiological parameters33 were calculated using the QikProp application of the Schrodinger software. Both the compounds followed Lipinski's rule of five and showed drug-like properties. The molecular weights of the compounds are below 500. The log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values of both the compounds are less than or equal to five. The hydrogen bond donor and hydrogen bond acceptor atoms of compound NV-1 are less than 5, whereas the hydrogen bond acceptor atoms of compound NV-9 are 5.75. The QPlogBB values of both the compounds lies within the optimum range of −3.0 to 1.2, and both show 100% human oral absorption.
P values of both the compounds are less than or equal to five. The hydrogen bond donor and hydrogen bond acceptor atoms of compound NV-1 are less than 5, whereas the hydrogen bond acceptor atoms of compound NV-9 are 5.75. The QPlogBB values of both the compounds lies within the optimum range of −3.0 to 1.2, and both show 100% human oral absorption.
Lipophilicity evaluation using an octanol-water partition coefficient study. Further, we experimentally, we experimentally evaluated the lipophilicity of the lead compound (NV-9) using an octanol/water partition coefficient study (Table S1 and Fig. S1–S3). An HPLC calibration curve was generated to enable accurate and reliable quantification of the compound in both hydrophilic and lipophilic phases. The compound was clearly detected in the octanol phase, whereas no peak was observed in the aqueous phase, indicating a strong preference of the compound for the lipophilic environment. These results support the computational calculation of the logP value of 4.42 reported in Table 2. The compound is insoluble in the aqueous phase (precipitated out at the aqueous–organic interface) and hence the logP value could not be calculated. Thus, NV-9 displayed strong binding affinity to AChE, MAO-A, and MAO-B, and in the lipophilicity evaluation studies, this compound was found to be highly lipophilic. A suitable salt formulation of this compound is expected to further improve the BBB permeability and drug-like characteristics.
Table 2 Physicochemical properties of the most potent compounds, NV-1 and NV-9a
| Compound |
Mol. wt |
LogP |
HB donor |
HB acceptor |
% Human oral absorption |
QPlogS |
QPlogHERG |
CNS |
QPlogBB (optimum range −3.0 to 1.2) |
Predicted BBB permeability |
| +ve = blood–brain barrier permeable and −ve = not permeable for blood–brain barrier. |
| NV-1 |
299.37 |
5.05 |
1.5 |
2.5 |
100 |
−5.91 |
−6.80 |
0 |
−0.085 |
+ve |
| NV-9 |
420.42 |
4.42 |
1.5 |
5.75 |
100 |
−6.18 |
−6.43 |
−2 |
−1.417 |
+ve |
Structure–activity relationship studies. A series of N/O-propargyl-substituted diarylpyrimidines were designed and synthesized. A total of 21 molecules were synthesized by varying the substitution pattern at both the aryl rings attached to the pyrimidine scaffold. Various electron-withdrawing and electron-donating substituents were attached to the aryl rings to develop a structure–activity relationship (SAR) profile (Fig. 9). The molecules were evaluated for their inhibitory potential against AChE, MAO-A, and MAO-B. Most of the compounds were active against the AChE enzyme, while selective activity was observed against the MAO-A and MAO-B isoforms. In general, there was no direct correlation between the substitution pattern and MAO-A inhibitory activity; however, N-propargylated pyrimidine derivatives were found to be more potent than the O-propargylated derivatives against the MAO-B isoform. On a similar note, no clear correlation was observed between AChE inhibitory activity and the presence of N/O-propargyl substituents on the pyrimidine scaffold. NV-1 with R1 as a para methyl substituent and R2 as H was found to be the most potent inhibitor of MAO-A and MAO-B isoforms with IC50 values of 0.232 µM and 0.949 µM, respectively. NV-1 displayed IC50 values of 1.30 µM against AChE enzymes. The diaryl rings were substituted with different substituents, such as CH3, OCH3, Cl, Br, and NO2 and no significant improvement in the activity was observed against the target enzymes. NV-9 with a 3,4,5-tri-OMe substituent on the first aryl ring and 3-NO2 group on the other aryl ring was found to be the most potent AChE inhibitor with IC50 value of 0.88 µM. However, the MAO-A and MAO-B inhibition activity of NV-9 was found to be reduced, as reported in Table 1. Substitution of either N- and O-propargylated groups on the pyrimidine ring did not show any direct impact on the AChE inhibitory activity; however, they influenced the MAO-B inhibitory potential of the compounds, whereas the N-propargylated derivatives were found to be more potent. Many of the O-propargylated compounds (NV-14, NV-15, and NV-17 to NV-21) were found to be inactive against the MAO-B isoform (IC50 > 25 µM). Molecules possessing planar or partially planar architectures can adopt extended conformations that enable simultaneous interaction with both the CAS and the PAS of AChE, while elongated substituents are expected to traverse the flexible gating residues and accommodate within the large substrate cavity of MAO-B. Cationic–π interactions and π–π stacking interactions with aromatic residues are essential for an enhanced activity profile, and both were observed for the leads in the docking studies. Thus, in this series, NV-1 and NV-9 were identified as the most potent derivatives with a balanced activity profile against AChE, MAO-A and MAO-B enzymes.
 |
| | Fig. 9 Heatmap depicting the variation in IC50 values of the synthesized derivatives for the structure activity relationship (SAR) against MAO and AChE enzymes. Blue and red colours indicate the most and least potent compounds with the lowest and highest IC50 values, respectively. White colour represents inactive compounds at the tested concentrations. | |
Conclusion
Alzheimer's disease is associated with multiple pathological hallmarks, and the exact aetiology of the disease is not yet known. It is considered a complex neurodegenerative condition that requires simultaneous inhibition of two or more targets to cure the disease effectively. AChE and MAO are two prime targets involved in the progression of the disease. We synthesized a series of N/O-propargylated diarylpyrimidines for the simultaneous inhibition of AChE and MAO enzymes. A total of 21 derivatives were synthesized by varying the substitution pattern with either electron-withdrawing or electron-donating groups at both the aryl rings. Amongst the synthesized compounds, NV-1 displayed the most potent MAO inhibitory activities with IC50 values of 0.232 µM and 0.949 µM against MAO-A and MAO-B, respectively. Similarly, NV-9 was found to be the most potent AChE inhibitor with IC50 value of 0.88 µM. It also showed inhibitory activity against MAO-A and MAO-B in the lower micromolar range. In the reversible inhibition studies, both compounds were found to be reversible inhibitors of AChE and MAO enzymes. The compounds are well accommodated within the active sites of enzymes. Although NV-1 and NV-9 indicate strong binding affinity to AChE, MAO-A, and MAO-B, based on the in silico prediction and lipophilicity evaluation, their BBB permeability could be further improved in future work for these compounds to be considered as drug candidates for AD treatment. In MD simulation studies, compounds NV-1 and NV-9 were found to be stable in the active cavities of AChE and MAO enzymes for 100 ns. Thus, the N/O-propargylated diarylpyrimidines have shown a multipotent activity profile and can be developed as drug candidates for the treatment of AD.
Experimental
Materials and methods
All the chemicals and reagents were purchased from Sigma Aldrich, Avra Synthesis Private Limited, Spectrochem and Qualikems Lifesciences Pvt. Ltd and used as received for the synthesis of the intermediates and target compounds. Glass silica plates coated with GF254 were used for thin-layer chromatography during the monitoring of the reaction. A 1:10 and 3:10 mixture of ethyl acetate in petroleum ether was used as the solvent for the chromatographic separation and purification of the compounds. For the MAO and AChE inhibition assays, Amplex Red and Ellman's assay kits were purchased from Sigma Aldrich and Molecular Probes, Invitrogen, Life Technologies, India, respectively. SHSY-5Y cell lines were obtained from the National Centre for Cell Science (NCCS), Pune, India. The mass spectra of the synthesized compounds were recorded using electrospray ionisation (ESI) combined with a Q-TOF high resolution mass analyser in a positive ion mode at the Central Instrumentation Laboratory in the Central University of Punjab, Bathinda. 1H and 13C NMR of the purified compounds were recorded in CDCl3 or DMSO at 400 MHz using Bruker's instrument at IIT Ropar and GNDU Amritsar. Tetramethylsilane, TMS (δ = 0), is used as the internal standard with the chemical shifts reported in parts per million (ppm) with respect to TMS. The HRMS of the synthesized compounds was recorded at the Central Instrumentation facility, GNDU Amritsar. A UV-vis spectrophotometer from Shimadzu was used for absorption studies, and a microplate reader from BioTek was used for fluorescence recording. Maestro (Schrodinger) software was utilised for molecular docking and modelling simulation studies.
Chemistry and biological studies
A total of 21 molecules were synthesized by following the synthetic procedure (refer SI) as earlier reported by our research group.34 All the biological methodology35–38 and characterization data, including NMR and HRMS, are also available in the SI.
Conflicts of interest
The authors declare no potential conflicts of interest.
Abbreviations
| AD | Alzheimer's disease |
| ACh | Acetylcholine |
| ADME | Absorption, distribution, metabolism, and excretion |
| APP | Amyloid precursor protein |
| Aβ | Amyloid beta |
| hMAO | Human monoamine oxidase |
| BuChE | Butyrylcholinesterase |
| BBB | Blood–brain barrier |
| CAS | Catalytic active site |
| ChEs | Cholinesterase |
| eeAChE | Electric eel acetylcholinesterase |
| DTNB | 5,5′-Dithiobis(2-Nitro Benzoic Acid) |
| FAD | Flavin adenine dinucleotide |
| FDA | Food and drug administration |
| MTDLs | Multi target-directed ligands |
| nM | Nanomolar |
| NMDA | N-Methyl-D-aspartate |
| MD | Molecular dynamics |
| PAS | Peripheral anionic site |
| IC50 | Half maximum inhibition conc |
| µM | Micromolar |
Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: a separate supplementary sheet is provided with the synthetic and biological procedures, along with NMR, HRMS data and graphs of the final compounds. See DOI: https://doi.org/10.1039/d5ra08679e.
Acknowledgements
Vinod Kumar is thankful to the Department of Science and Technology, New Delhi and STARS-IISc (MoE-STARS/STARS-2/2023-0040) for the research grants. NK and Vijay K. are thankful to the University Grants Commission for providing a Senior Research Fellowship. Vinay Kumar and TA are thankful to CSIR for an SRF. KJ is thankful to ICMR (file no. 45/29/2022-/BIO/BMS) for the fellowship grant. Bharti is thankful to DST-WISE for her fellowship. We are thankful to DST for the FIST grant (SR/FST/CS-I/2020/154) and the Central Instrumentation Laboratory of the Central University of Punjab.
References
- A. D. Korczyn and L. T. Grinberg, Is Alzheimer disease a disease?, Nat. Rev. Neurol., 2024, 20, 245–251 CrossRef PubMed.
- R. S. Wilson, S. E. Leurgans, P. A. Boyle and D. A. Bennett, Cognitive decline in prodromal Alzheimer disease and mild cognitive impairment, Arch. Neurol., 2011, 68, 351–356 CrossRef PubMed.
- P. Scheltens, B. De Strooper, M. Kivipelto, H. Holstege, G. Chételat, C. E. Teunissen, J. Cummings and W. M. van der Flier, Alzheimer's disease, Lancet, 2021, 397, 1577–1590 CrossRef CAS PubMed.
- J. Zhang, Y. Zhang, J. Wang, Y. Xia, J. Zhang and L. Chen, Recent advances in Alzheimer's disease: Mechanisms, clinical trials and new drug development strategies, Signal Transduct. Targeted Ther., 2024, 9, 211 CrossRef CAS PubMed.
- O. Sheppard and M. Coleman, Alzheimer's Disease: Etiology, Neuropathology and Pathogenesis, Exon Publications, 2020, pp. 1–21 Search PubMed.
- N. Kumar, V. Kumar, P. Anand, V. Kumar, A. R. Dwivedi and V. Kumar, Advancements in the development of multi-target directed ligands for the treatment of Alzheimer's disease, Bioorg. Med. Chem., 2022, 61, 116742 CrossRef CAS PubMed.
- I. Ferrer, Hypothesis review: Alzheimer's overture guidelines, Brain Pathol., 2023, 33, e13122 CrossRef CAS PubMed.
- C. Albertini, A. Salerno, P. de Sena Murteira Pinheiro and M. L. Bolognesi, From combinations to multitarget-directed ligands: A continuum in Alzheimer's disease polypharmacology, Med. Res. Rev., 2021, 41, 2606–2633 CrossRef PubMed.
- M. Sahu and N. Siddiqui, A review on biological importance of pyrimidines in the new era, Int. J. Res. Pharm. Sci., 2016, 8, 8–21 CAS.
- S. Singh, M. Dhanawat, S. Gupta, D. Kumar, S. Kakkar, A. Nair, I. Verma and P. Sharma, Naturally inspired pyrimidines analogues for alzheimer's disease, Curr. Neuropharmacol., 2021, 19, 136–151 CrossRef CAS PubMed.
- T. Mohamed, X. Zhao, L. K. Habib, J. Yang and P. P. Rao, Design, synthesis and structure–activity relationship (SAR) studies of 2, 4-disubstituted pyrimidine derivatives: Dual activity as cholinesterase and Aβ-aggregation inhibitors, Bioorg. Med. Chem., 2011, 19, 2269–2281 CrossRef CAS PubMed.
- T. Mohamed, J. C. Yeung and P. P. Rao, Development of 2-substituted-N-(naphth-1-ylmethyl) and N-benzhydrylpyrimidin-4-amines as dual cholinesterase and Aβ-aggregation inhibitors: Synthesis and biological evaluation, Bioorg. Med. Chem. Lett, 2011, 21, 5881–5887 CrossRef CAS PubMed.
- S. Ahmad, F. Iftikhar, F. Ullah, A. Sadiq and U. Rashid, Rational design and synthesis of dihydropyrimidine based dual binding site acetylcholinesterase inhibitors, Bioorg. Chem., 2016, 69, 91–101 CrossRef CAS PubMed.
- S. Manzoor, S. K. Prajapati, S. Majumdar, M. K. Raza, M. T. Gabr, S. Kumar, K. Pal, H. Rashid, S. Kumar and S. Krishnamurthy, Discovery of new phenyl sulfonyl-pyrimidine carboxylate derivatives as the potential multi-target drugs with effective anti-Alzheimer’s action: Design, synthesis, crystal structure and in-vitro biological evaluation, Eur. J. Med. Chem., 2021, 215, 113224 CrossRef CAS PubMed.
- J. Kumar, A. Gill, M. Shaikh, A. Singh, A. Shandilya, E. Jameel, N. Sharma, N. Mrinal, N. Hoda and B. Jayaram, Pyrimidine-Triazolopyrimidine and Pyrimidine-Pyridine Hybrids as Potential Acetylcholinesterase Inhibitors for Alzheimer's Disease, ChemistrySelect, 2018, 3, 736–747 CrossRef CAS.
- Y.-x. Xu, H. Wang, X.-k. Li, S.-n. Dong, W.-w. Liu, Q. Gong, Y. Tang, J. Zhu, J. Li and H.-y. Zhang, Discovery of novel propargylamine-modified 4-aminoalkyl imidazole substituted pyrimidinylthiourea derivatives as multifunctional
agents for the treatment of Alzheimer's disease, Eur. J. Med. Chem., 2018, 143, 33–47 CrossRef CAS PubMed.
- M. Shahid Nadeem, J. Azam Khan, I. Kazmi and U. Rashid, Design, synthesis, and bioevaluation of indole core containing 2-arylidine derivatives of thiazolopyrimidine as multitarget inhibitors of cholinesterases and monoamine oxidase A/B for the treatment of Alzheimer disease, ACS Omega, 2022, 7, 9369–9379 CrossRef CAS PubMed.
- M. A. Javed, M. S. Jan, A. M. Shbeer, M. Al-Ghorbani, A. Rauf, P. Wilairatana, A. Mannan, A. Sadiq, U. Farooq and U. Rashid, Evaluation of pyrimidine/pyrrolidine-sertraline based hybrids as multitarget anti-Alzheimer agents: In-vitro, in-vivo, and computational studies, Biomed. Pharmacother., 2023, 159, 114239 CrossRef CAS PubMed.
- S. Pant, R. Kumar K, P. Rana, T. Anthwal, S. M. Ali, M. Gupta, M. Chauhan and S. Nain, Novel substituted pyrimidine derivatives as potential anti-alzheimer’s agents: synthesis, biological, and molecular docking studies, ACS Chem. Neurosci., 2024, 15, 783–797 CrossRef CAS PubMed.
- B. Devi, K. Jangid, V. Kumar, T. Arora, N. Kumar, A. R. Dwivedi, J. Parkash and V. Kumar, Phenylstyrylpyrimidine derivatives as potential multipotent therapeutics for Alzheimer's disease, RSC Med. Chem., 2024, 15, 2922–2936 RSC.
- V. Kumar, K. Jangid, V. Kumar, N. Kumar, J. Mishra, T. Arora, A. R. Dwivedi, P. Kumar, J. S. Bhatti and J. Parkash, In vitro and in vivo Investigations of 4-Substituted 2-Phenylquinazoline derivatives as multipotent ligands for the treatment of Alzheimer's disease, Bioorg. Chem., 2025, 155, 108126 CrossRef CAS PubMed.
- B. Kumar, V. Kumar, V. Prashar, S. Saini, A. R. Dwivedi, B. Bajaj, D. Mehta, J. Parkash and V. Kumar, Dipropargyl substituted diphenylpyrimidines as dual inhibitors of monoamine oxidase and acetylcholinesterase, Eur. J. Med. Chem., 2019, 177, 221–234 CrossRef CAS PubMed.
- B. Kumar, A. R. Dwivedi, B. Sarkar, S. K. Gupta, S. Krishnamurthy, A. K. Mantha, J. Parkash and V. Kumar, 4, 6-Diphenylpyrimidine derivatives as dual inhibitors of monoamine oxidase and acetylcholinesterase for the treatment of Alzheimer's disease, ACS Chem. Neurosci., 2018, 10, 252–265 CrossRef PubMed.
- B. Kumar, A. R. Dwivedi, T. Arora, K. Raj, V. Prashar, V. Kumar, S. Singh, J. Prakash and V. Kumar, Design, synthesis, and pharmacological evaluation of N-propargylated diphenylpyrimidines as multitarget directed ligands for the treatment of Alzheimer's disease, ACS Chem. Neurosci., 2022, 13, 2122–2139 CrossRef CAS PubMed.
- Z. Özdemir, M. A. Alagöz, Ö. F. Bahçecioğlu and S. Gök, Monoamine oxidase-B (MAO-B) inhibitors in the treatment of Alzheimer's and Parkinson's disease, Curr. Med. Chem., 2021, 28, 6045–6065 Search PubMed.
- J. Reis and C. Binda, The peroxidase-coupled assay to measure MAO enzymatic activity, Monoamine oxidase, Methods Protoc., 2023, 23–34 CAS.
- S. V. Shetab-Boushehri, Ellman's method is still an appropriate method for measurement of cholinesterases activities, EXCLI J., 2018, 17, 798–799 Search PubMed.
- M. D. Lloyd, Steady-state enzyme kinetics, Biochemist, 2021, 43, 40–45 CrossRef CAS.
- X.-Y. Meng, H.-X. Zhang, M. Mezei and M. Cui, Molecular docking: a powerful approach for structure-based drug discovery, Curr. Comput. Aided Drug Des., 2011, 7, 146–157 CrossRef CAS PubMed.
- H. Dvir, I. Silman, M. Harel, T. L. Rosenberry and J. L. Sussman, Acetylcholinesterase: from 3D structure to function, Chem. Biol. Interact., 2010, 187, 10–22 CrossRef CAS PubMed.
- C. Binda, A. Mattevi and D. E. Edmondson, Structural properties of human
monoamine oxidases A and B, Int. Rev. Neurobiol., 2011, 100, 1–11 CrossRef CAS PubMed.
- M. De Vivo, M. Masetti, G. Bottegoni and A. Cavalli, Role of molecular dynamics and related methods in drug discovery, J. Med. Chem., 2016, 59, 4035–4061 CrossRef CAS PubMed.
- S. Q. Pantaleão, P. O. Fernandes, J. E. Gonçalves, V. G. Maltarollo and K. M. Honorio, Recent advances in the prediction of pharmacokinetics properties in drug design studies: a review, ChemMedChem, 2022, 17, e202100542 CrossRef PubMed.
- V. Kumar, P. P. Singh, A. R. Dwivedi, N. Kumar, S. C. Sahoo, S. Chakraborty and V. Kumar, Caesium carbonate promoted regioselective O-functionalization of 4, 6-diphenylpyrimidin-2 (1 H)-ones under mild conditions and mechanistic insight, RSC Adv., 2023, 13, 16899–16906 RSC.
- N. Kumar, K. Jangid, V. Kumar, R. P. Yadav, J. Mishra, S. Upadhayay, V. Kumar, B. Devi, V. Kumar and A. R. Dwivedi, In vitro and in vivo investigations of chromone derivatives as potential multitarget-directed ligands: Cognitive amelioration utilizing a scopolamine-induced zebrafish model, ACS Chem. Neurosci., 2024, 15, 2565–2585 CrossRef CAS PubMed.
- I. Ferah Okkay, U. Okkay, B. Cicek, A. Yilmaz, F. Yesilyurt, A. S. Mendil and A. Hacimuftuoglu, Neuroprotective effect of bromelain in 6-hydroxydopamine induced in vitro model of Parkinson's disease, Mol. Biol. Rep., 2021, 48, 7711–7717 CrossRef CAS PubMed.
- S. K. Yusufzai, M. S. Khan, O. Sulaiman, H. Osman and D. N. Lamjin, Molecular docking studies of coumarin hybrids as potential acetylcholinesterase, butyrylcholinesterase, monoamine oxidase A/B and β-amyloid inhibitors for Alzheimer's disease, Chem. Cent. J., 2018, 12, 1–57 Search PubMed.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. De Groot, H. Grubmüller and A. D. MacKerell Jr, CHARMM36m: an improved force field for folded and intrinsically disordered proteins, Nat. Methods, 2017, 14, 71–73 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Vijay Kumar
ab,
Vijay Kumar









